

Abstract
Ebola virus disease is a serious global health concern given its periodic occurrence, high lethality, and the lack of approved therapeutics. Certain drugs that alter intracellular calcium, particularly in endolysosomes, have been shown to inhibit Ebola virus infection; however, the underlying mechanism is unknown. Here, we provide evidence that Zaire ebolavirus (EBOV) infection is promoted in the presence of calcium as a result of the direct interaction of calcium with the EBOV fusion peptide (FP). We identify the glycoprotein residues D522 and E540 in the FP as functionally critical to EBOV’s interaction with calcium. We show using spectroscopic and biophysical assays that interactions of the fusion peptide with Ca2+ ions lead to lipid ordering in the host membrane during membrane fusion, and these changes are promoted at low pH and can be correlated with infectivity. We further demonstrate using circular dichroism spectroscopy that calcium interaction with the fusion peptide promotes α-helical structure of the fusion peptide, a conformational change that enhances membrane fusion, as validated using functional assays of membrane fusion. This study shows that calcium directly targets the Ebola virus fusion peptide and influences its conformation. As these residues are highly conserved across the Filoviridae, calcium’s impact on fusion, and subsequently infectivity, is a key interaction that can be leveraged for developing strategies to defend against Ebola infection. This mechanistic insight provides a rationale for the use of calcium-interfering drugs already approved by the FDA as therapeutics against Ebola and enables further development of novel drugs to combat the virus.
Keywords: calcium, Ebola, fusion peptide, virus entry
Graphical Abstract
Ebola virus disease, a hemorrhagic fever in humans and nonhuman primates, has an average lethality of ~50%.1 The most widespread epidemic to-date occurred in West Africa in 2014, with nearly 30,000 cases reported and over 11,000 deaths.2 Outbreaks happen periodically; in the current outbreak in the Democratic Republic of Congo, over 3000 individuals have contracted the disease.3 There are currently no therapies approved by the Food and Drug Administration specifically to treat the disease, though there are vaccines in clinical trials,4–7 and the development of antivirals against Ebola is ongoing.7–12 Given the disease’s importance in global health and the few countermeasures available, it is critical to understand the Ebola virus lifecycle to identify targets for therapeutic intervention.
Ebola virus disease is caused by Ebola virus, a single-stranded negative-sense RNA virus belonging to the Filoviridae family. The Ebola virus genome is enclosed in a lipid envelope derived from the host cell membrane. The Ebola virus spike glycoprotein (GP) is embedded in this envelope and mediates viral entry, particularly receptor binding and fusion. GP is a class I fusion protein and undergoes proteolytic priming by furin, likely during virus assembly, to cleave pre-GP into its mature form containing subunits GP1 and GP2. GP is not a prototypical class I fusion protein as it contains an “internal” fusion peptide (FP) incorporated within a larger disulfide-bonded loop (Figure 1d,e). After initial binding to a cell-surface receptor, the virus enters cells via macropinocytosis or endocytosis.13 Within the endosome, cathepsins cleave off a heavily glycosylated region of GP, known as the glycan cap.14,15 The virus is then able to bind its endosomal receptor, NPC1.16–19 After binding to NPC1, a subsequent trigger induces a conformational change in GP that allows the virus to fuse its envelope with the endosomal membrane,20 thereby releasing the viral genome. The exact nature of this fusion trigger remains unknown.
Figure 1.
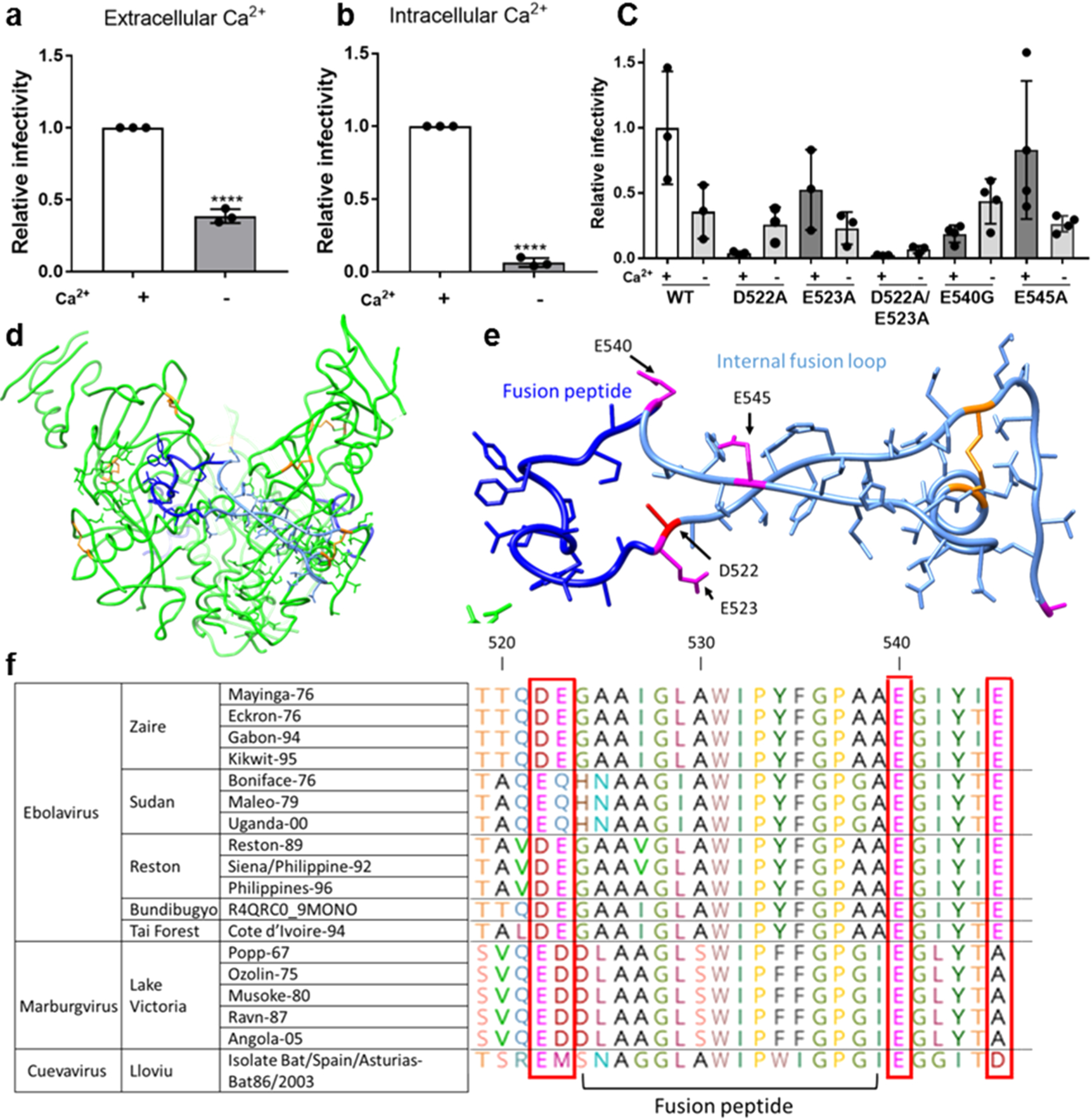
Ebolavirus entry is enhanced in the presence of extracellular and intracellular calcium. Viral particles pseudotyped with Zaire ebolavirus GP were added to (a) Vero E6 cells and allowed to internalize in calcium-free DMEM (−Ca2+) or DMEM with 1.8 mM calcium (+Ca2+) for 2 h to assess the impact of extracellular calcium and (b) Vero E6 cells pretreated with 50 μM of the chelator BAPTA AM or DMSO for 1 h. Pseudovirus particles were then added to cells with 50 μM BAPTA AM or DMSO and allowed to internalize for 2 h. Data was normalized so that infection +Ca2+ was equal to 1. Error bars represent the standard deviation for 3 independent experiments. (c) Infectivity of pseudotyped viral particles with WT or mutated EBOV GP. Data was normalized so that infectivity of WT EBOV in the presence of Ca2+ was 1. Error bars represent s.d. from 3 independent experiments, each with 3 technical replicates, with the exception of E545A and E54G, for which 4 independent experiments were conducted. Dots represent the mean of the technical replicates. p values were determined by the one-way ANOVA comparison to WT. (d) Trimer of Zaire EBOV GP with fusion peptide (dark blue), fusion loop (pale blue), and disulfide bonds (orange). (e) Zaire EBOV fusion loop and nearby negatively charged residues that may be involved in binding calcium. (f) Glycoprotein sequences of different filovirus strains were retrieved from UNIPROT and aligned using CLUSTAL. Horizontal lines demarcate different species.
