

Abstract
Fibroblast growth factor 23 (FGF23), one of the endocrine fibroblast growth factors, is a principal regulator in the maintenance of serum phosphorus concentration. Binding to its cofactor αKlotho and a fibroblast growth factor receptor is essential for its activity. Its regulation and interaction with other factors in the bone-parathyroid-kidney axis is complex. FGF23 reduces serum phosphorus concentration through decreased reabsorption of phosphorus in the kidney and by decreasing 1,25 dihydroxyvitamin D (1,25(OH)2D) concentrations. Various FGF23-mediated disorders of renal phosphate wasting share similar clinical and biochemical features. The most common of these is X-linked hypophosphatemia (XLH). Additional disorders of FGF23 excess include autosomal dominant hypophosphatemic rickets, autosomal recessive hypophosphatemic rickets, fibrous dysplasia, and tumor-induced osteomalacia. Treatment is challenging, requiring careful monitoring and titration of dosages to optimize effectiveness and to balance side effects. Conventional therapy for XLH and other disorders of FGF23-mediated hypophosphatemia involves multiple daily doses of oral phosphate salts and active vitamin D analogs, such as calcitriol or alfacalcidol. Additional treatments may be used to help address side effects of conventional therapy such as thiazides to address hypercalciuria or nephrocalcinosis, and calcimimetics to manage hyperparathyroidism. The recent development and approval of an anti-FGF23 antibody, burosumab, for use in XLH provides a novel treatment option.
Keywords: FGF23; Klotho; Phosphorus; 1,25(OH)2D; XLH; Burosumab; Rickets
Introduction
Fibroblast growth factor 23 (FGF23) is part of a family of fibroblast growth factors (FGFs) which are secreted signaling proteins (1). Found in several tissues, they serve essential functions in development and metabolism through all stages of life, beginning in the embryo and continuing through adulthood (2). There are three types of FGFs, categorized based on their mechanism of action: autocrine, paracrine, and endocrine (1). FGFs require heparin sulfate for receptor binding and signaling, enabling autocrine and paracrine functions (3). However, the endocrine FGFs are distinguished by their poor affinity for heparin sulfate, allowing release from the local extracellular matrix to circulate as endocrine hormones and bind receptors on distant cells (4). The endocrine FGFs play an important part in bile acid (FGF19), carbohydrate (FGF21), lipid (FGF21), and phosphate metabolism (FGF23) (2).
The FGF23 gene is located on human chromosome 12 (5). FGF23 is primarily produced in bone by osteocytes and is a 32 kDa protein containing 251 amino acids (5). The N-terminus contains a FGF homology region which binds to the FGF receptor and the C-terminus binds to the co-receptor αKlotho, ultimately creating a FGF receptor complex necessary for signaling at the FGF receptor (6).
In 1989, Meyer et al. suggested the presence of a phosphaturic factor (referred to as a “phosphatonin”) in Hyp mice, a mouse model of X-linked hypophosphatemic rickets (XLH) (7). Hyp mice produced this phosphaturic factor which could be transferred to normal mice through parabiosis experiments, resulting in an XLH phenotype (7). FGF23 was discovered in 2000 due to mutations in the FGF23 gene found in a kindred with autosomal dominant hypophosphatemic rickets (ADHR) (8), and was later identified as elevated in XLH and several other renal phosphate wasting disorders as the responsible phosphaturic factor. In our review, we explore the biological function and regulation of FGF23 and discuss hypophosphatemic disorders resulting from a state of FGF23 excess.
Cofactor and Receptors
The endocrine FGFs require a cofactor, αKlotho or βKlotho, in order to bind their respective receptors and provide tissue specificity (1). αKlotho or βKlotho are structurally related proteins consisting of approximately 1000 amino acids (2). FGF19 and FGF21 activate their receptor via βKlotho (1), while FGF23 activates its receptor via αKlotho (9). αKlotho binds to multiple fibroblast growth factor receptors (FGFR), forming a complex with greater affinity for FGF23 than αKlotho or the FGF receptor alone (10). Since many tissues express FGFR, the presence of klotho determines the target organs for the endocrine FGFs (11,12). The primary tissues expressing αklotho are the kidney’s proximal and distal tubules, the parathyroid glands, and the brain’s choroid plexus (13).
FGFRs are tyrosine kinases (2). Four of them are considered to be high-affinity receptors: FGFR1, FGFR2, FGFR3, and FGFR4. However, αKlotho does not bind strongly to FGFR2 (9,10). Alternative splicing creates different ‘b’ and ‘c’ isoforms of FGFR and αKlotho binds best to the ‘c’ isoform leading to signaling of FGF23 through FGFR1c, FGFR3c, or FGFR4c (9,10). FGF23 has the highest affinity for FGFR1c (9).
Regulation of FGF23
The regulation of FGF23 is complex, involving multiple components in the bone-parathyroid-kidney axis, including phosphorus, 1,25(OH)2D, parathyroid hormone (PTH), and calcium. FGF23 concentrations are increased by 1,25(OH)2D in humans and in animal models (14–20). In cell culture studies, 1,25(OH)2D increased FGF23 gene expression (17). Even in XLH, therapeutic treatment with phosphate and calcitriol led to a further significant increase in already elevated FGF23 levels despite persistent hypophosphatemia (19). In 30 adult dialysis patients with baseline elevated levels of FGF23 and secondary hyperparathyroidisim, FGF23 levels increased further after intravenous calcitriol (15). While phosphorus and 1,25(OH)2D both independently regulate FGF23 (17,18), phosphate binders may be able to block 1,25(OH)2D-induced FGF23 increases (16).
Phosphate intake increases FGF23 levels in studies of healthy adults and animal models (17,21–24). Dietary phosphate was shown to be a key regulator of serum FGF23 in healthy men and women (21,24). Oral phosphate loading significantly increased FGF23, while phosphate restriction led to a significant decrease (21,24). In healthy adult subjects, a diet high in both phosphate and calcium also significantly increased FGF23 levels (23).
The effect of PTH on FGF23 is not entirely clear. Animal models and cell culture studies indicate that PTH directly stimulates FGF23 production via the PTH/PTHrP receptor (25–29). However, human studies are conflicting. One study in healthy adults indicated after PTH (1–34) infusion, that FGF23 and phosphorus levels significantly decreased over 6 hours (30). In contrast, another study in healthy adult men receiving a PTH (1–34) infusion, found FGF23 increased significantly during an 18 hour period (31). In both studies, PTH (1–34) infusion increased 1,25(OH)2D.
Low serum calcium may act as a “brake” on FGF23 production, which may be an adaptive response to prevent further hypocalcemia. Low calcium levels have been shown to decrease FGF23 levels, which subsequently removes FGF23 suppression of 1,25(OH)2D (32,33). Using mutant mouse models, PTH and 1,25(OH)2D were unable to stimulate FGF23 in the setting of hypocalcemia (33). This may also explain why it is often difficult to normalize serum phosphorus in hypoparathyroidism, despite elevated FGF23 (34). In adult patients with hypoparathyroidism, treatment with 1,25(OH)2D increased serum calcium and FGF23 levels (20).
Additional regulation of FGF23 occurs through post-translational mechanisms. Proprotein convertases cleave FGF23 at the C-terminus between amino acids 179 and 180 (35). Mutations affecting this cleavage site result in excess FGF23 (36). FGF23 also requires O-glycosylation within the proprotein convertase cleavage site in order to secrete biologically active intact FGF23 (37). O-glycosylation is directed by the polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3) (37). Deficiency of GALNT3 activity prevents O-glycosylation, leading to increased cleavage and inactivation of FGF23 (37). Loss-of-function mutations in GALNT3 result in a deficiency of intact FGF23, and the resulting phenotype of hyperphosphatemic familial tumoral calcinosis (38). A kinase from the family with sequence similarity 20, member C (FAM20C) phosphorylates FGF23, preventing O-glycosylation, allowing for cleavage by proprotein convertases, such as furin (39). Deficiency of FAM20C leads to FGF23 excess and hypophosphatemia. FGF23 cleavage may be regulated specifically to maintain appropriate concentrations and normophosphatemia. Gene expression increases in the setting of iron deficiency (40), but generally intact FGF23 concentrations remain normal unless a mutation specifically impairs FGF23 cleavage (41). Phosphate regulating endopeptidase homolog X-linked (PHEX) and dentin matrix protein 1 (DMP1) are genes whose deficiency results in upregulation of FGF23 (42, 43). FGFR signaling pathways also regulate FGF23 through Ras-mitogen-activated protein kinase (MAPK), extracellular signal related kinase (ERK), and tyrosine kinase activity (44). Figure 1 lists positive and negative regulators of FGF23.
Figure 1.
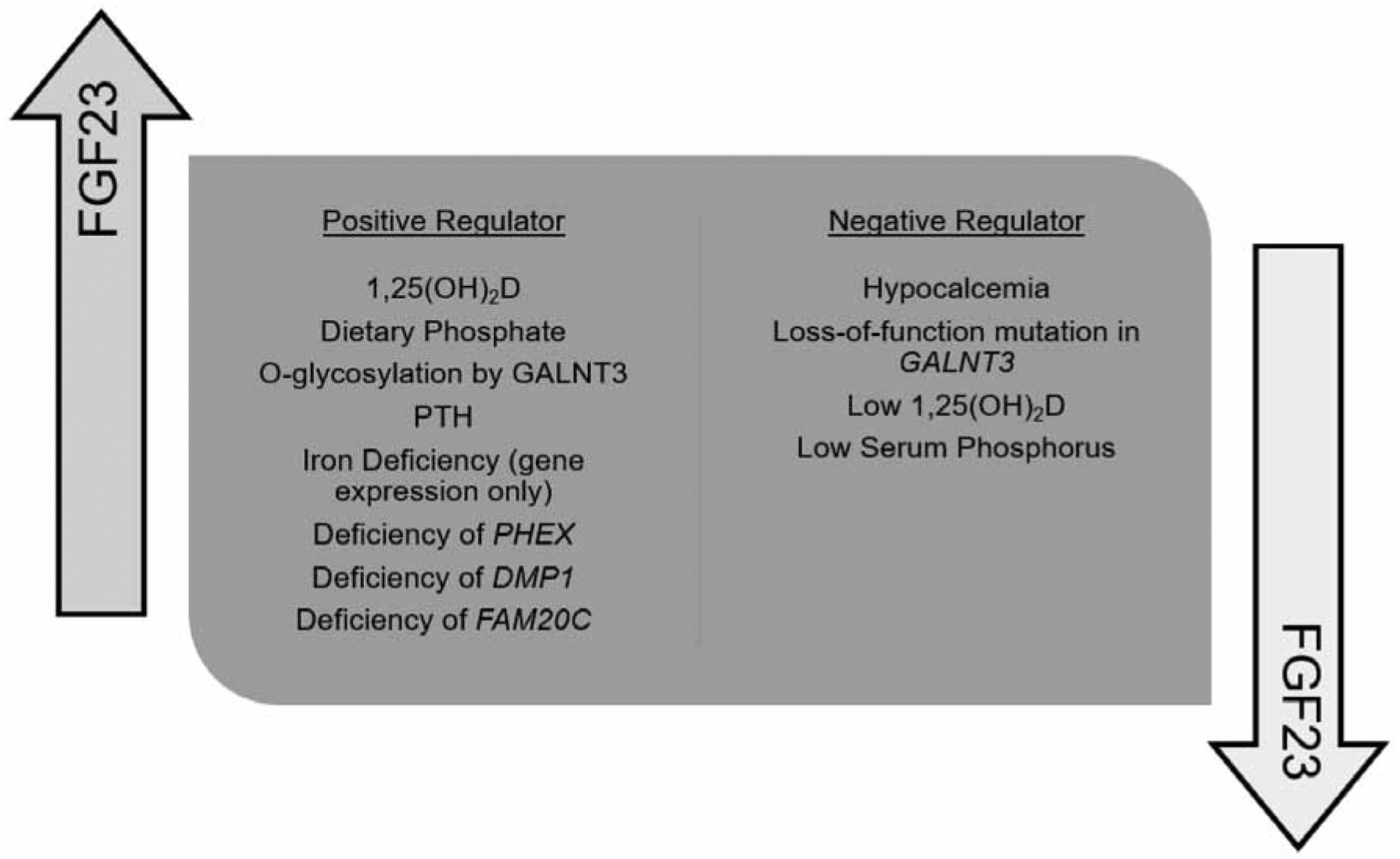
List of positive and negative regulators of FGF23
FGF23 Regulation of 1,25(OH)2D, Phosphorus and PTH
FGF23 is the principal regulator in the maintenance of serum phosphorus levels (figure 2). Administration of FGF23 in animal models decreases serum phosphorus due to a combination of effects on renal phosphate transport and vitamin D metabolism (45–48). Phosphorus is filtered by the glomerulus, but the vast majority is reabsorbed in the proximal convoluted tubule by renal sodium phosphate cotransporters type IIa (NaPi-IIa) and type IIc (NaPi-IIc) (49). FGF23 administration results in reduced brush border expression of the NaPi-IIa and NaPi-IIc (50,51). NaPi-IIa and NaPi-IIc are also down-regulated by PTH (52).
