

Abstract
The G2019S mutation in LRRK2 is one of the most common known genetic causes of neurodegeneration and Parkinson disease (PD). LRRK2 mutations are thought to enhance LRRK2 kinase activity. Efficacious small molecule LRRK2 kinase inhibitors with favorable drug properties have recently been developed for pre-clinical studies in rodent models, and inhibitors have advanced to safety trials in humans. Rats that express human G2019S-LRRK2 protein and G2019S-LRRK2 knock-in mice provide newly characterized models to better understand the ostensible target for inhibitors. Herein, we explore the relationships between LRRK2 kinase inhibition in the brain and the periphery to establish the link between LRRK2 kinase activity and protein stability, induction of lysosomal defects in kidney and lung, and how G2019S-LRRK2 expression impacts these phenotypes. Using a novel ultra-sensitive scalable assay based on protein capillary electrophoresis with LRRK2 kinase inhibitors included in-diet, G2019S-LRRK2 protein was resilient to inhibition compared to wild-type (WT)-LRRK2 protein, particularly in the brain. Whereas WT-LRRK2 kinase activity could be completed blocked without lowering LRRK2 protein levels, higher inhibitor concentrations were necessary to fully reduce G2019S-LRRK2 activity. G2019S-LRRK2 expression afforded robust protection from inhibitor-induced kidney lysosomal defects, suggesting a gain-of-function for the mutation in this phenotype. In rodents treated with inhibitors, parallel measurements of phospho-Rab10 revealed a poor correlation to phospho-LRRK2, likely due to cells that express Rab10 but poorly express LRRK2 in heterogenous tissues and cell isolates. In summary, our results highlight several challenges associated with the inhibition of the G2019S-LRRK2 kinase that might be considered in initial clinical efforts.
Keywords: Park8, Dardarin, Pharmacodynamics, Pharmacokinetics, Small-molecule inhibitor
Introduction
The leucine-rich repeat kinase 2 gene encodes LRRK2 protein that is expressed primarily in circulating leukocytes, kidney, lung, and the brain in humans (West, 2017). Genetic studies show that the pathogenic G2019S mutation in the LRRK2 kinase domain is one of the most frequent known genetic causes of neurodegeneration (Trinh et al., 2014). Initial in vitro studies in transfected cell lines revealed that G2019S-LRRK2 increased autophosphorylation activities as well as LRRK2 kinase activity towards generic peptide substrates, usually ~2–5 fold over endogenous wild-type (WT)-LRRK2. Analyses of LRRK2 protein harbored in extracellular exosomes purified from urine from LRRK2 mutation carriers with Parkinson’s disease (PD) also suggests a similar effect on LRRK2 autophosphorylation (Fraser et al., 2016a; Wang et al., 2017). Emerging evidence suggests that LRRK2 autophosphorylation or expression may be likewise increased in a proportion of idiopathic PD (Bliederhaeuser et al., 2016; Cook et al., 2017). Toxicity associated with G2019S-LRRK2 expression has been demonstrated in multiple models, for example viral-expression systems, to depend on LRRK2 kinase activity (Dusonchet et al., 2011; Greggio et al., 2006; Lee et al., 2010; Tsika et al., 2015). As such, intensive efforts are devoted towards the development of LRRK2 kinase inhibitors for the treatment of LRRK2-linked PD (West, 2017). Safety trials are underway with several LRRK2 kinase inhibitors of as-yet unknown identity (Hyland and Warners, 2017).
The G2019S mutation in LRRK2 protein alters the conserved DYG motif to DYS in the kinase activation loop, plausibly affecting metal binding and flexibility required for kinase activation (Nolen et al., 2004). While there is no high-resolution structure available for the LRRK2 kinase domain from higher-order eukaryotes, we previously used a library of thousands of ATP-competitive molecules to probe the ATP-binding pocket of WT- and G2019S-LRRK2 and identified molecules that could preferentially inhibit G2019S-LRRK2 versus WT-LRRK2 (Liu et al., 2014). Notably, several structurally distinct small molecule scaffolds have been described with very high specificity for LRRK2, where only weak binding to other protein kinases could be detected. We have attributed this property of some LRRK2 kinase inhibitors to the unique ATP-pocket and amino acid composition in human LRRK2 (Liu et al., 2014). Among ATP-competitive LRRK2 small molecule kinase inhibitors, the molecules MLi2 and PF-360 show low to sub-nanomolar binding in vitro and have outstanding selectivity profiles in blocking only LRRK2 kinase activity at lower concentrations out of hundreds of other kinases screened (Fell et al., 2015; Henderson et al., 2015; West, 2015).
To facilitate the development of successful LRRK2-targeting therapeutics, rats that express human G2019S-LRRK2 as well as mice with the mutation knocked into the genome have been developed (Daher et al., 2015; Volta et al., 2017). These rodent models together with potent small molecule inhibitors provide an excellent opportunity to explore pharmacodynamic responses related to LRRK2 kinase inhibition both in the brain and periphery. Some in vivo activity profiles have been reported in WT mice for MLi2 and in WT rats for PF-360 in separate studies (Andersen et al., 2018; Baptista et al., 2015; Fell et al., 2015; Scott et al., 2017), but LRRK2 inhibition profiles have been poorly described in the context of G2019S-LRRK2 expression. In rats and non-human primates, oral-dosing strategies that result in brief periods of time with high concentrations of LRRK2 kinase inhibitors result in mild lysosomal alterations in lung and kidney tissue, partly resembling LRRK2 knockout rodents (Baptista et al., 2013; Fuji et al., 2015). These phenotypes related to lysosome dysfunction have proved difficult to quantitatively measure in these past studies. Other studies in LRRK2 knockout mice hypothesize LRRK2-linked disease may relate better to loss-of-function phenotypes associated with LRRK2 mutations rather than gain-of-function phenotypes, as LRRK2 knockout mice may mimic some aspects of mice with LRRK2 mutations (Giaime et al., 2017). Further, some in vitro studies suggest that LRRK2 kinase activity controls LRRK2 protein levels (Skibinski et al., 2014), further obscuring the relationship between LRRK2 kinase inhibition and disease.
To explore these questions, we combine the most potent described LRRK2 tool kinase inhibitors MLi2 and PF-360 to probe LRRK2 kinase inhibition in the brain and periphery at different concentrations of compound in both WT and G2019S-LRRK2 expressing rats and mice. In deducing dose-response relationships for both LRRK2 inhibition and the development of potentially adverse phenotypes in the kidney and lung, we utilize in-diet drug regimens for both molecules able to stably sustain selected concentrations of compound between ~1 to ~100 nm in the brain and periphery. Further, we use a highly quantitative protein assay capable of measuring phospho-LRRK2 (pS935-LRRK2) in tissues and cells in the presence of strong detergents like sodium-dodecyl sulfate (SDS) and reducing agents like dithiothreitol (DTT) to ensure all LRRK2 protein is captured for analysis. Despite past reports, there is not a direct relationship between LRRK2 kinase activity and LRRK2 protein levels. Our observations in rats and mice expressing G2019S-LRRK2 demonstrate an unexpected resiliency of G2019S-LRRK2 protein to inhibition at controlled concentrations of drug. Tissue-specific effects add further complexity to G2019S-LRRK2 resiliency, with G2019S-LRRK2 in the brain more resilient than in the kidney or lung in both mice and rats. Inconsistent with a loss-of-function hypothesis for G2019S-LRRK2, mutant LRRK2 expression offers robust protection from lysosomal defects caused by higher concentrations of kinase inhibitors. Collectively, these results highlight some potentially unexpected challenges in the therapeutic targeting of G2019S-LRRK2 as well as provide additional support for a kinase-activation hypothesis for the G2019S mutation in PD.
Material and Methods
In vitro assessment of LRRK2 Kinase Activity-
Full length, recombinant human WT-LRRK2 (A15197/1779572) and G2019S-LRRK2 (A15200/1524191) kinase activity was measured using LanthaScreen kinase assays (ThermoFisher) as well as AlphaScreen assays (PerkinElmer) as previously described (Liu et al., 2014).
In silico docking models-
The LRRK2 ATP binding pocket was constructed based on the MST3 structure (4u8z) using homologue modeling on SWISS-MODEL, as described (Henderson et al., 2015). Molecule substitutions were created with Pymol Bulider script, and amino-acid substitutions made with the Mutagenesis script. Docking models were generated using iGEM dock.
Compounds-
MLi2 and PF-360 were synthesized in-house. Novel synthetic pathways are provided in Supplemental Files 1 and 2. Medicated chows were formulated at Research Diets.
Animals—
All animal protocols were approved by local Animal Care and Use Committees accredited by the AAALAC. Both male and female rodents, aged 8–12 weeks were used in this study. All rodents were housed on a 12 h light/dark cycle and given free access to food and water. Consumption of chow was monitored daily and body weight regularly measured. G2019S-LRRK2 human BAC rats (Sprague-Dawley, Taconic), LRRK2 knockout rats (Long-Evans, Sage), and G2019S-LRRK2 knock-in mice (C57Bl6, Taconic), as well as non-transgenic littermates were used in this study. Approximately equal numbers of males and females compose all experiments and observations.
Pharmacokinetics-
Male Sprague-Dawley rats (n=3 for all observations) were used with IV doses at 3 mg kg−1, with blood sampling at 5 min, 15 min, 30 min, 1, 2, 4, 8, and 24 h. PO dose was at 5 mg kg−1 with blood sampling at 15 min, 30 min, 1, 2, 4, 8, and 24 hr. No clinical symptoms were observed during the experiments. Samples were analyzed by HPLC. Microsomes were prepared from macaque liver and included, with or without NADPH, at 0.5 mg mL−1 (w/v). The final concentrations of test compounds in microsome stability assays were 2 μM.
Mass spectrometry-
Compound standards included 50% acetonitrile in water with known concentrations of compound (1–10,000 ng mL−1) combined with plasma (mouse or rat) obtained on the same day of analysis. Standards and experimental solutions were analyzed on an Applied Biosystems Sciex Triple Quad 5500, with online LC-30AD and Phenomenex Synergi 2.5 um Polar-RP 3×50 mm columns. Mobile phase included 5% acetonitrile in water with 0.1% Formic acid, and a solution of 95% acetonitrile in water with 0.1% formic acid.
Tissue and cell lysates-
Rat and mouse tissue was collected following transcardiac perfusion of cold PBS. Tissues were homogenized by sonication in a RIPA lysis buffer consisting of 50 mM Tris (pH7.4), 150 mM NaCl, 1% Triton, 0.1% SDS supplemented with 1x protease inhibitors (Fisher) and phosphatase inhibitors (Roche). Whole blood was collected into K2EDTA tubes (BD), diluted with equal amounts of PBS, and transferred to SepMate-50 tubes (StemCell) filled with 15 mL lymphocyte separation media (Corning). Peripheral blood mononuclear cells (PBMCs) were separated by centrifugation at 1200 xg for 15 min at room temperature. PBMCs were collected and washed twice with PBS before pelleted at 500 xg and lysis. Primary macrophages were obtained by intraperitoneal injection of 2 mL of thioglycollate broth (Sigma), and three days later harvesting cells by washing in the peritoneal cavity. Cells were cultured in DMEM media (Sigma) supplemented with 2 mM Glutamine (Sigma) and 10% FBS (Atlanta Biologicals) and 10 ng mL−1 GM-CSF (Peprotech). Cells were processed into RIPA buffer as described above. Protein concentrations were determined using BCA assay (Pierce). All lysates were further diluted 1:1 in 2x Laemelli buffer supplemented with 40 mM NaF and 10% DTT.