During membrane fusion, the hydrophobic FP buries into the endosomal membrane, which facilitates mixing of lipids between the viral and endosomal membranes. Further conformational changes in GP then allow a pore to form between the two membranes for genome escape. The Ebola virus FP is located within an unstructured loop and is flanked by negatively charged residues.21 NMR studies of Zaire ebolavirus (EBOV) FP indicate that it has a flexible conformation at both neutral and acidic pH, but the structures at pH 7 and 5.5 are different,22 which points to a possible role for pH in fusion.
It is well-appreciated that lumenal pH changes during endosome maturation; however, levels of other ions, including calcium (Ca2+), change as well.23,24 Initially, the Ca2+ concentration in endosomes drops rapidly,25 but it increases later in the endocytic pathway, reaching 2.5 μM in late endosomes26 and 400 μM in the lysosome.27 The current model of Ebola virus entry is that the virus undergoes fusion in endolysosomes containing both NPC1 and two-pore Ca2+ channels (TPC2),16 but the role of TPC2 or calcium ions in the infection process is unknown. Here, we elucidate this role and describe a mechanism that points to calcium ions directly targeting the fusion peptide during Ebola virus entry. As the search for antivirals against Ebola continues, our findings indicate that Ca2+ disruption is a powerful therapeutic tool and the mechanism we describe enables the rational design of drugs to exploit this Ca2+ dependence.
RESULTS
Depletion of Ca2+ Inhibits Ebola Virus Infection.
To investigate the impact of Ca2+ on EBOV infection of cultured cells, we measured the infectivity of EBOV pseudotyped virus containing a luciferase gene reporter while modulating Ca2+ in the extracellular and intracellular environment. First, cells were infected with Ebola GP-pseudotyped virus with or without extracellular Ca2+ present during the entry step. In the presence of external Ca2+, Ebola GP-pseudotyped virus was nearly 3 times more infectious than when in Ca2+-free media (Figure 1a).
To measure the impact of Ca2+ in the intracellular environment, we utilized 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (BAPTA-AM), a chelator that is only active intracellularly.28 Vero E6 cells were pretreated with 50 μM BAPTA-AM, a concentration that does not significantly impact their viability.29 EBOV infection was approximately 12 times higher in the presence of unaltered intracellular Ca2+ levels compared to when Ca2+ was chelated (Figure 1b). For a comparison of raw RLU values to the noninfectious control, see Figure S1.
Residues D522 and E540 Are Involved in Ca2+-Dependent Infection.
Ca2+ is known to play a role in the fusion of a few other enveloped viruses. Rubella virus depends on Ca2+ for proper orientation of its fusion loops.30 Specifically, Ca2+ coordinates with the residues N88 and D136 located on adjacent fusion loops within E1. Previously, we showed that viral entry of severe acute respiratory syndrome coronavirus (SARS-CoV) is Ca2+ dependent, and the FP cannot induce membrane ordering when chelator is present.29 The Ebola virus FP is flanked by negatively charged residues that may coordinate with Ca2+ ions (Figure 1e). Combined with evidence that drugs that interfere with cellular Ca2+ inhibit EBOV infection11,31,32 and the implication of TPC2 in EBOV entry, the presence of these negatively charged amino acids motivated us to explore whether Ca2+ plays a role in Ebola virus fusion via direct interaction with the FP, in a fashion similar to rubella virus and SARS-CoV.
We hypothesized that Ca2+ might be interacting with negatively charged amino acids near the viral FP and thus influencing the conformational changes necessary for fusion. The presence of negatively charged residues flanking the FP is highly conserved among the three genera of the Filoviridae: Cuevavirus, Marburgvirus, and Ebolavirus (Figure 5f). The presence of negatively charged residues at positions 522 and 540 is conserved, though the exact identity of the residue at position 522 varies between aspartate and glutamate from one genus to another. Within the Ebolavirus genus, the species Zaire, Reston, Bundibugyo, and Tȧï Forest contain an aspartate at position 522, whereas for the Sudan species, that position is occupied by glutamate. The FP is embedded in a loop, which brings residues D522, E523, E540, and E545 in close proximity to each other (Figure 1d,e). To investigate the activity of these conserved negatively charged residues, we used site-directed mutagenesis to generate pseudotyped virus with neutral residues in these positions. We generated the amino acid substitutions D522A, E523A, and E545A. Attempts to produce the mutation E540A were unsuccessful due to recombination within the bacteria used for plasmid amplification; because alanine and glycine have similar affinities for Ca2+,33 the mutant E540G was characterized instead. The presence of two negatively charged residues on each side of the FP implies possible redundancy, where a single mutation might still allow Ca2+ to coordinate with the remaining neighboring negatively charged residue. To test this possibility, we also generated the double mutant D522A/E523A. These mutations did not prevent the incorporation of GP into pseudotyped virus particles (Figure S2). In cell infectivity assays, pseudotyped virus containing D522A exhibited behavior drastically different than pseudotyped virus with wild type (WT) GP and yielded higher infectivity in the absence of extracellular Ca2+ (Figure 1c). E523A showed lower overall infectivity than WT but matched the WT Ca2+-dependent behavior, displaying higher infectivity in the presence of extracellular Ca2+. The double mutant D522A/E523A was infectious albeit at a very low level, as determined by comparison to a noninfectious negative control (Figure S3). D522A/E523A behaved similarly to D522A, with higher infectivity in the absence of extracellular Ca2+. This suggests that D522 plays a central role in Ebola’s response to Ca2+ and E523 likely does not serve a redundant function to D522. E540G had low infectivity and was relatively unaffected by extracellular Ca2+. E545A had similar overall infection and Ca2+-dependent behavior to the WT, indicating that it may not be critical in interacting with Ca2+. Of the mutations tested, D522A, D522A/E523A, and E540G exhibited altered or no Ca2+ dependence compared to the WT. Given the location of these residues adjacent to the FP, we reasoned that the altered entry behavior of the particles bearing mutated GPs might be related to fusion. Extracellular and intracellular Ca2+ may alter cellular processes beyond directly impacting viral entry, so it is hard to decouple these effects in interpreting the results of solely cell-based experiments. Therefore, we next employed biophysical approaches to isolate and further investigate the role of Ca2+ by examining the structure and fusion activity without the additional complications of concurrently occurring cellular pathways.
Ca2+ Increases EBOV Fusion in Vitro.
To further define the role of Ca2+ and isolate its impact on membrane fusion, we employed vesicle fusion assays that report lipid mixing. We used an E. coli-expressed EBOV FP construct containing residues 501–560 of GP (Figure 2a).22 Compared to the “short version” FP containing residues 524–539, this “long” FP includes the disulfide-bonded loop that is critical to the structure and function of the FP.22 In the lipid mixing experiment, the FP construct was mixed with large unilamellar vesicles (LUVs), 10% of which were fluorescently labeled with a FRET pair. LUVs were used to match their diameter similar to that of pseudoviral particles. Upon fusion of these labeled vesicles with unlabeled vesicles, FRET is relieved, resulting in a large increase in fluorescence intensity (Figure 2b). After adding the FP to the mixture, detergent was added to rupture the LUVs and fully release the dye. The resulting fluorescence intensity was set to 100% to normalize data from one trial to the next.
Figure 2.
EBOV FP mediated lipid mixing is Ca2+ dependent. (a) Sequence of the FP used in lipid mixing, ESR, and CD experiments with the core hydrophobic FP in bold. (b) Raw data from a representative lipid mixing experiment using the expressed long FP. (c) Normalized % lipid mixing induced by the fusion peptide or a scrambled peptide (Scr’d) from three independent experiments except for WT in the presence of Ca2+ where four independent experiments were conducted. Error bars indicate s.d. (d) Normalized lipid mixing where 0 is the basal lipid mixing level of the WT with 5 mM DTT (c, red) and 100% is the level of WT with 2 mM Ca2+ (c, black). Error bars represent s.d. from three independent experiments except for WT where four independent experiments were conducted. Dots represent the mean of the replicates. ns = p > 0.05, * = p < 0.05, ** = p < 0.01, and *** = p < 0.001 compared to WT (for (d), red indicates the comparison between +Ca2+ and −Ca2+ for each mutant), as calculated by the Student’s t test or ANOVA.