Figure 2.

Schematic representation of the regulation of serum phosphorus by FGF23
FGF23 decreases serum 1,25(OH)2D levels via suppressed expression of CYP27B1, limiting protein expression of 1α-hydroxylase (45,53), an enzyme necessary to convert 25-hydroxyvitamin D (25(OH)D) to its active form 1,25(OH)2D (53). FGF23 also increases expression of CYP24A1, increasing vitamin D 24-hydroxylase (45), which catabolizes 25(OH)D and 1,25(OH)2D into inactive metabolites (53). Since 1,25(OH)2D upregulates intestinal phosphate transport, decreasing 1,25(OH)2D also contributes to FGF23-mediated hypophosphatemia (50,51).
Conditions of FGF23 excess or deficiencies have expected effects on phosphorus and vitamin D metabolism. Transgenic mice expressing human FGF23 have reduced expression of NaPi-IIa, phosphaturia, and decreased serum 1,25(OH)2D with resultant hypophosphatemia and rachitic bone (50). FGF23 null mice had the opposite biochemical findings with elevated serum phosphorus levels, elevated serum 1,25(OH)2D, and increased renal phosphorus reabsorption (54). These phenotypes are recapitulated in the human diseases of XLH and hyperphosphatemic tumoral calcinosis, respectively.
The effect of FGF23 on PTH is not well understood. Evidence in vitro and from animal models suggests that FGF23 has an inhibitory effect on PTH, at least during short-term studies (11,55). FGF23 suppressed PTH in rats, but in the setting of hypocalcemia, the inhibition of PTH is lost (56). However, in human diseases or animal models of chronic FGF23 excess, hyperparathyroidism is common (51,57).
Effect of Age and Gender on FGF23
Multiple enzyme linked immunosorbent assays are available for measuring FGF23 in serum or plasma. An assay targeting the C-terminal end of FGF23 (cFGF23) will detect both C-terminal fragments and full length FGF23 (58). However only the intact form is biologically active. Intact FGF23 assays measure only the full-length intact FGF23 (iFGF23) (59). The cFGF23 is best measured in plasma, as serum values will be systematically lower (60). However, the Kainos iFGF23 assay provides similar results in plasma and serum (60).
Table 1 describes reported ranges of FGF23 in healthy populations. In general studies indicate higher values of cFGF23 in newborns and young children than in adults (58,61–67). However, reported iFGF23 ranges are generally similar across studies at different ages in healthy children and adults, though one study suggested lower iFGF23 in cord blood (59,61–63,65–67). While cFGF23 were mostly similar between boys and girls, some studies suggested higher iFGF23 in girls (62,63).
Table 1.
Reference values for cFGF23 and iFGF23 among age groups
| Age | Study Sample | Plasma or Serum | cFGF23 | cFGF23 Assay | iFGF23 | iFGF23 Assay | Study/Year |
|---|---|---|---|---|---|---|---|
| Neonate | 64-full term neonates | Plasma | Median 824 RU/mL (108–7508) Mean 1678 RU/mL (±1857) |
Immunotopics | Median <8.5 pg/mL (<8.5–135.4) Mean 16.7 pg/mL (±24.2) |
Immunotopics | Ali et al., 2016 (61) |
| 113 healthy infants | Cord blood serum at birth | Median Girls 536.2 RU/mL [731.7] Median Boys 605.9 RU/mL [842.9] |
Immunotopics | Median Girls 3.0 pg/mL [10.7] Median Boys 3.0 pg/mL [2.3] |
Kainos | Holmlund- Suila et al., 2016 (62) | |
| 3 months old | 113 healthy infants | Serum | Mean Girls 106.9 RU/mL (±64.0) Mean Boys 105.4 RU/mL (±52.1) |
Immunotopics | Median Girls 51.4 pg/mL [30.0] Median Boys 25.9 pg/mL [48] |
Kainos | Holmlund- Suila et al., 2016 (62) |
| 1 year old | 721 healthy children | Plasma | Median Girls 2.9 pmol/L [2.2–3.7] Median Boys 2.8 pmol/L [2.1–3.7] |
Biomedica Medizin-produkte GmbH & Co KG | Median Girls 44.4 pg/mL [36.8–51.9] Median Boys 40.9 pg/mL [34.5–49.0] |
Kainos | Holmlund- Suila et al., 2017 (63) |
| Childhood | 424 healthy youth and young adults (ages 0.1–21 years) | Plasma | Age ≤ 1 Median 105 RU/mL [75–153] Age 5 Median 68 RU/mL [53–89] Age 10 Median 71 RU/mL [56–88] Age 15 Median 76 RU/mL [59–95] Age ≥ 19 Median 50 RU/mL [39–61] |
Immunotopics | Fischer et al., 2012 (64) | ||
| 159 healthy children (mean age 8.78 ± 3.47 years) | Serum | Mean 51.14 RU/mL (±12.79) | Immunotopics | Median 35 pg/mL (8.8–120) | Kainos | Gkentzi et al., 2014 (65) | |
| Adulthood | 180 healthy adults | Plasma | Median 53.7 RU/mL (range not provided) | Immunotopics | Median 24.7 pg/mL (range not provided) | Immunotopics | Smith et al., 2012 (66) |
| 147 healthy adults | Plasma or serum | Mean Women 52.9 RU/mL (±20.8) Mean Men 42.0 RU/mL (±15.8) |
Immunotopics | Jonsson et al., 2003 (58) | |||
| 55 healthy adults | Plasma | Mean 61.0 RU/mL (±28.6) | Immunotopics | Mean 44.7 pg/mL (±14.9) | Kainos | Imel et al., 2007 (67) | |
| 104 healthy adults | Serum | Mean 28.9 ng/L (8.2–54.3) | Kainos | Yamazaki et al., 2002 (59) |
In a study of 180 healthy adults, cFGF23 had lower intra-individual variability, but higher inter-individual variability (66). Since iFGF23 had less inter-individual variability, it may be more clinically useful for diagnostic purposes (66), though currently the cFGF23 assay is more clinically available. Intact FGF23 levels >30 pg/mL using the Kainos intact assay (approximately the normal mean with this assay in some studies) during hypophosphatemia have been proposed as a cutoff for identifying FGF23-mediated hypophosphatemia (68). However, an analogous threshold with cFGF23 has not been determined.
FGF23-Mediated Disorders of Phosphate Wasting
The differential diagnosis for hypophosphatemia is quite broad, but etiologies largely include increased renal excretion (both FGF23-mediated and non-FGF23-mediated), impaired intake or intestinal absorption of phosphate, and transcellular shifts of phosphorus (69). This review concentrates on the FGF23-mediated causes, which include autosomal dominant hypophosphatemic rickets (ADHR), X-linked hypophosphatemic rickets (XLH), autosomal recessive hypophosphatemic rickets (ARHR), fibrous dysplasia (FD), and tumor-induced osteomalacia (TIO) (table 2). These FGF23-mediated hypophosphatemic disorders share common features which may include rickets or osteomalacia, bony deformities, short stature, and bone pain (8). Biochemically these conditions are characterized by low serum phosphorus, increased urinary phosphorus excretion [or decreased ratio of the maximum rate of tubular phosphate reabsorption to glomerular filtration rate (TmP/GFR)], normal serum and urine calcium, high alkaline phosphatase (ALP), normal PTH, normal 25(OH)D, decreased or inappropriately normal 1,25(OH)2D, and increased FGF23 (69).
Table 2.
Summary of FGF23-Mediated disorders of phosphate wasting and associated genetic & somatic mutations
| Disorder | Genetic Mutation |
|---|---|
| X-linked Hypophosphatemic Rickets | PHEX |
| Autosomal Recessive Hypophosphatemic Rickets | DMP1, ENPP1, FAM20C |
| Autosomal Dominant Hypophosphatemic Rickets | FGF23 |
| Fibrous Dysplasia (FD)/McCune-Albright Syndrome | GNAS |
| Tumor Induced Osteomalacia | FN1-FGFR1 fusion gene |
| Linear Nevus Sebaceous Syndrome | HRAS, KRAS, NRAS somatic mutations |
| Jansen’s Metaphyseal Chondrodysplasia | PTH/PTHrP receptor |
| Osteoglophonic Dysplasia | FGFR1 |
FGF23 Effect on Bone
FGF23 has both direct and indirect effects (through hypophosphatemia) on bone. Hypophosphatemia, secondary to excess FGF23, causes rickets due to arrested apoptosis of the hypertrophic chondrocytes of the growth plate and osteomalacia due to delayed mineral apposition rate of osteoid (70). In growing youth, prior to epiphyseal fusion, rickets and osteomalacia both occur, while in the adult, only osteomalacia occurs (70).
In mouse models, Murali et al. has recently shown that increased FGF23 also has direct autocrine and paracrine effects on the osteocyte, which occur independently of klotho and lead to suppression of osteocyte tissue nonspecific alkaline phosphatase (TNAP), contributing to impaired bone mineralization (71,72). Bone specific alkaline phosphatase hydrolyzes inorganic pyrophosphate (an inhibitor of mineralization) releasing inorganic phosphate, subsequently allowing for synthesis of hydroxyapatite (73). Its deficiency leads to impaired mineralization as seen in hypophosphatasia, while conversely, insufficient inorganic phosphate at the mineralization surface tends to increase alkaline phosphatase activity. In the Hyp mouse model of XLH, while the TNAP activity in the Hyp osteocyte is impaired secondary to increased FGF23 secretion, osteoblast TNAP is increased sufficient to lead to elevated serum ALP (72). ALP activity is also increased within growth plate cartilage during chondrocyte differentiation (74). Increased serum ALP activity is generally seen in human disease states of FGF23-mediated hypophosphatemia (69).
Autosomal Dominant Hypophosphatemic Rickets (ADHR)
In the year 2000, FGF23 was discovered due to the presence of missense mutations in kindreds with ADHR (8). These missense mutations decrease FGF23’s susceptibility to proteolytic cleavage, preventing its degradation, hence resulting in elevated circulating levels and hypophosphatemia (75). ADHR is a rare hypophosphatemic disorder with autosomal dominant inheritance pattern, but incomplete penetrance (76).
