

Abstract
Early-life exposure to arsenic (As) increases risks of respiratory diseases/infections in children. However, data on the ability of the innate immune system to combat bacterial infections in the respiratory tracts of As-exposed children is scarce. To evaluate whether persistent low-dose As exposure alters innate immune function among children younger than 5 yr-of-age, mothers and participating children (N = 51) that were members of the Health Effects of Arsenic Longitudinal Study (HEALS) cohort in rural Bangladesh were recruited. Household water As, past and concurrent maternal urinary As (U-As) as well as child U-As were all measured at enrollment. In addition, U-As metabolites were evaluated. Innate immune function was examined via measures of cathelicidin LL-37 in plasma, ex vivo monocyte-derived-macrophage (MDM)-mediated killing of Streptococcus pneumoniae (Spn), and serum bactericidal antibody (SBA) responses against Haemophilus influenzae type b (Hib). Cyto/chemokines produced by isolated peripheral blood mononuclear cells (PBMC) were assayed using a Multiplex system. Multivariable linear regression analyses revealed that maternal (p < 0.01) and child (p = 0.02) U-As were positively associated with plasma LL-37 levels. Decreased MDM-mediated Spn killing (p = 0.05) and SBA responses (p = 0.02) were seen to be each associated with fractions of mono-methylarsonic acid (MMA; a U-As metabolite) in the children. In addition, U-As levels were seen to be negatively associated with PBMC formation of fractalkine and IL-7, and positively associated with that for IL-13, IL-17 and MIP-1α. These findings suggested that early-life As exposure may disrupt the innate host defense pathway in these children. It is possible that such disruptions may have health consequences later in life.
Keywords: Arsenic, serum bactericidal antibody response, macrophage function, LL-37, respiratory pathogens
Introduction
Epidemiological studies have shown increased risk of morbidity in children due to chronic and early life arsenic (As) exposure, particularly related to respiratory tract infections (RTI) and diarrheal diseases (Raqib et al. 2009; Rahman et al. 2017; Sanchez et al. 2016). Emerging evidence indicates that As has multiple harmful effects on immune regulation and its surveillance system which potentially amplify susceptibility to many infectious diseases as well as other immune-related health outcomes (Dangleben et al. 2013; Ferrario et al. 2016). Arsenic can act as immuno-stimulant to cause an elevation/perpetuation of inflammatory responses harmful to the host. The immunosuppressive effects of As include modulation of the numbers, functions and survival of immune and hematopoietic cells, and impaired humoral and cell-mediated immunity (Dangleben et al. 2013; Ferrario et al. 2016). The majority of these findings are based on in vitro and experimental studies. Limited information is available on As-induced innate immune modulation in humans, particularly in children.
Macrophages are crucial regulators of innate immunity that bridge the innate and adaptive immune systems. Experimental and in vitro studies have shown that toxic effects of As on macrophages include the hindering of differentiation of monocytes into macrophages (Lemarie et al. 2006), as well as antigen-presenting capacity (Sikorski et al 1991), bactericidal (Bishayi et al. 2003) and phagocytic abilities (Sengupta et al. 2002) of macrophages. However, studies of macrophage function in humans exposed to As during early life are lacking. Evolutionarily conserved host defense peptides (HDP) are considered to be effectors of innate immunity and are secreted by various cells types including macrophages. There are two major classes in mammals, i.e., the defensins and the cathelicidins. Humans have only one cathelicidin, LL-37 that has broad spectrum antibacterial, chemotactic and immunomodulatory properties (van Harten et al. 2018). Beta-defensins and LL-37 play major roles in host responses against pulmonary pathogens related to bronchiectasis, recurrent airways infections, and in the pathogenesis of chronic obstructive pulmonary disease (COPD) (Dangleben et al. 2013; Persson et al 2017). Until now, only a single study reported an association between As exposure and beta-defensin-1 in humans (Hegedus et al. 2008).
Serum bactericidal antibody (SBA) are a major arm of the overall innate immune system. SBA responses measure functional antibody formation against various bacterial pathogens and are used to evaluate immunogenicity of bacterial vaccines as a correlate of protection (Jang et al. 2016; Shimanovich et al. 2017). In Bangladeshi patients with As-induced skin lesions, complement-mediated SBA and serum concentration of Complement 3 (C3) were found to be significantly low as compared to healthy unexposed controls (Islam et al. 2012). Higher expression of inflammatory cytokines have been found in individuals chronically-exposed to As (Ahmed et al. 2014; Dutta et al. 2015), although formation of T-cell cytokines was suppressed (Ahmed et al. 2014; Biswas et al. 2008; Martin-Chouly et al. 2011).
Against this backdrop, it was hypothesized that chronic As exposure modulates the induction of innate immune responses in young children. The study reported here determined the effect of early-life exposures to As on innate immunity in rural Bangladeshi children ≤ 5 yr-of-age who had been repeatedly exposed to As in their drinking water and food. The evaluations performed encompassed analyses of levels of As metabolites in urine, serum LL-37, as well as ex vivo measures of monocyte-derived-macrophage (MDM)-mediated Streptococcus pneumoniae (Spn) killing, serum bactericidal antibody (SBA) responses against Haemophilus influenzae type b (Hib), and cytokines production by isolated peripheral blood mononuclear cells (PBMC). Individuals who excrete relatively higher proportions of MMA and lower proportions of DMA exhibit a higher propensity for As related adverse health outcomes (Vahter et al. 2002) which may be a consequence of altered immune function.
Materials and Methods
Study area and subjects
Mothers of the participating children were members of the Health Effects of Arsenic Longitudinal Study (HEALS; established in 2000) cohort in Araihazar, Bangladesh. At baseline, the HEALS participants were chronically-exposed to a wide range of As levels in their drinking water (Chen et al. 2009), though exposure has declined dramatically since then (Huhmann et al. 2019). The purpose of the HEALS was to investigate health outcomes associated with As exposure through drinking water. All participants were followed-up for health assessments and personal interviews every 2–3 years via home visits by trained study personnel. Well water for each household was tested for As at baseline and during follow-up visits. Blood and urine samples were also collected from the participants using protocols outlined in Chen et al. (2009) and Huhmann et al. (2019).
The current study included 51 healthy children (≤ 5 yr-of-age) and their mothers who were already participants in the HEALS cohort. A total of 60 children were randomly identified from the HEALS central database. Written informed consent was obtained from all mothers of the participating children. Of the original 60, 3 were above the desired age range, 2 were ill, and 2 were unwilling to donate blood. In addition, 2 blood samples were discarded due to insufficient volume or inadequate numbers of isolatable PBMC. Body weight (in light clothes and barefoot) was measured to the nearest 0.1 kg (digital scale) and height was measured (stadiometer). These parameters were converted to height-for-age, weight-for-age, and body mass index-for-age Z-scores (SD scores), using the WHO growth reference for school-aged children and adolescents (de Onis et al. 2007). This study was approved by the Bangladesh Medical Research Council (BMRC); Columbia University also provided the ethical clearance for the study.
Water and urine sample collection and arsenic assessment
Household water samples that had been collected in trace element-free polyethylene bottles were analyzed by high-resolution inductively-coupled plasma mass spectrometry (HR ICP-MS) as previously described (van Geen et al. 2007). The detection limit of the method was 0.1 μg/L; the standard deviation of a single measurement was conservatively estimated at 4 μg/L.
