

Abstract
Mitochondrial disease is hugely diverse with respect to associated clinical presentations and underlying genetic causes, with pathogenic variants in over 300 disease genes currently described. Approximately half of these have been discovered in the last decade due to the increasingly widespread application of next generation sequencing technologies, in particular unbiased, whole exome—and latterly, whole genome sequencing. These technologies allow more genetic data to be collected from patients with mitochondrial disorders, continually improving the diagnostic success rate in a clinical setting. Despite these significant advances, some patients still remain without a definitive genetic diagnosis. Large datasets containing many variants of unknown significance have become a major challenge with next generation sequencing strategies and these require significant functional validation to confirm pathogenicity. This interface between diagnostics and research is critical in continuing to expand the list of known pathogenic variants and concomitantly enhance our knowledge of mitochondrial biology. The increasing use of whole exome sequencing, whole genome sequencing and other “omics” techniques such as transcriptomics and proteomics will generate even more data and allow further interrogation and validation of genetic causes, including those outside of coding regions. This will improve diagnostic yields still further and emphasizes the integral role that functional assessment of variant causality plays in this process—the overarching focus of this review.
Keywords: diagnosis, mitochondrial disease, molecular mechanisms, next generation sequencing
1. INTRODUCTION
1.1. Mitochondrial disease overview
Mitochondrial disorders are the most common group of inborn errors of metabolism with an estimated minimum disease prevalence in adults of ∼12.5 per 100 000,1 and ∼4.7 per 100 000 in children.2 “Mitochondrial disease” is a collective term for many different clinical disorders united by the common features of failure of mitochondrial function and aberrant energy metabolism.3 Mitochondrial disorders can present at any age and result from pathogenic variants in either the nuclear genome (nDNA) or mitochondrial genome (mtDNA). Due to this dual genetic control of mitochondrial function, these disorders can be inherited with any inheritance pattern: sporadic, maternal, autosomal dominant, autosomal recessive or X‐linked.
Mitochondria are present in all nucleated cell types and therefore, mitochondrial disease may affect any organ or tissue in the body. Some patients have an organ specific disease—“pure” myopathy, cardiomyopathy or optic neuropathy, while others have multisystem involvement at presentation or acquire this during the course of their progressive disease. While there may be some diagnostic, and potentially prognostic, utility in categorising the myriad clinical features as particular syndromes e.g. MELAS (Mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes); MERRF (Myoclonic epilepsy with ragged red fibres); LHON (Leber hereditary optic neuropathy); NARP (Neuropathy, ataxia, and retinitis pigmentosa); Leigh Syndrome and Pearson Syndrome, the reality is that many patients do not fit easily into this syndromic classification. A further complication is that genotype‐phenotype correlations in mitochondrial disease are often poor, even within these defined syndromes. For example, Leigh syndrome, which presents as a progressive neurodegenerative disorder in childhood, exhibits marked genetic variability and is associated with pathogenic variants in more than 75 different mtDNA or nDNA genes.4, 5 Conversely, a single genotype can present with a range of phenotypes; the most common heteroplasmic pathogenic mtDNA variant, m.3243A > G, can present with a classic MELAS phenotype, but also with MIDD (Maternally‐inherited diabetes and deafness), sensorineural hearing loss, myopathy, cardiomyopathy, seizures, migraine, ataxia, cognitive impairment, bowel dysmotility, short stature, diabetes, external ophthalmoplegia or Leigh syndrome and 9% of individuals are asymptomatic.6 The vast clinical and genetic heterogeneity of mitochondrial disease coupled with poor genotype‐phenotype correlations makes the genetic diagnosis of patients a challenging task.
1.2. Mitochondrial function and genetics
Mitochondria are dynamic organelles that are responsible for the generation of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS); approximately 90% of the energy requirement of the cell is met through hydrolysis of ATP produced this way.7 However, mitochondria are also involved in many other processes including, but not limited to, iron sulfur cluster formation,8 the citric acid cycle,9 regulation of apoptosis10 and calcium homeostasis in conjunction with the endoplasmic reticulum.11
Human mtDNA is a closed‐circular molecule of 16 569 bp and encodes 37 genes; 13 polypeptides, 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs).12 The mitochondrial genome is exclusively maternally‐inherited and is present in multiple copies within cells. Cells can be homoplasmic, where all mtDNA molecules are identical, or heteroplasmic, where two (or more) variant populations of mtDNA exist within one cell. Heteroplasmy levels are a factor in determining the aforementioned clinical heterogeneity in patients harboring the common pathogenic m.3243A > G variant.13 However, heteroplasmy does not fully explain the phenotypic variability with sex14 and heritable nuclear factors15 likely to play a role.
All 13 mtDNA‐encoded proteins are essential hydrophobic components of the OXPHOS system located in the inner mitochondrial membrane (IMM). The OXPHOS system comprises five multi‐subunit complexes and two electron carriers (ubiquinone and cytochrome c). Complexes I‐IV and the electron carriers constitute the electron transport chain, which establishes an electrochemical gradient across the IMM that is then dissipated via complex V (the F1FO ATP synthase) to synthesize ATP. Individual OXPHOS complexes can also combine into larger complexes; the crystal structures of some of these supercomplexes, including the mammalian respirasome, have recently been elucidated.16, 17
In addition to the 13 polypeptides encoded by mtDNA, more than 60 further nuclear‐encoded respiratory chain proteins are translated in the cytosol and imported into mitochondria prior to incorporation into the OXPHOS complexes. Indeed, the nuclear genome accounts for 99% of the mitochondrial proteome which is estimated to comprise 1158 total mitochondrial proteins.18 A large proportion of these proteins have important roles in OXPHOS, either directly as respiratory complex subunits, cofactors, assembly factors and substrate‐generating upstream pathways, or more indirectly, for example, factors involved in the expression of mtDNA‐encoded genes. Mitochondrial gene expression requires proteins involved in mtDNA maintenance, transcription, RNA processing/maturation, translation and post‐translational insertion into the IMM. Furthermore, all of these proteins need to be correctly targeted and imported into the mitochondria. Thus, any defects in mitochondrial protein import or the structure of the mitochondria, caused by aberrant cristae formation or abnormal membrane lipid composition, as well as factors affecting mitochondrial fission and fusion can also negatively impact the OXPHOS system. The genetic heterogeneity of mitochondrial disorders is a consequence of this wide range of proteins that impact OXPHOS function. Over 300 mitochondrial disease‐associated genes have been described to date (Figure 1)—a list that has grown enormously over the last decade largely due to the advent and application of next generation sequencing (NGS) technologies.
Figure 1.