In fact, disease activity fluctuates according to FGF23 levels in this condition, and there is waxing and waning of the biochemical and symptomatic phenotypes (36). Those with an onset in childhood develop hypophosphatemia, phosphate wasting, rickets, and lower extremity deformities. However, a large subgroup of patients has documented normal phosphate values for age in childhood, grows normally without rickets or leg deformities, and only later as adolescents or adults develops elevated iFGF23 and hypophosphatemia (36,41,76,77).
Those with late-onset of disease in adolescence or adulthood developed significant bone pain, weakness, and insufficiency fractures. Some of those with childhood-onset disease achieve spontaneous resolution of their renal phosphate-wasting defect. Similarly some with late-onset hypophosphatemia also spontaneously normalize their FGF23 and serum phosphorus concentrations, with associated resolution of symptoms (36,41).
Interestingly the FGF23 phenotype of ADHR appears to be driven by the consequences of iron deficiency. In the setting of iron deficiency, FGF23 gene expression increases (40). In healthy controls or wild type mice, this leads to elevated circulating concentrations of fragments (cFGF23), but biologically active iFGF23 remains normal, with normophosphatemia (40,41). However, in the setting of ADHR mutations, iFGF23 concentrations also become elevated during iron deficiency due to the effect of impaired FGF23 cleavage, and hypophosphatemia results (40,41). The observed waxing and waning of the ADHR biochemical phenotype corresponded to changes in iron status (41). We would propose that if a patient never became iron deficient, clinical features of ADHR might never manifest. In contrast, in XLH patients, iFGF23 is not related to serum iron (78).
The iron story is made somewhat more complicated by adverse effects of intravenous iron administration. In the setting of iron deficiency, patients without ADHR can sometimes be triggered by certain forms of intravenous iron to undergo sudden acute increases in iFGF23, even while their cFGF23 is normalizing (79, 80). This phenomenon can be severe enough to cause hypophosphatemia, and if repeated doses are necessary due to persistent iron deficiency, osteomalacia and insufficiency fractures may result. The mechanism is not certain but appears to involve a transient inability to effectively cleave iFGF23, even in patients without ADHR. This has been mainly reported with intravenous iron carboxymaltose and iron polymaltose (79–81). Thus, patients undergoing iron infusions should have serum phosphorus monitored.
X-Linked Hypophosphatemic Rickets (XLH)
The most common heritable form of rickets is XLH, with an estimated prevalence of 1 in 20,000 and accounting for about 80% of familial cases of hypophosphatemia (57). The inheritance pattern is X-linked dominant, indicating that a single allele will cause phenotypic expression in both males and females. Careful family history should identify the inheritance pattern and guide assessment of a genetic cause, as there are autosomal dominant and recessive disorders that clinically mimic XLH. A mutation in the PHEX gene causes XLH in humans (82), and in the Hyp mouse model as well (83). The PHEX gene is expressed in bone (osteocyte) and teeth (odontoblasts) (57). PHEX deficiency results in increased expression of FGF23, and consequent hypophosphatemia.
XLH has high penetrance, but clinical findings and severity vary widely among individuals, even within a kindred. Clinical features include short stature, lower-extremity deformities, osteomalacia, rickets, and bone pain (57). Features typically manifest around the time of walking, after age 1 to 2 years, when short stature and limb deformities become apparent (57). Frequent limb deformities include genu varum or valgum, tibial torsion, bowing of the tibia and femur, or windswept deformity (84). While rickets is a classic and common feature, its location and presence are variable among individuals (85). A distinctive histologic feature is hypomineralized periosteocytic lesions in cortical bone (86). In XLH, FGF23 is generally elevated. However, some patients may have high-normal levels which are still indicative of an FGF23-mediated cause of their hypophosphatemia (58,59,68).
Dental disease is very common including dental abscesses, caries, periodontal disease, and tooth loss (87–89). Phex and FGF23 are expressed in teeth, and patients with XLH have impaired mineralization of dentin and cementum layers (90–93). Osteopontin is involved in the intrinsic dental abnormalities(91), however hypophosphatemia likely plays a role as well, since treatment with calcitriol and phosphate is associated with fewer tooth abscesses, though they remain common despite treatment (88,89,92).
Osteoarthritis, enthesopathies, and residual lower-extremity long bone curvature are common in adults with XLH (94). Enthesopathies occur in several locations and are commonly found in the hands, feet, spine, hips, and sacroiliac joints and can become quite severe (95,96). These features greatly limit mobility and quality of life. Adults with PHEX mutations often need orthopedic procedures including joint replacement and spinal surgeries (87). With a mean age of 50 at the time of surgery, total knee and hip arthroplasties may benefit adult XLH patients with degenerative osteoarthritis (97). Up to half of adult patients may have pseudofractures (98–100), which are also a source of pain and decreased mobility, which often result in orthopedic procedures for stabilization.
Monitoring for neurologic symptoms is necessary throughout life as patients with XLH can develop neurologic complications due to several disease features. Skull malformations are common in patients with rickets which may sometimes be associated with neurologic consequences. In a study of 44 children with XLH, craniosynostosis, especially sagittal suture fusion, occurred in 59%, and Chiari type 1 malformation was found in 25%, 2 of which had neurological symptoms, and 4 required neurosurgical intervention (101). Hearing loss has been reported in 28.6% of XLH patients compared to 9.8% of unaffected family members (102), in 9% of children and in 48–82% of adults (103,104). Hearing loss is often sensorineural (104). The etiology may involve osteomalacia of the otic capsule bone, but in the Phex mutant mouse model, treatment with calcitriol and phosphate improved mineralization of the capsule bone, but did not prevent sensorineural hearing loss (105).
One study indicated that 12% of adults experience spinal complications including spinal stenosis, cord compression, and myelopathy (87), although lifetime rates may be higher, given the prominent involvement of the spine in enthesopathy, both at the anterior and posterior longitudinal ligament. Decompressive laminectomy may be needed. These features can contribute to disability. It is important to note that none of the medical treatments discussed below have ever been shown to alter the course of enthesopathy.
Autosomal Recessive Hypophosphatemic Rickets (ARHR)
Autosomal recessive hypophosphatemic rickets (ARHR) is another rare FGF23-mediated condition of renal phosphate wasting with multiple genetic causes. DMP1 mutations cause ARHR type 1 and FGF23 levels are elevated in these individuals and in the DMP1 null mouse (106,107), resulting in hypophosphatemia, severe rickets, and diffuse osteomalacia (43). Mutations in ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) cause ARHR type 2 (108). ENPP1 mutations are also known to cause generalized arterial calcification of infancy (GACI) which causes calcification and stenosis of medium and large-sized arteries (109,110) and is frequently lethal (111). In some individuals with GACI, hypophosphatemia due to renal phosphate wasting developed and was associated with survival past infancy (111), suggesting hypophosphatemia as a protective mechanism seen in milder phenotypes (109). However, in three individuals in a family with ARHR, ENPP1 mutations may also cause ARHR, without GACI (112). Rafaelsen et al. identified a nonlethal variant of Raine syndrome caused by a FAM20C mutation in 2 siblings with elevated FGF23 levels and hypophosphatemia, representing a third genetic form of ARHR (113).
Case reports of individuals with ARHR caused by DMP1 or ENPP1 mutations show a clinical phenotype of short stature, and skeletal deformities starting in early childhood, dental abnormalities such as hypoplasia, caries, and early tooth loss, bone and joint pain, contractures, ligamental calcification, enthesopathies, rickets, and osteomalacia similar to XLH (112,114–118). The siblings with a FAM20C mutation developed tooth decay, osteosclerosis of the long bones, ectopic brain calcifications, and mild facial and acral dysmorphic features (113).
Fibrous Dysplasia (FD)/McCune-Albright Syndrome (MAS)
Fibrous dysplasia (FD) of bone is characterized by replacement of normal bone and bone marrow by abnormal fibro-osseous tissue (119,120). Its occurrence is seen in individuals with McCune-Albright Syndrome (MAS), classically defined as the triad of fibrous dysplasia, café-au-lait macules, and precocious puberty, but can include other endocrinopathies such as hyperthyroidism, growth hormone excess, and Cushing syndrome (121). MAS is rare with an estimated prevalence between 1 in 100,000 and 1 in 1,000,000 (121). It is caused by a post-zygotic mutation in the guanine nucleotide binding protein, alpha stimulating (GNAS) gene, resulting in constitutive activation of the adenylyl cyclase system in affected cells (121,122). FD may also occur in individuals without other features of MAS.
Approximately 50% of individuals with MAS and FD have renal phosphate wasting (120,123), which correlates significantly with the degree of bone involvement (120). However, hypophosphatemic rickets is not common (124). FD lesions locally produce FGF23 and blood cFGF23 levels are increased in FD/MAS compared to normal controls and are significantly higher in FD/MAS with renal phosphate wasting compared to FD/MAS without renal phosphate wasting (123,125). Bone marrow stromal cells with the GNAS mutation have lower GALNT3, but higher furin activity, which ultimately leads to increased FGF23 cleavage causing a larger proportion of the increased FGF23 level being cFGF23, the non-biologically active form, which may explain why classic hypophosphatemic rickets may be less common in FD (126).
Tumor Induced Osteomalacia (TIO)
Tumor induced osteomalacia (TIO) is a rare, sporadic disorder that occurs in children and adults. The tumors are often small and slow growing, of mesenchymal origin, and located in the bone or soft tissue (127). They abundantly express FGF23 (48,128) resulting in elevated blood levels (58,59). Other factors, such as secreted frizzled-related-protein-4 (sFRP-4), fibroblast growth factor 7 (FGF7), and matrix extracellular phosphoglycoprotein (MEPE) are also produced by TIO-associated tumors (129–131), but have not clearly been linked to phosphate pathology. An FN1-FGFR1 fusion gene appears to cause some of these tumors (132). However, as ADHR can also cause late-onset hypophosphatemia, it should be considered in the differential diagnosis of TIO.
Patients often present with vague and long-standing symptoms of bone pain, muscle weakness, fractures, and fatigue (133). Localization of the tumor can be quite difficult, and multiple imaging modalities may need to be employed including ultrasound, computed tomography (CT), magnetic resonance imaging (MRI) (133), 111In-octreotide scan, and fluorodeoxyglucose (FDG) positron emission tomography (PET)/computed tomography (CT) (134). More recently, Dotatate PET/CT has been used. Selective venous sampling can sometimes detect local elevations in serum FGF-23 levels, allowing for localization of the responsible tumor (135–138). Complete tumor resection is the most effective approach (139) resulting in resolution of hypophosphatemia and a good prognosis in most (140). Post-operative recurrence can occur even many years later, especially when complete resection is not possible, so ongoing surveillance is necessary (139,140).
Other FGF23-Mediated Disorders of Phosphate Wasting
Other extremely rare causes of FGF23-mediated phosphate wasting include linear nevus sebaceous syndrome (or epidermal nevus syndrome), Jansen’s metaphyseal chondrodysplasia, and osteoglophonic dysplasia. Linear nevus sebaceous syndrome (LNSS) is a neurocutaneous disorder affecting multiple organ systems, but mainly the skeletal and central nervous systems (141,142). Postzygotic somatic mutations in HRAS, KRAS, and NRAS are described in LNSS (143,144). Hypophosphatemic rickets may occur with elevated FGF23 levels (145,146). Although some early reports suggested that excision of the nevus corrected the hypophosphatemia (145,147), there is growing evidence that the source is actually the skeleton and that excising these lesions is not beneficial (148,149).