In the pH 5 fusion buffer with 2 mM Ca2+, which is close to the calcium concentration of the ion found in typical cell culture media formulations and is on par with the concentration range (0–2 mM) used in previous literature,30 roughly 70% lipid mixing is observed (Figure 2b,c). In the fusion buffer without Ca2+ added and in the presence of 1 mM EGTA to chelate the trace Ca2+ (“calcium-less buffer”), the lipid mixing ratio is greatly reduced (39%). As a negative control, in the presence of 5 mM DTT, which removes the disulfide bond of the FP even though 2 mM Ca2+ was added, lipid mixing is greatly reduced (21%). The scrambled peptide, which has the same residues as the FP but in a shuffled order, has even lower lipid mixing activities (12%). Taken together, these data indicate that Ca2+ interacts with the FP to promote fusion. Execution of these same lipid mixing studies with pseudovirus yielded lipid mixing levels comparable to the DTT control, even in the presence of both low pH and calcium, indicating that the receptor NPC1 or some other fusion trigger is needed for lipid mixing (Figure S9) mediated by the full viral particle.
We produced modified FP constructs to match the mutations used in our cell-based assays. WT FP consistently yielded high levels of lipid mixing in the presence of Ca2+ and only half as much mixing when Ca2+ was removed (Figure 2c). FP with the D522A mutation yielded roughly 50% lipid mixing regardless of whether Ca2+ was present or absent (Figure 2d). In the infectivity studies (Figure 1c), this mutant did show behavior different from the WT but yielded less infection in the presence of extracellular Ca2+. The mutation E523A resulted in a modest decrease in lipid mixing compared to the WT but still induced more fusion when Ca2+ was present rather than absent (Figure 2d), agreeing with our cell infectivity observations. E540G was the least fusion-competent, agreeing with our infectivity studies where it was also the least infectious mutant tested, and was relatively unaffected by Ca2+. E545A yielded similarly low levels of lipid mixing and was also unaffected by the addition of Ca2+. This differs from our infectivity experiments, where Ca2+ did enhance the infection of pseudotyped virus with the mutation E545A. Given that these modified FPs no longer exhibit strong Ca2+ dependence, our data suggest that negatively charged residues at positions 522, 540, and 545 are important for coordinating with Ca2+ and that removal of these negatively charged amino acids is detrimental to fusion.
Ca2+ Increases EBOV FP Membrane Insertion and Lipid Ordering.
Our fusion assay experiments indicate that calcium is targeting the fusion peptide and, in turn, influencing membrane fusion. Because the fusion peptide inserts into the host membrane during the fusion process, we next focused on examining the impact of calcium on this insertion using electron spin resonance (ESR). In this technique, spin labels are incorporated into lipids at various positions, acting as depth probes. Multilamellar vesicles (MLVs) containing dipalmitoylphosphatidyl-tempo-choline (DPPTC) are spin labeled in the head region whereas those with phosphocholine are labeled in either the upper tail region (5PC) or the lower tail region (14PC) (Figure S4). The ordering parameter, S0, of each spin-labeled lipid was extracted from the ESR spectra using the NSSL program with the microscopic order–macroscopic disorder (MOMD) model.34 S0 is an indication of the amount of membrane ordering at a given depth and serves as a readout for FP membrane penetration depth.35 More importantly, FP-induced changes in S0 are thought to be functionally significant because changes in lipid ordering can lower the energy barrier between closely approaching membranes by dehydration, enabling fusion to occur.36 Previous ESR studies with other viral FPs indicate that membrane ordering by the viral FP is a critical step for viral membrane fusion.29,36–38 The S-shaped curves of S0 as a function of increasing WT peptide to lipid (P/L) ratio (Figure 3a–c) suggest cooperativity in membrane ordering, consistent with the requirement for class I and class II fusion proteins to oligomerize for efficient fusion to occur.
Figure 3.
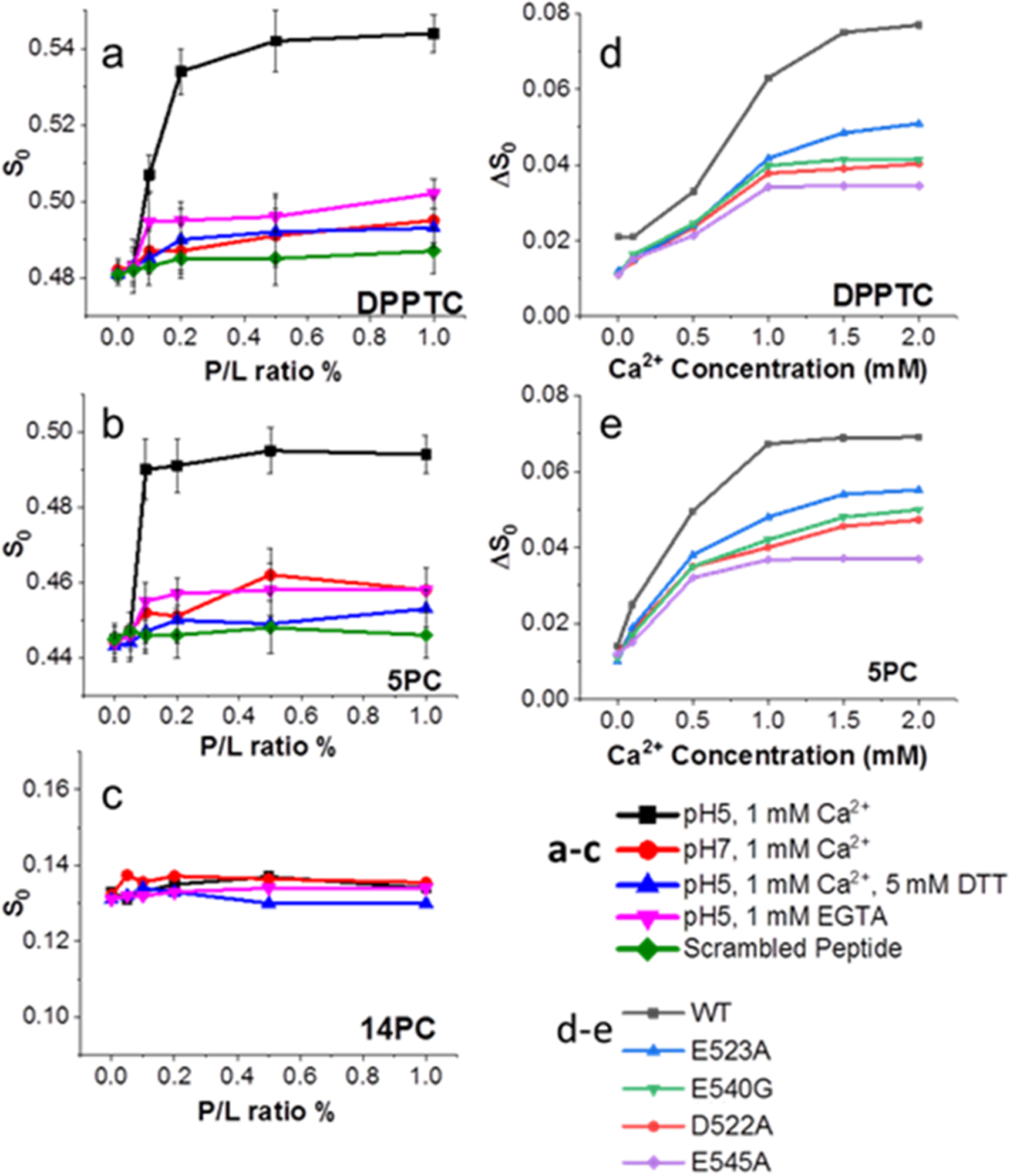
ESR spectroscopy shows FP induces membrane ordering in a Ca2+ dependent fashion. (a–c) Plots of order parameters (S0) of (a) DPPTC, (b) 5PC, and (c) 14PC versus peptide/lipid ratio (P/L ratio) of FP in POPC/POPG/Chol = 3:1:1 MLVs. (d, e) the plot of ΔS0 of (d) DPPTC and (e) 5PC versus Ca2+ concentration of the WT and mutant FPs at 1% P/L ratio. Each experiment was repeated two to three times. Typical uncertainties found for S0 are 6 × 10−3.36
We then tested the impact of calcium using 1 mM Ca2+ to match conditions consistent with our previous study on the SARS-CoV FP.29 The WT peptide induced the greatest ordering effect of the head and upper-tail regions at acidic pH with 1 mM Ca2+ (Figure 3a,b), but the deep hydrophobic region (14PC) was virtually unaffected (Figure 3c). The scrambled peptide has no ordering effect on the membrane, indicating the ordering effect is sequence specific. In the calcium-less buffer, the WT FP induced little lipid ordering and was similar to our negative control, the FP with the disulfide bond removed by DTT, which had virtually no lipid ordering effect (Figure 3a–c).