Spot urine samples were collected in 50-ml acid-washed tubes and stored at −80°C until shipped to Columbia University on dry ice. Urinary As analysis were performed by a graphite furnace atomic-absorption (GFAA) using a Perkin-Elmer Analyst 600 graphite furnace system as described earlier (Nixon et al. 1991). The detection limit for U-As was 2 μg/L. Urinary creatinine was analyzed by a colorimetric method based on the Jaffe reaction. To compensate for variations in hydration status, total U-As concentration was adjusted with creatinine and expressed as μg As/g creatinine (μg/g). Maternal U-As collected 2–4 yr prior was considered as past exposure and referred to as maternal U-Aspast and U-As during enrolment in the study as maternal U-Asconcurrent.
Inorganic As (iAs) is metabolized by a series of reduction and methylation reactions producing mono-methylarsonic acid (MMA) and di-methylarsinic acid (DMA). . Urinary As metabolites in samples were assayed by ICP-MS-DRC coupled to a high performance liquid chromatography (HPLC) system as previously described (Van Geen et al 2002). ICP-MS-DRC was used as a detector for As metabolites that had been chromato-graphically separated over a PRP-X100 Anion Exchange Column (Hamilton, Reno, Nevada, US) using 10 mM ammonium nitrate/ammonium phosphate solution (pH 9.1) as the mobile phase. The system allowed detection of inorganic As (i.e. As+3 and As+5), total MMA, and total DMA. Extent of As methylation efficiency was assessed by calculating relative amounts (%) of the metabolites in the children’s urine; total arsenic was calculated based on the concentration of the sum of iAs, MMA, and DMA in the urine.
Blood samples
Peripheral blood samples (5 ml) from children were collected in Li-Heparin-coated tubes (SARSTEDT, Nümbrecht, Germany), and were stored/transferred in cool boxes within 2–3 hr of collection to the Laboratory of the International Centre for Diarrheal Disease, Bangladesh (icddr,b) in Dhaka. Plasma and PBMC were separated from whole blood using Ficoll-Paque Plus density gradient centrifugation (GE Healthcare, Sigma, St. Louis, MO). Isolated plasma was stored in an ultralow freezer (−80°C) for later use in LL-37 analyses and bactericidal activity assessments. Isolated PBMC were cultured for use in evaluations of monocyte-derived-macro-phage (MDM) killing activities (see below).
LL-37 measurement
Human cathelicidin host defense peptide LL-37 acts as a first line of defense effector molecule in the innate immune system. Concentrations of LL-37 in the isolated plasma samples was measured by ELISA (Hycult Biotechnology, Uden, Netherlands), according to manufacturer protocols. The lower limit of detection of the kit was 0 14 ng/ml; the intra- and inter-assay CV was 4.96 and 7.89%, respectively. All samples were measured in duplicates.
Monocyte-derived-macrophage (MDM) culture
Bacteriolytic activity against a respiratory pathogen by MDM cultured from As-exposed children was described in Gordon et al. (Gordon et al. 2000). In the present study, Gram-positive Streptococcus pneumoniae (Spn), a major cause of community-acquired pneumonia and meningitis in children (DeAntonio et al. 2016), was the test pathogen. In brief, isolated PBMC were washed and suspended in enriched culture medium (RPMI-1640 supplemented with 10% autologous plasma, 1% L-glutamine, 1% sodium pyruvate, 1% penicillin-streptomycin) (all Gibco, Grand Island, NY). Thereafter, cells (5 × 106 cells/well) were plated into 4-well cell culture plates (NUNC, Roskilde, Denmark). After 2 hr incubation in 37°C in a 5% CO2 incubator, the supernatants containing non-adherent cells (mostly lymphocytes) were removed (see below for cytokine analysis), leaving the adherent cells (mostly monocytes) attached to the plastic surface. These cells were cultured for 7 d, and then treated (dose in well = 5 μg/ml) with bacterial lipopolysaccharide (LPS; from Escherichia coli 011:B4, Sigma). Other matched cells received vehicle only (no stimulant). After 48 hr of incubation, the MDM were then washed and incubated with enriched RPMI medium containing an additional 10% (w/v) bovine serum albumin (BSA, Sigma) for 30 min at 37°C. The supernatant in the well was removed and the MDM washed again with enriched media prior to use in the protocol below.
MDM-mediated killing assay
Type 1 Streptococcus pneumoniae (Spn) (ATCC 49616; Manassas, VA) was grown to mid-log phase in brain heart infusion (BHI) broth with 20% fetal calf serum (Thermo Fisher Scientific, Waltham, MA). A suspension of Spn was prepared to a concentration of 5 × 107 colony-forming units (CFU) at an absorbance of 0.6 at 600 nm and stored at −80°C for later use. For the assay, the bacterial cells were pelleted by centrifugation (10,000 rpm, 5 min), washed 3 times with RPMI, re-pelleted, and then opsonized by re-suspension in 10% autologous/pooled plasma or 10% phosphate-buffered saline (PBS, pH 7.4; control) and culturing in for 30 min at 37°C at 120 rpm, before being washed again. To evaluate killing activity, the MDM in the wells described above were infected with Spn at a multiplicity of infection of 100 (100 bacteria/one macrophage) and the plates were then incubated for 1 hr at 4°C. At that point, extracellular fluid (ECF) containing non-ingested bacteria was collected. The now-infected MDM were washed 3 times with warm RPMI and then further incubated in media with 10% autologous plasma for 20 min at 37°C. After this, the infected macrophages were lysed by addition of 2% saponin in RPMI to the wells to cause the release of all viable intracellular bacteria. Intracellular fluid (ICF) containing bacteria in the cell lysates in each well was collected with vigorous aspiration and then centrifuged 5 min at 10,000 rpm. Both ECF and ICF were cultured on chocolate agar plates overnight at 37°C and under 5% CO2; colonies (CFU) of the Spn were then counted.
The number of adhered Spn (bound to macrophage surface/ingested by cells) was calculated by subtracting the number of viable Spn in ECF from the number of Spn inoculated/well. The percentage of killing of internalized bacteria by MDM (% killing capacity) was determined as 100% x ([number adhered/ingested bacteria - number viable bacteria in ICF]/number of adhered/ingested bacteria). The ‘relative CFU count’ was calculated for each participant, i.e., ratio of CFU found with ICF of LPS-stimulated cells compared to that in ICF of unstimulated cells, to account for inter-subject variations in the baseline (unstimulated) killing activity of MDM (Gordon et al. 2000).
Serum bactericidal antibody (SBA) response
The SBA assay is an important in vitro method for measuring complement-mediated functionality of natural/vaccine-induced antibodies targeting pathogens (Ercoli et al. 2015). Gram-negative Haemophilus influenzae type b (Hib) was selected for evaluation of acquired SBA responses as all the study children were vaccinated during infancy with Hib through the Expanded Program on Immunization (EPI) in Bangladesh. Each serum sample was incubated at 56°C for 30 min to inactivate complement before being used in the assay. The exogenous complement source used in the assay was sera from healthy young rabbits, aged 4–6 weeks (Animal Resource Facility, icddr,b, Dhaka, Bangladesh).