Jansen’s metaphyseal chondrodysplasia is a rare form of short limbed dwarfism due to severe growth plate abnormalities with biochemical features similar to primary hyperparathyroidism with hypercalcemia and hypophosphatemia, however PTH is low or undetectable (150). This dysplasia occurs secondary to an activating mutation in the receptor for PTH and PTH-related peptide (PTHrP) (151). Elevated serum FGF23 levels were described in a case report of Jansen’s metaphyseal dysplasia and osteocyte expression of this mutation causes elevating FGF23 levels (29,152).
Osteoglophonic dysplasia (OD) is a rare skeletal dysplasia with findings of disproportionate dwarfism, craniofacial defects, and non-ossifying bone lesions caused by an activating mutation in the FGFR1 receptor (153–155). Hypophosphatemia can be seen associated with an elevated FGF23 level, perhaps from local production within bone lesions (155).
Treatment
Conventional therapy for XLH involves multiple daily dosing of oral phosphate supplementation and active vitamin D analogs, such as calcitriol or alfacalcidol (156). Phosphate salts should never be given without an active form of Vitamin D in XLH both because of a lack of effectiveness as monotherapy and due to the effect of phosphate to induce development of secondary and tertiary hyperparathyroidism (157). Conversely some patients can be managed with calcitriol alone (158).
There is no consensus on the optimal doses, and given the extreme variability between patients with XLH, doses need to be individualized (159). Typically published dose recommendations range from 20–60 mg/kg/day of oral phosphate divided into three to five doses per day, and either calcitriol 20–30 ng/kg/day divided into two to three doses per day or alfacalcidol 40–60 ng/kg/day (57,156,160,161). Doses up to 80 mg/kg/day of phosphate and 60 ng/kg/day of calcitriol or higher have also been described in various studies (57,160–162). To our knowledge no systematic study has compared different doses to define an optimal dose level, and most trials typically just report the doses that were administered and are underpowered to compare magnitude of effectiveness of different dose regimens. However, given variability in response, with some patients responding well to lower doses, while others requiring higher doses to achieve effect, it is important to individualize therapy.
Laboratory monitoring, especially of calcium, phosphorus, creatinine, alkaline phosphatase and PTH, as well as of urine calcium and urine creatinine, should be conducted every 3–6 months. Overall, doses should be carefully titrated to achieve a decrease in serum ALP activity while paying careful attention to serum and urine calcium concentrations and serum PTH (160). It is important to know that the primary goal of conventional therapy is not to normalize the serum phosphorus, but rather to improve skeletal outcomes including growth and deformity. In this regard, normalizing the alkaline phosphatase as a marker of osteomalacia is an important goal of therapy and failure to normalize alkaline phosphatase indicates a need to modify therapy and confirm compliance.
However, careful monitoring is also necessary to avoid or manage the clinically important complications of therapy. Gastrointestinal symptoms from the laxative effects of phosphate can often be managed by titrating the dose slowly, but these can be limiting for some patients and complicate adherence. Other clinical complications of conventional therapy include hypercalciuria, nephrocalcinosis, and secondary (or often tertiary) hyperparathyroidism (57,163), which may be related to higher doses (164–166). Consequently, limiting doses or adding adjunctive therapies may be required in some individuals to minimize risk or address occurrence of complications. In particular high doses of phosphate >100 mg/kg/day are associated with higher risk for tertiary hyperparathyroidism (164), though this also is observed with lower doses, especially if accompanied by insufficient dosing of active form of vitamin D.
PTH should be monitored every 3–6 months in children and every 6 months in adults receiving therapy for XLH (57,159). Increasing doses of the active form of vitamin D can ameliorate or normalize the PTH in secondary hyperparathyroidism, though lowering phosphate doses can sometimes be necessary (57,159). Secondary hyperparathyroidism is common, occurring in 83.3% of patients with XLH, leading to tertiary hyperparathyroidism in 16.7%, including some adolescents (167). There is little data on the outcomes of treatment for tertiary hyperparathyroidism in XLH, which is mostly based on case reports and series. In a recent case series, 75% of XLH patients having parathyroidectomy had recurrence or persistence of tertiary hyperparathyroidism (167).
In a well done study in children with XLH, short term treatment with cinacalcet increased TmP/GFR and serum phosphate, while decreasing PTH levels (168). In an interesting case report, a single patient with XLH was managed with cinacalcet, calcitriol, and hydrochlorothiazide without phosphate salts, demonstrating improvement of rickets (169). Of note cinacalcet use in children is off-label. Several authors have reported success managing the secondary or tertiary hyperparathyroidism of XLH patients with the calcimimetic, cinacalcet (167,170–172). However the responses are variable and patients often still require parathyroidectomy (167).
Nephrocalcinosis is very common on conventional therapy. At baseline, prior to randomization, nephrocalcinosis was present in 23% of the children and 54% of the adults recruited into the recent burosumab clinical trials (98,162). Some studies have found associations of nephrocalcinosis with higher doses of conventional therapy (165,166), while others did not (173). However, these studies also indicated that episodes of hypercalciuria may be associated with nephrocalcinosis risk in XLH. Thus, monitoring urine calcium excretion is important. The long-term consequences of nephrocalcinosis in XLH are uncertain regarding renal function, though CKD is reported in about 8–9 % of patients with XLH (167,174), and end stage renal disease has been reported (167). Renal ultrasounds are recommended every 1–2 years during treatment of XLH with either conventional therapy or burosumab (159).
Thiazides have been used to decrease urinary calcium excretion in XLH patients with hypercalciuria or nephrocalcinosis. In 11 children with XLH on therapy with calcitriol and phosphate, adding the thiazide diuretic, hydrochlorothiazide decreased urinary calcium excretion and while nephrocalcinosis did not resolve, further progression was prevented (175). In another series, the use of thiazides resulted in resolution of nephrocalcinosis in two patients with XLH (176).
Treatment of XLH is required in children to allow for growth and adequate bone mineralization (156), and outcomes are improved when initiated in infancy as opposed to later in childhood (177). However, despite treatment, and even with good adherence, many children have suboptimal growth, and persistent leg deformities with need for surgical correction (177). In particular, Zivicnjak et al. highlight the growth deficits that worsen during puberty even during conventional therapy (178). Therapy has often been stopped at the end of growth in an attempt to balance risks versus benefits of ongoing therapy. During this time period many XLH patients are lost to follow-up until a time when symptoms lead to seeking additional care. However, in adult patients, therapy is typically restarted or continued in symptomatic adults having bone pain or fractures due to osteomalacia. ADHR and ARHR, like XLH, are also treated with oral phosphate and active vitamin D analogs. However, emerging evidence suggests ADHR could be treated with oral iron (41,179) instead of with phosphate and vitamin D, though this approach would be ineffective for XLH (78).
Several studies have evaluated the use of recombinant human growth hormone (GH) in XLH. Uncontrolled studies noted improvement in linear growth in children with short stature and XLH (180–182). Two years of treatment with growth hormone (GH) improved height SDS, with a better response in prepubertal compared to pubertal children (181). GH monotherapy also improved the serum phosphate and 1,25(OH)2D while normalizing PTH in a study of 10 children with XLH (180). A randomized controlled trial in children with XLH and short stature suggested benefit of adding GH to conventional therapy as linear growth significantly improved, although mean height SDS did not differ compared to controls at 3 years (183). However, when these same subjects were followed to final adult height, there was no difference in height between GH-treated patients and controls with XLH, although the sample size was small (184). Another important finding of the controlled study was that GH did not appear to worsen the body disproportion that is seen in XLH.
Recent advances in XLH therapy include regulatory approval of burosumab, an anti-FGF23 antibody, as monotherapy by the Food and Drug Administration and European Medicines Agency. Anti-FGF23 antibodies corrected hypophosphatemia and improved rickets and bone length in Hyp mice (185). Burosumab (previously termed KRN23) is a human anti-FGF23 monoclonal antibody and has been shown to significantly increase serum phosphorus, TmP/GFR, and 1,25(OH)2D in adults and children (186–189). The biochemical pattern after injections results in peak and trough effects over a 4-week dosing cycle in adults, with peak 1,25(OH)2D about 3–7 days after injection and peak phosphorus about 7 days after injection (186). In an adult randomized controlled trial, 134 adults randomized to burosumab every 4 weeks for 24 weeks, demonstrated clear improvements in serum phosphorus versus placebo (98). In this trial the burosumab group demonstrated greater healing of fractures/pseudofractures (43.1% vs 7.7%) during this time period, and improved stiffness scores.
Open label dose-finding phase 2 clinical trials also demonstrated improvements in phosphorus, alkaline phosphatase and rickets severity in 52 children ages 5–12 years (188) and in 13 children ages 1–4 years (189). Children age 5–12 years demonstrated improvements in physical function as well (188). Modest improvements in height Z-score (+0.15 ± 0.04) were noted (188).
Only one randomized controlled trial has directly compared conventional therapy to burosumab. This phase 3 open-label randomized controlled trial was conducted in 61 children ages 1–12 years with XLH (162). Children who had persistent rickets despite a mean of 3.3–4.3 years of prior conventional therapy were randomized to switch to burosumab (0.8 mg/kg every 2 weeks) or continue conventional therapy (oral phosphate 20–60 mg/kg/day, and calcitriol 20–30 ng/kg/day or alfacalcidiol 40–60 ng/kg/day, titrated based on clinical parameters). By the end of the study most burosumab patients were still receiving 0.8 mg/kg burosumab, though some increased to 1.2 mg/kg, while the control group’s mean phosphate dose was 46 mg/kg/day, calcitriol 27 mg/kg/day and alfacalcidiol 86.5 ng/kg/day (162) Clinical improvements in rickets were seen in both groups as rated by radiologists blinded to treatment group using a radiographic global impression of change scale (190), where negative scores indicated worsening, 0 indicated no change +1 minimal healing +2 substantial healing and + 3 complete healing. This study demonstrated superior improvements with burosumab. At the primary outcome of 40 weeks (72.4% of those in the burosumab group achieved substantial healing of rickets by RGI-C of ≥+2 versus only 6.3% in the conventional therapy group). At 64 weeks the mean RGI-C score after burosumab was +2.1 compared to a compared to +1 in the conventional therapy arm. Other statistically significant improvements were seen in serum phosphorus, TmP/GFR, alkaline phosphatase, linear growth, and mobility in the burosumab group compared to the conventional therapy group.
These trials also show a favorable safety profile, with the most common side effects being transient injection site reactions (186–188). There were no signals of increased risk for nephrocalcinosis. However, some subjects in the adult burosumab trial did require dose reductions due to hyperphosphatemia. Consequently, monitoring serum phosphorus remains important to avoid hyperphosphatemia, which could carry a risk of nephrocalcinosis or other ectopic calcifications. Tooth abscesses were numerically higher in the burosumab group for the controlled trials. It is not clear what the long-term impact of burosumab on tooth abscesses, nephrocalcinosis or hyperparathyroidism will be.
Burosumab was approved as monotherapy with dosing in adults of 1 mg/kg every 4 weeks subcutaneously and in children 0.8 to 1.2 mg/kg every 2 weeks, with a maximum dose of 90 mg. Serum phosphorus is targeted within the low-normal range at trough, and we would recommend avoiding high or high-normal values anywhere in the dose cycle. However, clinical monitoring for safety remains important, including monitoring phosphorus, calcium, creatinine, PTH, urine calcium excretion, and renal ultrasounds. Monitoring for efficacy includes measures of ALP and radiographic imaging to monitor rachitic changes and lower limb deformities, or to monitor healing of pseudofractures. It is as yet unknown what the impact of burosumab will be on the need for corrective leg surgeries, final adult height, enthesopathy, or other long-term XLH complications.