If Ca2+’s effect on the WT peptide was strictly due to mediating the electrostatic repulsion between the negative lipids and negative charges in the FP, we would expect that, when mixed with DTT, the FP would still be able to induce ordering because the charges of the amino acids have not been altered. The observation that DTT is detrimental to peptide-induced lipid ordering indicates that Ca2+ is not simply promoting peptide insertion by screening charge, as has been suggested in other work.39 To further test the effect of Ca2+ on S0, we increased the Ca2+ concentration while fixing the P/L ratio at 1% (Figure 3d,e). The effects of any ordering caused by Ca2+ interacting with the liposomes rather than the fusion peptide were removed by subtracting S0 of a liposome and Ca2+ only (no peptide) mixture from the measurement of S0 when all three components (liposomes, peptides, and Ca2+) were present, yielding ΔS0. This ΔS0 increased with increasing Ca2+ indicating that there is more FP-induced membrane ordering at higher Ca2+ concentrations in the lipid head (Figure 3d) and midtail (Figure 3e) regions. Our prior observations of influenza HA, which does not show increased membrane interaction in the presence of Ca2+,29 indicate that Ca2+ does not act as a blanket promoter of viral FP insertion into target membranes. ESR measurements of mutant peptides all showed the induction of lipid ordering with increasing P/L ratio but less so than the WT peptide (Figure S6). In the calcium-less buffer, the ordering effect for all peptides was minimal. Increasing Ca2+ increased the FP-induced membrane ordering in head (Figure 3d) and midtail (Figure 2e) regions but also had less of an effect on the mutated peptides than the WT. Of the mutants tested, D522A and E545A had the least membrane ordering at all Ca2+ concentrations tested. These results imply that the ability of the mutated peptides to induce membrane ordering is impaired due to their reduced ability to interact with Ca2+.
Ca2+ Promotes Insertion of Viral GP into the Target Membrane.
The N-terminal FPs for viruses such as influenza and HIV are anchored to the rest of the fusion protein at only one end of the FP, so they are easily able to form the functional domain by themselves. However, as the GP FP is internal (i.e., both of its ends are connected to the remainder of GP), it is unknown whether isolated FP can recapitulate the fusion functionality of the complete GP. To determine whether the Ca2+-dependent membrane ordering effects we observed for isolated FP were also true within the context of complete GP, we used the technique of time-resolved ESR to monitor the change of S0 during fusion of small unilamellar vesicles (SUVs) and pseudovirus particles (PPs). The PPs were pretreated with thermolysin to remove the glycan cap from GP, and when the PPs were mixed with the SUVs containing DPPTC, the GP FP was free to insert into the SUV membrane during the docking of the PPs on the SUV.
In the time-resolved ESR experiments, we reduced the conversion time and scanning times, which shortened the collection time and allowed collection of the spectra every 30 s. Noisier signals were denoised using the Wavelet Denoising Package40 before NLSL analysis if necessary. As shown in Figure S7a, the shape of the ESR signal changes during the time course, indicating a change of membrane structure. After denoising (Figure S7b), the details of the difference are more obvious. As shown in Figure 4a, the S0 increases in the presence of Ca2+ at pH 5, and the half maximal ordering parameter occurs at about 3.5 min. When either the Ca2+ or the GP is removed, S0 does not increase over time. At lower PP/SUV ratios, S0 increases over time, though it takes longer to reach the half maximal ordering parameter (Figure S7c). This can be explained simply on the basis of concentration: as more GP is available, more docking events happen. Figure 4b shows the effect of the PP/SUV ratio on S0 at 10.5 min postmixing. The S-shaped pattern similar to the isolated FP data (Figure 3a) is observed also only in the presence of Ca2+ and at pH 5. The SUV–PP lipid mixing experiments show very low mixing ratios (Figure S9), indicating no fusion in this condition. Thus, the membrane ordering is due to docking of the PPs with the SUVs, not fusion between the two. If the function of the FP in the context of the entire GP is similar to that of the isolated one, then the membrane ordering effect should be observed in these experiments too, and they are. In summary, we report FP-induced membrane ordering with the time-resolved method and in a “biological” context for the first time. This result supports that the FP can induce lipid ordering both alone and in the context of the whole GP.
Figure 4.
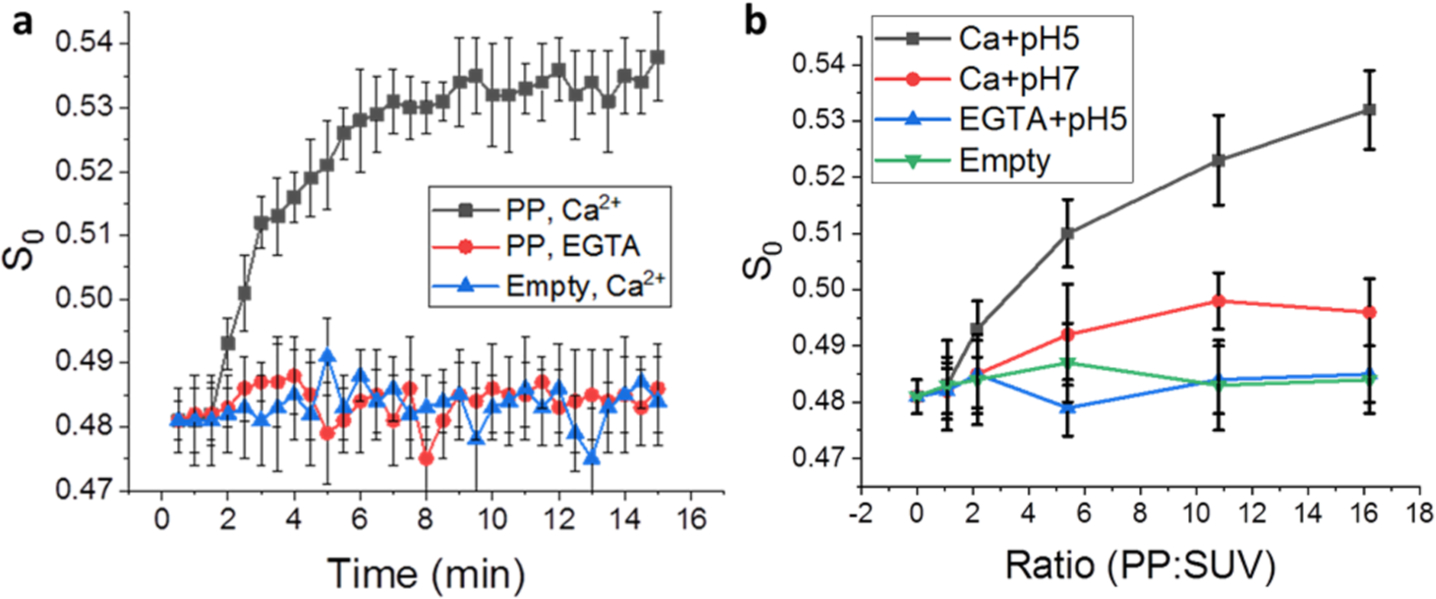
Time-dependent ESR experiments on DPPTC in the pseudoviral particle–SUV interaction. (a) The plot of order parameters of DPPTC changes during the time course of the measurement. The ratio of PP/SUV is about 15:1. Black, with 1 mM Ca2+; red, with 1 mM EGTA; blue, using the empty particles instead of the pseudoviral particles. (b) The plot of order parameter of DPPTC in different PP/SUV ratios. The order parameters are extracted from the curves collected at 10.5 min after activation.
Ca2+ Increases FP Helical Content.
To better understand the structural importance of the negatively charged amino acids on FP conformation, we used circular dichroism (CD) spectroscopy to examine the secondary structures of mutant and WT FPs in membranes with and without Ca2+ present (Figure 5). We used SUVs because the LUVs generate a significant scattering effect in our instrument, which makes the signals noisy. In our control condition, where the disulfide bond of the fusion loop is removed by the addition of DTT, the peptide adopts a random coil structure. The WT FP exhibits a well-formed α-helical structure in the presence of SUVs at pH 5 in 1 mM Ca2+ (22.7 ± 1.8%). At higher pH with Ca2+, there is a decrease in helical content (12.1 ± 2.2%). In the pH 5 calcium-less buffer, the helical content is even lower (10.8 ± 1.7%). From the helical content vs Ca2+ concentration (Figure S10), we estimated that about 0.8 mM Ca2+ is required to induce 50% conformational change of the FP. All mutants exhibit less helical structure than the WT, indicating that these negatively charged amino acids are important for maintaining the structure of the FP. The mutant E545A exhibits the least helical structure, indicating E545 is important for maintaining the FP structure. When interacting with lipid bilayers, the mutants have less formed secondary structure. Combined with our functional studies, these results indicate that Ca2+ influences FP structure, which impacts its interaction with the membranes, and thereby its membrane fusion function.