From chocolate agar plates, 20–30 colonies of Hib type B (ATCC 49247) that had been grown at 37°C after receipt, were inoculated into Brain Heart Infusion (BHI) broth supplemented with hemin (10 μg/ml) and NAD (10 μg/ml) (both Sigma), and incubated for 2 hr at 37°C. Bacteria in mid-log phase were then harvested by centrifugation (12,000 x g, 4°C) and washed twice with PBS. The final pellet was suspended in Hanks Buffer Salt Solution (HBSS) containing CaCl2 and MgCl2 with 2% BHI. The optical density of the suspension was adjusted to 0.41 (at 600 nm) which corresponded to 2.5 × 108 CFU/ml. The bacterial solution was then opsonized with rabbit complement. After washing, the bacteria were prepared for introduction (at 103 CFU/well) into sera and diluent media-containing wells of MaxiSorp flat bottom microtiter plates (Sigma) kept on ice. The plates were incubated for 75 min at 37°C in a 5% CO2 incubator and then the absorbance in each well was taken at 595 nm. Pooled sera was used as a positive control. As other controls, bacteria without sera, and sera plus complement without bacteria, were used.
Cytokines and chemokines in non-adherent cell culture supernatant
A subset of cell samples (n = 16) was assayed for cytokine production. The non-adherent cells (mostly lymphocytes) originally separated from the PBMC adherent cells were plated into 96-well plates at 1×106 cells/well and then treated with 5 μg phytohemagglutinin/ml (PHA; Sigma) or cultured medium only (control). The cells were then cultured for 48 hr at 37°C in a 5% CO2 incubator. Thereafter, culture supernatants were collected for assessment of cytokines and chemokines secreted by diverse cell types such as monocytes, dendritic cells, neutrophils, NK cells as well as Th1 and Th2 cells (Fractalkine, GM-CSF, IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 (p70), IL-13, IL-17A, IL-21, IL-23, CXCL11, MIP1α, MIP1β, MIP-3α, TNFα) using a MILLIPLEX® MAP 384-Well High Sensitivity Human Cytokine Magnetic Bead Panel (MeRCK, Darmstadt, Germany). Cytokine data was expressed as the ratio of PHA-stimulated levels to control levels.
Statistical analyses
Statistical Package for Social Science (SPSS) for Windows (v.20; IBM, Armonk, NY) and Stata/IC (v.13, Stata Corp., Houston, TX) were used for data analysis. Demographic characteristic of the study participants was calculated based on child age, sex, body mass index (BMI), z-scores, and mother’s age and BMI. Urinary arsenic, LL-37, MDM killing was log transformed since data did not follow the Gaussian distribution. Associations between exposures, outcomes, and covariates were initially evaluated using Spearman’s rank correlation coefficient (for continuous variables), Mann-Whitney U test, analysis of variance, or Kruskal-Wallis test (for categorical variables), as appropriate.
To evaluate As effects on participant LL-37, cytokines, MDM killing capacity, and SBA, multivariate linear regression was applied. The association between %MMA U-As and water As and outcomes (LL-37, SBA and MDM killing) was not linear. To evaluate potential effects of % MMA U-As and water arsenic on LL-37, SBA and MDM killing, the multivariate-adjusted regression analysis was restricted to the lowest tertile of As metabolites (reference) and comparisons made with the second or third tertiles. The associations between %MMA and LL-37, MDM killing and SBA were measured by using multivariate linear regression. All the regression models were adjusted for covariates that were significantly associated with both exposure and outcomes or changed the effect estimate by 5% or more. These covariates were age, sex, weight for age z-score and mother’s BMI. A p-value < 0.05 was considered significant.
Results
Study subjects
The mean age of the children was 4.15 [± 0.54] years, with an almost equal number of boys and girls (26 and 25, respectively). The children were mildly-malnourished (Table 1). Compared to the World Health Organization (WHO) reference standard, about 24% (n = 12) and 30% (n = 15) children were underweight and stunted, respectively. All children were examined by a physician and found to be healthy without any recent history of illness during enrolment. The average age of the mothers at time of enrollment was 24 years. The median BMI of the mothers was 20.2 (15.9, 32.3) kg/m2, with 27% being undernourished and 16% overweight/obese. About 10% of the mothers completed at least 5 years of school and about 65% had high school level education; 14% were illiterate.
Table 1.
Demographic and exposure characteristics of study children and mothers (N = 51).
| Children | |
|---|---|
| Age, years | 4.15±0.54 |
| BMI, kg/m2 | 14.39±1.89 |
| WAZ | −1.32 (−1.98, −0.84)) |
| HAZ | −1.11(−2.32, −0.35) |
| BAZ | −0.73(−1.29, −0.19) |
| Underweight, n (%) | 12 (23.5%) |
| Stunted, n (%) | 15 (29.4%) |
| Mothers | |
| BMI, kg/m2 | 20.88±3.36 |
| Age, years | 23.67±5.68 |
| Mothers’ education, n (%) | |
| Illiterate | 7 (13.73%) |
| Primary | 5 (9.80%) |
| High school | 33 (64.71%) |
| Graduate | 6 (11.76%) |
| Arsenic concentrations | |
| Water-As, μg/L, median | 43.0 (5.0, 66.0) |
| All children, μg/g, median | 39.0 (16.0, 102.0) |
| Boys U-As, μg/g | 37.0 (24.0, 111.0) |
| Girls U-As, μg/g | 45.0 (16.0, 80.0) |
| Mother U-Asconcurrent μg/g | 114.3 (44.0, 141.5) |
| Mother U-Aspast μg/g | 197.1 (81.9, 299.5) |
Note. BMI body mass index; WAZ, weight-for-age Z-scores; HAZ, height-for-age Z-scores; BAZ, body-mass-index-for-age Z-scores; U-As, urinary arsenic; HHW-As, household water arsenic.
Data are given as mean± standard deviation or in numbers with percentage in brackets; data for anthropometric indices and urinary arsenic are given as median with inter-quartile range within brackets.
Relationship of arsenic concentration in water and urine
The median household W-As concentration (43.0 μg/L) found was below the Bangladesh maximum contaminant level of 50 μg/L; the range varied from 0.6 – 326 μg/L. About 55% (n = 28) of the household had W-As levels were ≤ 50 μg/L; 45% (n = 23) were > 50 μg/L. Household W-As concentration was strongly associated with maternal U-Asconcurrent (r = 0.51, p = 0.0001), U-Aspast (r = 0.56, p = 0.000), and child U-As (r = 0.37, p = 0.006), indicating drinking water was the major source of As of the mothers and to a lesser extent the children.
Maternal U-Aspast was as high as 197 μg/g (median) and declined to a median concentration of 114.3 μg/g at the time of the present study (p < 0.001). In a majority of the mothers (79%), concurrent U-As concentrations decreased while in about 1/5th (21%) of the mothers the concentration increased. Child U-As concentration (median with interquartile range, 39 (16, 102) μg/g) was significantly lower than maternal U-Asconcurrent (p = 0.01) and U-Aspast (p < 0.001) which reflected considerably reduced extent of As exposure among children under 5 yr-of-age.