Children in the randomized controlled trial were those that had persistent evidence of significant rickets despite prior conventional therapy. Out of 122 screened, 55 (45%) were ineligible due to lesser rickets severity, consistent with the known benefits of conventional therapy. Persistent rickets in these patients could be due to prior compliance or dosing or inherent underlying resistance of their disease to therapy (191,192). Compliance is challenging for patients with multiple daily dosing of medications and patients often find conventional therapy burdensome (192). Furthermore, compliant patients are highly variable in the response to conventional therapy, with some recovering completely and some persisting with severe deformities. These concerns also highlight the difficulties and challenges managing patients with conventional therapy (191). Thus, patients who are responding well to conventional therapy may continue to do well on conventional therapy, while patients with persistent rickets clearly benefited from switching to burosumab.
One commentary raised a concern that patients on higher doses of conventional therapy might have responded better, acknowledging that higher doses of phosphate might also lead to elevated PTH levels which can also contribute to phosphaturia (191). The recent guideline has recommended a somewhat higher dose range for conventional therapy, as cited above (159). A subanalysis of the responses by either pre-trial or on-trial dose range has not been conducted, but patients in the upper quartile of dosing during the trial were within ranges similar to these new guidelines, and in some individuals much higher. However, it is not clear that the very large magnitude of differences in rickets responses in this randomized controlled trial can be explained solely by differences in the conventional therapy dose (at week 40: 72% burosumab vs 6 % conventional therapy having substantial healing or greater with RGI-C of ≥+2; and at week 64: 87% vs 19%).
While a Hyp mouse study comparing a different FGF23 antibody to enormous doses of calcitriol monotherapy found greater improvements in bone parameters with calcitriol (193,194), we would recommend caution in this comparison. That mouse study used doses of calcitriol that were several fold higher than the highest doses recommended in humans even from recent guidelines (159), raising concern for risks of hypercalciuria and nephrocalcinosis. However, a clinical trial is underway (ClinicalTrials.gov Identifier: ) which will provide useful information on calcitriol monotherapy but does not include comparison with other treatment regimens.
One proposed approach, given the expense of burosumab is to initiate patients on conventional therapy and advance to burosumab if inadequate skeletal outcomes are seen (191,192). However, there may also be benefit to initiating burosumab early in severely affected patients (191), while the randomized controlled trial would indicate that patients who have been on conventional therapy for years with insufficient improvement are less likely to improve further remaining on conventional therapy (162). Those with pseudofractures, especially while on conventional therapy are also likely to benefit from burosumab (98). As with conventional therapy, decisions regarding burosumab treatment for children and adults should be individualized, taking into account the risks and benefits of therapy, and both approaches require careful monitoring.
Corrective Surgery
Conventional therapy improves limb deformities in most patients, though not necessarily with complete correction. The benefits of therapy on skeletal deformities are likely greatest when therapy is started early (177). Children do frequently still require orthopedic surgery to correct long bone deformities to straighten the lower limbs. Such procedures include osteotomies with internal fixation or external fixation to address bowing and torsional abnormalities, and guided growth procedures using plate across the medial or lateral physis (of the distal femur for example) to limit growth on that side of the physis, while allowing the opposite side to grow, to straighten the deformity (159,195). The timing of surgery is variable. One retrospective study found that patients having initial surgery at younger ages had more total surgeries than those having their first surgery later (195). Though this may have been confounded by severity of the initial deformity, it remains an important consideration and supports that osteotomies may be better performed at later ages when growth is complete or near complete. However, guided growth procedures must be completed while the patient still has at least 2 years of growth remaining in order for the desired effect (159). The optimal timing of surgery must be individualized based on several factors including the severity of the deformity and its functional impact on the developing child, which may indicate earlier surgery. Prior to elective skeletal surgery, medical therapy should be optimized. There is also a risk of overcorrection of a deformity after surgery, or of recurrence of the original deformity, thus patients require continued monitoring. Continued medical therapy is important to promote bone healing after surgery and with a goal of decreasing risk for new additional deformities and optimizing growth. It should be noted that whether burosumab will alter the need for corrective surgery or not has not yet been established in clinical trials.
Conclusion
FGF23 functions as an endocrine factor with its cofactor αKlotho and is a principal regulator in phosphorus homeostasis, its primary action being reduction of phosphorus levels and regulation of vitamin D metabolism. It is one of multiple factors involved in the bone-parathyroid-kidney axis and its interaction with these factors is quite complex. FGF23-mediated disorders of phosphate wasting share similar clinical and biochemical features. Conventional treatment involves multiple daily doses of oral phosphate salts and active vitamin D analogs. Recent development of an anti-FGF23 antibody, burosumab, for use in XLH has shown promising results, but more research is needed, especially regarding long-term outcomes.
Disclosure
This work was supported by NIH grants T32DK065549 and by P30AR072581. EAI has received research funding from Ultragenyx Pharmaceuticals and participated in advisory boards.
References
- 1.Itoh N, Ohta H, Konishi M. Endocrine FGFs: evolution, physiology, pathophysiology, and pharmacotherapy. Frontiers in endocrinology 2015;6:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdisciplinary Reviews: Developmental Biology 2015;4(3):215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991;64(4):841–848 [DOI] [PubMed] [Google Scholar]
- 4.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Molecular and cellular biology 2007;27(9):3417–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Quarles LD. How fibroblast growth factor 23 works. Journal of the American Society of Nephrology 2007;18(6):1637–1647 [DOI] [PubMed] [Google Scholar]
- 6.Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, Fujita T, Kuroki R, Yamashita T, Fukumoto S. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. Journal of Bone and Mineral Research 2008;23(9):1509–1518 [DOI] [PubMed] [Google Scholar]
- 7.Meyer RA Jr, Meyer MH, Gray RW. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 1989;4(4):493–500 [DOI] [PubMed] [Google Scholar]
- 8.White KE, Evans WE, O’Riordan JL, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature genetics 2000;26(3):345. [DOI] [PubMed] [Google Scholar]
- 9.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006;444(7120):770. [DOI] [PubMed] [Google Scholar]
- 10.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW. Regulation of fibroblast growth factor-23 signaling by klotho. Journal of Biological Chemistry 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. The Journal of clinical investigation 2007;117(12):4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiological reviews 2012;92(1):131–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erben RG, Andrukhova O. FGF23-Klotho signaling axis in the kidney. Bone 2017;100:62–68 [DOI] [PubMed] [Google Scholar]
- 14.Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1α, 25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. American Journal of Physiology-Gastrointestinal and Liver Physiology 2005;289(6):G1036–G1042 [DOI] [PubMed] [Google Scholar]
- 15.Nishi H, Nii-Kono T, Nakanishi S, Yamazaki Y, Yamashita T, Fukumoto S, Ikeda K, Fujimori A, Fukagawa M. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism. Nephron Clinical Practice 2005;101(2):c94–c99 [DOI] [PubMed] [Google Scholar]
- 16.Georgiadou E, Marketou H, Trovas G, Dontas I, Papaioannou N, Makris K, Galanos A, Papavassiliou AG. Effect of calcitriol on FGF23 level in healthy adults and its dependence on phosphate level. in vivo 2017;31(1):145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito H, Maeda A, Ohtomo SI, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto KI, Fukushima N. Circulating FGF-23 is regulated by 1α, 25-dihydroxyvitamin D3 and phosphorus in vivo. Journal of Biological Chemistry 2005;280(4):2543–2549 [DOI] [PubMed] [Google Scholar]
- 18.Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate-and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone 2005;36(6):971–977 [DOI] [PubMed] [Google Scholar]
- 19.Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ. Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. The Journal of Clinical Endocrinology & Metabolism 2010;95(4):1846–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins MT, Lindsay JR, Jain A, Kelly MH, Cutler CM, Weinstein LS, Liu J, Fedarko NS, Winer KK. Fibroblast Growth Factor-23 Is Regulated by 1α, 25-Dihydroxyvitamin D. Journal of bone and mineral research 2005;20(11):1944–1950 [DOI] [PubMed] [Google Scholar]
- 21.Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. The Journal of Clinical Endocrinology & Metabolism 2005;90(3):1519–1524 [DOI] [PubMed] [Google Scholar]
- 22.Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. The Journal of Clinical Endocrinology & Metabolism 2006;91(8):3144–3149 [DOI] [PubMed] [Google Scholar]
- 23.Vervloet MG, van Ittersum FJ, Büttler RM, Heijboer AC, Blankenstein MA, ter Wee PM. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clinical Journal of the American Society of Nephrology 2011;6(2):383–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burnett SAM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. Journal of Bone and Mineral Research 2006;21(8):1187–1196 [DOI] [PubMed] [Google Scholar]
- 25.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. Journal of the American Society of Nephrology 2007;18(10):2683–2688 [DOI] [PubMed] [Google Scholar]
- 26.Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, Naveh-Many T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney international 2014;86(6):1106–1115 [DOI] [PubMed] [Google Scholar]
- 27.López I, Rodríguez-Ortiz ME, Almadén Y, Guerrero F, De Oca AM, Pineda C, Shalhoub V, Rodríguez M, Aguilera-Tejero E. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney international 2011;80(5):475–482 [DOI] [PubMed] [Google Scholar]
- 28.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. American Journal of Physiology-Renal Physiology 2010;299(4):F882–F889 [DOI] [PubMed] [Google Scholar]