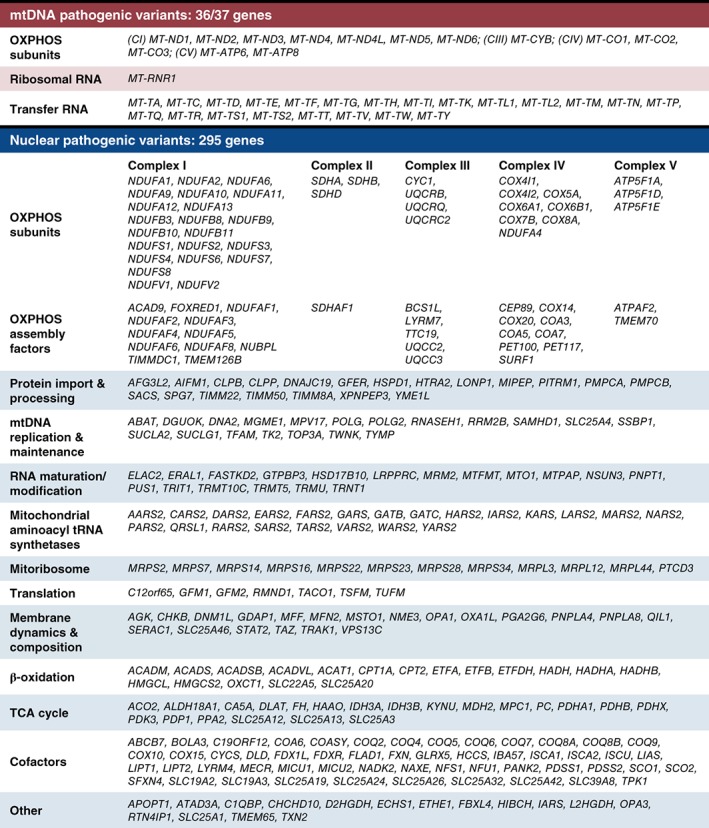
List of genes currently associated with mitochondrial disease sorted according to function. Some genes have more than one mitochondrial function, so we have used broad categories to ensure their most appropriate assignment. Our selection criteria necessitated causative genes have a primary or secondary impact on OXPHOS and does not include genes where variants have been described in cancer, but not a mitochondrial disorder (eg, SDHC). Over 150 genes linked to mitochondrial disease have been discovered since the implementation of next generation sequencing (NGS) in 2010. Today, pathogenic variants in 36/37 mitochondrial‐encoded genes and 295 nuclear‐encoded mitochondrial genes have been shown to affect mitochondrial energy metabolism, highlighting the impact NGS has had in the identification of causative genes that are associated with a wide range of mitochondrial functions
The impact of NGS in identifying novel disease genes can be illustrated using examples related to mitochondrial protein synthesis. Before the advent of widespread NGS, surprisingly few of the genes encoding proteins involved in mitochondrial protein synthesis were associated with disease. The first to be described was a variant in MRPS16 encoding a subunit of the mitoribosome.19 Only one other mitoribosomal component (MRPS22) was associated with disease20 before NGS was introduced, but since then pathogenic variants in MRPL3, 21 MRPL44, 22 MRPL12, 23 MRPS7, 24 MRPS23, 25 MRPS34, 26 MRPS2, 27 MRPS14, 28 MRPS28 29 and MRPS39 30 have been identified in patients. All were discovered by NGS methods with the exception of MRPL12 which was identified using microsatellite genotyping and Sanger sequencing.23 Similarly, prior to use of NGS, only three mitochondrial aminoacyl tRNA synthetases were established as disease genes; the first described was the aspartyl tRNA synthetase (DARS2 31) followed by the arginyl (RARS232) and tyrosyl (YARS233) aminoacyl tRNA synthetases. The remaining 14 mitochondrial tRNA synthetases (AARS2, CARS2, EARS2, FARS2, HARS2, IARS2, LARS2, MARS2, NARS2, PARS2, SARS2, TARS2, VARS2 and WARS2) plus GARS and KARS, which encode synthetases used in both the cytosol and the mitochondrion, were all identified as disease genes by NGS, with the last one to be found being WARS2.34, 35, 36 Mitochondria do not contain a glutaminyl tRNA synthetase, instead tRNAGln is first charged with Glu by EARS2 before a transamidation reaction converts the Glu‐mt‐tRNAGln to Gln‐mt‐tRNAGln. This reaction is catalyzed by GatCAB, the glutamyl‐tRNAGln amidotransferase protein complex, which consists of three proteins encoded by QRSL1, GATB and GATC respectively. Until recently, only variants in QRSL1 were associated with disease,25 but the first pathogenic variants in GATB and GATC have now been identified in patients.37 This completes the list of genes involved in mitochondrial tRNA aminoacylation associated with mitochondrial disorders.
This clearly demonstrates the impact of next generation sequencing in terms of expanding the spectrum of genes associated with disease; advances in sequencing technology will facilitate further gene discovery and this list is likely to continue expanding for some time yet.38
2. GENETIC DIAGNOSIS OF MITOCHONDRIAL DISORDERS: SEQUENCING STRATEGIES
Traditionally, initial suspicion of mitochondrial disease relies upon varied clinical observations, metabolic changes, such as increased plasma or cerebrospinal fluid (CSF) lactate or urinary 3‐methylglutaconic acid,39 and neuroimaging.40 All of these can be indicators of a mitochondrial etiology, but are not in themselves unique to mitochondrial patients; the complexity of these disorders means that a clear diagnostic algorithm can be difficult to implement. This raises a number of questions concerning diagnosis in an era when NGS is so prevalent. When to perform a muscle biopsy and integrate functional testing? Which sequencing strategy should be used? Is a “multi‐omics” approach the future of mitochondrial diagnostics? Which experiments are necessary to affirm pathogenicity of novel gene variants or candidate disease genes? Here, we will dissect the importance and application of NGS technologies and discuss the functional validation of novel disease genes. Figure 2 outlines an overview of the various stages and techniques involved in the genetic diagnosis of mitochondrial disorders and will be expanded upon throughout the remainder of this review.
Figure 2.
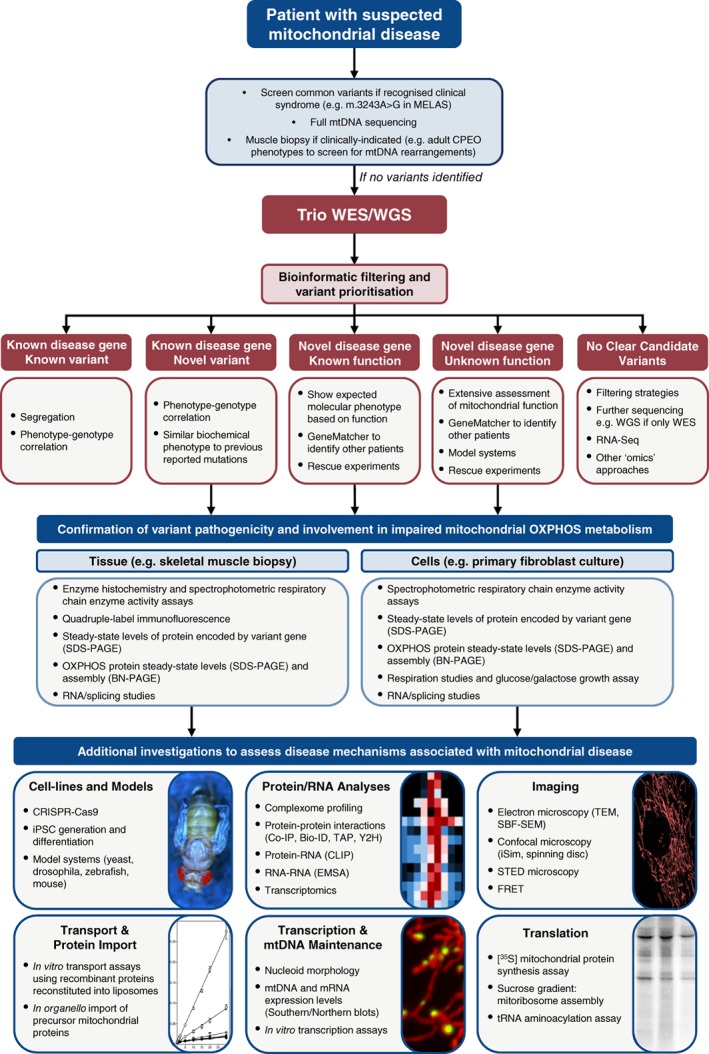
An overview of the workflow utilized to identify and validate variants associated with mitochondrial disease. First, clinical information is vital to inform appropriate genetic testing. If no mitochondrial DNA (mtDNA) or syndrome‐associated nuclear variants are identified, we advocate the use of trio whole‐exome (WES) or whole‐genome (WGS) sequencing. For each of the outcomes of WES/WGS, different levels of investigation are required to prove pathogenicity. Then, we have outlined some of the basic techniques that can be used to investigate the impact of those variants on OXPHOS metabolism using patient tissue or cells. In cases where disease mechanisms are poorly understood, these materials alongside cell and animal models can aid investigations. There is a plethora of techniques available, and instead of providing an exhaustive list we have highlighted those most commonly used, as well as gene function‐specific investigations, some of which are expanded upon in the text. Abbreviations: Co‐IP (co‐immunoprecipitation); EMSA (electrophoretic mobility shift assay); FRET (fluorescence resonance energy transfer); iPSC (induced pluripotent stem cells); MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes); MIDD (maternally inherited diabetes and deafness); OXPHOS (oxidative phosphorylation); SBF‐SEM (serial block‐face scanning electron microscopy); STED (stimulated emission depletion); TAP (transporter associated with antigen processing); TEM (transmission electron microscopy); WES (whole‐exome sequencing); WGS (whole‐genome sequencing); Y2H (yeast two‐hybrid)
Historically, genetic diagnosis of mitochondrial disease was achieved through candidate gene studies guided by histochemical and biochemical phenotyping of patient tissue, usually collected from a skeletal muscle biopsy. This has been described as a “biopsy first” or “from function to gene” approach.41 Only in cases with very clear syndromic presentations would a genetic test be carried out prior to a muscle biopsy, for example, a child presenting with MELAS would be tested for the common m.3243A > G MT‐TL1 pathogenic variant. The advent of NGS has since revolutionized the diagnosis of many rare genetic disorders, especially heterogeneous disorders such as mitochondrial disease. Over the past decade a number of NGS approaches have been successful including whole mtDNA sequencing,42 targeted gene panels (eg, complex I),43 targeted exome sequencing (“MitoExome”),44, 45 whole exome sequencing (WES),25, 46, 47, 48 whole genome sequencing (WGS)49 and RNA‐Seq.50 We will further discuss the impact and merits of each strategy below.