Association of arsenic exposure with cathelicidin LL-37 in plasma
The mean plasma concentration of LL-37 in children was 25.6 [± 14.2] ng/ml. Multivariable adjusted linear regression analysis showed that both maternal U-Asconcurrent (β coefficient = 0.20; 95% confidence interval CI = 0.03, 0.37; p = 0.02) and U-Aspast (β = 0.27; 95% CI = 0.06, 0.49; p = 0.01) and child U-As (β = 0.17; 95% CI = 0.02, 0.32; p = 0.02) were positively associated with child plasma LL-37. In stratified analysis by sex, height and weight, the association was found to be stronger among boys, particularly in children with normal weight and height (Table 2). The household W-As of children was also positively associated with plasma LL-37.
Table 2.
Multivariable adjusted linear regression analysis of the association between urinary/water As and plasma LL-37, stratified by sex and anthropometric Z-score.
| LL-37 (ng/ml) | All childrena (n = 50) |
Boysb (n = 24) |
Girlsb (n = 26) |
|---|---|---|---|
| Child U-As (μg/g) | **0.17 (0.02, 0.32) | **0.21 (0.04, 0.39) | 0.06 (−0.22, 0.35) |
| Mother U-Asconcurrent |
*0.20 (0.03, 0.37) | 0.20 (−0.05, 0.44) | 0.17 (−0.12, 0.47) |
|
1Mother U-Aspast |
**0.27 (0.06, 0.49) | †0.25 (−0.05, 0.54) | 0.27 (−0.09, 0.63) |
| Water As (μg/L) | †0.11 (−0.01, 0.22) | *0.16 (0.001, 0.32) | 0.04 (−0.14, 0.22) |
| LL-37 (ng/ml) | Normal weightc (n = 39) |
Under weightc (n = 12) |
Normal heightc (n = 36) |
Stuntingc (n = 15) |
|---|---|---|---|---|
| Child U-As (μg/g) | **0.22 (0.06, 0.38) | 0.03 (−0.45, 0.50) | **0.21 (0.04, 0.37) | 0.01 (−0.49, 0.51) |
| Mother U-Asconcurrent |
**0.21 (0.03, 0.39) | 0.01 (−0.82, 0.84) | †0.20 (−0.01, 0.41) | 0.0001 (−0.50, 0.50) |
|
1Mother U-Aspast |
**0.26 (0.02, 0.51) | 0.11 (−0.76, 0.98) | **0.28 (0.02, 0.54) | 0.25 (−0.37, 0.87) |
| Water As (μg/L) | †0.11 (−0.02, 0.24) | 0.08 (−0.15, 0.31) | 0.10 (−0.05, 0.24) | †0.25 (−0.04, 0.54) |
Note. Data presented as beta (β) coefficient and 95% confidence interval (CI). U-As, urinary arsenic; HAZ, height-for-age Z-score; WAZ, weight-for-age Z-score.
U-As (past), collected 2–4 years prior.
Models adjusted for child age, sex, WAZ and mothers’ BMI.
Adjusted by child age, sex, WAZ and Mother BMI;
Adjusted by child age, WAZ and Mother BMI.
Adjusted for child age, sex and Mother BMI.
p < 0.05,
p < 0.005,
p ≤ 0.08
To evaluate the influence of child As metabolites in urine (iAs, MMA, or DMA) with plasma LL-37 levels, each metabolite was categorized into tertiles. Compared to the lowest tertile (reference) of %MMA, plasma LL-37 concentrations in the second tertile increased significantly mainly in girls (p= 0.05) (Figure 1A). This suggested a possible non-monotonic relationship between As and %MMA.
Figure 1.

Association of tertiles of %MMA with (A) plasma LL-37, (B) MDM killing, and (C) Serum Bactericidal antibody (SBA) response, stratified by sex. “p” was determined by multivariate adjusted regression analysis and the model was adjusted by child age, sex, WAZ and mother BMI.
MMA, mono-methylarsonic acid; MDM, monocyte-derived macrophage; SBA, serum bactericidal antibody response; WAZ, weight for age z-score; and, BMI, body mass index
Monocyte-derived macrophage (MDM) mediated killing of Spn
No association was found between MDM-mediated killing of Spn and As exposure (W-As, maternal U-As and child U-As). There was no influence from nutritional status (stunting or underweight) or sex in the association between As exposure and MDM-mediated killing. However, when the association by child %MMA was stratified in tertiles, MDM-mediated Spn killing capacity was significantly reduced (p=0.05) in the highest fraction of MMA compared to the lowest (Figure 1B).
Serum bactericidal antibody (SBA) response against Hib
In the multivariable adjusted linear regression analysis, a significant positive association was obtained between child SBA response and household W-As (p = 0.04) and SBA in boys with maternal U-Asconcurrent (p = 0.02). No association was found with current exposure of children (Figure 2). However, a positive association between W-As and SBA was pronounced among boys and in children with normal weight. When the associations between As metabolites and SBA were stratified by child %MMA in tertiles, SBA response in the second tertile of %MMA decreased considerably compared to the first tertile (β =- 26.05, 95% CI = −48.83, −3.26; p = 0.02) (Figure 1C) and the impact was stronger in the girls (β = −44.60, 95% CI = −85.34, −3.86; p = 0.03).
Figure 2.
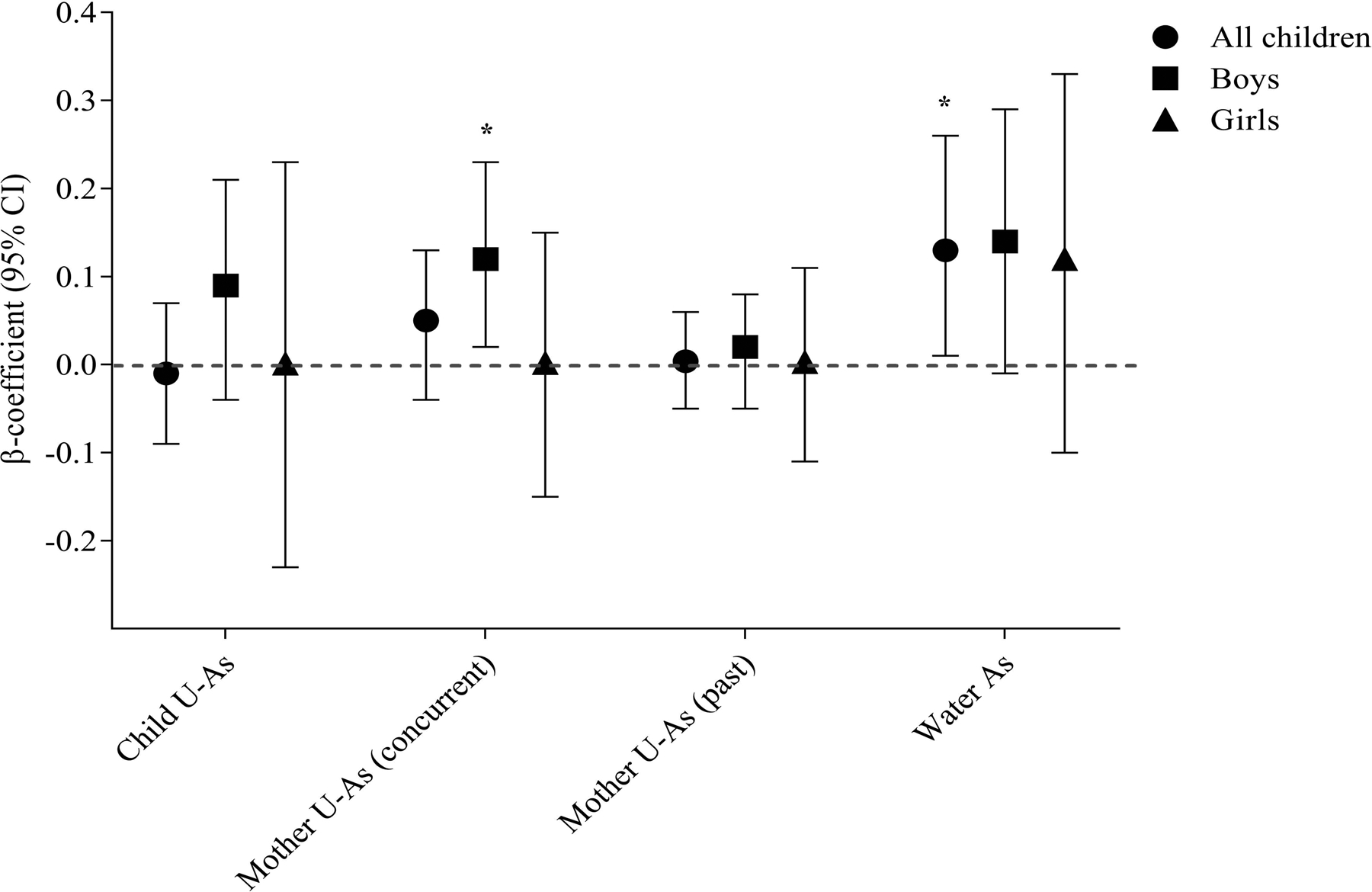
Association of arsenic exposure (child U-As, mother’s concurrent U-As, mother’s past U-As and household water arsenic) with serum bactericidal antibody (SBA) response. Analysis was performed stratifying by sex. “p” was determined by multivariate adjusted regression analysis and the model was adjusted by child age, sex, WAZ and mother BMI. WAZ, weight for age z-score and BMI, body mass index