- 29.Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, Bellido T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011;49(4):636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutiérrez OM, Smith KT, Barchi-Chung A, Patel NM, Isakova T, Wolf M. (1–34) Parathyroid hormone infusion acutely lowers fibroblast growth factor 23 concentrations in adult volunteers. Clinical Journal of the American Society of Nephrology 2012;7(1):139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burnett-Bowie SAM, Henao MP, Dere ME, Lee H, Leder BZ. Effects of hPTH (1–34) infusion on circulating serum phosphate, 1, 25-dihydroxyvitamin D, and FGF23 levels in healthy men. Journal of bone and mineral research 2009;24(10):1681–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Ortiz ME, Lopez I, Muñoz-Castañeda JR, Martinez-Moreno JM, Ramírez AP, Pineda C, Canalejo A, Jaeger P, Aguilera-Tejero E, Rodriguez M. Calcium deficiency reduces circulating levels of FGF23. Journal of the American Society of Nephrology 2012:ASN. 2011101006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium regulates FGF-23 expression in bone. Endocrinology 2013;154(12):4469–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta A, Winer K, Econs M, Marx S, Collins M. FGF-23 is elevated by chronic hyperphosphatemia. The Journal of Clinical Endocrinology & Metabolism 2004;89(9):4489–4492 [DOI] [PubMed] [Google Scholar]
- 35.Benet-Pagès A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004;35(2):455–462 [DOI] [PubMed] [Google Scholar]
- 36.Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. Journal of Bone and Mineral Research 2007;22(4):520–526 [DOI] [PubMed] [Google Scholar]
- 37.Kato K, Jeanneau C, Tarp MA, Benet-Pagès A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNActransferase T3 and familial tumoral calcinosis Secretion of fibroblast growth factor 23 requires O-glycosylation. Journal of Biological Chemistry 2006;281(27):18370–18377 [DOI] [PubMed] [Google Scholar]
- 38.Bergwitz C, Banerjee S, Abu-Zahra H, Kaji H, Miyauchi A, Sugimoto T, Jüppner H. Defective O-glycosylation due to a novel homozygous S129P mutation is associated with lack of fibroblast growth factor 23 secretion and tumoral calcinosis. The Journal of Clinical Endocrinology & Metabolism 2009;94(11):4267–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Appaiah HN, Koller A, Nizet V, White KE, Dixon JE. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proceedings of the National Academy of Sciences 2014:201402218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, Robling AG, Stayrook KR, Jideonwo V, Magers MJ. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proceedings of the National Academy of Sciences 2011:201110905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. The Journal of Clinical Endocrinology & Metabolism 2011;96(11):3541–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. American Journal of Physiology-Endocrinology and Metabolism 2006;291(1):E38–E49 [DOI] [PubMed] [Google Scholar]
- 43.Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD. Pathogenic role of Fgf23 in Dmp1-null mice. American Journal of Physiology-Endocrinology and Metabolism 2008;295(2):E254–E261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu X, White KE. Fibroblast growth factor 23 and its receptors. Therapeutic Apheresis and Dialysis 2005;9(4):308–312 [DOI] [PubMed] [Google Scholar]
- 45.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. Journal of Bone and Mineral Research 2004;19(3):429–435 [DOI] [PubMed] [Google Scholar]
- 46.Baum M, Schiavi S, Dwarakanath V, Quigley R. Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney international 2005;68(3):1148–1153 [DOI] [PubMed] [Google Scholar]
- 47.Perwad F, Zhang MY, Tenenhouse HS, Portale AA. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1α-hydroxylase expression in vitro. American Journal of Physiology-Renal Physiology 2007;293(5):F1577–F1583 [DOI] [PubMed] [Google Scholar]
- 48.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proceedings of the National Academy of Sciences 2001;98(11):6500–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflügers Archiv 2004;447(5):763–767 [DOI] [PubMed] [Google Scholar]
- 50.Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochemical and biophysical research communications 2004;314(2):409–414 [DOI] [PubMed] [Google Scholar]
- 51.Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren Os, Tenenhouse HS, Jüppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1 (I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004;145(7):3087–3094 [DOI] [PubMed] [Google Scholar]
- 52.Kempson SA, Lotscher M, Kaissling B, Biber J, Murer H, Levi M. Parathyroid hormone action on phosphate transporter mRNA and protein in rat renal proximal tubules. American Journal of Physiology-Renal Physiology 1995;268(4):F784–F791 [DOI] [PubMed] [Google Scholar]
- 53.Chanakul A, Zhang MY, Louw A, Armbrecht HJ, Miller WL, Portale AA, Perwad F. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PloS one 2013;8(9):e72816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. The Journal of clinical investigation 2004;113(4):561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krajisnik T, Björklund P, Marsell R, Ljunggren Ö, Åkerström G, Jonsson KB, Westin G, Larsson TE. Fibroblast growth factor-23 regulates parathyroid hormone and 1α-hydroxylase expression in cultured bovine parathyroid cells. Journal of Endocrinology 2007;195(1):125–131 [DOI] [PubMed] [Google Scholar]
- 56.Mace ML, Gravesen E, Nordholm A, Olgaard K, Lewin E. Fibroblast Growth Factor (FGF) 23 Regulates the Plasma Levels of Parathyroid Hormone In Vivo Through the FGF Receptor in Normocalcemia, But Not in Hypocalcemia. Calcified tissue international 2018;102(1):85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. Journal of Bone and Mineral Research 2011;26(7):1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren Ö. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. New England Journal of Medicine 2003;348(17):1656–1663 [DOI] [PubMed] [Google Scholar]
- 59.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. The Journal of Clinical Endocrinology & Metabolism 2002;87(11):4957–4960 [DOI] [PubMed] [Google Scholar]
- 60.El-Maouche D, Dumitrescu C, Andreopoulou P, Gafni R, Brillante B, Bhattacharyya N, Fedarko NS, Collins M. Stability and degradation of fibroblast growth factor 23 (FGF23): the effect of time and temperature and assay type. Osteoporosis International 2016;27(7):2345–2353 [DOI] [PubMed] [Google Scholar]
- 61.Ali FN, Josefson J, Mendez AJ, Mestan K, Wolf M. Cord blood ferritin and fibroblast growth factor-23 levels in neonates. The Journal of Clinical Endocrinology & Metabolism 2016;101(4):1673–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holmlund-Suila E, Viljakainen H, Ljunggren Ö, Hytinantti T, Andersson S, Mäkitie O. Fibroblast growth factor 23 concentrations reflect sex differences in mineral metabolism and growth in early infancy. Hormone research in paediatrics 2016;85(4):232–241 [DOI] [PubMed] [Google Scholar]
- 63.Holmlund-Suila E, Enlund-Cerullo M, Valkama S, Hauta-alus H, Rosendahl J, Helve O, Hytinantti T, Viljakainen H, Andersson S, Mäkitie O. Sex and Iron Modify Fibroblast Growth Factor 23 Concentration in 1-Year-Old Children. The Journal of Clinical Endocrinology & Metabolism 2017;102(12):4526–4533 [DOI] [PubMed] [Google Scholar]
- 64.Fischer DC, Mischek A, Wolf S, Rahn A, Salweski B, Kundt G, Haffner D. Paediatric reference values for the C-terminal fragment of fibroblast-growth factor-23, sclerostin, bone-specific alkaline phosphatase and isoform 5b of tartrate-resistant acid phosphatase. Annals of clinical biochemistry 2012;49(6):546–553 [DOI] [PubMed] [Google Scholar]
- 65.Gkentzi D, Efthymiadou A, Kritikou D, Chrysis D. Fibroblast growth factor 23 and Klotho serum levels in healthy children. Bone 2014;66:8–14 [DOI] [PubMed] [Google Scholar]
- 66.Smith ER, Cai MM, McMahon LP, Holt SG. Biological variability of plasma intact and C-terminal FGF23 measurements. The Journal of Clinical Endocrinology & Metabolism 2012;97(9):3357–3365 [DOI] [PubMed] [Google Scholar]
- 67.Imel EA, Hui SL, Ecibs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2007;22(4):520–526 [DOI] [PubMed] [Google Scholar]
- 68.Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, Minagawa M, Sugimoto T, Yamauchi M, Michigami T. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone 2008;42(6):1235–1239 [DOI] [PubMed] [Google Scholar]
- 69.Imel EA, Econs MJ. Approach to the hypophosphatemic patient. The Journal of Clinical Endocrinology & Metabolism 2012;97(3):696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tiosano D, Hochberg Ze. Hypophosphatemia: the common denominator of all rickets. Journal of bone and mineral metabolism 2009;27(4):392–401 [DOI] [PubMed] [Google Scholar]
- 71.Murali SK, Roschger P, Zeitz U, Klaushofer K, Andrukhova O, Erben RG. FGF23 Regulates Bone Mineralization in a 1, 25 (OH) 2D3 and Klotho-Independent Manner. Journal of Bone and Mineral Research 2016;31(1):129–142 [DOI] [PubMed] [Google Scholar]
- 72.Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp mice. PLoS biology 2016;14(4):e1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma U, Pal D, Prasad R. Alkaline phosphatase: an overview. Indian journal of clinical biochemistry: IJCB 2014;29(3):269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishikawa Y, Valhmu WB, Wuthier RE. Induction of alkaline phosphatase in primary cultures of epiphyseal growth plate chondrocytes by a serum-derived factor. Journal of cellular physiology 1987;133(2):344–350 [DOI] [PubMed] [Google Scholar]
- 75.White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney international 2001;60(6):2079–2086 [DOI] [PubMed] [Google Scholar]
- 76.Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. The Journal of Clinical Endocrinology & Metabolism 1997;82(2):674–681 [DOI] [PubMed] [Google Scholar]
- 77.Sun Y, Wang O, Xia W, Jiang Y, Li M, Xing X, Hu Y, Liu H, Meng X, Zhou X. FGF23 analysis of a Chinese family with autosomal dominant hypophosphatemic rickets. Journal of bone and mineral metabolism 2012;30(1):78–84 [DOI] [PubMed] [Google Scholar]
- 78.Imel EA, Gray AK, Padgett LR, Econs MJ. Iron and fibroblast growth factor 23 in X-linked hypophosphatemia. Bone 2014;60:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. Journal of Bone and Mineral Research 2013;28(8):1793–1803 [DOI] [PubMed] [Google Scholar]
- 80.Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. The Journal of Clinical Endocrinology & Metabolism 2009;94(7):2332–2337 [DOI] [PubMed] [Google Scholar]
- 81.Schaefer B, Würtinger P, Finkenstedt A, Braithwaite V, Viveiros A, Effenberger M, Sulzbacher I, Moschen A, Griesmacher A, Tilg H. Choice of high-dose intravenous iron preparation determines hypophosphatemia risk. PLoS One 2016;11(12):e0167146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Francis F, Hennig S, Korn B, Reinhardt R, De Jong P, Poustka A, Lehrach H, Rowe P, Goulding J, Summerfield T. A gene (PEX) with homologies to endopeptidases is mutated in patients with X–linked hypophosphatemic rickets. Nature genetics 1995;11(2):130. [DOI] [PubMed] [Google Scholar]
- 83.Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, Tenenhouse HS. Pex/PEX tissue distribution and evidence for a deletion in the 3’region of the Pex gene in X-linked hypophosphatemic mice. The Journal of clinical investigation 1997;99(6):1200–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pavone V, Testa G, Iachino SG, Evola FR, Avondo S, Sessa G. Hypophosphatemic rickets: etiology, clinical features and treatment. European Journal of Orthopaedic Surgery & Traumatology 2015;25(2):221–226 [DOI] [PubMed] [Google Scholar]
- 85.Econs MJ, Feussner JR, Samsa GP, Effman EL, Vogler JB, Martinez S, Friedman NE, Quarles LD, Drezner MK. X-linked hypophosphatemic rickets without “rickets”. Skeletal radiology 1991;20(2):109–114 [DOI] [PubMed] [Google Scholar]
- 86.Marie PJ, Glorieux FH. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D-resistant rickets. Calcified tissue international 1983;35(1):443–448 [DOI] [PubMed] [Google Scholar]
- 87.Chesher D, Oddy M, Darbar U, Sayal P, Casey A, Ryan A, Sechi A, Simister C, Waters A, Wedatilake Y, Lachmann RH, Murphy E. Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations. J Inherit Metab Dis 2018;41(5):865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Connor J, Olear EA, Insogna KL, Katz L, Baker S, Kaur R, Simpson CA, Sterpka J, Dubrow R, Zhang JH, Carpenter TO. Conventional Therapy in Adults With X-Linked Hypophosphatemia: Effects on Enthesopathy and Dental Disease. J Clin Endocrinol Metab 2015;100(10):3625–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, Briot K, Linglart A, Chaussain C. Phosphate and Vitamin D Prevent Periodontitis in X-Linked Hypophosphatemia. J Dent Res 2017;96(4):388–395 [DOI] [PubMed] [Google Scholar]