Western blots and antibodies—
Protein lysates were electrophoresed on a 7.5% Mini-Protean TGX stain- free gel (BioRad) and then transferred onto Immobilon-FL membranes (Millipore) and blocked with Odyssey Blocking Buffer (LiCOR). Membranes were cut in half below the high-molecular weight markers. Primary antibodies included mouse anti-LRRK2 clone N241A/34 (1: 2000, Antibodies, Inc.), rabbit anti-LRRK2 antibody MJFF2 (1:2000, Abcam), Ser(P)-935 (1: 2000, Abcam), Ser(P)-1292 (1:2000, Abcam) HSPA8 (1:5000, Cell Signaling), phospho-T73-Rab10 antibody (1:1000, MJF-R21, Abcam), and total Rab10 (1:1000, clone D36C4, Cell Signaling). Secondary antibodies were IRdye 680LT donkey anti-mouse and IRdye 800CW donkey anti-rabbit (both 1:10000, LiCOR). Membranes were scanned using a LiCOR CLx with 685-nm and 785-nm lasers and band intensities were quantified using the LiCOR-Odyssey system.
Protein Capillary Electrophoresis and antibodies-
Protein lysates were combined with High and Low Molecular weight protein kits (ProteinSimple). Samples were prepared and analyzed following the manufacturer protocol. Diluted lysates were mixed with 5x fluorescent master mix and loaded with blocking reagent, primary and secondary antibodies, chemiluminescent substrate, and wash buffer. The microplate was then loaded into the Wes instrument (ProteinSimple) with separation and immunodetection performed automatically using default hardware and software settings. Data were analyzed using Compass SW software. Primary antibodies were as above except for HSPA8 which was substituted for mouse anti-HSPA8 (StressMarq).
Kidney and Lung analysis-
Kidneys were collected following transcardiac perfusion with cold PBS. Lungs were inflated prior to fixing in 70% alcoholic formalin overnight. The lungs were then transferred to a tissue cassette add submerged in 10% formalin until they were paraffin embedded. Kidneys were dissected into medial sections comprised of cortex and medulla and tissue placed in a cassette in formalin overnight then transferred to 70% ethanol. Lung and kidney sections were cut at 5 μm with a cryostat and processed with Haemotoxylin and Eosin dyes. For autofluorescence analysis, kidneys were perfused with 1x PBS, fixed in 2% paraformaldehyde (PFA) for 2 hrs at 4°C, and then soaked in 20% sucrose solution until floating. 5 μm thick sections were mounted to slides with Vectashield HardSet Antifade Mounting Medium with DAPI (Vector Laboratories). Some sections were treated with Sudan Black B dye (EMD Millipore) per manufacturer’s instructions. Images were obtained using a 488-line laser and 500–550 nm emission and signals analyzed with Leica Application Suite Advanced Fluorescence (LAS AF) software.
Results
Common properties of two structurally distinct LRRK2 kinase inhibitors
The G2019S-LRRK2 molecule is considered the initial therapeutic target of clinical-candidate small molecule LRRK2 kinase inhibitors (West, 2017). To study the characteristics of G2019S-LRRK2 versus WT-LRRK2 inhibition, two structurally distinct LRRK2 kinase inhibitors would produce more reliable conclusions than those surmised from a single small molecule. Of the numerous LRRK2 small molecule inhibitors described, two mixed ATP-competitors known as MLi2 and PF-360 demonstrate low nanomolar potencies in blocking LRRK2 kinase activity in cells and tissue (Fell et al., 2015; Henderson et al., 2015; Scott et al., 2017; Volpicelli-Daley et al., 2016). These two tool-compound molecules have recently been studied in non-human primates (Baptista et al., 2015). We synthesized MLi2 and PF-360 to high purity (i.e., >97.4%) as assessed by nuclear-magnetic resonance traces and mass spectrometry (Supplemental Files 1 and 2). MLi2 and PF-360 are structurally different but with similar polar surface areas (Figure 1). Aqueous solubility of MLi2 is poor compared to the PF-360 compound that has lower lipophilicity. Previously we developed an assay to measure LRRK2 autophosphorylation in vitro using AlphaScreen technology (Liu et al., 2014). Using this assay, we assessed a variety of concentrations of MLi2 and PF-360 at Km,app(ATP) for WT and G2019S-LRRK2 in the autophosphorylation of the Thr1503 residue in the Rab-like ROC (Ras-of-complex) GTPase domain. MLi2 and PF-360 have similar potencies of 4 and 6 nM IC50, respectively, for blocking WT-LRRK2 autophosphorylation. Both molecules are considerably more potent (~1 order of magnitude) in blocking LRRK2-mediated phosphorylation of a ROC-kinase like generic peptide (LRRKtide) in LanthaScreen, also at the Km,app(ATP) of that assay. Whereas MLi2 blocks WT and G2019S-LRRK2 kinase activity with similar potencies, PF-360 is considerably weaker in blocking G2019S-LRRK2 autophosphorylation (~6-fold less potent) as well as G2019S-LRRK2 mediated LRRKtide phosphorylation (~4-fold less potent).
Figure 1. Drug properties and LRRK2 inhibition profiles of MLi2 and PF-360 with respect to wild-type and G2019S-human LRRK2 protein.
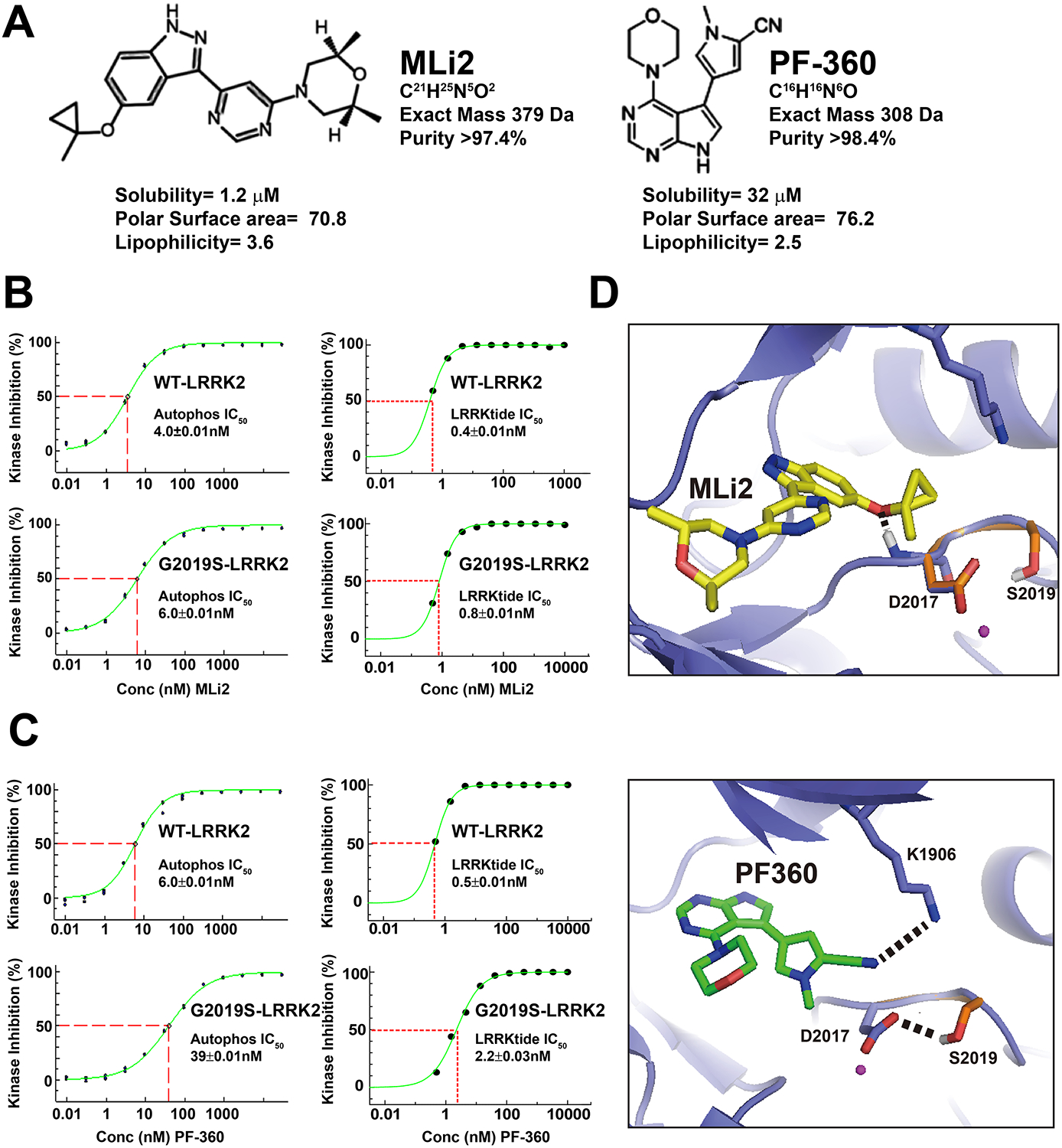
A, structurally distinct compounds MLi2 and PF-360 have comparable drug like properties. B, Kinase inhibition profiles are given with respect to inhibition of LRRK2 kinase activity with MLi2 or C, PF-360, in autophosphorylation that occurs in vitro at the pThr-1503 residue, or inhibition of LRRKtide, with WT or G2019S-LRRK2 recombinant protein. In these assays, previously measured ATPKm,App was used for each condition (Liu et al., 2014). This includes 9.5 and 7.4 uM ATP for WT- and G2019S-LRRK2 (respectively) in the autophosphorylation assay, respectively, and 57 and 134 μM ATP for WT- and G2019S-LRRK2 (respectively) in the LRRK2tide assay. D, Model of MLi2 of PF-360 binding with LRRK2 ATP binding pocket based on the crystal structure of MST3 in complex with PF-06447475 (PDB ID 4u8z). MLi2 is shown in yellow (top) and PF-360 is shown in green (bottom). LRRK2 ATP binding pocket is shown in slate. The DYS motif of G2019S LRRK2 is shown in orange. Predicted hydrogen bonds are shown in dashed lines. Atoms are color coded (white: hydrogen, red: oxygen, blue: nitrogen, magenta: magnesium).
Although a high-resolution structure of the LRRK2 ATP-binding pocket from higher-order eukaryotes has not been described, homology models based on the ATP pocket in the MST3 kinase share some homology and have been used to study LRRK2-kinase inhibitor docking (Henderson et al., 2015). The DYG motif of WT-LRRK2 is predicted to be highly flexible and undergoes conformational changes upon activation (altered to DYS with the G2019S mutation). The G2019S mutation may stabilize the DYS motif in an activated conformation through hydrogen bonds (Gilsbach et al., 2012; Liu et al., 2013) and facilitates the electrostatic interaction between the highly conserved D2017 and K1906 residues. Docking simulations show that PF-360 forms potential hydrogen bonds between the cyanophenyl group and K1906 residue. The G2019S mutation stabilizes the negatively charged D2017 residue in the activated form and could potentially interfere with the hydrogen bonding between cyanophenyl group and K1906, thereby reducing the binding affinity. In contrast, MLi2 is unlikely to directly contact the side chain of Lys1906 / Asp2017. These results suggest that the pyrrole ring in PF-360 may interact with the LRRK2 activation loop in a manner different from MLi2. In contrast, the purine-like heterocyclic structures common to MLi2 and PF-360 are predicted to occupy near overlapping spaces in the LRRK2 ATP pocket.