2.1. Whole mtDNA sequencing
Using NGS for whole mtDNA sequencing allows any mtDNA variant to be identified and gives accurate assessment of heteroplasmy levels.42 It remains common practice in many mitochondrial diagnostic centers to first sequence the mtDNA to exclude mitochondrial variants before performing WES or WGS. In adult‐onset cases, a mtDNA etiology is far more common, so whole mtDNA sequencing remains a more pragmatic option than going directly to WES/WGS. It is important to remember that many pathogenic mtDNA variants are restricted to clinically‐affected tissues such as skeletal muscle.51
2.2. Targeted gene panels
Initial application of NGS methods in mitochondrial disorders tended to sequence targeted mitochondrial gene panels that included a spectrum of genes encoding respiratory chain components and known disease‐associated genes52, 53 or more expansive panels, known as the “MitoExome,” which included all genes listed in the MitoCarta inventory.44, 45 Smaller, more focussed, gene panels have also been successful in identifying variants in known genes related to a specific clinical phenotype and OXPHOS presentation. For example, we have used a custom Ampliseq panel for genes involved in Complex I function to identify pathogenic variants in TMEM126B and NDUFA6.43, 54 The effectiveness of this approach relies upon having biochemical evidence of an isolated complex I defect, and with continued improvements in turnaround times and decreasing sequencing costs of WES, using an approach that is not restricted to a specific subset of predetermined genes would seem sensible given the number of possible disease genes associated with mitochondrial dysfunction. Indeed, at least 15 genes now associated with mitochondrial disease are not included on the wide‐ranging “MitoExome” panel55 highlighting the advantages of non‐targeted, unbiased approaches such as WES. This will no doubt change again as WGS costs are already decreasing to allow this to become more commonly used.
2.3. Gene agnostic approaches (WES and WGS)
In mitochondrial disease WES has been hugely successful improving diagnostic yields in patients with nuclear gene defects and identifying variants in novel disease genes. Many centers now report that approximately 60% of patients receive a genetic diagnosis.46, 47, 48 This success has led towards a “genetics first” approach to diagnosis which may avoid the requirement for skin or muscle biopsies entirely. The benefits of such an approach are clear if WES or WGS identifies a known pathogenic variant in a known disease gene, but this is only one of many potential outcomes and the only one that gives a firm diagnosis using sequencing alone.
Other outcomes from WES/WGS include identifying: (1) a novel variant in a known disease gene, (2) a novel variant in a known mitochondrial protein of known function that has not been previously associated with disease (3) a predicted pathogenic variant in a protein of unknown function or (4) no clear candidate variants (Figure 2). With an ever‐increasing number of variants of unknown significance (VUS) being identified, functional validation of pathogenicity is vital. Indeed, the high diagnostic rates of the aforementioned publications are partly due to WES being applied to biochemically well‐characterized cases of mitochondrial disease, which aided the prioritization of variants. For example, in one study with more heterogeneous cohorts, the diagnostic yield was less than 39% compared to 57% in the subgroup with the highest suspicion of mitochondrial disease.48
Despite the revolutionary impact of WES on the genetic diagnosis of mitochondrial disease, a significant proportion of cases (~40%) remain unresolved. This may be due to the variant being detected, but not prioritized by current bioinformatic pipelines or may be because the causative variant does not reside in the coding regions of the genome. WGS is able to detect all genetic variants and therefore has the potential to further increase the diagnostic yield. However, with the increased number of VUS identified and incomplete coverage of inherited disease genes,56 variant prioritization is a major challenge. There have been many studies assessing the annotation of variants, with examples of previously described pathogenic variants shown to be present in healthy individuals,57, 58 calling into question the accuracy of disease gene annotation. Furthermore, WGS technologies are still improving and use of technical benchmarks are required to ensure accurate interpretation of variant calls.59
Variant prioritization is key and most in‐house bioinformatic filtering pipelines should take into consideration the following: the rarity or presence of the variant in databases such as gnomAD (http://gnomad.broadinstitute.org) or ExAC60; conservation of the amino acid; modeling the amino acid change in the protein (https://zhanglab.ccmb.med.umich.edu/I-TASSER/); use of in silico tools such as SIFT61 and PolyPhen262 to predict pathogenicity of the variant. There are online tools which can take into account these factors such as Ensembl's Variant Effect Predictor.63 Bioinformatic tools will continue to improve and inclusion of such data may allow for better assessment into the authenticity of variants.
Guidelines from the American College of Medical Genetics (ACMG) aim to standardize interpretation of variants into five classifications (“pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign”) and there are five criteria listed as either “very strong” or “strong” evidence for classifying novel variants as pathogenic: (1) null variant, for example, nonsense, frameshift, splice site etc., (2) a variant resulting in the same amino acid change as an established pathogenic variant, (3) a de novo variant, where paternity and maternity have been established, (4) well‐established functional studies show damaging effect on the gene or gene product, (5) the prevalence of the variant in affected individuals is significantly higher than in controls.64 There are also six criteria listed as “moderate” and five listed as “supporting” evidence of pathogenicity which can be found in the ACMG standards and guidelines.64
When using WES or WGS, we advocate the sequencing of the family trio (ie, the patient and both unaffected parents) whenever possible. Trio sequencing is particularly powerful as it enables prioritization of de novo variants based on knowledge of segregation within the family (listed as criteria 3 of the ACMG guidelines above). Recently, there have been increasing reports of de novo dominant causes of mitochondrial disorders in cases suspected of having a recessive etiology, including variants in SLC25A4, 65, 66 ATAD3A, 67 SLC25A24, 68, 69 DNM1L, 70, 71, 72 CTBP1 73, 74 and ISCU.75 In the case of de novo SLC25A4 variants, sequencing the family trio was vital as the clinical phenotype of the patients did not resemble any of the previously reported patients with either autosomal dominant or recessive variants in SLC24A4.66 Thus, the variant had failed to be prioritized in cases where only the proband was sequenced.