Figure 5.
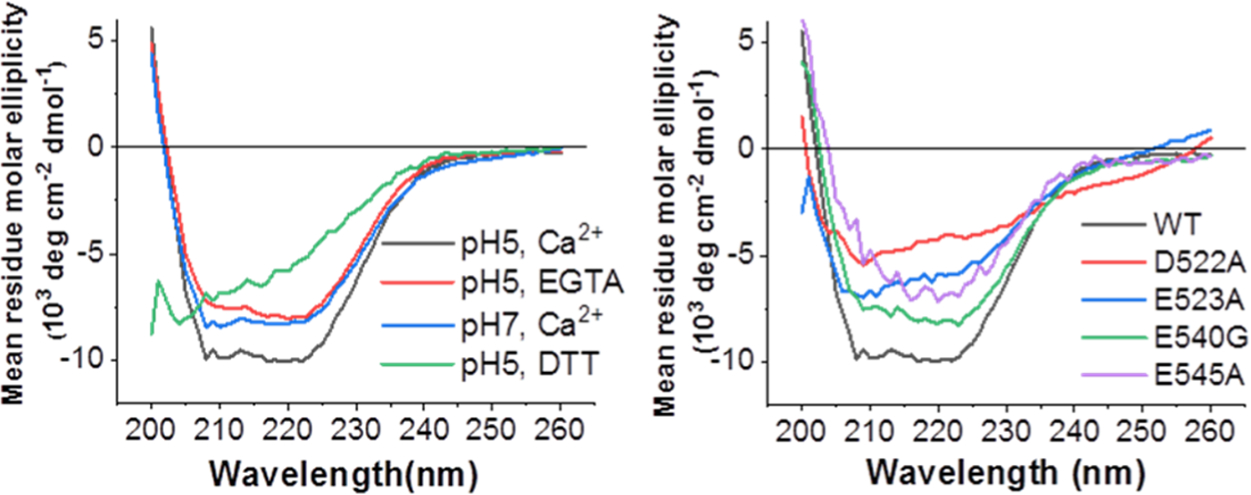
CD spectra of EBOV FPs show that Ca2+ and the mutation of negatively charged residues alter FP structure. (a) WT FPs at acidic or neutral pH and ± Ca2+. (b) Mutated FPs at pH 5, 1 mM Ca2+ and WT (black) for reference. Two to three spectra were collected independently for each sample; representative spectra are shown.
DISCUSSION
The direct role of Ca2+ in Ebola virus fusion has not been previously identified, although there have been previous links between potential therapeutics and intracellular Ca2+ levels. Screens of potential filovirus therapeutics indicated that ion channel inhibitors, such as amiodarone, dronedarone, bepridil, lomerizine, verapamil, and tetrandrine,11,31,41 and drugs that induce Ca2+ efflux from endosomes, such as U18666A, tamoxifen, and clomiphene,32 are able to inhibit EBOV infection. Expression of the endosomal EBOV receptor, NPC1, is also linked to Ca2+ levels in the endosomal lumen. Removal of NPC1 results in Ca2+ depletion in late endosomes and lysosomes,42 in addition to blocking EBOV entry.17,19 Sakurai et al. demonstrated that blocking or knocking out the calcium transport channel TPC2 can also inhibit infection of Ebola virus.31 The anti-EBOV nature of these Ca2+-inhibiting drugs agrees with our results and indicates that further development of similar therapeutics may be fruitful. Our studies indicate that, from a therapeutic standpoint, modulation of both extracellular and intracellular Ca2+ is an attractive target.
We identified residues D522 and E540 as key players in the dependence of Ebola virus infection on Ca2+. Mutation of other nearby negatively charged residues did not alter calcium dependence, indicating that D522 and E540 play specific roles in the interaction with the ion, rather than calcium dependence being driven by the overall charge of GP. Previous studies of E523 and E540 found these residues to be detrimental to fusion in the context of the hydrophobic core fusion peptide alone.43 In ESR, the “long” FP, which extends beyond the core and includes the disulfide bond, is more efficient in inducing membrane ordering than the short hydrophobic core alone (Figure S5), indicating that the larger context of these residues within the peptide is functionally important. Our work with a longer peptide clarifies the roles of these residues in mediating FP conformation and the interaction with Ca2+ as well as their effect on virion infectivity. Others have observed reduced binding of the mutant D522A to the membrane proximal external region of GP.44 Our work links this mutation to the reduced interaction with Ca2+ and thereby reduced fusion competence due to alteration in the direct interaction of the FP with the membrane, adding new insight into the role of D522 in fusion. Given that D522 and E540 are implicated in the Ca2+-dependent entry of EBOV and that negative residues at those sites are conserved across the Filoviridae, Ca2+-targeting drugs might be effective against all viruses within the family. Indeed, the drugs toremifene and clomiphene increase Ca2+ release from endosomes32 and are able to inhibit multiple strains of both EBOV and MARV.10
As the endosome matures, the endosomal membrane becomes enriched with anionic lipids.45 Others have observed that negatively charged lipids are necessary for partitioning of the Ebola FP into the target membrane43 and suggested that calcium alters the hydrogen-bonding network at the surface of the membrane rather than targeting the fusion peptide itself.39,43 Our spectroscopic data combined with our mutant studies suggest a wholly orthogonal mechanism to what this previous work suggested. Our work shows that the conformational changes in FP are due to its interaction with the ion, which we further show leads to specific changes in lipid ordering in the host membrane and an increase in fusion activity. Our work thus sheds further insight into how these conformational changes are functionally important within the context of viral fusion and entry and links these biophysical observations to the mechanism that enables Ca2+-perturbing drugs to inhibit filovirus infection. As pH decreases, the FP forms an α-helical fist-like structure.22 Our studies indicate that Ca2+ may act to stabilize or promote that α-helical structure. This results in higher extents of fusion as Ca2+ concentration increases, aligned with higher level infections of the virus in the presence of both extracellular and intracellular Ca2+.
Our previous ESR work with the FPs of influenza virus and HIV (both are N-terminal FP of class I glycoproteins) and dengue virus (an internal FP of a class II glycoprotein) suggested that FP-induced membrane ordering is a result of dehydration due to FP insertion, which is a prerequisite step for reducing repulsive forces between two opposing membranes to initiate membrane fusion.36,46 Our current study extends these concepts to the Ebola FP, an internal FP within a class I glycoprotein. We show that Ebola FP also induces membrane ordering, while the functionally impaired mutated peptides have significantly lower membrane ordering capacity. This result upholds the theory that membrane ordering induced by the FP is a common prerequisite for viral entry. Taken together, the Ca2+-dependent EBOV FP conformational changes, membrane ordering effect, and subsequent modifications in fusion activity found here point to the FP being at least one of the direct targets of Ca2+ in the entry of EBOV.
Blocking two-pore channels (TPC2), which are involved in calcium efflux from the endosome,47 inhibits Ebola infection.31 Perhaps when the channels are functional, they create a locally high concentration of calcium near the membrane, encouraging fusion and thereby infection. However, the exact role of TPC2 in Ebola infection remains unclear.
In our studies, low pH and calcium seem to act synergistically to promote the insertion of GP into the target membrane. During endosomal maturation, the concentration of calcium within the endosome initially drops from 2 mM extracellularly to 3 μM25 within the early endosome. However, the calcium concentration then goes back up to 400–600 μM27 as lysosomes merge with late endosomes. Simultaneously, the pH drops from 6.8 in the early endosome to 4.8 in the late endosome.48 Given that Ebola virus fuses in late endosomes,16,20 after binding NPC1, the virus may undergo fusion in response to these dual environmental cues of low pH and high calcium. Our ESR studies with both fusion peptide and pseudovirus indicate that low pH and calcium together promote the insertion of the fusion peptide. We saw little lipid mixing between pseudovirus and liposomes, indicating that even with the glycan cap removed from GP an additional factor beyond low pH and Ca2+, such as NPC1 binding, is needed for insertion of the fusion peptide into the host membrane to progress to full fusion.