Associations of As exposures with ex vivo cytokine/chemokine formation by lymphocytes/PBMC
PHA stimulation of isolated lymphocytes (predominantly non-adherent cells as described above) from the children led to significant induction of IL-10, IL-13, IL-17A, IL-2, IL-21, IL-5 and MIP-1α compared to unstimulated controls (data not shown). Multivariable regression analysis of As exposure and cytokine/chemokine concentrations in culture supernatants showed that there were negative associations between child U-As and concentrations of fractalkine (CX3CL) (β =−0.47; 95% CI = −0.88, −0.05; p = 0.03) as well as tumor necrosis factor (TNF)-α (β = −3.30, 95% CI = −7.20, 0.43; p = 0.08). Inverse associations were also detected between household W-As or maternal U-Aspast and interleukin (IL)-7 concentrations (Table 3). On the other hand, strong positive associations were observed between maternal U-Asconcurrent and IL-13, IL-17A and MIP-1α concentrations. No significant associations were noted between As exposure and other cytokines.
Table 3.
Association of As exposure with cytok-/chemokine levels in culture supernatant (n = 16).
| Cytokines/chemokines | β-coefficient (95% CI) | p-value |
|---|---|---|
| Fractalkine | ||
| Household water As | −0.06(−0.39, 0.28) | 0.72 |
| Child U-As | *−0.47(−0.88, −0.05) | 0.03 |
| Mother U-Asconcurrent | −0.28(−1.61, 1.04) | 0.63 |
| Mother U-Aspast | −0.24(−0.90, 0.41) | 0.42 |
| IL-13 | ||
| Household water As | 38.64(−95.17, 172.45) | 0.53 |
| Child U-As | −6.97(−212.26, 198.31) | 0.94 |
| Mother U-Asconcurrent | *463.64(102.20, 825.07) | 0.01 |
| Mother U-Aspast | 130.58(−124.01, 385.16) | 0.28 |
| IL-17A | ||
| Household water As | 1.35(−3.60, 6.30) | 0.56 |
| Child U-As | 1.95(−5.52, 9.42) | 0.57 |
| Mother U-Asconcurrent | *19.31(7.03, 31.59) | 0.007 |
| Mother U-Aspast | 4.72(−4.70, 14.14) | 0.29 |
| IL-7 | ||
| Household water As | *−0.14(−0.26, −0.03) | 0.02 |
| Child U-As | −0.14(−0.34, 0.07) | 0.17 |
| Mother U-Asconcurrent | −0.14(−0.66, 0.38) | 0.55 |
| Mother U-Aspast | *−0.27(−0.50, −0.04) | 0.03 |
| MIP-1α | ||
| Household water As | 0.04(−7.56, 7.64) | 0.99 |
| Child U-As | 3.84(−7.32, 15.01) | 0.47 |
| Mother U-Asconcurrent | *24.35(3.52, 45.18) | 0.03 |
| Mother U-Aspast | 5.84(−8.66, 20.33) | 0.40 |
| TNF-α | ||
| Household water As | −1.95(−4.51, 0.61) | 0.12 |
| Child U-As | †−3.30(−7.20, 0.43) | 0.08 |
| Mother U-Asconcurrent | −5.15(−15.23, 4.92) | 0.27 |
| Mother U-Aspast | −2.55(−7.96, 2.85) | 0.32 |
Regression model adjusted by child age, sex, and BMI.
p < 0.05,
p ≤ 0.08
Discussion
The findings of this study suggest that early childhood As exposure dysregulates fundamental processes of innate immunity in these children. Serum bactericidal antibody responses and LL-37 levels increased with increasing As exposure, but without any apparent change in macrophage-mediated killing activities. Expression of selected cytokines was also impacted by the As exposure. In addition, poor As methylation efficiency was seen to be associated with reduced innate immune function in these children.
A recent long-term follow-up study conducted in rural Bangladesh with widespread As contamination of groundwater reported higher risk of deaths due to respiratory, cardiovascular, and cerebro-vascular diseases (Rahman et al. 2018). Accumulating evidence suggests that As exposure disrupts immune function and contribute to chronic lung diseases (Ahmed et al. 2017; Parvez et al. 2008). Inflammation orchestrated by inflammatory and other immune cells is a crucial component of pulmonary diseases such as asthma, COPD, tuberculosis, and lung cancers (Coussens et al. 2002; Gorska et al. 2017; Leepiyasakulchai et al. 2012). In turn, each of these pathologies can be exacerbated by As-induced inflammatory responses (Olivas-Calderon et al. 2015).
An important role for the defense peptide LL-37 in the pathophysiology of chronic lung diseases is gradually being recognized ; and has been linked with increased risk of airway inflammation, lung infections, and acute exacerbation of COPD (Jiang et al. 2012; Persson et al. 2017). Recent studies suggested LL-37 could promote release of inflammatory IL-8 and proliferation of non-specifically activated CD4+ T-cells, actions that may ultimately induce apoptosis of bronchial and alveolar epithelial cells (Jiang et al. 2012; Thomi et al. 2018). These findings are relevant in the context of inflammation-mediated chronic lung diseases. In contrast, another study reported high risk of frequent exacerbations in COPD patients with low plasma LL-37 levels (Yang et al. 2015).