- 90.Onishi T, Umemura S, Shintani S, Ooshima T. Phex mutation causes overexpression of FGF23 in teeth. Arch Oral Biol 2008;53(2):99–104 [DOI] [PubMed] [Google Scholar]
- 91.Boukpessi T, Hoac B, Coyac BR, Leger T, Garcia C, Wicart P, Whyte MP, Glorieux FH, Linglart A, Chaussain C, McKee MD. Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone 2017;95:151–161 [DOI] [PubMed] [Google Scholar]
- 92.Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabedian M. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr 2003;142(3):324–331 [DOI] [PubMed] [Google Scholar]
- 93.Fong H, Chu EY, Tompkins KA, Foster BL, Sitara D, Lanske B, Somerman MJ. Aberrant cementum phenotype associated with the hypophosphatemic hyp mouse. J Periodontol 2009;80(8):1348–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hardy D, Murphy W, Siegel B, Reid I, Whyte M. X-linked hypophosphatemia in adults: prevalence of skeletal radiographic and scintigraphic features. Radiology 1989;171(2):403–414 [DOI] [PubMed] [Google Scholar]
- 95.Polisson RP, Martinez S, Khoury M, Harrell RM, Lyles KW, Friedman N, Harrelson JM, Reisner E, Drezner MK. Calcification of entheses associated with X-linked hypophosphatemic osteomalacia. New England Journal of Medicine 1985;313(1):1–6 [DOI] [PubMed] [Google Scholar]
- 96.Liang G, Katz LD, Insogna KL, Carpenter TO, Macica CM. Survey of the enthesopathy of X-linked hypophosphatemia and its characterization in Hyp mice. Calcified tissue international 2009;85(3):235–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mills ES, Iorio L, Feinn RS, Duignan KM, Macica CM. Joint replacement in X-linked hypophosphatemia. Journal of orthopaedics 2019;16(1):55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Insogna KL, Briot K, Imel EA, Kamenický P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2018;33(8):1383–1393 [DOI] [PubMed] [Google Scholar]
- 99.Beck-Nielsen SS, Brusgaard K, Rasmussen LM, Brixen K, Brock-Jacobsen B, Poulsen MR, Vestergaard P, Ralston SH, Albagha OM, Poulsen S, Haubek D, Gjorup H, Hintze H, Andersen MG, Heickendorff L, Hjelmborg J, Gram J. Phenotype presentation of hypophosphatemic rickets in adults. Calcified tissue international 2010;87(2):108–119 [DOI] [PubMed] [Google Scholar]
- 100.Reid IR, Hardy DC, Murphy WA, Teitelbaum SL, Bergfeld MA, Whyte MP. X-linked hypophosphatemia: a clinical, biochemical, and histopathologic assessment of morbidity in adults. Medicine (Baltimore) 1989;68(6):336–352 [PubMed] [Google Scholar]
- 101.Rothenbuhler A, Fadel N, Debza Y, Bacchetta J, Diallo MT, Adamsbaum C, Linglart A, Di Rocco F. High Incidence of Cranial Synostosis and Chiari I Malformation in Children With X-Linked Hypophosphatemic Rickets (XLHR). Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2019;34(3):490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Econs MJ, Samsa GP, Monger M, Drezner MK, Feussner JR. X-Linked Hypophosphatemic Rickets - a Disease Often Unknown to Affected Patients. Bone and mineral 1994;24(1):17–24 [DOI] [PubMed] [Google Scholar]
- 103.Davies M, Kane R, Valentine J. Impaired hearing in X-linked hypophosphataemic (vitamin-D-resistant) osteomalacia. Ann Intern Med 1984;100(2):230–232 [DOI] [PubMed] [Google Scholar]
- 104.Fishman G, Miller-Hansen D, Jacobsen C, Singhal VK, Alon US. Hearing impairment in familial X-linked hypophosphatemic rickets. European journal of pediatrics 2004;163(10):622–623 [DOI] [PubMed] [Google Scholar]
- 105.Wick CC, Lin SJ, Yu H, Megerian CA, Zheng QY. Treatment of ear and bone disease in the Phex mouse mutant with dietary supplementation. American journal of otolaryngology 2017;38(1):44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nature genetics 2006;38(11):1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lorenz-Depiereux B, Bastepe M, Benet-Pagès A, Amyere M, Wagenstaller J, Müller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nature genetics 2006;38(11):1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. The American Journal of Human Genetics 2010;86(2):267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruf N, Uhlenberg B, Terkeltaub R, Nürnberg P, Rutsch F. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Human mutation 2005;25(1):98. [DOI] [PubMed] [Google Scholar]
- 110.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D. Mutations in ENPP1 are associated with’idiopathic’infantile arterial calcification. Nature genetics 2003;34(4):379. [DOI] [PubMed] [Google Scholar]
- 111.Rutsch F, Böyer P, Nitschke Y, Ruf N, Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ, Mughal Z, Gregory JW. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circulation: Genomic and Precision Medicine 2008;1(2):133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. The American Journal of Human Genetics 2010;86(2):273–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rafaelsen SH, Ræder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23–related hypophosphatemia, dental anomalies, and ectopic calcification. Journal of Bone and Mineral Research 2013;28(6):1378–1385 [DOI] [PubMed] [Google Scholar]
- 114.Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, Fukumoto S, Fujitab T. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49(4):913–916 [DOI] [PubMed] [Google Scholar]
- 115.Mehta P, Mitchell A, Tysoe C, Caswell R, Owens M, Vincent T. Novel compound heterozygous mutations in ENPP1 cause hypophosphataemic rickets with anterior spinal ligament ossification. Rheumatology 2012;51(10):1919–1921 [DOI] [PubMed] [Google Scholar]
- 116.Turan S, Aydin C, Bereket A, Akcay T, Güran T, Yaralioglu BA, Bastepe M, Jüppner H. Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia. Bone 2010;46(2):402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mäkitie O, Pereira RC, Kaitila I, Turan S, Bastepe M, Laine T, Kröger H, Cole WG, Jüppner H. Long-term clinical outcome and carrier phenotype in autosomal recessive hypophosphatemia caused by a novel DMP1 mutation. Journal of Bone and Mineral Research 2010;25(10):2165–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Koshida R, Yamaguchi H, Yamasaki K, Tsuchimochi W, Yonekawa T, Nakazato M. A novel nonsense mutation in the DMP1 gene in a Japanese family with autosomal recessive hypophosphatemic rickets. Journal of bone and mineral metabolism 2010;28(5):585–590 [DOI] [PubMed] [Google Scholar]
- 119.Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, Feuillan P, Leet AI, Kushner H, Robey PG. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. Journal of bone and mineral research 2007;22(9):1468–1474 [DOI] [PubMed] [Google Scholar]
- 120.Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, Wientroub S, Bianco P, Robey PG. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. Journal of Bone and Mineral Research 2001;16(5):806–813 [DOI] [PubMed] [Google Scholar]
- 121.Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet journal of rare diseases 2008;3(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune–Albright syndrome. New England Journal of Medicine 1991;325(24):1688–1695 [DOI] [PubMed] [Google Scholar]
- 123.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. The Journal of clinical investigation 2003;112(5):683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Imel EA, Econs MJ. Fibrous dysplasia, phosphate wasting and fibroblast growth factor 23. PER 2007;4:434–439 [PubMed] [Google Scholar]
- 125.Kobayashi K, Imanishi Y, Koshiyama H, Miyauchi A, Wakasa K, Kawata T, Goto H, Ohashi H, Koyano HM, Mochizuki R. Expression of FGF23 is correlated with serum phosphate level in isolated fibrous dysplasia. Life sciences 2006;78(20):2295–2301 [DOI] [PubMed] [Google Scholar]
- 126.Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, Gafni RI, Cherman N, Cho M, Hager GL. Mechanism of FGF23 processing in fibrous dysplasia. Journal of bone and mineral research 2012;27(5):1132–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Drezner MK. Tumor-induced osteomalacia. Reviews in Endocrine and Metabolic disorders 2001;2(2):175–186 [DOI] [PubMed] [Google Scholar]
- 128.White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren Os. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. The Journal of Clinical Endocrinology & Metabolism 2001;86(2):497–500 [DOI] [PubMed] [Google Scholar]
- 129.Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, De Beur SMJ, Schiavi SC, Kumar R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. The Journal of clinical investigation 2003;112(5):785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. The Journal of Clinical Endocrinology & Metabolism 2005;90(2):1012–1020 [DOI] [PubMed] [Google Scholar]
- 131.Rowe PS, De Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 2000;67(1):54–68 [DOI] [PubMed] [Google Scholar]
- 132.Lee JC, Jeng YM, Su SY, Wu CT, Tsai KS, Lee CH, Lin CY, Carter JM, Huang JW, Chen SH. Identification of a novel FN1–FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. The Journal of pathology 2015;235(4):539–545 [DOI] [PubMed] [Google Scholar]
- 133.Jiang Y, Xia Wb, Xing Xp, Silva BC, Li M, Wang O, Zhang Hb, Li F, Jing Hl, Zhong. Tumor-induced osteomalacia: An important cause of adult-onset hypophosphatemic osteomalacia in China: Report of 39 cases and review of the literature. Journal of Bone and Mineral Research 2012;27(9):1967–1975 [DOI] [PubMed] [Google Scholar]
- 134.Chong WH, Yavuz S, Patel SM, Chen CC, Collins MT. The importance of whole body imaging in tumor-induced osteomalacia. The Journal of Clinical Endocrinology & Metabolism 2011;96(12):3599–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Andreopoulou P, Dumitrescu CE, Kelly MH, Brillante BA, Cutler Peck CM, Wodajo FM, Chang R, Collins MT. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. Journal of Bone and Mineral Research 2011;26(6):1295–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nasu T, Kurisu S, Matsuno S, Tatsumi K, Kakimoto T, Kobayashi M, Nakano Y, Wakasaki H, Furuta H, Nishi M. Tumor-induced hypophosphatemic osteomalacia diagnosed by the combinatory procedures of magnetic resonance imaging and venous sampling for FGF23. Internal Medicine 2008;47(10):957–961 [DOI] [PubMed] [Google Scholar]
- 137.Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, Miyamoto Y, Okazaki H, Nakamura K, Nakahara K. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. The Journal of Clinical Endocrinology & Metabolism 2004;89(8):3979–3982 [DOI] [PubMed] [Google Scholar]
- 138.Westerberg PA, Olauson H, Toss G, Wikström B, Morales O, Linde T, Jonsson K, Ljunggren Ö, Larsson T. Preoperative tumor localization by means of venous sampling for fibroblast growth factor-23 in a patient with tumor-induced osteomalacia. Endocrine Practice 2008;14(3):362–367 [DOI] [PubMed] [Google Scholar]
- 139.Sun Zj, Jin J, Qiu Gx, Gao P, Liu Y. Surgical treatment of tumor-induced osteomalacia: a retrospective review of 40 cases with extremity tumors. BMC musculoskeletal disorders 2015;16(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zuo Qy, Wang H, Li W, Niu Xh, Huang Yh, Chen J, You Yh, Liu By, Cui Am, Deng W. Treatment and outcomes of tumor-induced osteomalacia associated with phosphaturic mesenchymal tumors: retrospective review of 12 patients. BMC musculoskeletal disorders 2017;18(1):403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Grebe TA, Rimsza ME, Richter SF, Hansen RC, Hoyme HE. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. American journal of medical genetics 1993;47(1):24–30 [DOI] [PubMed] [Google Scholar]
- 142.Menascu S, Donner EJ. Linear nevus sebaceous syndrome: case reports and review of the literature. Pediatric neurology 2008;38(3):207–210 [DOI] [PubMed] [Google Scholar]