Ca2+ has been implicated in the fusion of a few other enveloped viruses. Rubella virus, which utilizes the class II fusion protein E1, depends on Ca2+ for proper orientation of its fusion loops.30 Specifically, Ca2+ coordinates with the residues N88 and D136 located on adjacent fusion loops within E1. Previously, we showed that viral entry of SARS-CoV is Ca2+ dependent and the FP cannot induce membrane ordering when chelator is present.29 Our work adds to this nascent understanding of the role of Ca2+ in viral fusion and points to Ca2+ as an attractive host factor for antiviral therapeutic intervention.
EXPERIMENTAL SECTION
Peptides.
The EBOV FP was expressed using a vector developed by the Tamm Lab.22 The mutant FPs were generated using a USB Change-IT site directed mutagenesis kit (Affymetrix). The protocol of expression and purification also follows published procedures.22 Briefly, the relevant plasmids were transformed in BL21(DE3) Escherichia coli competent cells and grown at 37 °C to an optical density of ~0.8. Protein expression was induced for 3 h at 30 °C by 0.5 mM IPTG. The harvested cells were lysed by sonication and clarified by centrifugation at 40 000 rpm for 45 min. The supernatant containing the His-tagged fusion protein was transferred to a pre-equilibrated Ni-NTA agarose resin column, and the supernatant and resin were incubated for 2 h at 4 °C on a rotator in wash buffer (containing 25 mM Tris, 500 mM NaCl, 20 mM imidazole, 5% glycerol, 5 mM β-ME, and 10 mM CHAPS, pH 8). The resin was then rinsed with digestion buffer (containing 25 mM Tris, 50 mM NaCl, 5 mM CaCl2, and 5% glycerol, pH 7.5). 125 μL of Factor Xa (1 mg/mL) in 15 mL of digestion buffer was added to the resin and incubated overnight at room temperature. The proteins were eluted using 50 mL of wash buffer, dialyzed against dialysis buffer (25 mM Tris, 50 mM NaCl, 5% glycerol, pH 8.5, with 1 g/L Chelex 100 resin), and purified using a Superdex Peptide 10/300 GL gel-filtration column (GE Healthcare) using gel-filtration buffer (containing 30 mM sodium phosphate and 200 mM NaCl, pH 7.4). The sample was either exchanged to the fusion buffer (5 mM HEPES, 10 mM MES, 150 mM NaCl, pH 7 or pH 5) for measurement or lyophilized for storage. The fusion buffers are free of calcium unless CaCl2 was intentionally added. As a further safeguard, 1 mM EGTA was added to chelate the trace Ca2+ and thus generate the “calcium-less buffer”. We have compared ESR as well as CD spectra in the fusion buffer and the “calcium-less buffer”; the differences were minimal.
Cell Lines and Plasmids.
HEK 293T (accession no.: CRL-11268, ATCC) cells and Vero E6 cells (accession no.: CRL-1586, ATCC) were grown in complete DMEM [DMEM with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 10 μg/mL streptomycin, and 1% HEPES]. Cells were maintained at 37 °C and 5% CO2. pcDNA3.1-EBOV-GP (BEI Resources) encodes Zaire ebolavirus GP. The mutations D522A, E523A, E540G, E545A, and D522A/E523A were produced by site directed mutagenesis of pcDNA3.1-EBOV-GP using the QuickChange XL site-directed mutagenesis kit (Agilent) with primers designed using the Agilent online tool.
Lipid and Vesicle Preparation.
The lipids POPC and POPG, the chain spin labels 5PC and 14PC, and a headgroup spin label DPPTC (Figure S4) were purchased or synthesized by our laboratory. The composition of membranes used in this study is consistent with our previous study.46 The desired amounts of POPC, POPG, cholesterol, and 0.5% (mol/mol) spin-labeled lipids in chloroform were mixed well and dried by N2 flow. The mixture was evacuated in a vacuum drier overnight to remove any trace of chloroform. To prepare MLVs, the lipids were resuspended and fully hydrated using 1 mL of pH 7 or pH 5 buffer (5 mM HEPES, 10 mM MES, 150 mM NaCl, pH 7 or pH 5) at room temperature for 2 h. To prepare SUVs for CD measurements, the lipids were resuspended in fusion buffer and sonicated in an ice bath for 20 min or when the suspension became clear. The SUV solution was then further clarified by ultracentrifugation at 13 000 rpm for 10 min.
Sequence Analysis.
Sequences of Filoviridae spike proteins were aligned using CLUSTAL with Geneious software and the following UniProt accession numbers: Zaire ebolavirus: Mayinga-76 (Q05320), Gabon-94 (O11457), Eckron-76 (P87671), and Kikwit-95 (P87666); Reston ebolavirus: Reston-89 (Q66799), Philippines-96 (Q91DD8), Siena/Philippine-92 (Q89853), and Uganda-00 (Q7T9D9); Sudan ebolavirus: Boniface-76 (Q66814) and Maleo-79 (Q66798); Tȧï Forest ebolavirus: Cote d’Ivoire-94 (Q66810); Lake Victoria marburgvirus: Musoke-80 (P35253), Ozolin-75 (Q6UY66), Popp-67 (P35254), Ravn-87 (Q1PDC7), and Angola/2005 (Q1PD50); Lloviu cuevavirus isolate Bat/Spain/Asturias-Bat86/2003 (G8EFI5); Bundibugyo ebolavirus (R4QRC0).
Pseudotyped Virus Production.
Pseudotyped viruses were produced following published protocols.49,50 Briefly, particles were produced by transfecting HEK 293T cells with plasmids encoding for luciferase, Murine Leukemia Virus gag and polymerase, and the viral spike protein of interest using Lipofectamine 2000 following the manufacturer’s instructions and subsequently maintained in DMEM with 10% fetal bovine serum (FBS) and 1% HEPES. Supernatant was harvested 48 h post-transfection, filtered through a 0.45 μm membrane, and stored at −80 °C. To remove the glycan cap, pseudovirus particles were pelleted by ultracentrifugation at 42 000 rpm, 4 °C, for 2 h in a TLA 55 rotor (Beckman Coulter) and then resuspended in a buffer (50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% Brij, pH 7.3) containing 0.167 mg/mL thermolysin. The particles were then placed at 37 °C overnight. Removal of the glycan cap was confirmed by Western Blot (Figure S8) and is consistent with published literature.14
Infection.
Vero E6 cells were seeded at 5 × 105 cells/mL in a 24-well plate. Twenty-four hours after seeding, cells were rinsed three times with Ca2+-free PBS and then incubated with 200 μL of pseudotyped virus and 300 μL of RPMI supplemented with 0.2% BSA and 10 mM HEPES at pH 7 on ice at 4 °C with rocking for 1.5 h. Cells were washed with Ca2+-free PBS to remove unbound viral particles and incubated at 37 °C for 2 h in 200 μL of DMEM with the specified concentration of Ca2+ for internalization. Complete DMEM with 20 mM NH4Cl was added to the cells to block further infection, and 48 h later, luciferase activity was measured using the Luciferase Assay System (Promega) and Glomax 20/20 luminometer.
BATPA-AM Treatment.
Vero E6 cells were seeded at 5 × 105 cells/mL in a 24-well plate. Twenty-four hours after seeding, cells were pretreated with 50 μM BATPA-AM or an equivalent volume of DMSO diluted in DMEM with 2% FBS for 1 h at 37 °C. Cells were then infected with 200 μL of pseudotyped virus at 37 °C in the presence of 50 μM BATPA-AM or an equivalent volume of DMSO. After 2 h, complete DMEM was added and the cells were incubated for 72 h. Luciferase activity was measured as described above.
Lipid Mixing Assays.
Fluorescently labeled LUVs (2.5 μM final concentration) containing 1% 7-nitrobenz-2-oxa-1,3-diazole (NBD)-egg-POPE and 1% Rhodamine-egg-POPE and unlabeled LUV (22.5 μM, final concentration) were mixed in 1 mL of pH 5 fusion buffer. Fusion peptides were then added from concentrated stock solutions to give a 1 μM final concentration of each peptide. 10% Triton X-100 was added to achieve a 1% final concentration after fusion reactions were completed. The fluorescence spectra were collected on a Varian Cary Eclipse Fluorescence spectrometer. Fluorescence intensities of the samples before the addition of fusion peptides and after the addition of Triton X-100 were used to set the baseline (0%) and 100% fusion levels, respectively. The fluorescence yields of the experimental samples were normalized to these levels to determine % lipid mixing. Fluorescence intensity variations due to volume changes were corrected in each case. All experiments were performed at least 3 times, and representative curves are shown.
ESR Spectroscopy and Nonlinear Least-Squares Fit of ESR Spectra.