In the present study, plasma LL-37 levels increased with increasing As exposure, predominantly in children of normal weight and height. It is likely that with chronic As exposure, LL-37 levels may remain elevated in the systemic circulation owing to its role in innate immune surveillance and chemoattractant activity. Elevated LL-37 levels, in turn, may promote inflammation and have long-lasting consequences for decreased lung function (Zhang et al. 2014) or chronic lung disease later in life (Rahman et al. 2017; Sanchez et al. 2016). The stimulating effects of As exposure on LL-37 levels may be masked by multiple stress factors in growth-retarded children, thus hindering the optimum response. However, this hypothesis needs to be tested in larger cohorts of children with both a wider age range and As exposure levels.
The SBA is as an innate host defense mechanism against pathogens; however, recently it is being increasingly used as a tool to assess functional antibody responses against infection or vaccination (2014; Jang et al. 2016; Rahman et al. 2005; Shimanovich et al. 2017). An earlier study from our group showed that there was a reduced antibody response to live attenuated mumps vaccination among children with high vs. low levels of As exposure (Raqib et al. 2017). In an NHANES study among adults, chronic As exposure was associated with lower protective humoral immunity to herpes zoster (Cardenas et al. 2015). However, another NHANES study showed positive association between U-As and total anti-hepatitis A virus seroprevalence (Cardenas et al. 2016). In an earlier study, no significant difference in SBA responses was found against oral cholera vaccine in 2–5-yr-old Bangladeshi children with high As exposure (mean U-As, 292μg/L) compared to those with low exposure (Saha et al. 2013). However, in these same children, diphtheria and tetanus vaccine-specific IgG titers were higher in the high As exposure group. The current findings are not directly comparable with other studies as those were mostly used for vaccine efficacy and there were methodological differences. However, it is conceivable that to compensate for suppressed cellular immunity as a result of chronic As exposure (Ahmed et al. 2014; Mannan et al. 2018; Yu et al. 2018) humoral immunity is disproportionately activated, as seen in HIV-infected individuals (Shearer et al. 2000).
Experimental studies reveal that low-to-moderate levels of As influence monocyte differentiation, compromise antigen presentation, reduce phagocytic efficiency, and alter the chemotactic indices of macrophages (Abdul et al. 2015; Ferrario et al. 2016; Lemarie et al. 2006 & 2015; Sengupta et al. 2002). There are limited studies on in vivo As exposure effects on human macrophage function (Banerjee et al. 2009), especially the intracellular killing capacity. Inefficient As metabolism has been associated with As-related adverse health effects in adults and altered immune function in children (Ahmed et al. 2017; Mannan et al. 2018; Tseng et al. 2009). Similarly, poor As methylation efficiency was related to reduced MDM-mediated Spn killing capacity and SBA responses in 2–5-yr-old Bangladeshi children.
The findings of imbalanced CX3CL expression due to chronic As exposure may reflect perturbed immunostasis and impaired recruitment or survival of immune and/or inflammatory cells to counteract an infection or toxic insult (Liu et al. 2016). Reduced plasma IL-7 levels in relation to As exposure was seen earlier in individuals living in rural areas with As-contaminated drinking water (Raqib et al. 2009). The IL-7 results are supported by other studies showing toxic effects of MMA+3 and As+3 in suppressing early T-cell development in the thymus through inhibition of IL-7 signaling pathways (Xu et al. 2016), especially at low-to-moderate levels of As (Xu et al. 2017).
A central role of IL-13 in mediating airway hyper-reactivity and mucus expression in asthma is well known (Newcomb et al. 2012). Studies have also suggested a link between increased IL-17A production and severe asthma as well as lung infections; it is believed IL-17A contributes to asthma pathophysiology by increasing the capacity of IL-13 to activate intracellular signaling pathways (Hall et al. 2017). Our recent work (Parvez et al. 2019) has found a similar effect of As on IL-17A among adults from the same study area in Bangladesh. Whether increased expression of IL-13 and IL-17A in response to high As exposure also contributes to respiratory illness in the As-exposed children remains to be determined. Lastly, MIP-1α is also important in the pathogeneses of various inflammatory diseases (Bhavsar et al. 2015; Serody et al. 2000). Accordingly, chronic As exposure may lead to a state of persistent inflammation through imbalanced cytokine expression by various cell types and contribute to the pathogenesis of chronic lung diseases. However, no changes in Th1 and Th2 cytokines in relation to As exposure were noted in the present study.
The current study has a few limitations. Due to its pilot nature, the sample size was small and so inferring a conclusion is not feasible. However, the findings do suggest directions for further research. One limitation was that in utero exposure data of children was not available, though data from the mothers’ past exposures (2–4 years back) suggested a continued persistent exposure of their children to As. In addition, the range of As exposures was relatively small (median 39 μg/g) compared to that in other as well as our own studies (>79 μg/L) (Ahmed et al. 2014 & 2017; Islam et al. 2012; Olivas-Calderon et al. 2015; Raqib et al. 2017; Saha et al. 2013).
In summary, the present findings suggest that early-life exposure to As disrupts innate host defense pathway in children ≤ 5 yr-of-age by affecting SBA responses and plasma LL-37 levels. These findings, albeit preliminary, indicate that children exposed to As may face long-term respiratory health consequences. Poor As methylation efficiency in the children was also found to adversely affect SBA and macrophage-mediated killing of respiratory pathogens. The study also indicated that persistent As exposure may compromise host defenses and innate immune surveillance including formation of key peptides needed against infections. Findings from this study warrant further studies of respiratory infections and chronic lung diseases in As-exposed populations.
Acknowledgement
This work was supported by the funds from University of Chicago and icddr,b. icddr,b is also grateful to the governments of Bangladesh, Canada, Sweden, and the UK for providing core/unrestricted support. The authors would like to thank all HEALS staff and fieldworkers for their ongoing commitment to the study. FP acknowledge the grant US NIH grants support R01 ES019968S1 and R01 ES023888.
Footnotes
Declaration of interest
The authors declare no conflicts of interest. The authors alone are responsible for the content of this manuscript.