Of the five ACMG criteria we have listed above (“very strong” or “strong”) at least two are usually required to classify the variant as “pathogenic” rather than “likely pathogenic” or “unknown significance” (see ACMG standards and guidelines for full classification rules64). Criteria 4 (functional studies) is the only one of these “strong” criteria where there is scope to provide additional information on the consequences of the variant that may promote its classification to a clinically actionable level. Undertaking such studies is therefore immensely valuable and central to the genetic diagnosis of many mitochondrial diseases.
3. FUNCTIONAL VALIDATION
WES is currently more widely used than WGS, but we expect this to change in the near future. There have been calls for a first‐line WGS based approach in mitochondrial disease.76 We agree that trio WGS should form a central role in the diagnostic process, but that the majority of cases will likely require some degree of functional validation (Figure 2). When a known pathogenic variant explaining the clinical phenotype has not been identified by WES/WGS, skin and skeletal muscle biopsies can be crucial. Their use has enabled identification of many novel genetic associations where functional assessment was essential for affirming pathogenicity. It is in this niche where skeletal muscle biopsy continues to prove invaluable in the diagnosis of mitochondrial disease.
3.1. Variants in mtDNA
In the case of mtDNA variants it is helpful to assess the mutation load if the variant is not homoplasmic, but it is important to do so in an appropriate tissue since mutation load can differ between tissues.13, 51 Furthermore, for novel mtDNA variants of unknown significance, the “gold standard” approach for verifying pathogenicity of a mtDNA variant is to demonstrate an observable correlation between higher mutant loads and severity of biochemical phenotype; this is typically done, for novel mt‐tRNA gene variants, by assessing heteroplasmy in individual COX‐positive and COX‐deficient fibers to demonstrate a functional threshold.77, 78, 79
3.2. Variants in nuclear DNA
3.2.1. Novel variants in a known disease gene
The functional workup varies on a case by case basis, particularly when variants identified via WES are novel. When those variants appear in a gene that has previously been associated with disease, the functional workup requires confirmation of segregation in the family and demonstration of a biochemical phenotype, where appropriate, in patient samples that is similar to the phenotype previously described in other patients with variants in the same gene. If the clinical and biochemical phenotypes are similar and the variant segregates with disease, then there is little doubt as to the pathogenicity of the variant.
3.2.2. Novel variants in a known mitochondrial protein not associated with disease
In cases where the results from WES have indicated a likely pathogenic variant in a gene not previously linked to disease then additional work may be required. If the function of the gene is known, then it is important to design experiments to test for an expected phenotype. For example, if the protein is known to be involved in RNA processing then demonstrating aberrant processing and increased levels of RNA precursors in patient samples can strengthen the case for the variant being pathogenic, as was demonstrated in cell lines harboring pathogenic TRMT10C (MRPP1) variants.80 The gold standard for proving pathogenicity is to perform rescue experiments by introducing a wild‐type copy of the gene into patient fibroblasts, often using a viral delivery system. Restoration of the biochemical phenotype to control levels upon expression of the wild‐type gene allows confirmation of pathogenicity.
3.2.3. Novel variants in a gene encoding a protein of unknown function
The approaches used to validate novel pathogenic variants in genes of unknown function identified by NGS approaches are similar to those described above with rescue experiments being particularly important. In these cases, patient samples can be instrumental in implicating a mitochondrial role for a gene of unknown function (for example, RMND1, 81 MGME1, 82 FBXL4 83) or elucidating a previously unknown mitochondrial function of a gene (for example, TRMT5 84 and TOP3A).85
3.2.4. No clear candidate pathogenic variants
One aspect of functional assessment that has grown with the advent of WGS is transcriptomics and proteomic approaches to complement WGS data and aid in variant prioritization. The potential of using transcriptomics (RNA sequencing [RNA‐Seq]) to tackle undiagnosed cases of mitochondrial disease has recently been assessed.50 Of 48 cases that WES had previously failed to diagnose, RNA‐Seq yielded a genetic diagnosis in five patients and candidate variants in the remaining 43, including identification of novel disease gene TIMMDC1 which encodes a complex I assembly factor.50 Additionally, Cummings and colleagues successfully diagnosed 35% of 50 unsolved rare muscle disease cases using RNA‐Seq; this approach compared patient RNA‐seq data to RNA‐seq data from 184 control skeletal muscle samples, illustrating the power required to identify significant variations.86 They also highlighted the importance of acquiring pathologically‐relevant tissue; analysis of tissue from the Genotype‐Tissue Exppression (GTEx) Consortium87 revealed that many of the most common muscle‐disease genes are associated with significantly lower expression in blood and fibroblasts compared to skeletal muscle, rendering them underpowered. Implementation of RNA‐Seq can therefore improve diagnostic rates when utilized alongside WGS when no clear variants are initially identified, but the tissue‐specificity of many disorders may render the use of RNA‐Seq case‐limited and highlights another instance where skeletal muscle biopsy may be essential.
4. METHODS TO FUNCTIONALLY VALIDATE PATHOGENICITY AND DISSECT MOLECULAR MECHANISMS
The techniques used to functionally characterize variants identified by WES also varies on a case by case basis, but there are some common approaches. Following Sanger sequencing confirmation and demonstration that the variant segregates with disease in the family, one of the first functional tests performed is western blotting of proteins from patient and control tissue samples. it is important to assess the steady‐state levels of the protein encoded by the variant gene. ACMG guidelines consider frameshift variants and those affecting splicing as loss‐of‐function alleles which are defined as pathogenic with the protein expected to be absent from affected patient tissues. Decreased steady‐state protein levels are often observed if there is a missense change, either homozygous or compound heterozygous (with another missense or nonsense variant), indicating either lower expression levels or increased turnover of the mutant protein and demonstrates a functional consequence of the identified variant. However, this is not always the case as variants can cause a decrease in function without a decrease in expression.
4.1. Protein studies
Since our definition of mitochondrial disorders generally focusses on impaired energy production, standard experiments assess the steady‐state levels of various OXPHOS complex subunits via SDS‐PAGE and immunoblotting and assembly of each complex by blue‐native PAGE. More recent advances include complexome profiling, which uses mass spectrometric analysis of 2D BN/SDS‐PAGE before hierarchical clustering to analyze the assembly of complexes and their subunits. This technique can be used to assess respiratory chain complexes43, 88 and the mitoribosome, recently showing that MRPS2‐deficiency leads to aberrant small mitoribosomal subunit formation.27 The aforementioned techniques can be achieved using either muscle biopsy or skin fibroblasts. Skeletal muscle often shows a more severe molecular phenotype and can be used to further demonstrate the pathogenic nature of variants, using oxidative enzyme histochemistry and quantitative immunohistochemistry89 but does not provide a renewable source of material for further investigation into disease mechanisms, as is the case with skin fibroblasts. For example, fibroblasts provide an opportunity to study mitochondrial translation defects that underlie an OXPHOS deficiency using [35S] labeled methionine and cysteine incorporation to assess nascent mitochondrial protein synthesis, and when mitoribosomal defects are suspected, sucrose gradients to analyze their assembly. These techniques recently revealed that homozygous MRPS14 variants diminished translation, but did not affect mitoribosomal assembly.28 An observable biochemical defect in patient fibroblasts also allows rescue experiments to be performed and, as stated earlier, these are crucial for functionally confirming the pathogenicity of novel variants in novel disease genes.