To prepare the samples for lipid ESR study, a stock solution of the FP (1 mg/mL) was added to the lipid POPC/POPG/Chol = 3:1:1 MLV dispersion (above) at the experimentally indicated ratios. After 20 min of incubation, the dispersion was centrifuged at 13 000 rpm for 10 min to concentrate the vesicles for better signal-to-noise ratio, which is consistent with our previous protocol.29,36–38 The pellet was transferred to a quartz capillary tube for ESR measurement with FPs.
For the SUV–PP interaction, SUV was prepared by sonication, and the concentrations of the SUVs and PPs were determined using a Malvern Nanosight NS300 (Malvern, UK). A recent denoising algorithm developed in our lab40 increases the signal-to-noise ratio by signal processing and enables us to use SUV instead of MLV for these experiments, despite the lower signal-to-noise ratio for the former. The desired ratios of vesicles were mixed and then acidified immediately before the acquisition. ESR spectra were collected on an ELEXSYS ESR spectrometer at X-band (9.5 GHz) at 25 °C using a N2 Temperature Controller. The ESR spectra from the labeled lipids were analyzed using the NLLS fitting program based on the stochastic Liouville equation34 using the MOMD (microscopic order–macroscopic disorder) model as in previous studies.29,34,51 The fitting strategy is the same as previously reported.36 S0 is defined as follows: S0 = 〈D2,00〉 = 〈1/2(3 cos2θ − 1)〉, where D2,00 is the Wigner rotation matrix element and θ is the polar angle for the orientation of the rotating axes of the nitroxide bonded to the lipid relative to the director of the bilayer; i.e., the preferential orientation of lipid molecules and the angular brackets imply ensemble averaging. S0 indicates how well the chain segment to which the nitroxide is attached is aligned along the normal to the lipid bilayer.
CD Spectroscopy.
Fusion peptides (0.2 mg/mL in pH 5 solution) were mixed with SUVs composed of POPC/POPG/Chol = 3:1:1 at a ratio of 1:100 peptide/lipid at room temperature for 10 min before measurement. The CD spectra were collected at 25 °C on an AVIV Model 202–01 Circular Dichroism spectrometer. The signals from pure SUVs or pure solution were subtracted from the sample spectra as blanks. The CD spectra were analyzed using K2D3.52
Statistical Analysis.
Exact numbers of replicates are defined in each figure legend. All p values were determined using a two-tailed Student’s t test unless otherwise noted in the figure legend. All error bars indicate standard deviation among independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the National Science Foundation 1504846 (to S.D. and G.R.W.) and National Institutes of Health P41GM103521 and R01GM123779 (to J.H.F.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. L.N. would like to thank Tiffany Tang and Dr. Judith White for helpful discussions. The plasmid pcDNA3.1 containing the sequence for Ebolavirus, Zaire Glycoprotein, NR-19814 was obtained through the Biodefense and Emerging Infections Research Resources Repository, National Institute of Allergy and Infectious Diseases, National Institutes of Health. Molecular graphics were generated with the Chimera package developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by the National Institute of General Medical Sciences P41-GM103311). L.N. acknowledges support from a Samuel C. Fleming Family Graduate Fellowship and a National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1650441. A.L.L. acknowledges Dr. Lukas Tamm and Dr. Sonia Gregory for the Ebola FP construct plasmid and advice. A.L.L. also acknowledges Dr. Brian Crane and Dr. Richard Cerione for access to instrumentation. We thank Dr. David Eliezer for useful discussions.
ABBREVIATIONS
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis
- CD
circular dichroism
- DPPTC
dipalmitoylphosphatidyl-tempo-choline
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EBOV
Ebola virus
- EGTA
ethylene glycol tetraacetic acid
- ESR
electron spin resonance
- FP
fusion peptide
- GP
glycoprotein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- LUV
large unilamellar vesicle
- MARV
Marburg virus
- MES
2-(N-morpholino) ethanesulfonic acid
- MOMD
microscopic order–macroscopic disorder
- NBD
7-nitrobenz-2-oxa-1,3-diazole
- NPC1
Niemann-Pick disease, type C1
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- PPs
pseudovirus particles
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SUV
small unilamellar vesicle
- TPC2
two-pore calcium channel
- WT
wild type
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.9b00296.
(1) Raw data from infectivity studies for WT EBOV pseudovirus and noninfectious control; (2) Western blot confirming incorporation of WT GP and mutated GP into pseudovirus; (3) raw data from infectivity study comparing WT and mutated GP; (4) structure of spin-labeled lipids; (5) ordering effects of long vs short FP; (6) ordering effects of WT FP vs mutated FP as a function of peptide to lipid ratio; (7) ESR spectra of pseudovirus/lipid mixture; (8) Western blot confirming removal of mucin cap from pseudovirus GP; (9) lipid mixing data from pseudovirus/lipid mixture; (10) helical contents (%) vs Ca2+ concentration based on CD spectroscopy (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Feldmann H, and Geisbert TW (2011) Ebola Haemorrhagic Fever. Lancet 377 (9768), 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Centers for Disease Control and Prevention. (accessed May 24, 2018) Years of Ebola Virus Disease Outbreaks, https://www.cdc.gov/vhf/ebola/history/chronology.html.
- (3).World Health Organization. Ebola virus disease: Fact sheet, http://www.who.int/mediacentre/factsheets/fs103/en/.
- (4).Regules JA, Beigel JH, Paolino KM, Voell J, Castellano AR, Hu Z, Muñoz P, Moon JE, Ruck RC, Bennett JW, et al. (2017) A Recombinant Vesicular Stomatitis Virus Ebola Vaccine. N. Engl. J. Med 376 (4), 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ledgerwood JE, DeZure AD, Stanley DA, Coates EE, Novik L, Enama ME, Berkowitz NM, Hu Z, Joshi G, Ploquin A, et al. (2017) Chimpanzee Adenovirus Vector Ebola Vaccine. N. Engl. J. Med 376 (10), 928–938. [DOI] [PubMed] [Google Scholar]
- (6).Milligan ID, Gibani MM, Sewell R, Clutterbuck EA, Campbell D, Plested E, Nuthall E, Voysey M, Silva-Reyes L, McElrath MJ, et al. (2016) Safety and Immunogenicity of Novel Adenovirus Type 26-and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized Clinical Trial. JAMA −. J. Am. Med. Assoc 315 (15), 1610–1623. [DOI] [PubMed] [Google Scholar]
- (7).Cross RW, Mire CE, Feldmann H, and Geisbert TW (2018) Post-Exposure Treatments for Ebola and Marburg Virus Infections. Nat. Rev. Drug Discovery 17, 413–434. [DOI] [PubMed] [Google Scholar]
- (8).Sweiti H, Ekwunife O, Jaschinski T, and Lhachimi SK (2017) Repurposed Therapeutic Agents Targeting the Ebola Virus: A Systematic Review. Curr. Ther. Res 84, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hayden FG, Friede M, and Bausch DG (2017) Experimental Therapies for Ebola Virus Disease: What Have We Learned? J. Infect. Dis, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, Dewald LE, Schornberg KL, Scully C, et al. (2013) FDA-Approved Selective Estrogen Receptor Modulators Inhibit Ebola Virus Infection. Sci. Transl. Med 5 (190), 190ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Johansen LM, DeWald LE, Shoemaker CJ, Hoffstrom BG, Lear-Rooney CM, Stossel A, Nelson E, Delos SE, Simmons JA, Grenier JM, et al. (2015) A Screen of Approved Drugs and Molecular Probes Identifies Therapeutics with Anti-Ebola Virus Activity. Sci. Transl. Med 7 (290), 290ra89. [DOI] [PubMed] [Google Scholar]
- (12).Madrid PB, Panchal RG, Warren TK, Shurtleff AC, Endsley AN, Green CE, Kolokoltsov A, Davey R, Manger ID, Gilfillan L, et al. (2015) Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis 1 (7), 317–326. [DOI] [PubMed] [Google Scholar]