Common pharmacokinetic properties of LRRK2 inhibitors MLi2 and PF-360
Few published studies have reported successful chronic pharmacological inhibition of LRRK2 kinase activity in rodents. So-called first and second generation LRRK2 inhibitors suffer from very short half-lives and first-pass metabolism liabilities together with limited brain penetration, limiting most LRRK2 kinase inhibitors to single-dose or in vitro experiments (West, 2017). Microsomal stability tests of MLi2 and PF-360 show that both compounds have short half-lives with no significant concentrations of compound left after one-hour incubation in the presence of NADPH (Figure 2A). Intravenous dosing experiments in non-transgenic rats with MLi2 and PF-360 support similar stability profiles of the two molecules with t½= ~1.3 h after a 5 mg kg−1 oral administration (Figure 2B). Both molecules show good bioavailability of ~40% in oral dosing with both molecules administered in a common 1% methylcellulose vehicle. As expected, total measures of these compounds in plasma closely correlates to brain concentrations (r=0.99 and 0.89 for MLi2 and PF-360, respectively) with a brain-to-plasma ratio (total compound) of 0.75±0.07 for MLi2 and 0.48±0.03 for PF-360. Based on these data, we conclude that both MLi2 and PF-360 molecules have similar pharmacokinetic properties in rats, but short half-lives may preclude standard oral administration, and limited solubility obviate osmotic pump routes for chronic treatment strategies.
Figure 2. MLi2 and PF-360 have similar pharmacokinetic properties.
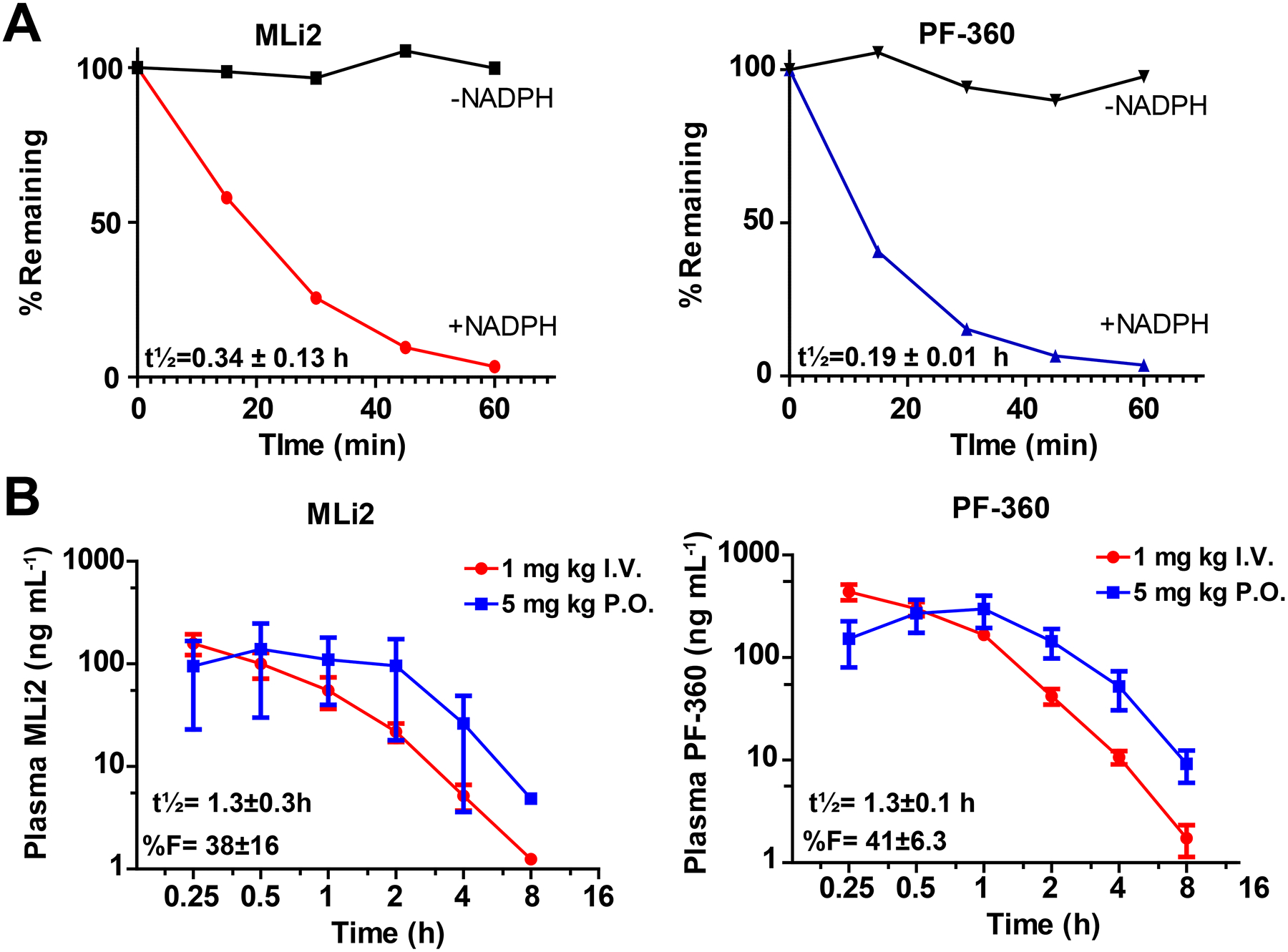
A, in vitro microsome stability assays. Representative traces from pooled male monkey liver microsomes are shown. Compound stability without NADPH (black lines) are presented as controls. B, Turnover of compounds in plasma for MLi2 and PF-360 in non-transgenic rats, with representative I.V. and P.O. experiments shown, at 1 mg kg−1 and 5 mg kg−1 single-dose experiments, respectively. Error is given as S.E.M. from at least three replicate experiments.
LRRK2-inhibitor mediated degradation of LRRK2 protein in the brain
Previous studies in vitro and in cultured cells demonstrated that LRRK2 kinase activity may be required for LRRK2 stability and thus small molecule LRRK2 kinase inhibitors rapidly induce the degradation of LRRK2 (Lobbestael et al., 2016; Skibinski et al., 2014; Zhao et al., 2015). Yet, in vivo studies with MLi2 introduced in-diet for treatment of mice demonstrated the strong potential of this route of drug administration for chronic inhibition of LRRK2 without apparent lowering of LRRK2 protein levels (Fell et al., 2015). Similar to past MLi2 studies, we find that measures of plasma concentrations with the in-diet strategy results in stable drug levels during a 24-hour period (Figure 3A) compared to traditional oral gavage strategies we have used in the past with LRRK2 kinase inhibitors (Figure 2, and (Daher et al., 2015)).
Figure 3. Continuous compound exposure with in-diet dosing regimen.
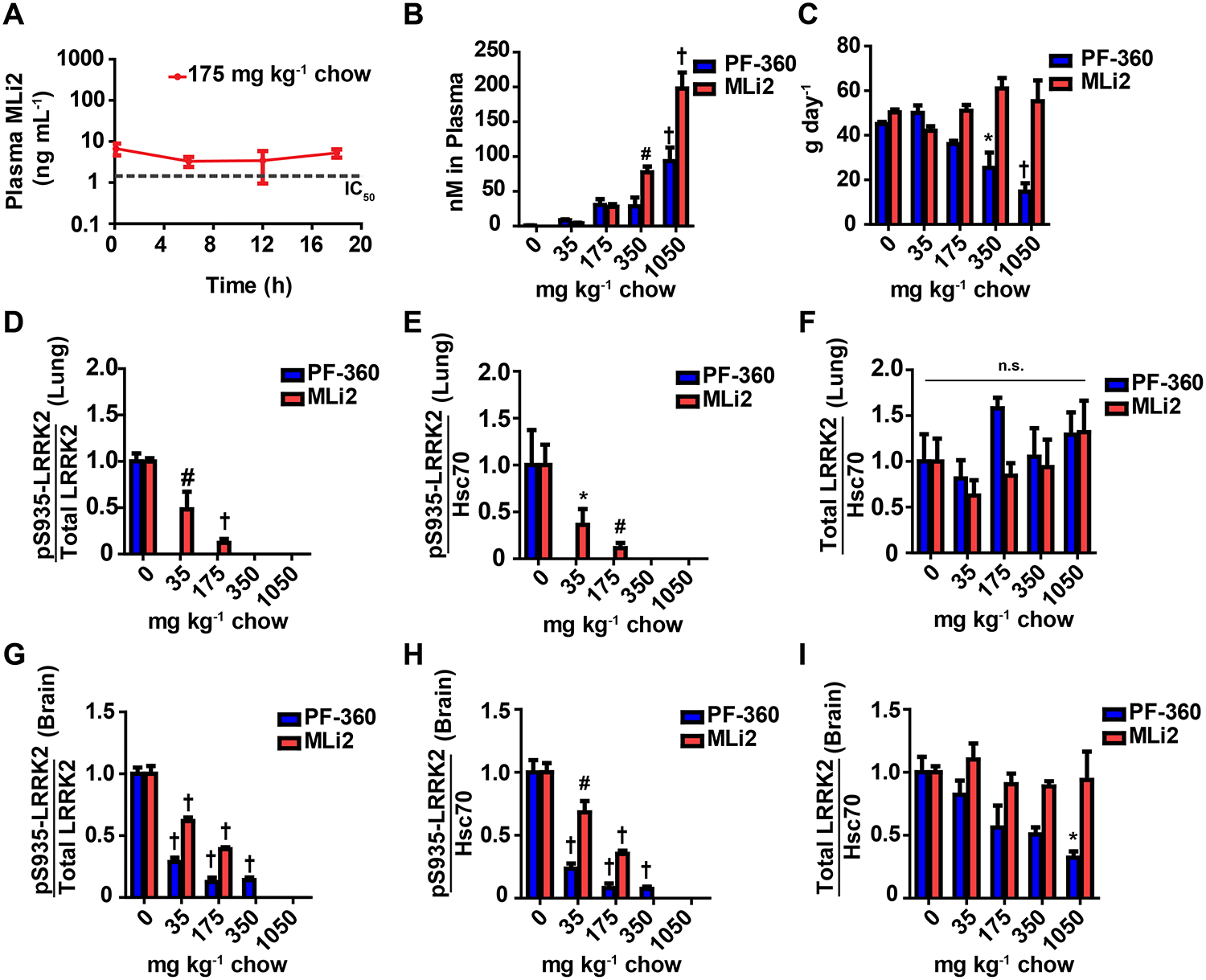
Non-transgenic rats were co-housed in groups of three with ad libitum access to food containing 35, 175, 350, or 1050 milligrams of compound (red: MLi2 or blue: PF-360) per kilogram of chow. Rats were dosed for three days and sacrificed two hours into their light cycle. A, steady-state dosing was attained using in-diet treatment compared to oral gavage (Figure 2B). Drug plasma levels remain constant above IC50 (Figure 1B) and are maintained over a 24 hour sampling period. B, The concentration of compound in circulation increases as diet concentrations increase as shown by plasma levels determined by LC/MS/MS analysis. C, Overall the compounds were well-tolerated as indicated by the total amount of chow consumed daily. Higher concentrations of PF-360 chow (350 and 1050 mg kg−1) were consumed less compared to the control diet. No significant changes in body weight were observed (data not shown). LRRK2 inhibition profiles for MLi2 and PF-360 were characterized by quantifying by the ratio of pS935-LRRK2 to total LRRK2, phosphorylated LRRK2 levels (pSer935), and total LRRK2 protein levels in whole lung and brain lysate. D, E, F LRRK2 inhibition characterization in the lung. Lung protein lysates were analyzed via Western blot. HSC70 was used as a loading control for normalization. G, H, I LRRK2 inhibition characterization in the brain. Brain protein lysates were analyzed using protein capillary electrophoresis, using 2 μg of total protein. Significance reported as treatment compared to untreated control * p<0.05, #p<0.01,† p<0.0001, as calculated by one-way analysis of variance with Dunnet’s post-hoc test (B, D, E, F, G, H, I). All other group comparisons were not significant (p>0.05) compared with control groups. Data are presented as means ± S.E.M.