4.2. Imaging studies
There are a multitude of imaging techniques that can help further our understanding of the pathological role of novel disease genes, including transmission electron microscopy (TEM) and high‐resolution confocal imaging. These techniques in combination helped characterize the mitochondrial abnormalities associated with SLC25A46 variants in Leigh Syndrome and demonstrated the function of SLC25A46 within a mitochondrial/ER pathway involved in lipid transfer.90 Similar imaging studies were employed in the characterization of a mitochondrial neurodegenerative disorder caused by NME3 variants and demonstrated that the dual functions of NME3 (NDP‐kinase activity and a role in mitochondrial fusion) contributed to the disease mechanism.91 High‐resolution confocal microscopy in particular is playing a major role in elucidating the roles of novel mitochondrial genes in mitochondrial dynamics. Although many of these genes have not yet been described in disease etiology, a better understanding of their functions will aid prioritization of variants in genetically undiagnosed mitochondrial disease patients. Furthermore, the integration of improvements to more recently developed techniques such as serial block‐face scanning electron microscopy (SBF‐SEM) in skeletal muscle will help further our understanding of mitochondrial disease genes and disease mechanisms.92
4.3. Modeling putative disease variants
If a biochemical defect is only detectable in skeletal muscle, modeling experiments might be required to obtain more evidence of pathogenicity. One such example is the recent identification of de novo variants in SLC25A4 encoding the skeletal muscle isoform of the mitochondrial ATP/ADP carrier.65, 66 The biochemical defect was only observable in skeletal muscle, so the equivalent mutations were modeled in yeast and recombinant proteins were expressed in a bacterial system to allow assessment of ADP/ATP transport and demonstrate pathogenicity of the variants.
Limited availability of patient samples mean that other model systems including cell line models (eg, CRISPR, iPSCs) or whole organisms (eg, Drosophila, zebrafish and mice) may be used to provide supportive evidence of pathogenicity or to further investigate the molecular mechanisms of disease to further the knowledge of mitochondrial biology.
4.4. Discovery of novel genes linked to mitochondrial metabolism via genome wide CRISPR/Cas9 screening
The CRISPR/Cas9 genome editing system is a powerful tool that has accelerated our ability to discover and annotate gene functions, particularly when patient cells harboring pathogenic variants are not available. The phenotypic expression and tissue‐specificity of mitochondrial disorders also restrict our understanding of disease mechanisms occurring in different cell types and tissues, making it difficult to dissect exact functions of mitochondrially‐destined proteins. Therefore, generating either iPSC‐based disease models93 or animal models,94, 95 using genome engineering tools is important for a complete functional characterization of mitochondrial disease genes.
The discovery of new genes linked to mitochondrial metabolism through the use of functional genetic screening approaches, including genome‐wide CRISPR/Cas9 screens can aid the interpretation of variants identified by WES/WGS. A genome‐wide CRISPR/Cas9 knockout screen has been recently employed to identify genes essential for human mitochondrial oxidative phosphorylation.96 The screening strategy relied on a “death screening” using the cell death marker Annexin V and uncovered 191 high confidence genes necessary for survival in galactose rich media where cells entirely rely on OXPHOS. In addition to the known 72 OXPHOS disease genes, the screen also identified two genes absent from the MitoCarta 2.0 (TMEM261 and N6AMT1). Another high‐throughput genetic screen of 2231 genes using a CRISPR interference (CRISPRi) library identified 136 mitochondrial genes involved in mitochondrial bioenergetics.97 Additionally, the screen uncovered 20 non‐mitochondrial genes whose knockdown led to a decrease in real‐time ATP levels measured by a combined Fluorescence Resonance Energy Transfer (FRET)‐based biosensor and Fluorescence‐activated Cell Sorting (FACS) system.
The identification of a catalogue of new genes associated with mitochondrial metabolism will allow these to be factored into bioinformatic pipelines when analyzing WES/WGS data and may also provide further biochemical evidence of the mitochondrial defect, contributing to improved patient diagnosis.
4.5. Animal models
A variety of animal models (commonly including Caenorhabditis elegans, Drosophila melanogaster, zebrafish and mice) can be utilized to assess tissue specificity, disease progression and mechanisms associated with mitochondrial dysfunction. For example, in humans, pathogenic variants in TRMU (encoding the mitochondrial tRNA 5‐methylaminomethyl‐2‐thiouridylate methyltransferase) are associated with deafness‐associated mitochondrial disease but the pathophysiology was initially poorly understood.98 A CRISPR‐generated Mtu1 (homologue of human TRMU) knockout zebrafish model offered novel insights into disease mechanisms underlying the deafness seen in patients—a phenotype that was recapitulated in the fish model.95 A recent report of the first case of mitochondrial disease due to OXA1L variants utilized RNA interference of the Drosophila homologue, CG6404, to show defects in complexes I, IV, and V assembly in flies, which was consistent with what was observed in patient tissues.99 Despite the ease of use of zebrafish and Drosophila and their ability to recapitulate mitochondrial disease in certain cases, mice remain the most commonly developed models for studying mitochondrial pathophysiology and dysfunction due to the genetic and physiological similarities to humans and the potential for testing novel therapies.100 Some mouse models recapitulate human disease effectively, for example, mitochondrial translation optimization protein 1 (MTO1) knockout replicates the cardiomyopathy displayed in humans.101 The mtDNA helicase Twinkle knock‐in mouse phenotype correlates strongly with human disease presentation including progressive neurodegeneration and epileptic seizures.102 However, tissue‐specific presentations are not always recapitulated; for example, the NDUFS4 mouse model reproduces a Leigh‐like phenotype similar to that displayed by human patients, but not the characteristic basal ganglia changes.103, 104 Furthermore, some mouse models do not demonstrate corresponding biochemical phenotypes to those observed in human patients; for example, human SURF1 variants are associated with severe mitochondrial COX defects and early lethality whereas SURF1 −/− mice demonstrate increased longevity in the absence of many of the mitochondrial phenotypes associated with the human disease.105, 106 The aforementioned genes are all nuclear‐encoded highlighting the historic difficulties associated with generating faithful models of human disease related to pathogenic, heteroplasmic mtDNA variants. However, using a random mutagenesis and a phenotype‐driven approach, mice with a mutation (m.5024C > T) in the mitochondrial tRNA ALA gene have been generated,107 and more recently used to test experimental therapeutic strategies.108, 109
These studies, among many others, demonstrate animal models can be a powerful tool to study new disease genes, particularly when there is only a single patient or patient samples cannot be obtained. Moving forwards, high throughput screening using targeted CRISPR/Cas9 in zebrafish and collaborations such as the International Mouse Phenotyping Consortium will enable functional and pathobiological investigations of many genes.110, 111 This will lead to the characterization of novel phenotypes and provide candidate genes for many genetically undiagnosed clinical diseases.