REFERENCES
- Abdul KS, Jayasinghe SS, Chandana EP, Jayasumana C and De Silva PM (2015). “Arsenic and human health effects: A review.” Environ Toxicol Pharmacol 40(3): 828–846. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Akhtar E, Roy A, von Ehrenstein OS, Vahter M, Wagatsuma Y and Raqib R (2017). “Arsenic exposure alters lung function and airway inflammation in children: A cohort study in rural Bangladesh.” Environ Int 101: 108–116. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Moore SE, Kippler M, Gardner R, Hawlader MD, Wagatsuma Y, Raqib R and Vahter M (2014). “Arsenic exposure and cell-mediated immunity in pre-school children in rural Bangladesh.” Toxicol Sci 141(1): 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Banerjee S, Sen R, Bandyopadhyay A, Sarma N, Majumder P, Das JK, Chatterjee M, Kabir SN and Giri AK (2009). “Chronic arsenic exposure impairs macrophage functions in the exposed individuals.” J Clin Immunol 29(5): 582–594. [DOI] [PubMed] [Google Scholar]
- Bhavsar I MC, Sabbagh MA, (2015). “Macrophage Inflammatory Protein-1 Alpha (MIP-1 alpha)/CCL3: As a Biomarker.” Biomarkers in Disease: Methods, Discoveries and Applications 223–249 [Google Scholar]
- Bishayi B and Sengupta M (2003). “Intracellular survival of Staphylococcus aureus due to alteration of cellular activity in arsenic and lead intoxicated mature Swiss albino mice.” Toxicology 184(1): 31–39. [DOI] [PubMed] [Google Scholar]
- Biswas R, Ghosh P, Banerjee N, Das JK, Sau T, Banerjee A, Roy S, Ganguly S, Chatterjee M, Mukherjee A and Giri AK (2008). “Analysis of T-cell proliferation and cytokine secretion in the individuals exposed to arsenic.” Hum Exp Toxicol 27(5): 381–386. [DOI] [PubMed] [Google Scholar]
- Cardenas A, Smit E, Bethel JW, Houseman EA and Kile ML (2016). “Arsenic exposure and the seroprevalence of total hepatitis A antibodies in the US population: NHANES, 2003–2012.” Epidemiol Infect 144(8): 1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas A, Smit E, Houseman EA, Kerkvliet NI, Bethel JW and Kile ML (2015). “Arsenic exposure and prevalence of the varicella zoster virus in the United States: NHANES (2003–2004 and 2009–2010).” Environ Health Perspect 123(6): 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, Graziano JH and Ahsan H (2009). “Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh.” Toxicol Appl Pharmacol 239(2): 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM and Werb Z (2002). “Inflammation and cancer.” Nature 420(6917): 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangleben NL, Skibola CF and Smith MT (2013). “Arsenic immunotoxicity: a review.” Environ Health 12(1): 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C and Siekmann J (2007). “Development of a WHO growth reference for school-aged children and adolescents.” Bull World Health Organ 85(9): 660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAntonio R, Yarzabal JP, Cruz JP, Schmidt JE and Kleijnen J (2016). “Epidemiology of community-acquired pneumonia and implications for vaccination of children living in developing and newly industrialized countries: A systematic literature review.” Hum Vaccin Immunother 12(9): 2422–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta K, Prasad P and Sinha D (2015). “Chronic low level arsenic exposure evokes inflammatory responses and DNA damage.” Int J Hyg Environ Health 218(6): 564–574. [DOI] [PubMed] [Google Scholar]
- Ercoli G, Baddal B, Alessandra G, Marchi S, Petracca R, Arico B, Pizza M, Soriani M and Rossi-Paccani S (2015). “Development of a serological assay to predict antibody bactericidal activity against non-typeable Haemophilus influenzae.” BMC Microbiol 15: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario D, Gribaldo L and Hartung T (2016). “Arsenic Exposure and Immunotoxicity: a Review Including the Possible Influence of Age and Sex.” Curr Environ Health Rep 3(1): 1–12. [DOI] [PubMed] [Google Scholar]
- Gordon SB, Irving GR, Lawson RA, Lee ME and Read RC (2000). “Intracellular trafficking and killing of Streptococcus pneumoniae by human alveolar macrophages are influenced by opsonins.” Infect Immun 68(4): 2286–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska K, Paplinska-Goryca M, Nejman-Gryz P, Goryca K and Krenke R (2017). “Eosinophilic and Neutrophilic Airway Inflammation in the Phenotyping of Mild-to-Moderate Asthma and Chronic Obstructive Pulmonary Disease.” COPD 14(2): 181–189. [DOI] [PubMed] [Google Scholar]
- Hall SL, Baker T, Lajoie S, Richgels PK, Yang Y, McAlees JW, van Lier A, Wills-Karp M, Sivaprasad U, Acciani TH, LeCras TD, Myers JB, Kovacic MB and Lewkowich IP (2017). “IL-17A enhances IL-13 activity by enhancing IL-13-induced signal transducer and activator of transcription 6 activation.” J Allergy Clin Immunol 139(2): 462–471 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus CM, Skibola CF, Warner M, Skibola DR, Alexander D, Lim S, Dangleben NL, Zhang L, Clark M, Pfeiffer RM, Steinmaus C, Smith AH, Smith MT and Moore LE (2008). “Decreased urinary beta-defensin-1 expression as a biomarker of response to arsenic.” Toxicol Sci 106(1): 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhmann BL, Harvey CF, Navas-Acien A, Graziano J, Parvez F, Chen Y, Argos M, Ahmed A, Hasan A, Ahsan H and van Geen A (2019). “Changes in arsenic exposure in Araihazar, Bangladesh from 2001 through 2015 following a blanket well testing and education campaign.” Environ Int 125: 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam LN, Zahid MS, Nabi AH and Hossain M (2012). “Function of serum complement in drinking water arsenic toxicity.” J Toxicol 2012: 302817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MS, Sahastrabuddhe S, Yun CH, Han SH and Yang JS (2016). “Serum bactericidal assay for the evaluation of typhoid vaccine using a semi-automated colony-counting method.” Microb Pathog 97: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YY, Xiao W, Zhu MX, Yang ZH, Pan XJ, Zhang Y, Sun CC and Xing Y (2012). “The effect of human antibacterial peptide LL-37 in the pathogenesis of chronic obstructive pulmonary disease.” Respir Med 106(12): 1680–1689. [DOI] [PubMed] [Google Scholar]
- Leepiyasakulchai C, Ignatowicz L, Pawlowski A, Kallenius G and Skold M (2012). “Failure to recruit anti-inflammatory CD103+ dendritic cells and a diminished CD4+ Foxp3+ regulatory T cell pool in mice that display excessive lung inflammation and increased susceptibility to Mycobacterium tuberculosis.” Infect Immun 80(3): 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire M, Negro Silva LF, Lemarie CA, Bolt AM, Flores Molina M, Krohn RM, Smits JE, Lehoux S and Mann KK (2015). “Arsenic Exposure Increases Monocyte Adhesion to the Vascular Endothelium, a Pro-Atherogenic Mechanism.” PLoS One 10(9): e0136592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarie A, Morzadec C, Bourdonnay E, Fardel O and Vernhet L (2006). “Human macrophages constitute targets for immunotoxic inorganic arsenic.” J Immunol 177(5): 3019–3027. [DOI] [PubMed] [Google Scholar]
- Mannan T, Ahmed S, Akhtar E, Ahsan KB, Haq A, Kippler M, Vahter M and Raqib R (2018). “Associations of Arsenic Exposure With Telomere Length and Naive T Cells in Childhood-A Birth Cohort Study.” Toxicol Sci 164(2): 539–549. [DOI] [PubMed] [Google Scholar]