Based on the similar drug-properties of MLi2 and PF-360, we designed a series of medicated chows with both compounds incorporated at various concentrations for use in both non-transgenic mice and rats expressing endogenous WT-LRRK2 as well as in G2019S-LRRK2-expressing rodents. In non-transgenic rats, levels of drug in the plasma increase with increasing amounts of compound in chow (Figure 3B) as expected, although at the highest concentrations of PF-360 the rats consume less chow than expected. During this period, rats did not consume more chow than would deliver more than 20 mg of PF-360 per day, whereas MLi2 was ingested up to our highest concentration (estimated ~60 mg per day) without changes in the amount of chow consumed (Figure 3C).
In protein lysates, a proportion of LRRK2 is soluble with non-ionic detergents, but a significant proportion of LRRK2 protein is highly insoluble in non-ionic detergents and requires SDS and DTT for extraction and analysis (Biskup et al., 2007; Davies et al., 2013). LRRK2 kinase inhibition has been reported to decrease the solubility of LRRK2 (Sen et al., 2009), thereby potentially mimicking a loss of total LRRK2 protein when analyzing the soluble fraction. To measure proteins in total protein lysates that include LRRK2, phospho-LRRK2, and housekeepers, we developed a scalable so-called medium-throughput protein capillary electrophoresis assay compatible with high concentrations of SDS and DTT (Supplemental Figure 1). Consistent with our in vitro assays for inhibition of LRRK2 autophosphorylation, drug concentrations in the plasma greater than 10 nM of either MLi2 or PF-360 correspond to completely dephosphorylated LRRK2 protein in the lung in SDS-solubilized total protein lysates (i.e., pS935-LRRK2, Figure 3D). Different from past in vitro studies, total LRRK2 protein in the lung, as normalized to HSC70 as a housekeeping protein, did not reduce over three days of drug treatment (Figure 3F). In the brain, similar inhibition profiles for LRRK2 were observed, albeit higher plasma levels of drug (i.e., 50 nM) were required for the complete dephosphorylation of LRRK2 in the brain, an unexpected result given the in vitro binding data and that MLi2 is highly (~75%) brain penetrant (Figure 3G). In contrast to LRRK2 expressed in the lung that can be fully dephosphorylated, higher concentrations of PF-360 administration reduce total LRRK2 protein in the brain nearly to the same extent as reducing the amount of phosphorylated LRRK2 protein. In contrast, MLi2 compound did not show these trends (Figure 3I). Of note, we did not achieve concentrations of MLi2 or PF-360 higher than 200 or 100 nM, respectively, an order-of-magnitude more than Cmax concentrations associated with oral gavage strategies. These results show that WT-LRRK2 kinase activity can be blocked without reducing levels of total protein (i.e., in the lung).
G2019S-LRRK2 is resilient to kinase inhibition
Previously we described a transgenic rat developed by the Michael J. Fox Foundation that expresses human G2019S-LRRK2 protein from a human BAC construct, with neuronal expression patterns that closely mimic the distribution of LRRK2 protein in human brains but greatly differs from endogenous rat LRRK2 distribution (West et al., 2014). G2019S-LRRK2 rats show ~10-fold overexpression of LRRK2 compared to endogenous rat LRRK2, although transgenic expression is detected in many neurons throughout the brain that lack any endogenous rat LRRK2 owing to the human LRRK2 promoter (West et al., 2014). To explore this model as a surrogate for G2019S-LRRK2 protein in humans that carry the G2019S-LRRK2 mutation, we included the MLi2 and PF-360 medicated chows for three-days in cohorts of G2019S-LRRK2 expressing and non-transgenic littermate rats (Figure 4). As expected, plasma levels of drug in G2019S-LRRK2 rats treated with the different chows was identical to that of non-transgenic rats.
Figure 4. G2019S-LRRK2 is resilient to chronic kinase inhibition in rats.
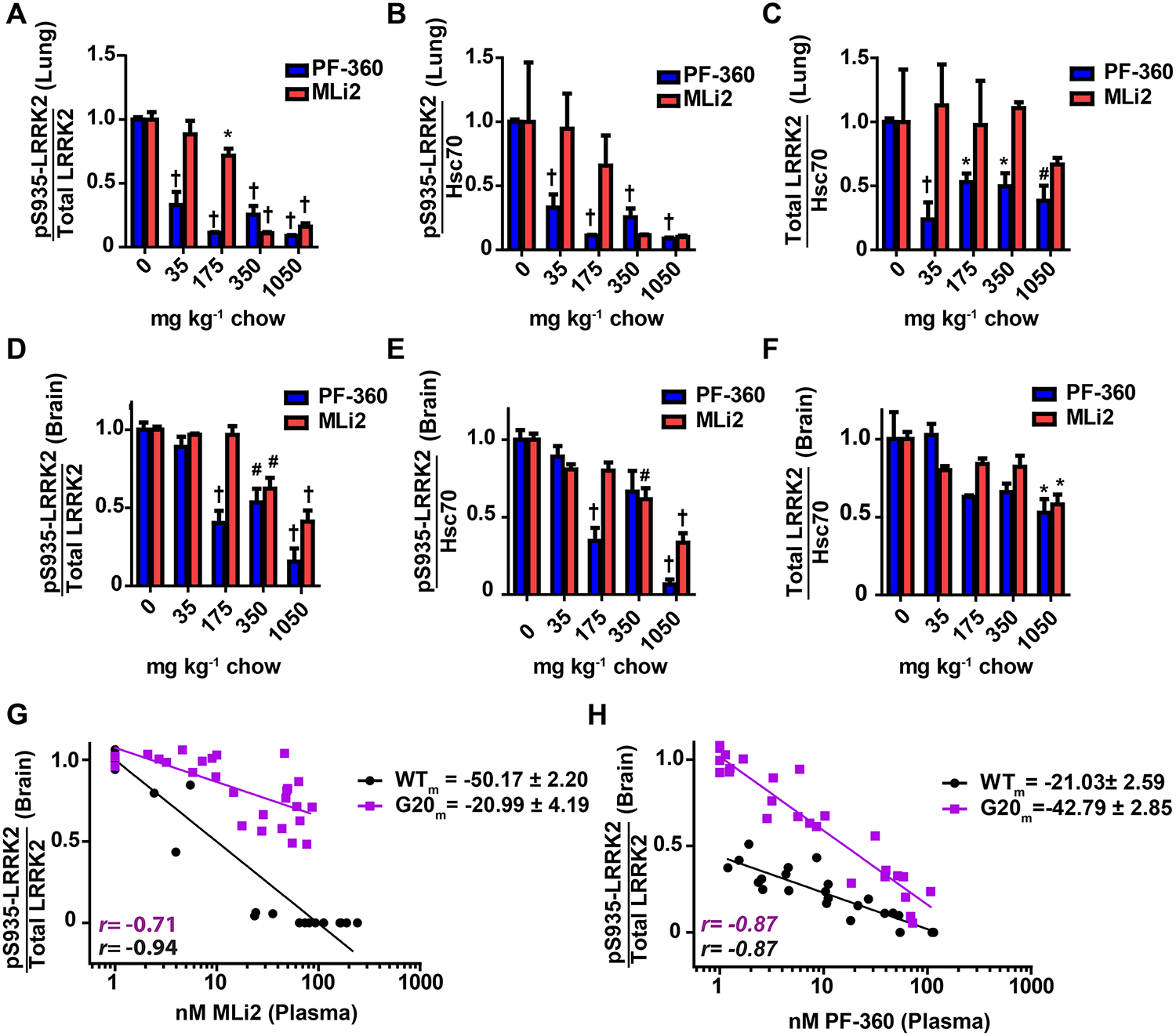
G2019S-LRRK2 expressing rats were cohoused in groups of three and had ad libitum access to food containing 35, 175, 350, or 1050 milligram of compound (red: MLi2 or blue: PF-360) per kilogram of chow. Animals were dosed for three days and sacrificed two hours into their light cycle. Whole lung and brain were collected and processed immediately into protein lysate. Quantification of A, the ratio of pS935-LRRK2 to total LRRK2, B, pS935-LRRK2 levels, and C, total LRRK2 in the lung was determined by western blot. Quantification of D, the ratio of pS935-LRRK2 to total LRRK2, E, pS935-LRRK2 levels, and F, total LRRK2 in the brain was determined by protein capillary electrophoresis. Hsc70 was used as a loading control for normalization. Significance reported as treatment compared to untreated control * p<0.05, #p<0.01,† p<0.0001, as calculated by one-way analysis of variance with Dunnets’s post-hoc test (A, B, C, D, E, F). All other group comparisons were not significant (p>0.05) compared with control. Data are means ± S.E.M. The correlation between plasma drug levels and LRRK2 kinase inhibition (ratio of pS935-LRRK2 to total LRRK2) illustrates genotypic differences in kinase inhibition profiles for G, MLi2 and H, PF-360. Slopes for each correlation are reported and Pearson correlation coefficients are shown with corresponding r values (black: WT-LRRK2, purple: G2019S-LRRK2).
G2019S-LRRK2 expressed in lung demonstrated similar inhibition profiles as WT-LRRK2 in non-transgenic rats, although a very small (<5%) signal for pS935-G2019S-LRRK2 was detected even at the highest drug concentrations (Figure 4A). Total G2019S-LRRK2 expression in the transgenic rats varies much more than endogenous LRRK2 in the non-transgenic rats, and a trend towards lower LRRK2 protein expression in the lung in PF-360 treated rats was noted but the effect did not appear dose-responsive (Figure 4C). In contrast to G2019S-LRRK2 inhibition in the lung, G2019S-LRRK2 inhibition in the brain was unexpectedly weak compared to the levels of WT-LRRK2 inhibition observed in the brain in non-transgenic rats treated with the same chows (Figure 4D). At the highest levels of drug (e.g., >50 nM), significant levels of pS935-LRRK2 protein remained in both MLi2 and PF-360 treated G2019S-LRRK2 rats. MLi2 poorly blocked G2019S-LRRK2 compared to PF-360. As PF-360 inhibited more G2019S-LRRK2 than MLi2, in vitro predictions of weaker PF-360 binding to G2019S-LRRK2 compared to MLi2 (Figure 1) fail to manifest in vivo. Unexpectedly, the highest concentrations of MLi2 measured in the plasma with the 1050-mg kg−1 chow (i.e., ~ 200 nM, or at least 10-times IC50 values, Figure 1) results in less than half the inhibition of pS935-LRRK2 levels in G2019S-LRRK2 total protein lysates. In contrast, pS935-LRRK2 is easily ablated in non-transgenic rats treated with the same chows. PF-360 treatments thus fared better in blocking G2019S-LRRK2 at the highest concentrations but again was considerably less efficient compared to action in non-transgenic rats (Figure 4H). Overall, these results indicate that G2019S-LRRK2 protein in the brain is more resilient to inhibition compared to WT-LRRK2 protein in non-transgenic rats.