4.6. “Multi‐omic” approaches to investigate gene functions
As discussed earlier, the identification of causal variants in mitochondrial disease is moving slowly towards a “bi‐omic” approach that analyses the genome and transcriptome.50, 86 However, truly “multi‐omic” approaches are emerging in a research setting that focus on investigating basic mitochondrial biology. It is easy to envisage this approach yielding elucidations of novel disease mechanisms, especially when new disease genes of unknown function are identified. A recent study used analysis of mRNA, proteins, lipids and metabolites to identify over 90 targets of the RNA‐binding protein Puf3p, and delineate its role in coordinating Coenzyme Q and OXPHOS biogenesis in Saccharomyces cerevisiae.112 Previous efforts to identify Puf3p targets delivered unclear results, as it was difficult to identify truly productive binding events; the integration of these four “omics” strategies highlight the power of a “multi‐omics” approach in elucidating the function of a protein in certain situations and we expect these approaches to be used more commonly in the future, including investigating the function of novel mitochondrial disease genes.
5. CONCLUDING REMARKS
The introduction of NGS into mainstream genetics, combined with the large number of mitochondrial disease gene candidates, means that putative pathogenic variants are now identified in many different scenarios. We recognize the many difficulties associated with assignation of pathogenicity and, in our experiences, the collaborative integration of accredited diagnostic pathways and research activity has proven vital. Clinical information can be extremely important in directing appropriate genetic testing, but we often advocate the use of WES or WGS on the basis of speed, comprehensive coverage of nuclear and mitochondrial DNA variants and simultaneous assessment of heteroplasmy. It must be reiterated that in such cases it is vital that trios are sequenced whenever possible to ensure rapid variant prioritization. Then, when novel disease genes are identified, case‐specific techniques should be undertaken to further understand disease mechanisms as outlined (Figure 2). Rescue experiments either in patient cell lines or CRISPR‐generated cell lines are particularly important in proving pathogenicity of a novel variant.
It is also worth highlighting the importance of international collaboration within the mitochondrial disease field. Identification of additional cases with similar clinical phenotypes is hugely beneficial in being able to confirm pathogenicity of variants in novel disease genes. Online tools such as GeneMatcher113 allow cases to be brought together and aid in the collaborative effort to report new genes associated with disease. Collaboration has also facilitated studies that complement those in humans, as demonstrated by the International Mouse Phenotyping Consortium.
The main output measure in a diagnostic setting should relate to the help offered to families affected by mitochondrial disease. There is no cure for mitochondrial disease so consistent and rapid diagnosis, best delivered using NGS, will ensure that these families are able to make informed decisions regarding provision of care of affected members and for many, reproductive options. It is apparent that a “multi‐omics” approach (eg, WES/WGS in addition to transcriptomics, proteomics, metabolomics, etc.) has the potential to increase even further the diagnostic yield and will no doubt result in the identification of new disease genes which will further enhance the understanding of mitochondrial biology and disease mechanisms.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Author contributions
K.T., J.J.C.: Review concept, drafting and critical revision of the manuscript, and preparation of tables and figures. R.I.C.G., F.M.R., A.P., E.L.B., C.L.A., M.O.: Drafting and critical revision of the manuscript. R.M., R.W.T.: Review concept, drafting and critical revision of the manuscript.
ACKNOWLEDGMENTS
Work in our laboratory is supported by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the Medical Research Council (MRC) Centre for Translational Research in Neuromuscular Disease, Mitochondrial Disease Patient Cohort (UK) (G0800674), the Lily Foundation, the UK NIHR Biomedical Research Centre for Aging and Age‐related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust, the MRC/EPSRC Molecular Pathology Node and the UK NHS Highly Specialised Service for Rare Mitochondrial Disorders of Adults and Children. CLA holds a National Institute for Health Research (NIHR) doctoral fellowship (NIHR‐HCS‐D12‐03‐04).
Thompson K, Collier JJ, Glasgow RIC, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis. 2020;43:36–50. 10.1002/jimd.12104
Communicating Editor: Verena Peters
Funding information Medical Research Council, Grant/Award Number: G0800674; National Institute for Health Research, Grant/Award Number: NIHR‐HCS‐D12‐03‐04; The Lily Foundation; UK NHS Highly Specialised Service for Rare Mitochondrial Disorders; Wellcome Trust, Grant/Award Number: 203105/Z/16/Z
Kyle Thompson and Jack J. Collier contributed equally to this study.
REFERENCES
- 1. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753‐759. 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905‐1912. 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 3. Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 4. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190‐203. 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- 5. Rahman S, Thorburn D. Nuclear gene‐encoded Leigh syndrome overview. In: GeneReviews® , ed. Seattle, WA: University of Washington; 2015. [Google Scholar]
- 6. Nesbitt V, Pitceathly RDS, Turnbull DM, et al. The UKMRC mitochondrial disease patient cohort study: clinical phenotypes associated with the m.3243A>G mutation—implications for diagnosis and management. J Neurol Neurosurg Psychiatr. 2013;84:936‐938. 10.1136/jnnp-2012-303528. [DOI] [PubMed] [Google Scholar]
- 7. Harris DA, Das AM. Control of Mitochondrial ATP Synthesis in the Heart. Biochem J. 1991;280:561‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rawat S, Stemmler TL. Key players and their role during mitochondrial iron‐sulfur cluster biosynthesis. Chemistry. 2011;17:746‐753. 10.1002/chem.201002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rustin P, Bourgeron T, Parfait B, Chretien D. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim Biophys Acta. 1997;22;1361(2):185‐97. [DOI] [PubMed] [Google Scholar]
- 10. Chipuk JE, Bouchier‐Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396‐1402. 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 11. de Brito OM, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 2010;29:2715‐2723. 10.1038/emboj.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457‐465. [DOI] [PubMed] [Google Scholar]
- 13. Grady JP, Pickett SJ, Ng YS, et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med. 2018;10:e8262 10.15252/emmm.201708262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mancuso M, Orsucci D, Angelini C, et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J Neurol. 2014;261:504‐510. 10.1007/s00415-013-7225-3. [DOI] [PubMed] [Google Scholar]
- 15. Pickett SJ, Grady JP, Ng YS, et al. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: the role of nuclear factors. Ann Clin Transl Neurol. 2018;5:333‐345. 10.1002/acn3.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gu J, Wu M, Guo R, et al. The architecture of the mammalian respirasome. Nature. 2016;537:639‐643. 10.1038/nature19359. [DOI] [PubMed] [Google Scholar]
- 17. Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature. 2016;537:644‐648. 10.1038/nature19774. [DOI] [PubMed] [Google Scholar]
- 18. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251‐D1257. 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller C, Saada A, Shaul N, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56:734‐738. 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- 20. Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784‐786. 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galmiche L, Serre V, Beinat M, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat. 2011;32:1225‐1231. 10.1002/humu.21562. [DOI] [PubMed] [Google Scholar]
- 22. Carroll CJ, Isohanni P, Pöyhönen R, et al. Whole‐exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet. 2013;50:151‐159. 10.1136/jmedgenet-2012-101375. [DOI] [PubMed] [Google Scholar]
- 23. Serre V, Rozanska A, Beinat M, et al. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochimica et Biophysica Acta (BBA)—Mol Basis Dis. 2013;1832:1304‐1312. 10.1016/j.bbadis.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Menezes MJ, Guo Y, Zhang J, et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum Mol Genet. 2015;24:2297‐2307. 10.1093/hmg/ddu747. [DOI] [PubMed] [Google Scholar]