- (13).Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, and Schnittler H-J (2011) Ebola Virus Enters Host Cells by Macropinocytosis and Clathrin-Mediated Endocytosis. J. Infect. Dis 204, S957–S967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kaletsky RL, Simmons G, and Bates P (2007) Proteolysis of the Ebola Virus Glycoproteins Enhances Virus Binding and Infectivity. J. Virol 81 (24), 13378–13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, and White J (2006) Role of Endosomal Cathepsins in Entry Mediated by the Ebola Virus Glycoprotein. J. Virol 80, 4174–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Simmons JA, D’Souza RS, Ruas M, Galione A, Casanova JE, and White JM (2016) Ebolavirus Glycoprotein Directs Fusion through NPC1+ Endolysosomes. J. Virol 90 (1), 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. (2011) Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature 477 (7364), 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, et al. (2012) Ebola Virus Entry Requires the Host-Programmed Recognition of an Intracellular Receptor. EMBO J. 31 (8), 1947–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, et al. (2011) Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 477 (7364), 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Spence JS, Krause TB, Mittler E, Jangra RK, and Chandran K (2016) Direct Visualization of Ebola Virus Fusion Triggering in the Endocytic Pathway. mBio 7 (1), e01857–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, and Saphire EO (2008) Structure of the Ebola Virus Glycoprotein Bound to an Antibody from a Human Survivor. Nature 454 (7201), 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gregory SM, Harada E, Liang B, Delos SE, White JM, and Tamm LK (2011) Structure and Function of the Complete Internal Fusion Loop from Ebolavirus Glycoprotein 2. Proc. Natl. Acad. Sci. U. S. A 108 (27), 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Scott CC, and Gruenberg J (2011) Ion Flux and the Function of Endosomes and Lysosomes: pH Is Just the Start. BioEssays 33 (2), 103–110. [DOI] [PubMed] [Google Scholar]
- (24).Huotari J, and Helenius A (2011) Endosome Maturation. EMBO J. 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gerasimenko JV, Tepikin AV, Petersen OH, and Gerasimenko OV (1998) Calcium Uptake via Endocytosis with Rapid Release from Acidifying Endosomes. Curr. Biol 8 (24), 1335–1338. [DOI] [PubMed] [Google Scholar]
- (26).Albrecht T, Zhao Y, Nguyen TH, Campbell RE, and Johnson JD (2015) Fluorescent Biosensors Illuminate Calcium Levels within Defined Beta-Cell Endosome Subpopulations. Cell Calcium 57 (4), 263–274. [DOI] [PubMed] [Google Scholar]
- (27).Christensen KA, Myers JT, and Swanson JA (2002) pH-Dependent Regulation of Lysosomal Calcium in Macrophages. J. Cell Sci. 115 (Pt 3), 599–607. [DOI] [PubMed] [Google Scholar]
- (28).Tymianski M, Spigelman I, Zhang L, Carlen PL, Tator CH, Charlton MP, and Wallace MC (1994) Mechanism of Action and Persistence of Neuroprotection by Cell-Permeant Ca2+ Chelators. J. Cereb. Blood Flow Metab 14 (6), 911–923. [DOI] [PubMed] [Google Scholar]
- (29).Lai AL, Millet JK, Daniel S, Freed JH, and Whittaker GR (2017) The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform That Perturbs Membrane Order in a Calcium-Dependent Manner. J. Mol. Biol 429 (24), 3875–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dubé M, Rey FA, and Kielian M (2014) Rubella Virus: First Calcium-Requiring Viral Fusion Protein. PLoS Pathog. 10 (12), No. e1004530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sakurai Y, Kolokoltsov AA, Chen C-C, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl-Schott C, Biel M, and Davey RA (2015) Ebola Virus. Two-Pore Channels Control Ebola Virus Host Cell Entry and Are Drug Targets for Disease Treatment. Science 347 (6225), 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Fan H, Du X, Zhang J, Zheng H, Lu X, Wu Q, Li H, Wang H, Shi Y, and Gao G (2017) Selective Inhibition of Ebola Entry with Selective Estrogen Receptor Modulators by Disrupting the Endolysosomal Calcium. Sci. Rep 7, 41226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tang N, and Skibsted LH (2016) Calcium Binding to Amino Acids and Small Glycine Peptides in Aqueous Solution: Toward Peptide Design for Better Calcium Bioavailability. J. Agric. Food Chem 64 (21), 4376–4389. [DOI] [PubMed] [Google Scholar]
- (34).Budil DE, Lee S, Saxena S, and Freed JH (1996) Nonlinear-Least-Squares Analysis of Slow-Motion EPR Spectra in One and Two Dimensions Using a Modified Levenberg-Marquardt Algorithm. J. Magn. Reson., Ser. A 120 (2), 155–189. [Google Scholar]
- (35).Lou Y, Ge M, and Freed J (2001) A Multifrequency ESR Study of the Complex Dynamics of Membranes. J. Phys. Chem. B 105 (45), 11053–11056. [Google Scholar]
- (36).Lai AL, and Freed JH (2015) Interaction between the Influenza HA Fusion Peptide and Transmembrane Domain Affects Membrane Structure. Biophys. J 109 (12), 2523–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lai AL, Moorthy AE, Li Y, and Tamm LK (2012) Fusion Activity of HIV gp41 Fusion Domain Is Related to Its Secondary Structure and Depth of Membrane Insertion in a Cholesterol-Dependent Fashion. J. Mol. Biol 418 (1–2), 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pinello JF, Lai AL, Millet JK, Cassidy-Hanley D, Freed JH, and Clark TG (2017) Structure-Function Studies Link Class II Viral Fusogens with the Ancestral Gamete Fusion Protein HAP2. Curr. Biol 27 (5), 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Suárez T, Gómara MJ, Goñi FM, Mingarro I, Muga A, Pérez-Payá E, and Nieva JL (2003) Calcium-Dependent Conformational Changes of Membrane-Bound Ebola Fusion Peptide Drive Vesicle Fusion. FEBS Lett. 535 (1–3), 23–28. [DOI] [PubMed] [Google Scholar]
- (40).Srivastava M, Anderson CL, and Freed JH (2016) A New Wavelet Denoising Method for Selecting Decomposition Levels and Noise Thresholds. IEEE Access 4, 3862–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gehring G, Rohrmann K, Atenchong N, Mittler E, Becker S, Dahlmann F, Pöhlmann S, Vondran FWR, David S, Manns MP, et al. (2014) The Clinically Approved Drugs Amiodarone, Dronedarone and Verapamil Inhibit Filovirus Cell Entry. J. Antimicrob. Chemother 69 (8), 2123–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, and Platt FM (2008) Niemann-Pick Disease Type C1 Is a Sphingosine Storage Disease That Causes Deregulation of Lysosomal Calcium. Nat. Med 14 (11), 1247–1255. [DOI] [PubMed] [Google Scholar]
- (43).Ruiz-Argüello MB, Goñi FM, Pereira FB, and Nieva JL (1998) Phosphatidylinositol-Dependent Membrane Fusion Induced by a Putative Fusogenic Sequence of Ebola Virus. J. Virol 72 (3), 1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lee J, Nyenhuis DA, Nelson EA, Cafiso DS, White JM, and Tamm LK (2017) Structure of the Ebola Virus Envelope Protein MPER/TM Domain and Its Interaction with the Fusion Loop Explains Their Fusion Activity. Proc. Natl. Acad. Sci. U. S. A 114 (38), E7987–E7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Gruenberg J, et al. (2002) Separation and Characterization of Late Endosomal Membrane Domains. J. Biol. Chem 277 (35), 32157–32164. [DOI] [PubMed] [Google Scholar]
- (46).Lai AL, and Freed JH (2014) HIV gp41 Fusion Peptide Increases Membrane Ordering in a Cholesterol-Dependent Fashion. Biophys. J 106 (1), 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, et al. (2009) NAADP Mobilizes Calcium from Acidic Organelles through Two-Pore Channels. Nature 459 (7246), 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Maxfield FR, and Yamashiro DJ (1987) Endosome Acidification and the Pathways of Receptor-Mediated Endocytosis In Immunobiology of Proteins and Peptides IV, pp 189–198, Springer, DOI: 10.1007/978-1-4684-5442-0_16. [DOI] [PubMed] [Google Scholar]
- (49).Bartosch B, Dubuisson J, and Cosset F-L (2003) Infectious Hepatitis C Virus Pseudo-Particles Containing Functional E1–E2 Envelope Protein Complexes. J. Exp. Med 197 (5), 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Millet JK, and Whittaker GR (2016) Murine Leukemia Virus (MLV)-Based Coronavirus Spike-Pseudotyped Particle Production and Infection. Bio-protocol 6 (23), e2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ge M, and Freed JH (2003) Hydration, Structure, and Molecular Interactions in the Headgroup Region of Dioleoylphosphatidylcholine Bilayers: An Electron Spin Resonance Study. Biophys. J 85 (6), 4023–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Louis-Jeune C, Andrade-Navarro MA, and Perez-Iratxeta C (2012) Prediction of Protein Secondary Structure from Circular Dichroism Using Theoretically Derived Spectra. Proteins: Struct., Funct., Genet. 80 (2), 374–381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.