Through post-mortem examinations of these treated rats, we noticed that non-transgenic rats treated with MLi2 or PF-360 chows that offered continuous plasma concentrations of compounds greater than ~50 nM (e.g., >IC90) discolored the kidney cortex and medulla after several weeks of exposure (21 days). In contrast, none of the G2019S-LRRK2 expressing rats in our study had a perceivable discoloration phenotype (Figure 5A). Previously we and others identified vacuoles in renal tubule cells and pigmented lipofuscin and blood-product deposits in LRRK2 knockout rats (Boddu et al., 2015). This phenotype is robust in rats and appears shortly after weaning, whereas the phenotype takes months or more than a year to develop in mice (Baptista et al., 2013; Herzig et al., 2011; Tong et al., 2012; Tong et al., 2010). Similar to the lung lysates analyzed from non-transgenic and G2019S-LRRK2 rats in Figure 4, at the higher concentrations of the LRRK2 inhibitors MLi2 and PF-360, there was essentially no phospho-LRRK2 remaining in total protein lysates from kidney from both non-transgenic and G2019S-LRRK2 rats (Figure 5B,C). Quantitative analysis of fluorescent lipofuscin and blood product deposition revealed a similar abnormal brown/red coloration in rats treated with medicated chow as compared to comparably aged LRRK2 knockout rats (Figure 5E). Manual counts of abnormal vacuoles within the tubule cells revealed that nearly all LRRK2 knockout rat tubule cells are abnormal in comparison to ~25% of non-transgenic rats treated with a LRRK2 inhibitor (Figure 5F). Counts of the number of tubule cells demonstrating deposits of brown pigment nearby the tubule cells show that ~50% of cells from LRRK2 knockout rats are positive compared to ~15% of non-transgenic rats treated with inhibitor (Figure 5E). In these phenotypes, G2019S-LRRK2 expressing rats were fully resistant to the effects of the LRRK2 kinase inhibitors.
Figure 5. G2019S-LRRK2 protects from kinase inhibitor-induced lysosomal defects.
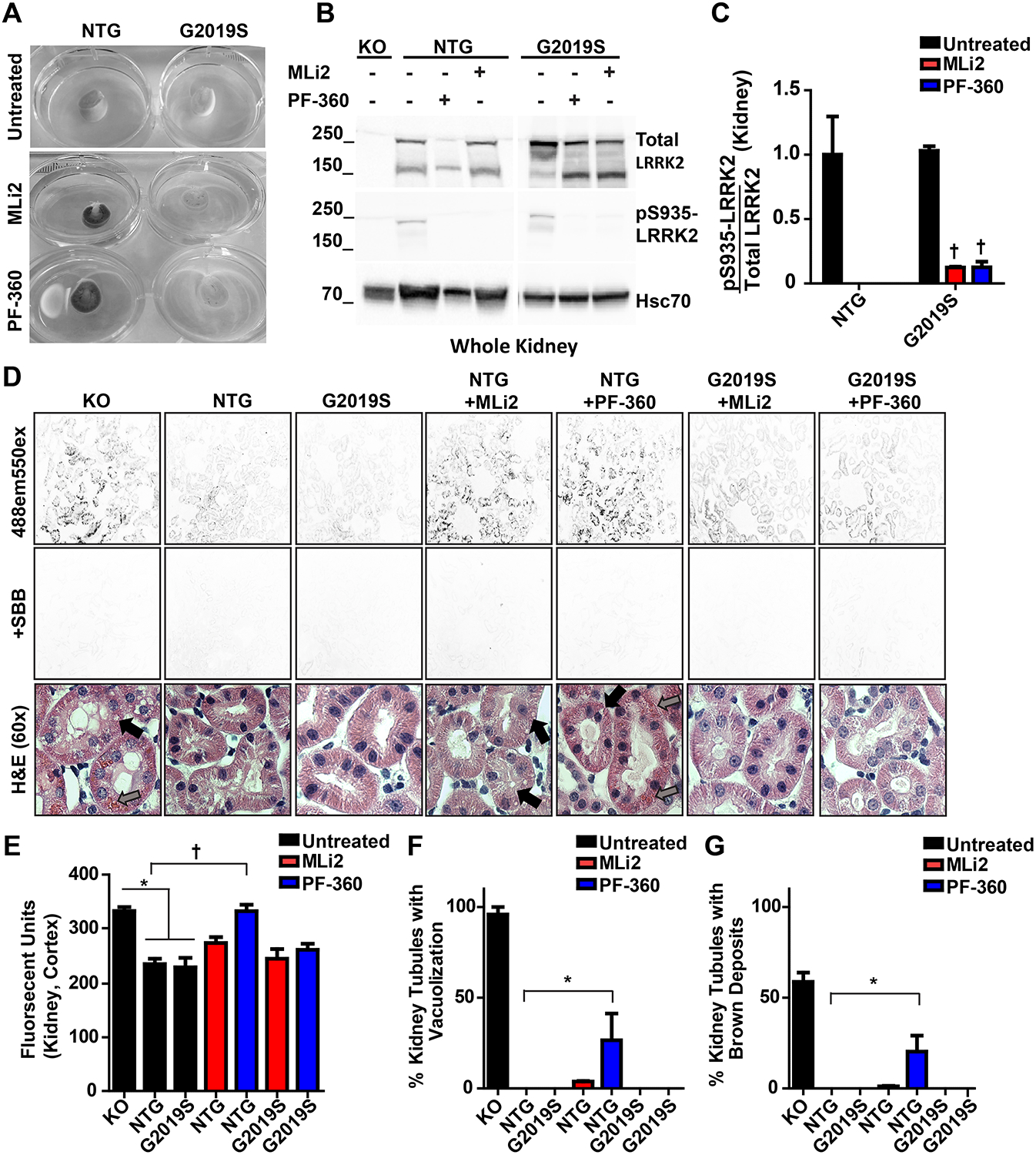
A, Cross-sectional view of kidneys from non-transgenic and G2019S-LRRK2 rats chronically exposed to MLi2 (red) and PF-360 (blue). B, representative western blot and C, quantification of LRRK2 kinase inhibition in kidneys of rats exposed to 175 mg kg−1 PF-360 chow for three weeks. D, representative images of kidney lipofuscin autofluorescence and H&E stain. The autofluorescence of lipofuscin was completely blocked in all conditions by Sudan Black B dye. Images were obtained with a 488 nm laser and 520–540 nm emission collection, and quantified using Image J. F, counts of vacuolization (black arrows) and brown deposits (grey arrows) in kidney tubules observed during histological analysis. Ten images were acquired per sample with an average of 12 tubules analyzed per frame. *p<0.05,† p<0.0001 calculated by one-way analysis of variance with Dunnet’s post-hoc test (C) and Tukey’s post-hoc test (E, F, G). All other group comparisons were not significant (p>0.05) compared with control groups. Data are presented as means ± S.E.M.
One caveat to interpreting the G2019S-LRRK2 expressing rats compared to littermate WT-LRRK2 controls is that G2019S-LRRK2 expression is driven from the human LRRK2 promoter and expresses much higher than endogenous LRRK2 protein in rats in many cells in the brain (West et al., 2014). While higher levels of protein kinases are not thought to necessitate higher levels of drugs for inhibition (Knight and Shokat, 2005), recently, G2019S-LRRK2 knock-in (KI) mice have become available and appear to have comparable LRRK2 distribution and expression compared to littermate controls (Matikainen-Ankney et al., 2016). We applied the same chows used in the rats in homozygous G2019S-LRRK2 KI mice, congenic on a C57BL6J background, and compared inhibition profiles to those generated in C57BL6J mice. A complete loss of phospho-LRRK2 protein levels at the highest concentrations of drug in WT-LRRK2 could be observed, similar to observations in non-transgenic rats (Figure 6A). However, in contrast to WT-LRRK2 in the rat brain, a loss of WT-LRRK2 protein was not detected in mouse brain, consistent with results reported in other studies using MLi2 in-diet in mice (Fell et al., 2015). In mice homozygous for G2019S-LRRK2, similar to G2019S-LRRK2 rats, concentrations of MLi2 that resulted in the complete ablation of brain pS935-LRRK2 levels in WT-LRRK2-expressing mice significantly but incompletely lowered pS935-LRRK2 levels with our highest concentration of chow (Figure 6D). These results support the relative resiliency of G2019S-LRRK2 to full inhibition at these concentrations of drug. Also consistent with observations in rats, PF-360 treatment was more effective at blocking pS935-LRRK2 levels in G2019S-LRRK2 KI mice despite the lower affinity PF-360 shows for binding to G2019S-LRRK2 in vitro, with residual pS935-LRRK2 levels detected in G2019S-LRRK2 KI mice at the highest levels of compounds (Figure 6D). Although there were trends towards a reduction of total levels of LRRK2 protein, these results were modest and not statistically significant given the normal variability of LRRK2 expression between animals (Figure 6). In sum, G2019S-LRRK2 protein in the brain demonstrates resiliency to kinase inhibition in two different species with two different inhibitors. While LRRK2 knockout rats robustly develop discolored kidneys early, mice appear to be more resistant to this phenotype with darkened kidneys not apparent until a year or longer in age (Baptista et al., 2013; Boddu et al., 2015; Tong et al., 2012). In contrast, young LRRK2 knockout mice develop vacuoles (i.e., lamellar bodies) in lung type II pneumocytes whereas LRRK2 knockout rats do not develop the same phenotype, or at least to the same extent, with several years of aging (Baptista et al., 2013; Herzig et al., 2011). We evaluated lung tissue from mice treated with LRRK2 inhibitors in both C57BL6J and homozygous G2019S-LRRK2 KI mice. As a control, age-matched LRRK2 knockout mice were analyzed in parallel. Abnormal lamellar body accumulations could be detected in ~30% of lung epithelial cells in 12-week old LRRK2 knockout mice. Similarly-aged non-transgenic or G2019S-LRRK2 mice treated with compounds for up to 21 days did not result in the development of any apparent lung phenotype (Figure 6G). Further, the kidneys in mice treated with inhibitor did not show any evidence of darkening or tubule cell vacuoles.
Figure 6. Lack of LRRK2 inhibitors in causing lung lysosomal defects in mice.