- 25. Kohda M, Tokuzawa Y, Kishita Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12:e1005679 10.1371/journal.pgen.1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lake NJ, Webb BD, Stroud DA, et al. Biallelic mutations in MRPS34 Lead to instability of the small Mitoribosomal subunit and Leigh syndrome. Am J Hum Genet. 2017;101:239‐254. 10.1016/j.ajhg.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gardeitchik T, Mohamed M, Ruzzenente B, et al. Bi‐allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am J Hum Genet. 2018;102:685‐695. 10.1016/j.ajhg.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jackson CB, Huemer M, Bolognini R, et al. A variant in MRPS14 (uS14m) causes perinatal hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features and neurological involvement. Hum Mol Genet. 2018;28:639‐649. 10.1093/hmg/ddy374. [DOI] [PubMed] [Google Scholar]
- 29. Pulman J, Ruzzenente B, Bianchi L, et al. Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum Mol Genet. 2019. May 1;28(9):1445‐1462. 10.1093/hmg/ddy441. [DOI] [PubMed] [Google Scholar]
- 30. Borna NN, Kishita Y, Kohda M, et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics. 2019;20:1‐17. 10.1007/s10048-018-0561-9. [DOI] [PubMed] [Google Scholar]
- 31. Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl‐tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534‐539. 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 32. Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl‐transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857‐862. 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl‐tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am J Hum Genet. 2010;87:52‐59. 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burke EA, Frucht SJ, Thompson K, et al. Biallelic mutations in mitochondrial tryptophanyl‐tRNA synthetase cause Levodopa‐responsive infantile‐onset Parkinsonism. Clin Genet. 2018;93:712‐718. 10.1111/cge.13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Musante L, Püttmann L, Kahrizi K, et al. Mutations of the aminoacyl‐tRNA‐synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum Mutat. 2017;38:621‐636. 10.1002/humu.23205. [DOI] [PubMed] [Google Scholar]
- 36. Theisen BE, Rumyantseva A, Cohen JS, et al. Deficiency of WARS2, encoding mitochondrial tryptophanyl tRNA synthetase, causes severe infantile onset leukoencephalopathy. Am J Med Genet A. 2017;34:604‐2510. 10.1002/ajmg.a.38339. [DOI] [PubMed] [Google Scholar]
- 37. Friederich MW, Timal S, Powell CA, et al. Pathogenic variants in glutamyl‐tRNAGln amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat Commun. 2018;9:4065 10.1038/s41467-018-06250-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms and clues to pathology. J Biol Chem. 2019;294:5386‐5395. 10.1074/jbc.R117.809194. Epub 2017 Dec 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wortmann SB, Kluijtmans LAJ, Rodenburg RJ, et al. 3‐Methylglutaconic aciduria—lessons from 50 genes and 977 patients. J Inherit Metab Dis. 2013;36:913‐921. 10.1007/s10545-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 40. de Beaurepaire I, Grévent D, Rio M, et al. High predictive value of brain MRI imaging in primary mitochondrial respiratory chain deficiency. J Med Genet. 2018;55:378‐383. 10.1136/jmedgenet-2017-105094. [DOI] [PubMed] [Google Scholar]
- 41. Wortmann SB, Mayr JA, Nuoffer J‐M, et al. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics. 2017;48:309‐314. 10.1055/s-0037-1603776. [DOI] [PubMed] [Google Scholar]
- 42. Tang S, Wang J, Zhang VW, et al. Transition to next generation analysis of the whole mitochondrial genome: a summary of molecular defects. Hum Mutat. 2013;34:882‐893. 10.1002/humu.22307. [DOI] [PubMed] [Google Scholar]
- 43. Alston CL, Compton AG, Formosa LE, et al. Biallelic mutations in TMEM126B cause severe complex I deficiency with a variable clinical phenotype. Am J Hum Genet. 2016;99:217‐227. 10.1016/j.ajhg.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next‐generation sequencing. Sci Transl Med. 2012;4:118ra10 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80:1762‐1770. 10.1212/WNL.0b013e3182918c40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pronicka E, Piekutowska‐Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: two years' experience with whole‐exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174 10.1186/s12967-016-0930-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taylor RW, Pyle A, Griffin H, et al. Use of whole‐exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. Jama. 2014;312:68‐77. 10.1001/jama.2014.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38:437‐443. 10.1007/s10545-015-9823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hartmannová H, Piherová L, Tauchmannová K, et al. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non‐coding mutation in complex I assembly factor NDUFAF6. Hum Mol Genet. 2016;25:4062‐4079. 10.1093/hmg/ddw245. [DOI] [PubMed] [Google Scholar]
- 50. Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824 10.1038/ncomms15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sue CM, Quigley A, Katsabanis S, et al. Detection of MELAS A3243G point mutation in muscle, blood and hair follicles. J Neurol Sci. 1998;161:36‐39. [DOI] [PubMed] [Google Scholar]
- 52. DaRe JT, Vasta V, Penn J, et al. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med Genet. 2013;14:118 10.1186/1471-2350-14-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Legati A, Reyes A, Nasca A, et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim Biophys Acta. 2016;1857:1326‐1335. 10.1016/j.bbabio.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 54. Alston CL, Heidler J, Dibley MG, et al. Bi‐allelic mutations in NDUFA6 establish its role in early‐onset isolated mitochondrial complex I deficiency. Am J Hum Genet. 2018;103:592‐601. 10.1016/j.ajhg.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stenton SL, Prokisch H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018;62:399‐408. 10.1042/EBC20170110. [DOI] [PubMed] [Google Scholar]
- 56. Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole‐genome sequencing. Jama. 2014;311:1035‐1045. 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haer‐Wigman L, van der Schoot V, Feenstra I, Vulto‐van Silfhout AT, Gilissen C, Brunner HG, Vissers LELM, Yntema HG (2018) 1 in 38 individuals at risk of a dominant medically actionable disease. Eur J Hum Genet 15:565. doi: 10.1038/s41431-018-0284-2, 325, 330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xue Y, Chen Y, Ayub Q, et al. Deleterious‐ and disease‐allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population‐scale resequencing. Am J Hum Genet. 2012;91:1022‐1032. 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goldfeder RL, Priest JR, Zook JM, et al. Medical implications of technical accuracy in genome sequencing. Genome Med. 2016;8:24 10.1186/s13073-016-0269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436‐446. 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 76:7.20.1‐7.20.41. 2013. 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Richards S, Aziz N, Bale S, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐24. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. King MS, Thompson K, Hopton S, et al. Expanding the phenotype of de novo SLC25A4‐linked mitochondrial disease to include mild myopathy. Neurol Genet. 2018;4:e256 10.1212/NXG.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thompson K, Majd H, Dallabona C, et al. Recurrent De novo dominant mutations in SLC25A4 cause severe early‐onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet. 2016;99:860‐876. 10.1016/j.ajhg.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harel T, Yoon WH, Garone C, et al. Recurrent De novo and Biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am J Hum Genet. 2016;99:831‐845. 10.1016/j.ajhg.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ehmke N, Graul‐Neumann L, Smorag L, et al. De novo mutations in SLC25A24 cause a Craniosynostosis syndrome with hypertrichosis, Progeroid appearance, and mitochondrial dysfunction. Am J Hum Genet. 2017;101:833‐843. 10.1016/j.ajhg.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Writzl K, Maver A, Kovačič L, et al. De novo mutations in SLC25A24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am J Hum Genet. 2017;101:844‐855. 10.1016/j.ajhg.2017.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A. 2016;170:2002‐2011. 10.1002/ajmg.a.37721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ladds E, Whitney A, Dombi E, et al. De novo DNM1L mutation associated with mitochondrial epilepsy syndrome with fever sensitivity. Neurol Genet. 2018;4:e258 10.1212/NXG.0000000000000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sheffer R, Douiev L, Edvardson S, et al. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am J Med Genet A. 2016;170:1603‐1607. 10.1002/ajmg.a.37624. [DOI] [PubMed] [Google Scholar]