- 143.Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, Langmann T, Singer S, Klingseisen L, Schneider-Brachert W. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nature genetics 2012;44(7):783. [DOI] [PubMed] [Google Scholar]
- 144.Lim YH, Ovejero D, Sugarman JS, DeKlotz CM, Maruri A, Eichenfield LF, Kelley PK, Jüppner H, Gottschalk M, Tifft CJ. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Human molecular genetics 2013;23(2):397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hoffman WH, Jueppner HW, DeYoung BR, O’Dorisio MS, Given KS. Elevated fibroblast growth factor-23 in hypophosphatemic linear nevus sebaceous syndrome. American Journal of Medical Genetics Part A 2005;134(3):233–236 [DOI] [PubMed] [Google Scholar]
- 146.Skovby F, Svejgaard E, Møller J. Hypophosphatemic rickets in linear sebaceous nevus sequence. The Journal of pediatrics 1987;111(6):855–857 [DOI] [PubMed] [Google Scholar]
- 147.Sethi SK, Hari P, Bagga A. Elevated FGF-23 and parathormone in linear nevus sebaceous syndrome with resistant rickets. Pediatric Nephrology 2010;25(8):1577–1578 [DOI] [PubMed] [Google Scholar]
- 148.Heike CL, Cunningham ML, Steiner RD, Wenkert D, Hornung RL, Gruss JS, Gannon FH, McAlister WH, Mumm S, Whyte MP. Skeletal changes in epidermal nevus syndrome: does focal bone disease harbor clues concerning pathogenesis? American Journal of Medical Genetics Part A 2005;139(2):67–77 [DOI] [PubMed] [Google Scholar]
- 149.Ovejero D, Lim Y, Boyce A, Gafni R, McCarthy E, Nguyen T, Eichenfield L, DeKlotz C, Guthrie L, Tosi LL. Cutaneous skeletal hypophosphatemia syndrome: clinical spectrum, natural history, and treatment. Osteoporosis International 2016;27(12):3615–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jüppner H Jansen’s metaphyseal chondrodysplasia: a disorder due to a PTH/PTHrP receptor gene mutation. Trends in Endocrinology & Metabolism 1996;7(5):157–162 [DOI] [PubMed] [Google Scholar]
- 151.Schipani E, Langman C, Parfitt A, Jensen G, Kikuchi S, Kooh S, Cole W, Jüppner H. Constitutively activated receptors for parathyroid hormone and parathyroid hormone–related peptide in Jansen’s metaphyseal chondrodysplasia. New England Journal of Medicine 1996;335(10):708–714 [DOI] [PubMed] [Google Scholar]
- 152.Brown WW, Jüppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. The Journal of Clinical Endocrinology & Metabolism 2009;94(1):17–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kuthiroly S, Yesodharan D, Ghosh A, White KE, Nampoothiri S. Osteoglophonic Dysplasia: Phenotypic and Radiological Clues. Journal of pediatric genetics 2017;6(04):247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brooks SS, Kassner G, Qazi Q, Keogh M, Gorlin R. Osteoglophonic dysplasia: review and further delineation of the syndrome. American journal of medical genetics 1996;66(2):154–162 [DOI] [PubMed] [Google Scholar]
- 155.White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. The American Journal of Human Genetics 2005;76(2):361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prié D, Rothenbuhler A. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocrine connections 2014;3(1):R13–R30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Penido MG, Alon US. Hypophosphatemic rickets due to perturbations in renal tubular function. Pediatric nephrology (Berlin, Germany) 2014;29(3):361–373 [DOI] [PubMed] [Google Scholar]
- 158.Drezner MK, Lyles KW, Haussler MR, Harrelson JM. Evaluation of a role for 1,25-dihydroxyvitamin D3 in the pathogenesis and treatment of X-linked hypophosphatemic rickets and osteomalacia. J Clin Invest 1980;66(5):1020–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nature reviews Nephrology 2019;15(7):435–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nature reviews Nephrology 2019;15(7):435–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wilson DM, Alon U. Renal Hypophosphatemia In: Alon U, Chan JC, editors. Phosphate in Pediatric Health and Disease: CRC Press; 1993; pp. 160–192 [Google Scholar]
- 162.Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, Simmons JH, Padidela R, Namba N, Cheong HI, Pitukcheewanont P, Sochett E, Hogler W, Muroya K, Tanaka H, Gottesman GS, Biggin A, Perwad F, Mao M, Chen CY, Skrinar A, San Martin J, Portale AA. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet (London, England) 2019;393(10189):2416–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M. Effects of therapy in X-linked hypophosphatemic rickets. New England Journal of Medicine 1991;325(26):1843–1848 [DOI] [PubMed] [Google Scholar]
- 164.Makitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol (Oxf) 2003;58(2):163–168 [DOI] [PubMed] [Google Scholar]
- 165.Goodyer PR, Kronick JB, Jequier S, Reade TM, Scriver CR. Nephrocalcinosis and its relationship to treatment of hereditary rickets. J Pediatr 1987;111(5):700–704 [DOI] [PubMed] [Google Scholar]
- 166.Reusz GS, Hoyer PF, Lucas M, Krohn HP, Ehrich JH, Brodehl J. X linked hypophosphataemia: treatment, height gain, and nephrocalcinosis. Archives of disease in childhood 1990;65(10):1125–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.DeLacey S, Liu Z, Broyles A, El-Azab SA, Guandique CF, James BC, Imel EA. Hyperparathyroidism and parathyroidectomy in X-linked hypophosphatemia patients. Bone 2019;127:386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clinical journal of the American Society of Nephrology. CJASN 2008;3(3):658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Alon US, Jarka D, Monachino PJ, Sebestyen VanSickle J, Srivastava T. Cinacalcet as an alternative to phosphate therapy in X-linked hypophosphataemic rickets. Clin Endocrinol (Oxf) 2017;87(1):114–116 [DOI] [PubMed] [Google Scholar]
- 170.Grove-Laugesen D, Rejnmark L. Three-year successful cinacalcet treatment of secondary hyperparathyroidism in a patient with x-linked dominant hypophosphatemic rickets: a case report. Case reports in endocrinology 2014;2014:479641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yavropoulou MP, Kotsa K, Gotzamani Psarrakou A, Papazisi A, Tranga T, Ventis S, Yovos JG. Cinacalcet in hyperparathyroidism secondary to X-linked hypophosphatemic rickets: case report and brief literature review. Hormones (Athens, Greece) 2010;9(3):274–278 [DOI] [PubMed] [Google Scholar]
- 172.Raeder H, Shaw N, Netelenbos C, Bjerknes R. A case of X-linked hypophosphatemic rickets: complications and the therapeutic use of cinacalcet. Eur J Endocrinol 2008;159(Suppl 1):S101–S105 [DOI] [PubMed] [Google Scholar]
- 173.Kooh SW, Binet A, Daneman A. Nephrocalcinosis in X-linked hypophosphataemic rickets: its relationship to treatment, kidney function, and growth. Clin Invest Med 1994;17(2):123–130 [PubMed] [Google Scholar]
- 174.Nakamura Y, Takagi M, Takeda R, Miyai K, Hasegawa Y. Hypertension is a characteristic complication of X-linked hypophosphatemia. Endocr J 2017;64(3):283–289 [DOI] [PubMed] [Google Scholar]
- 175.Seikaly MG, Baum M. Thiazide diuretics arrest the progression of nephrocalcinosis in children with X-linked hypophosphatemia. Pediatrics 2001;108(1):E6. [DOI] [PubMed] [Google Scholar]
- 176.Auron A, Alon US. Resolution of medullary nephrocalcinosis in children with metabolic bone disorders. Pediatric nephrology (Berlin, Germany) 2005;20(8):1143–1145 [DOI] [PubMed] [Google Scholar]
- 177.Makitie O, Doria A, Kooh S, Cole W, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. The Journal of Clinical Endocrinology & Metabolism 2003;88(8):3591–3597 [DOI] [PubMed] [Google Scholar]
- 178.Zivicnjak M, Schnabel D, Billing H, Staude H, Filler G, Querfeld U, Schumacher M, Pyper A, Schroder C, Bramswig J, Haffner D, Hypophosphatemic Rickets Study Group of Arbeitsgemeinschaft fur Padiatrische E, Gesellschaft fur Padiatrische N. Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 2011;26(2):223–231 [DOI] [PubMed] [Google Scholar]
- 179.Imel Erik A., Liu Z, Coffman M, Acton D, Econs MJ. Oral Iron Therapy Normalizes Fibroblast Growth Factor 23 (FGF23) in Patients with Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res 2018;33(S1):S56(LB-1170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Makitie O, Toiviainen-Salo S, Marttinen E, Kaitila I, Sochett E, Sipila I. Metabolic control and growth during exclusive growth hormone treatment in X-linked hypophosphatemic rickets. Hormone research 2008;69(4):212–220 [DOI] [PubMed] [Google Scholar]
- 181.Rothenbuhler A, Esterle L, Gueorguieva I, Salles JP, Mignot B, Colle M, Linglart A. Two-year recombinant human growth hormone (rhGH) treatment is more effective in pre-pubertal compared to pubertal short children with X-linked hypophosphatemic rickets (XLHR). Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society 2017;36:11–15 [DOI] [PubMed] [Google Scholar]
- 182.Seikaly MG, Brown R, Baum M. The effect of recombinant human growth hormone in children with X-linked hypophosphatemia. Pediatrics 1997;100(5):879–884 [DOI] [PubMed] [Google Scholar]
- 183.Zivicnjak M, Schnabel D, Staude H, Even G, Marx M, Beetz R, Holder M, Billing H, Fischer DC, Rabl W, Schumacher M, Hiort O, Haffner D. Three-year growth hormone treatment in short children with X-linked hypophosphatemic rickets: effects on linear growth and body disproportion. The Journal of clinical endocrinology and metabolism 2011;96(12):E2097–E2105 [DOI] [PubMed] [Google Scholar]
- 184.Meyerhoff N, Haffner D, Staude H, Wuhl E, Marx M, Beetz R, Querfeld U, Holder M, Billing H, Rabl W, Schroder C, Hiort O, Bramswig JH, Richter-Unruh A, Schnabel D, Zivicnjak M, Hypophosphatemic Rickets Study Group of the “Deutsche Gesellschaft fur Kinderendokrinologie und d, Gesellschaft fur Padiatrische N. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol 2018;33(3):447–456 [DOI] [PubMed] [Google Scholar]
- 185.Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. Journal of Bone and Mineral Research 2009;24(11):1879–1888 [DOI] [PubMed] [Google Scholar]
- 186.Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. The Journal of Clinical Endocrinology & Metabolism 2015;100(7):2565–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. The Journal of clinical investigation 2014;124(4):1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, Padidela R, van’t Hoff W, Mao M, Chen CY. burosumab Therapy in Children with X-linked Hypophosphatemia. New England Journal of Medicine 2018;378(21):1987–1998 [DOI] [PubMed] [Google Scholar]
- 189.Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, Imel EA. Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. The Lancet Diabetes & Endocrinology 2019;7(3):189–199 [DOI] [PubMed] [Google Scholar]
- 190.Whyte MP, Fujita KP, Moseley S, Thompson DD, McAlister WH. Validation of a Novel Scoring System for Changes in Skeletal Manifestations of Hypophosphatasia in Newborns, Infants, and Children: The Radiographic Global Impression of Change Scale. J Bone Miner Res 2018 [DOI] [PubMed] [Google Scholar]
- 191.Gordon RJ, Levine MA. Burosumab treatment of children with X-linked hypophosphataemic rickets. The Lancet 2019;393(10189):2364–2366 [DOI] [PubMed] [Google Scholar]
- 192.Collins M Burosumab: At Long Last, an Effective Treatment for FGF23-Associated Hypophosphatemia. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2018;33(8):1381–1382 [DOI] [PubMed] [Google Scholar]
- 193.Liu ES, Martins JS, Raimann A, Chae BT, Brooks DJ, Jorgetti V, Bouxsein ML, Demay MB. 1,25-Dihydroxyvitamin D Alone Improves Skeletal Growth, Microarchitecture, and Strength in a Murine Model of XLH, Despite Enhanced FGF23 Expression. J Bone Miner Res 2016;31(5):929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ovejero D, Gafni RI, Collins MT. 1,25-Dihydroxyvitamin D as Monotherapy for XLH: Back to the Future? Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2016;31(5):925–928 [DOI] [PubMed] [Google Scholar]
- 195.Gizard A, Rothenbuhler A, Pejin Z, Finidori G, Glorion C, de Billy B, Linglart A, Wicart P. Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets (XLHR). Endocr Connect 2017;6(8):566–573 [DOI] [PMC free article] [PubMed] [Google Scholar]