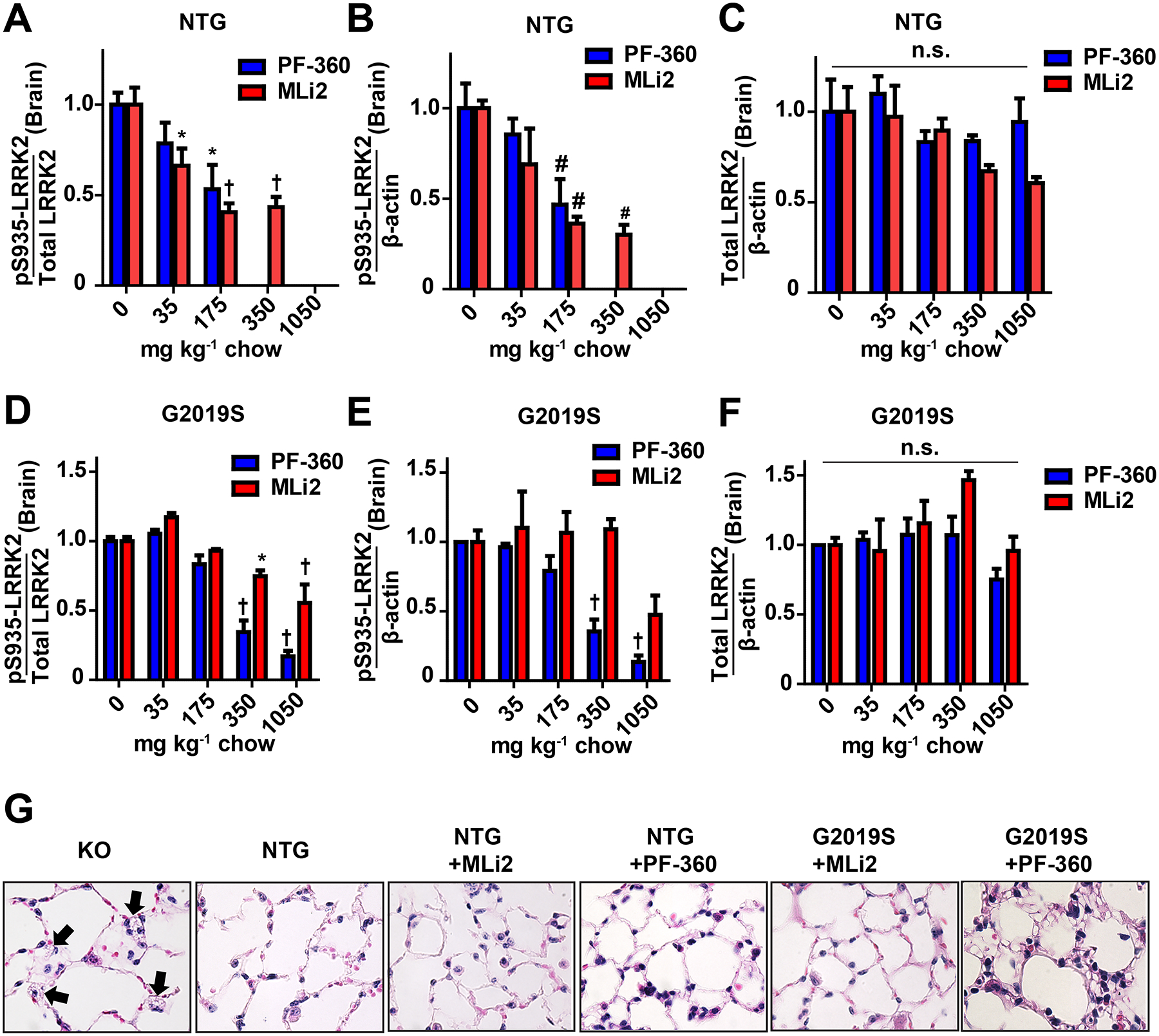
Brain LRRK2 inhibition profiles for G2019S-LRRK2 KI mice and non-transgenic C57BL6J mice, assessed using in-diet dosing (n=3 per group). A, the ratio of pS935-LRRK2 to total LRRK2, B, phosphorylated LRRK2 levels, and C, total LRRK2 protein levels in wild-type mice. D, the ratio of pS935-LRRK2 to total LRRK2, E, phosphorylated LRRK2 levels, and F, total LRRK2 protein levels in G2019S knock-in mice. G, representative 60x images of H&E stained inflated lung tissue from mice chronically exposed to 350 mg kg−1 MLi2 (red) and 175 mg kg−1 PF-360 (blue) for a month. KO mice (~12 weeks) exhibit micro-vacuolization in type II pneumocytes (black arrows) to a limited extent whereas control and treated mice do not. * p<0.05, #p<0.01,† p<0.0001, calculated by one-way analysis of variance with Dunnet’s post-hoc test (A, B, C, D, E, F). All other group comparisons were not significant (p>0.05) compared with control groups. Data are presented as means ± S.E.M.
LRRK2 control of pT73-Rab10 levels: limitations of pharmacodynamic profiles in G2019S-LRRK2 expressing rats and mice
Although pS935-LRRK2 (i.e., phospho-LRRK2) levels track closely with inhibition with all known LRRK2 kinase inhibitors, the pS935 phosphorylation site is not a LRRK2 autophosphorylation site but can reflect a loss of 14-3-3 binding caused by a loss of LRRK2 kinase activity. We wondered whether G2019S-LRRK2 resiliency could also be demonstrated through measures of LRRK2 autophosphorylation and a trans-LRRK2 substrate such as Rab10. Previously, we characterized the specificity and utility of measuring LRRK2 autophosphorylation via pS1292-LRRK2 in cell lines, primary cells, and clinical samples (Fraser et al., 2016b; Wang et al., 2017). Unfortunately, in our rat lysates, the monoclonal antibody we previously characterized detected a very strong signal in the LRRK2 knockout lysates at near the same molecular weight as LRRK2, obviating any utility in rats. Further, in G2019S-LRRK2 knock-in mice, a very strong non-specific band was present in all lysates analyzed corresponding to a protein only slightly larger than LRRK2 (Supplemental Figure 2). In measuring this pS1292-LRRK2 band in these lysates, we observed a very close (r>0.9) relationship with pS935-LRRK2. We were not able to resolve specific pS1292-LRRK2 signal using the protein capillary electrophoresis technique, or signal from non-transgenic rats or mice that lack G2019S-LRRK2.
In Rab10, LRRK2 controls phosphorylation of pT73 in an effector loop and pRab10 has been proposed as a powerful means to measure LRRK2 activity and inhibition (Eyers, 2018). Using the same monoclonal antibody previously described (Lis et al., 2018), we isolated several types of immune cells known to express LRRK2 and treated them with the MLi2 inhibitor ex vivo (100 nM, or ~20x IC50) for two hours prior to the procurement of protein lysates. Specifically, we isolated mouse macrophage cells that we previously demonstrated express very high LRRK2 levels (Moehle et al., 2015), as well as human neutrophils as described by others (Fan et al., 2018; Thirstrup et al., 2017). Consistent with these past reports, in both macrophages and neutrophils, treatment with MLi2 virtually eliminated pT73-Rab10 signal. However, in the mixed population of peripheral blood mononuclear cells (PBMC) from the same blood isolation that includes T-cells with little or no LRRK2 expression (Thévenet et al., 2011), we could not observe a reliable reduction in pRab10 after ex vivo MLi2 treatment (Figure 7A). We also analyzed pRab10 immunofluorescent signal via confocal analysis of mouse macrophages cultured for one-week and treated with MLi2 or control and observed a complete elimination of pRab10 signal, consistent with pRab10 dependence on LRRK2 kinase activity in these cells (Figure 7B). As PBMC cell pellets are routinely procured for biomarker exploration and are considered for target engagement purposes for LRRK2-targeting therapies in the clinic (Hyland and Warners, 2017; West, 2017), we wondered whether our rats and mice, both G2019S-LRRK2 and WT-LRRK2, chronically treated with LRRK2 kinase inhibitors would demonstrate lowered levels of pRab10 protein in PBMCs. We did not observe a reduction of pRab10 in any treatment condition in any of the strains of rodents in the mixed PBMC cell population (Figure 7C,E), in comparison to reductions of pS935-LRRK2 in the same PBMC lysate (Figure 7D,F). Based on the mixed-cell populations in PBMCs, these results suggest that cell-type heterogeneity may confound interpretation of LRRK2 kinase inhibition with pRab10 signals as other protein kinases in cells of unknown composition apparently phosphorylate the same site in Rab10.
Figure 7. pT73-Rab10 as a pharmacodynamic marker of LRRK2 inhibition in rodent peripheral cells and tissue.
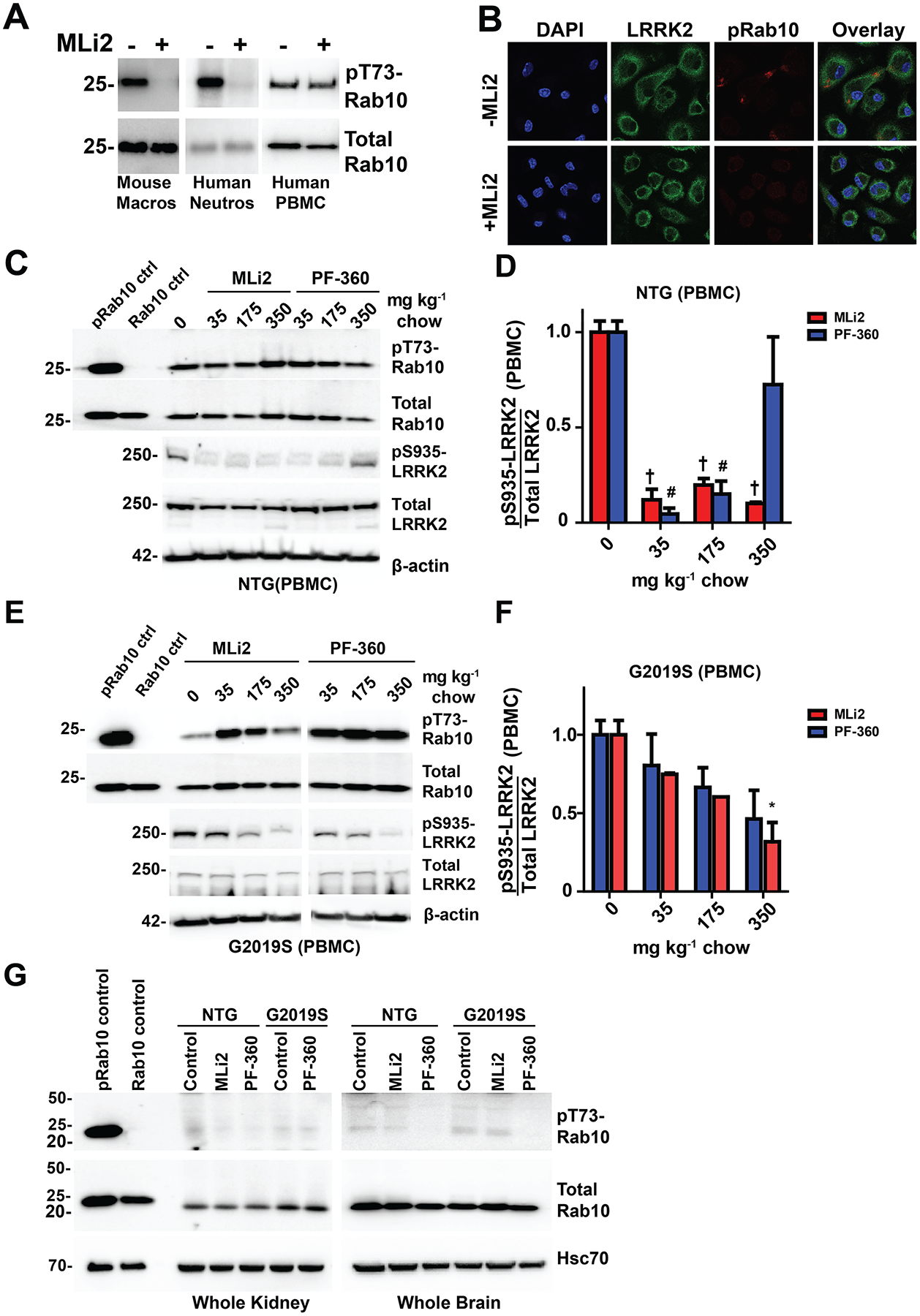
A, western blots of pT73-Rab10 and total Rab10 signal in mouse macrophages, human neutrophils, and human PBMCs. Ex vivo treatment of cells with 100 nM MLi2 ablates pT73-Rab10 in mouse macrophages and human neutrophils, but not human PBMCs. B, representative confocal images (60x) of LRRK2 and pT73-Rab10 signal in mouse macrophages treated with or without 100 nM MLi2. C, representative western blot of Rab10 and LRRK2 signal in PBMCs isolated from wild-type LRRK2 rats treated with MLi2 and PF-360 chow. D, quantification of the ratio of pS935-LRRK2 to total LRRK2 in PBMCs of wild-type rats. E, representative western blot of phosphorylated and total Rab10 and LRRK2 signal in PBMCs isolated from G2019S-LRRK2 knock-in mice treated with MLi2 and PF-360 chow. F, quantification of the ratio of pS935-LRRK2 to total LRRK2 in PBMCs of G2019S knock-in mice. G, representative blot of phospho-Rab10 and total Rab10 signal in kidney and brain tissue of wild-type and G2019S-LRRK2 rats. The pRab10 control is HEK cell lysate co-transfected with G2019S-LRRK2 and Rab10. The Rab10 control is HEK cell lysate co-transfected with G2019S-LRRK2 and Rab10 treated with 300 nM MLi2 for 2 hours. * p<0.05, #p<0.01,† p<0.000, calculated by one-way analysis of variance with Dunnet’s post-hoc test (D, F). All other group comparisons were not significant (p>0.05) compared with control groups. Data are presented as means ± S.E.M.