- 73. Beck DB, Cho MT, Millan F, et al. A recurrent de novo CTBP1 mutation is associated with developmental delay, hypotonia, ataxia, and tooth enamel defects. Neurogenetics. 2016;17:173‐178. 10.1007/s10048-016-0482-4. [DOI] [PubMed] [Google Scholar]
- 74. Sommerville EW, Alston CL, Pyle A, et al. De novo CTBP1 variant is associated with decreased mitochondrial respiratory chain activities. Neurol Genet. 2017;3:e187 10.1212/NXG.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Legati A, Reyes A, Ceccatelli Berti C, et al. A novel de novo dominant mutation in ISCU associated with mitochondrial myopathy. J Med Genet. 2017;54:815‐824. 10.1136/jmedgenet-2017-104822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raymond FL, Horvath R, Chinnery PF. First‐line genomic diagnosis of mitochondrial disorders. Nat Rev Genet. 2018;19:399‐400. 10.1038/s41576-018-0022-1. [DOI] [PubMed] [Google Scholar]
- 77. Blakely EL, Yarham JW, Alston CL, et al. Pathogenic mitochondrial tRNA point mutations: nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum Mutat. 2013;34:1260‐1268. 10.1002/humu.22358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hardy SA, Blakely EL, Purvis AI, et al. Pathogenic mtDNA mutations causing mitochondrial myopathy: the need for muscle biopsy. Neurol Genet. 2016;2:e82 10.1212/NXG.0000000000000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yarham JW, Al‐Dosary M, Blakely EL, et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. 2011;32:1319‐1325. 10.1002/humu.21575. [DOI] [PubMed] [Google Scholar]
- 80. Metodiev MD, Thompson K, Alston CL, et al. Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet. 2016;98:993‐1000. 10.1016/j.ajhg.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Janer A, Antonicka H, Lalonde E, et al. An RMND1 mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet. 2012;91:737‐743. 10.1016/j.ajhg.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kornblum C, Nicholls TJ, Haack TB, et al. Loss‐of‐function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet. 2013;45:214‐219. 10.1038/ng.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bonnen PE, Yarham JW, Besse A, et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93:471‐481. 10.1016/j.ajhg.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Powell CA, Kopajtich R, D'Souza AR, et al. TRMT5 mutations cause a defect in post‐transcriptional modification of mitochondrial tRNA associated with multiple respiratory‐chain deficiencies. Am J Hum Genet. 2015;97:319‐328. 10.1016/j.ajhg.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nicholls TJ, Nadalutti CA, Motori E, et al. Topoisomerase 3α is required for Decatenation and segregation of human mtDNA. Mol Cell. 2018;69:9‐23.e6. 10.1016/j.molcel.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9:eaal5209 10.1126/scitranslmed.aal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. GTEx Consortium . Human genomics. The genotype‐tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648‐660. 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heide H, Bleier L, Steger M, et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538‐549. 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 89. Rocha MC, Grady JP, Grünewald A, et al. A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: understanding mechanisms and improving diagnosis. Sci Rep. 2015;5:15037 10.1038/srep15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Janer A, Prudent J, Paupe V, et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol Med. 2016;8:1019‐1038. 10.15252/emmm.201506159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen C‐W, Wang H‐L, Huang C‐W, et al. Two separate functions of NME3 critical for cell survival underlie a neurodegenerative disorder. Proc Natl Acad Sci USA. 2019;116:566‐574. 10.1073/pnas.1818629116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vincent AE, White K, Davey T, et al. Quantitative 3D mapping of the human skeletal muscle mitochondrial network. Cell Rep. 2019;26:996‐1009.e4. 10.1016/j.celrep.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Romero‐Moya D, Castaño J, Santos‐Ocaña C, Navas P, Menendez P. Generation, genome edition and characterization of iPSC lines from a patient with coenzyme Q10 deficiency harboring a heterozygous mutation in COQ4 gene. Stem Cell Res. 2017;24:144‐147. 10.1016/j.scr.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 94. Perks KL, Rossetti G, Kuznetsova I, et al. PTCD1 is required for 16S rRNA maturation complex stability and mitochondrial ribosome assembly. Cell Rep. 2018;23:127‐142. 10.1016/j.celrep.2018.03.033. [DOI] [PubMed] [Google Scholar]
- 95. Zhang Q, Zhang L, Chen D, et al. Deletion of Mtu1 (Trmu) in zebrafish revealed the essential role of tRNA modification in mitochondrial biogenesis and hearing function. Nucleic Acids Res. 2018;46:10930‐10945. 10.1093/nar/gky758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Arroyo JD, Jourdain AA, Calvo SE, et al. A genome‐wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 2016;24:875‐885. 10.1016/j.cmet.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mendelsohn BA, Bennett NK, Darch MA, et al. A high‐throughput screen of real‐time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol. 2018;16:e2004624 10.1371/journal.pbio.2004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Guan M‐X, Yan Q, Li X, et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness‐associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291‐302. 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Thompson K, Mai N, Oláhová M, et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol Med. 2018;10:e9060 10.15252/emmm.201809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ruzzenente B, Rötig A, Metodiev MD. Mouse models for mitochondrial diseases. Hum Mol Genet. 2016;25:R115‐R122. 10.1093/hmg/ddw176. [DOI] [PubMed] [Google Scholar]
- 101. Becker L, Kling E, Schiller E, et al. MTO1‐deficient mouse model mirrors the human phenotype showing complex I defect and cardiomyopathy. PLoS ONE. 2014;9:e114918 10.1371/journal.pone.0114918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tyynismaa H, Mjosund KP, Wanrooij S, et al. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late‐onset mitochondrial disease in mice. Proc Natl Acad Sci USA. 2005;102:17687‐17692. 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312‐320. 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Torraco A, Peralta S, Iommarini L, Diaz F. Mitochondrial diseases part I: mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion. 2015;21:76‐91. 10.1016/j.mito.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pulliam DA, Deepa SS, Liu Y, et al. Complex IV‐deficient Surf1(−/−) mice initiate mitochondrial stress responses. Biochem J. 2014;462:359‐371. 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhu Z, Yao J, Johns T, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337‐343. 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 107. Kauppila JHK, Baines HL, Bratic A, et al. A phenotype‐driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016;16:2980‐2990. 10.1016/j.celrep.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bacman SR, Kauppila JHK, Pereira CV, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 2018;24:1696‐1700. 10.1038/s41591-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gammage PA, Viscomi C, Simard M‐L, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med. 2018;24:1691‐1695. 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meehan TF, Conte N, West DB, et al. Disease model discovery from 3,328 gene knockouts by the international mouse phenotyping consortium. Nat Genet. 2017;49:1231‐1238. 10.1038/ng.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Varshney GK, Carrington B, Pei W, et al. A high‐throughput functional genomics workflow based on CRISPR/Cas9‐mediated targeted mutagenesis in zebrafish. Nat Protoc. 2016;11:2357‐2375. 10.1038/nprot.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lapointe CP, Stefely JA, Jochem A, et al. Multi‐omics reveal specific targets of the RNA‐binding protein Puf3p and its orchestration of mitochondrial biogenesis. Cell Syst. 2018;6:125‐135.e6. 10.1016/j.cels.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928‐930. 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]