- Martin-Chouly C, Morzadec C, Bonvalet M, Galibert MD, Fardel O and Vernhet L (2011). “Inorganic arsenic alters expression of immune and stress response genes in activated primary human T lymphocytes.” Mol Immunol 48(6–7): 956–965. [DOI] [PubMed] [Google Scholar]
- Newcomb DC, Boswell MG, Huckabee MM, Goleniewska K, Dulek DE, Reiss S, Lukacs NW, Kolls JK and Peebles RS Jr. (2012). “IL-13 regulates Th17 secretion of IL-17A in an IL-10-dependent manner.” J Immunol 188(3): 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon DE, Mussmann GV, Eckdahl SJ and Moyer TP (1991). “Total arsenic in urine: palladium-persulfate vs nickel as a matrix modifier for graphite furnace atomic absorption spectrophotometry.” Clin Chem 37(9): 1575–1579. [PubMed] [Google Scholar]
- Olivas-Calderon E, Recio-Vega R, Gandolfi AJ, Lantz RC, Gonzalez-Cortes T, Gonzalez-De Alba C, Froines JR and Espinosa-Fematt JA (2015). “Lung inflammation biomarkers and lung function in children chronically exposed to arsenic.” Toxicol Appl Pharmacol 287(2): 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez F, Chen Y, Brandt-Rauf PW, Bernard A, Dumont X, Slavkovich V, Argos M, D’Armiento J, Foronjy R, Hasan MR, Eunus HE, Graziano JH and Ahsan H (2008). “Nonmalignant respiratory effects of chronic arsenic exposure from drinking water among never-smokers in Bangladesh.” Environ Health Perspect 116(2): 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez F, Lauer FT, Factor-Litvak P, Liu X, Santella RM, Islam T, Eunus M, Alam N, Sarwar G, Rahman M, Ahsan H, Graziano J and Burchiel SW (2019). “Assessment of arsenic and polycyclic aromatic hydrocarbon (PAH) exposures on immune function among males in Bangladesh.” PLoS One 14(5): e0216662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson LJ, Aanerud M, Hardie JA, Miodini Nilsen R, Bakke PS, Eagan TM and Hiemstra PS (2017). “Antimicrobial peptide levels are linked to airway inflammation, bacterial colonisation and exacerbations in chronic obstructive pulmonary disease.” Eur Respir J 49(3). [DOI] [PubMed] [Google Scholar]
- Rahman A, Granberg C and Persson LA (2017). “Early life arsenic exposure, infant and child growth, and morbidity: a systematic review.” Arch Toxicol 91(11): 3459–3467. [DOI] [PubMed] [Google Scholar]
- Rahman M, Sohel N, Yunus FM, Alam N, Nahar Q, Streatfield PK and Yunus M (2018). “Arsenic exposure and young adult’s mortality risk: A 13-year follow-up study in Matlab, Bangladesh.” Environ Int 123: 358–367. [DOI] [PubMed] [Google Scholar]
- Raqib R, Ahmed S, Sultana R, Wagatsuma Y, Mondal D, Hoque AM, Nermell B, Yunus M, Roy S, Persson LA, Arifeen SE, Moore S and Vahter M (2009). “Effects of in utero arsenic exposure on child immunity and morbidity in rural Bangladesh.” Toxicol Lett 185(3): 197–202. [DOI] [PubMed] [Google Scholar]
- Raqib R, Ahmed S, Ahsan KB, Kippler M, Akhtar E, Roy AK, Lu Y, Arifeen SE, Wagatsuma Y and Vahter M (2017). “Humoral Immunity in Arsenic-Exposed Children in Rural Bangladesh: Total Immunoglobulins and Vaccine-Specific Antibodies.” Environ Health Perspect 125(6): 067006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio-Vega R, Gonzalez-Cortes T, Olivas-Calderon E, Lantz RC, Gandolfi AJ and Gonzalez-De Alba C (2015). “In utero and early childhood exposure to arsenic decreases lung function in children.” J Appl Toxicol 35(4): 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Chowdhury MI, Nazim M, Alam MM, Ahmed T, Hossain MB, Hore SK, Sultana GN, Svennerholm AM and Qadri F (2013). “Vaccine specific immune response to an inactivated oral cholera vaccine and EPI vaccines in a high and low arsenic area in Bangladeshi children.” Vaccine 31(4): 647–652. [DOI] [PubMed] [Google Scholar]
- Sanchez TR, Perzanowski M and Graziano JH (2016). “Inorganic arsenic and respiratory health, from early life exposure to sex-specific effects: A systematic review.” Environ Res 147: 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta M and Bishayi B (2002). “Effect of lead and arsenic on murine macrophage response.” Drug Chem Toxicol 25(4): 459–472. [DOI] [PubMed] [Google Scholar]
- Serody JS, Burkett SE, Panoskaltsis-Mortari A, Ng-Cashin J, McMahon E, Matsushima GK, Lira SA, Cook DN and Blazar BR (2000). “T-lymphocyte production of macrophage inflammatory protein-1alpha is critical to the recruitment of CD8(+) T cells to the liver, lung, and spleen during graft-versus-host disease.” Blood 96(9): 2973–2980. [PubMed] [Google Scholar]
- Shearer WT, Easley KA, Goldfarb J, Jenson HB, Rosenblatt HM, Kovacs A, McIntosh K and Group PCHS (2000). “Evaluation of immune survival factors in pediatric HIV-1 infection.” Ann N Y Acad Sci 918: 298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimanovich AA, Buskirk AD, Heine SJ, Blackwelder WC, Wahid R, Kotloff KL and Pasetti MF (2017). “Functional and Antigen-Specific Serum Antibody Levels as Correlates of Protection against Shigellosis in a Controlled Human Challenge Study.” Clin Vaccine Immunol 24(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski EE, Burns LA, McCoy KL, Stern M and Munson AE (1991). “Suppression of splenic accessory cell function in mice exposed to gallium arsenide.” Toxicol Appl Pharmacol 110(1): 143–156. [DOI] [PubMed] [Google Scholar]
- Tseng CH (2009). “A review on environmental factors regulating arsenic methylation in humans.” Toxicol Appl Pharmacol 235(3): 338–350. [DOI] [PubMed] [Google Scholar]
- Van Geen A, Ahsan H, Horneman AH, Dhar RK, Zheng Y, Hussain I, Ahmed KM, Gelman A, Stute M, Simpson HJ, Wallace S, Small C, Parvez F, Slavkovich V, Loiacono NJ, Becker M, Cheng Z, Momotaj H, Shahnewaz M, Seddique AA and Graziano JH (2002). “Promotion of well-switching to mitigate the current arsenic crisis in Bangladesh.” Bull World Health Organ 80(9): 732–737. [PMC free article] [PubMed] [Google Scholar]
- van Geen A, Cheng Z, Jia Q, Seddique AA, Rahman MW, Rahman MM and Ahmed KM (2007). “Monitoring 51 community wells in Araihazar, Bangladesh, for up to 5 years: implications for arsenic mitigation.” J Environ Sci Health A Tox Hazard Subst Environ Eng 42(12): 1729–1740. [DOI] [PubMed] [Google Scholar]
- van Harten RM, van Woudenbergh E, van Dijk A and Haagsman HP (2018). “Cathelicidins: Immunomodulatory Antimicrobials.” Vaccines (Basel) 6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Lauer FT, Liu KJ, Hudson LG and Burchiel SW (2016). “Environmentally relevant concentrations of arsenite and monomethylarsonous acid inhibit IL-7/STAT5 cytokine signaling pathways in mouse CD3+CD4-CD8- double negative thymus cells.” Toxicol Lett 247: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Medina S, Lauer FT, Douillet C, Liu KJ, Styblo M and Burchiel SW (2017). “Genotoxicity induced by monomethylarsonous acid (MMA(+3)) in mouse thymic developing T cells.” Toxicol Lett 279: 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Liao WT, Lee CH, Chai CY, Yu CL and Yu HS (2018). “Immunological dysfunction in chronic arsenic exposure: From subclinical condition to skin cancer.” J Dermatol 45(11): 1271–1277. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jiang Y, Sun C, Wang Q, Yang Z, Pan X, Zhu M and Xiao W (2014). “The human cathelicidin LL-37 enhances airway mucus production in chronic obstructive pulmonary disease.” Biochem Biophys Res Commun 443(1): 103–109. [DOI] [PubMed] [Google Scholar]