Previous reports suggested high endogenous Rab10 in peripheral organs like kidney as well as the brain, and that pT73-Rab10 could be measured in these tissues (Lis et al., 2018). We can confirm very robust total Rab10 levels in both the kidney and brain of rats and mice, but reliable pT73-Rab10 signal of the expected molecular weight (in comparison to control lysates expressing pRab10) was not obtained in either our kidney or brain lysates analyzed for pS935-LRRK2 at the same time (Figure 7G). We conclude that although ex-vivo treatments of isolated cells validate pRab10 as a bone fide LRRK2 kinase substrate, measures of pRab10 in total protein lysates composed of heterogenous cells and tissues may have limited utility in interpretation of pharmacodynamic LRRK2 responses.
Discussion
As LRRK2-targeting therapeutics, particularly LRRK2 kinase inhibitors, make their way towards efficacy studies for G2019S-LRRK2 carriers in the clinic, pre-clinical models and tool compound LRRK2 kinase inhibitors may have predictive value in identifying potential problems and solutions. Towards this end, in this study, our conclusions center on four observations made with the latest small molecules and rodent models. First, we found that in-diet dosing in both rats and mice offers a convenient compound delivery method for both MLi2 and PF-360 that achieves more stable concentrations of compounds over time that allow more precise observations of dose-response relationships previously attributed to LRRK2 inhibitors. We selected MLi2 and PF-360 for these studies based on their similar drug-like properties and potencies. Plasma concentrations of both compounds can be controlled in the range of 1 to ~100 nM based on the amounts engineered into the diet. Second, we conclude that LRRK2 can be chronically and fully inhibited without reducing total LRRK2 protein levels. These results clearly decouple LRRK2 kinase activity from protein turnover. Third, we find that G2019S-LRRK2 is resistant to inhibition and kinase-inhibitor induced phenotypes. In both rats and mice, with both MLi2 and PF-360, higher compound concentrations are required to cause the dephosphorylation of G2019S-LRRK2 to a similar degree as WT-LRRK2. Finally, we conclude that LRRK2 is not the exclusive kinase that regulates phospho-Rab10. LRRK2 robustly regulates pRab10 in immune cells where LRRK2 is highly expressed, but in tissues and other cell mixtures with differential (and not ubiquitous) LRRK2 expression (PBMCs, brain, kidneys), pRab10 does not correlate with phospho-LRRK2 in response to LRRK2 kinase inhibitors.
LRRK2 is linked to several complex diseases including Parkinson disease, Crohn’s disease, and leprosy. Model systems often require chronic treatments over long durations. So far, only a few studies have reported successful chronic pharmacological LRRK2 kinase inhibition, namely our own studies with PF-475 in twice daily oral gavage strategies in rats (PF-475 a close analog of PF-360 used here), and MLi2 engineered into chow used with mouse models of mitochondria mutation and degeneration (Daher et al., 2015; Fell et al., 2015). Besides MLi2 and PF-360, many other reported LRRK2 kinase inhibitors suffer from either a lack of brain penetration or inhibition of LRRK2 in the brain (West, 2017), or for those that get to the brain, a very short half-life and selectivity problems. Past seminal studies with MLi2 show that in-diet dosing is a viable strategy to solve rapid-turnover issues in mice, without evidence of toxic compound accumulations over time. We extend these observations to rats, as well as use the same strategy for a more novel compound developed by Pfizer from the described pyrrolopyrimidine class, dubbed PF-360. The in-diet dosing descriptions here can be deployed in any variety of chronic model systems where feeding behavior is not grossly perturbed.
Through observations born from two structurally unique compounds that interact in the LRRK2 ATP pocket in slightly different ways, we can be more confident in our conclusions. Studies in cell culture with earlier series of LRRK2 kinase inhibitors (e.g., second-generation inhibitors, (Deng et al., 2011; Doggett et al., 2012)) suggest that LRRK2 kinase activity exquisitely controls LRRK2 expression levels. In vivo this appears to not be true. At best, the highest levels of PF-360 compound are associated with nominal decreases in total LRRK2 protein levels but the effects were not seen with MLi2. In silico predictions demonstrate that interaction with the flexible loop segment may distinguish PF-360 from MLi2, and we hypothesize blocking the flexibility of the activation loop may stabilize a conformation of LRRK2 protein more prone to turn-over. Further studies are required to test these predictions using larger numbers of molecules that differentially interact in the pocket in similar ways. However, with MLi2 as well as lower concentrations of PF-360, we can conclude that LRRK2 kinase activity is by and large decoupled from LRRK2 protein turnover.
Our past oral dosing strategies, such as those we described for PF-475 (Daher et al., 2015), result in peaks of compound concentrations in the micromolar range that rapidly drop to low nanomolar levels over the course of hours. At the micromolar levels, all ATP-competitive compounds have larger probabilities for off-target binding and unpredictable effects (Knight and Shokat, 2005). Through stabilized compound concentrations from in-diet dosing, we can make a number of conclusions of dose-response relationships that would be difficult or impossible to make with traditional oral dosing. The most important of these relationships that were uncovered here show that the G2019S-LRRK2 protein is more resilient to inhibition with both MLi2 and PF-360. As PF-360 shows weaker affinity to G2019S-LRRK2 protein, differential binding affinity to the LRRK2 ATP-pocket could explain these results. However, MLi2 is agnostic (in vitro) to binding G2019S versus WT-LRRK2 and performs worse than PF-360 in inhibiting G2019S-LRRK2 in every tissue and cell type measured. It is possible that the G2019S-LRRK2 mutation, occurring in the ATP-pocket, may stabilize a conformation or endogenous protein complex not present in vitro that shows lower affinity towards MLi2 and PF-360. Additional studies that explore mechanisms of LRRK2 kinase activation such as dimerization and complex formation in the endolysosomal system may reveal the mechanisms of G2019S-LRRK2 differential sensitivity to inhibition.
The G2019S-LRRK2 mutation occurs in the heterozygous state in humans susceptible to PD. Because there are no means yet to separate G2019S-LRRK2 from WT-LRRK2 inhibition when both proteins are expressed (i.e., heterozygous G2019S), we utilized homozygous knock-in mice that only express G2019S-LRRK2, and rats that primarily express G2019S-LRRK2 (>90% of total LRRK2) in comparisons to WT-LRRK2 found in non-transgenic rodents. The rats, as opposed to the mice, over-express LRRK2 protein as a cost of the BAC system and human isoform driven by the human LRRK2 promoter. Relevant to the conclusions made here in rats, the potency of a kinase inhibitor is generally independent of the concentration of the kinase in cells and tissue, as cells and tissue that have more kinase expression do not typically need more drug to block the target (Knight and Shokat, 2005). Even in extreme differences of protein kinase expression, the binding of soluble and diffusible inhibitors to abundant proteins will drive normalization of the concentration gradient of unbound inhibitor. Thus, we attribute pharmacodynamic differences between rat WT-LRRK2 and G2019S-LRRK2 to either the presence of the mutation or the human isoform of LRRK2. As results in the rats were largely corroborated with the G2019S-LRRK2 mouse knock-in model, we can reasonably attribute differences to the presence of the G2019S-LRRK2 mutation.
An important implication of our results is that in measures of G2019S-LRRK2 in the heterozygous state, WT-LRRK2 might be fully inhibited with only partial inhibition of G2019S-LRRK2. Therefore, higher concentrations of inhibitor may be required to inhibit G2019S-LRRK2 for the same duration as WT-LRRK2. Even though tools do not yet exist to separate G2019S-LRRK2 from WT-LRRK2 in pharmacodynamic profiles, we predict that careful observations of phospho-LRRK2 protein in PBMCs and exosomes from cerebral spinal fluid and urinary exosomes will give some assurances that at least partial G2019S-LRRK2 inhibition can be obtained, especially when phospho-LRRK2 levels dip below 50% of baseline.
A difference in our study versus past reports is that we heavily utilize a protein quantification strategy that measures all SDS-solubilized protein available in cells, not just the protein fraction associated with high solubility often required in some antibody-capture methods (e.g., ELISA). The protein capillary electrophoresis methodology we developed here is one of few known methodologies available (others often reliant on mass spectrometry) that are compatible with the higher concentrations of SDS and DTT required to measure insoluble protein fractions. As the technique is scalable to the 96-well format, our data indicate the method could be adapted to a clinical setting utilizing difficult-to digest exosomes and viscous PBMC lysates as abundant and reliable sources of LRRK2 protein from as little as a few milliliters of blood or biofluid. With measures of total LRRK2, more confidence can be attributed to pharmacodynamic studies and whether thresholds for kinase inhibition were met.
Recent controversies have developed over the safety of targeting LRRK2. Defects in lysosomes in kidneys (in LRRK2 knockout rats) and lungs (in LRRK2 knockout mice) have been described. During the course of our studies, a report described the development of darkened kidneys in rats treated via oral gavage with PF-360 but did not quantify the differences (Andersen et al., 2018). We recapitulated this observation in WT-LRRK2 but not G2019S-LRRK2 expressing rats using three quantitative assays: autofluorescence associated with oxidized lipids and blood products, proportion of abnormal vacuolization of tubule cells, and number of tubule cells juxtaposed to deposits of brown/red pigment. Consistent with measures of phospho-LRRK2 in other tissues that suggests G2019S-LRRK2 is resilient to LRRK2 kinase inhibition, the expression of mutant G2019S-LRRK2 in the rats completely prevented these phenotypes in the kidney. These results may imply that safety profiles may be enhanced in LRRK2 mutation carriers versus controls. Specifically, we predict that mutation carriers may tolerate the on-target effects of LRRK2 kinase inhibitors better than non-mutation carriers.
Lastly, we explore the utility of phospho-Rab10 as well as pS1292-LRRK2 as possible pharmacodynamic replacements for pS935-LRRK2 measures which are indirect. In the tissue lysates used here, pS1292-LRRK2 antibodies unfortunately demonstrated off-target binding to proteins similarly sized to LRRK2, limiting utility. Rab10 is an attractive target as a bone fide trans LRRK2 substrate that may reflect biological pathways relevant to disease. Our results in multiple cells and tissues show that phospho-Rab10 may be an overall unreliable protein to detect LRRK2 kinase inhibition, plausibly due to the more ubiquitous and much stronger expression of Rab10 compared to weak and cell type restricted expression of LRRK2. In macrophages and neutrophils, LRRK2 expression and activity are high and kinase inhibition completely blocked phospho-Rab10. However, results in PBMCs as well as brain and kidney lysates were disappointing. We can conclude that there are other kinases that must phosphorylate Rab10, especially in cells that lack LRRK2 expression. These results are partially consistent with those that show LRRK2 inhibitors partially and not fully reduce pRab10 levels in both isolated neutrophils, monocytes, and tissue sources of LRRK2 (Bliederhaeuser et al., 2016; Thirstrup et al., 2017). We suggest caution in interpretation of pRab10 reductions in understanding LRRK2 kinase inhibition.
This study highlights how in vitro assays of LRRK2 binding characteristics and inhibition in cell lines does not necessarily predict in vivo performance. Through more careful approaches and methodical and adaptive designs, successful (at least partial) engagement of G2019S-LRRK2 seems possible. Further studies in model systems of disease, potentially using the tools and observations developed here, can help resolve dose-response relationships in achieving beneficial phenotypes as well as uncover the pathogenic effects of the G2019S mutation.
Supplementary Material
Highlights.
Distinct LRRK2 inhibitors MLi2 and PF-360 show similar PK and potency
Chronic LRRK2 inhibition is achieved using in-diet dosing in rats and mice
LRRK2 can be chronically and fully inhibited without reducing total LRRK2 levels
G2019S-LRRK2 is resilient to inhibition and blocks inhibitor induced phenotypes
Acknowledgements
The authors thank Xianzhen Hu and Yijian Zhang for rat and mouse husbandry and genotyping.
Funding
This work was supported by R33 NS097643 from the National Institute of Neurological Disorders and Stroke, Division of Translational Research, and R01 NS064934. Additional support was provided by the Michael J. Fox Foundation and the Alabama Drug Discovery Alliance.
Abbreviations
- LRRK2
Leucine-rich repeat kinase 2
- PD
Parkinson disease
- ATP
adenosine-tri phosphate
- DTT
dithiothreitol
- HSC70
Heat-shock cognate 70
- Rab
Ras gene from rat brain, or Ras-related in brain
References
- Andersen MA, Wegener KM, Larsen S, Badolo L, Smith GP, Jeggo R, Jensen PH, Sotty F, Christensen KV, Thougaard A, 2018. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology 395, 15–22. [DOI] [PubMed] [Google Scholar]
- Baptista M, Merchant K, Bharghava S, Bryce D, Ellis M, Estrada A, Fell M, Fuji R, Galatsis P, Hill S, Hirst W, Houle C, Kennedy M, Liu X, Maddess M, Markgraf C, Mei H, Needle E, Steyn S, Yi Z, Yu H, Fiske B, Sherer T, 2015. LRRK2 Kinase Inhibitors of Different Structural Classes Induce Abnormal Accumulation of Lamellar Bodies in Type II Pneumocytes in Non-Human Primates but are Reversible and Without Pulmonary Functional Consequences. 45th International Society for Neuroscience Meeting 763.02. [Google Scholar]
- Baptista MA, Dave KD, Frasier MA, Sherer TB, Greeley M, Beck MJ, Varsho JS, Parker GA, Moore C, Churchill MJ, Meshul CK, Fiske BK, 2013. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PloS one 8, e80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB, 2007. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC neuroscience 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliederhaeuser C, Zondler L, Grozdanov V, Ruf WP, Brenner D, Melrose HL, Bauer P, Ludolph AC, Gillardon F, Kassubek J, Weishaupt JH, Danzer KM, 2016. LRRK2 contributes to monocyte dysregulation in Parkinson’s disease. Acta Neuropathol Commun 4, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddu R, Hull TD, Bolisetty S, Hu X, Moehle MS, Daher JPL, Kamal AI, Joseph R, George JF, Agarwal A, Curtis LM, West AB, 2015. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Human molecular genetics 24, 4078–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, Grigoryan N, Factor SA, West AB, Boss JM, Tansey MG, 2017. LRRK2 levels in immune cells are increased in Parkinson’s disease. npj Parkinson’s Disease 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, Hirst WD, West AB, 2015. Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates alpha-Synuclein Gene-induced Neurodegeneration. J Biol Chem 290, 19433–19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, Hinkle KM, Sukar NN, Sepulveda B, Mesias R, Serrano G, Alessi DR, Beach TG, Benson DL, White CL, Cowell RM, Das SS, West AB, Melrose HL, 2013. Comprehensive characterization and optimization of anti-LRRK2 (leucine-rich repeat kinase 2) monoclonal antibodies. The Biochemical journal 453, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, Gray NS, 2011. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nature chemical biology 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett EA, Zhao J, Mork CN, Hu D, Nichols RJ, 2012. Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinson’s disease mutations and LRRK2 pharmacological inhibition. Journal of neurochemistry 120, 37–45. [DOI] [PubMed] [Google Scholar]
- Dusonchet J, Kochubey O, Stafa K, Young SM Jr., Zufferey R, Moore DJ, Schneider BL, Aebischer P, 2011. A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J Neurosci 31, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers PA, 2018. Back to the future: new target-validated Rab antibodies for evaluating LRRK2 signalling in cell biology and Parkinson’s disease. The Biochemical journal 475, 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Howden AJM, Sarhan AR, Lis P, Ito G, Martinez TN, Brockmann K, Gasser T, Alessi DR, Sammler EM, 2018. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. The Biochemical journal 475, 23–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, Hyde LA, Lin Y, Markgraf CG, Mei H, Miller M, Poulet FM, Scott JD, Smith MD, Yin Z, Zhou X, Parker EM, Kennedy ME, Morrow JA, 2015. MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. The Journal of pharmacology and experimental therapeutics 355, 397–409. [DOI] [PubMed] [Google Scholar]
- Fraser KB, Moehle MS, Alcalay RN, West AB, Consortium LC, 2016a. Urinary LRRK2 phosphorylation predicts parkinsonian phenotypes in G2019S LRRK2 carriers. Neurology 86, 994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser KB, Rawlins AB, Clark RG, Alcalay RN, Standaert DG, Liu N, West AB, 2016b. Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 31, 1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuji RN, Flagella M, Baca M, Baptista MA, Brodbeck J, Chan BK, Fiske BK, Honigberg L, Jubb AM, Katavolos P, Lee DW, Lewin-Koh SC, Lin T, Liu X, Liu S, Lyssikatos JP, O’Mahony J, Reichelt M, Roose-Girma M, Sheng Z, Sherer T, Smith A, Solon M, Sweeney ZK, Tarrant J, Urkowitz A, Warming S, Yaylaoglu M, Zhang S, Zhu H, Estrada AA, Watts RJ, 2015. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Science translational medicine 7, 273ra215. [DOI] [PubMed] [Google Scholar]
- Giaime E, Tong Y, Wagner LK, Yuan Y, Huang G, Shen J, 2017. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 96, 796–807 e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach BK, Ho FY, Vetter IR, van Haastert PJM, Wittinghofer A, Kortholt A, 2012. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc Natl Acad Sci USA 109, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR, 2006. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23, 329–341. [DOI] [PubMed] [Google Scholar]
- Henderson JL, Kormos BL, Hayward MM, Coffman KJ, Jasti J, Kurumbail RG, Wager TT, Verhoest PR, Noell GS, Chen Y, Needle E, Berger Z, Steyn SJ, Houle C, Hirst WD, Galatsis P, 2015. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. Journal of medicinal chemistry 58, 419–432. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR, 2011. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Human molecular genetics 20, 4209–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland L, Warners M, 2017. Denali Therapeutics Announces Advancement and Expansion of Its LRRK2 Inhibitor Clinical Program for Parkinson’s Disease. globenewswire.com, https://globenewswire.com/news-release/2017/12/20/1267310/0/en/Denali-Therapeutics-Announces-Advancement-and-Expansion-of-Its-LRRK2-Inhibitor-Clinical-Program-for-Parkinson-s-Disease.html. [Google Scholar]
- Knight ZA, Shokat KM, 2005. Features of selective kinase inhibitors. Chem Biol 12, 621–637. [DOI] [PubMed] [Google Scholar]
- Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM, 2010. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med 16, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, Tonelli F, Holton JL, Ho PW, Ho SL, Chou MY, Polinski NK, Martinez TN, Davies P, Alessi DR, 2018. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. The Biochemical journal 475, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Bender SA, Cuny GD, Sherman W, Glicksman M, Ray SS, 2013. Type II kinase inhibitors show an unexpected inhibition mode against Parkinson’s disease-linked LRRK2 mutant G2019S. Biochemistry 52, 1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Galemmo RA Jr., Fraser KB, Moehle MS, Sen S, Volpicelli-Daley LA, DeLucas LJ, Ross LJ, Valiyaveettil J, Moukha-Chafiq O, Pathak AK, Ananthan S, Kezar H, White EL, Gupta V, Maddry JA, Suto MJ, West AB, 2014. Unique functional and structural properties of the LRRK2 protein ATP-binding pocket. The Journal of biological chemistry 289, 32937–32951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobbestael E, Civiero L, De Wit T, Taymans JM, Greggio E, Baekelandt V, 2016. Pharmacological LRRK2 kinase inhibition induces LRRK2 protein destabilization and proteasomal degradation. Scientific reports 6, 33897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, Benson DL, 2016. Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J Neurosci 36, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle MS, Daher JP, Hull TD, Boddu R, Abdelmotilib HA, Mobley J, Kannarkat GT, Tansey MG, West AB, 2015. The G2019S LRRK2 mutation increases myeloid cell chemotactic responses and enhances LRRK2 binding to actin-regulatory proteins. Human molecular genetics 24, 4250–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G, 2004. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15, 661–675. [DOI] [PubMed] [Google Scholar]
- Scott JD, DeMong DE, Greshock TJ, Basu K, Dai X, Harris J, Hruza A, Li SW, Lin SI, Liu H, Macala MK, Hu Z, Mei H, Zhang H, Walsh P, Poirier M, Shi ZC, Xiao L, Agnihotri G, Baptista MA, Columbus J, Fell MJ, Hyde LA, Kuvelkar R, Lin Y, Mirescu C, Morrow JA, Yin Z, Zhang X, Zhou X, Chang RK, Embrey MW, Sanders JM, Tiscia HE, Drolet RE, Kern JT, Sur SM, Renger JJ, Bilodeau MT, Kennedy ME, Parker EM, Stamford AW, Nargund R, McCauley JA, Miller MW, 2017. Discovery of a 3-(4-Pyrimidinyl) Indazole (MLi-2), an Orally Available and Selective Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitor that Reduces Brain Kinase Activity. Journal of medicinal chemistry 60, 2983–2992. [DOI] [PubMed] [Google Scholar]
- Sen S, Webber PJ, West AB, 2009. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. The Journal of biological chemistry 284, 36346–36356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S, 2014. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ, 2011. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PloS one 6, e21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirstrup K, Dächsel JC, Oppermann FS, Williamson DS, Smith GP, Fog K, Christensen KV, 2017. Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Scientific reports 7, 10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J, 2012. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Molecular neurodegeneration 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, Shen J, 2010. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proceedings of the National Academy of Sciences of the United States of America 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Guella I, Farrer MJ, 2014. Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol 71, 1535–1539. [DOI] [PubMed] [Google Scholar]
- Tsika E, Nguyen AP, Dusonchet J, Colin P, Schneider BL, Moore DJ, 2015. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis 77, 49–61. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE, West AB, 2016. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta M, Beccano-Kelly DA, Paschall SA, Cataldi S, MacIsaac SE, Kuhlmann N, Kadgien CA, Tatarnikov I, Fox J, Khinda J, Mitchell E, Bergeron S, Melrose H, Farrer MJ, Milnerwood AJ, 2017. Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu Z, Ye T, Mabrouk OS, Maltbie T, Aasly J, West AB, 2017. Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol Commun 5, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, 2015. Ten years and counting: moving leucine-rich repeat kinase 2 inhibitors to the clinic. Movement disorders : official journal of the Movement Disorder Society 30, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, 2017. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp Neurol 298, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Cowell RM, Daher JP, Moehle MS, Hinkle KM, Melrose HL, Standaert DG, Volpicelli-Daley LA, 2014. Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. The Journal of comparative neurology 522, 2465–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Molitor TP, Langston JW, Nichols RJ, 2015. LRRK2 dephosphorylation increases its ubiquitination. The Biochemical journal 469, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.