

Abstract
Pattern recognition receptors (PRRs) are potent triggers of tissue injury following renal ischemia/reperfusion injury (IRI). Specific PRRs, such as the toll-like receptor 2 (TLR2) and the nucleotide-binding oligomerization domain-like receptors (NLRs) NOD1 and NOD2 are promising targets to abrogate inflammatory injury associated with renal IRI. Several recent reports have shown there is crosstalk between TLRs and NODs, which might boost inflammatory responses to tissue injury. This study examined the relative roles of TLR2 and NODs 1 and 2 in activation of myeloid cells that contribute to inflammation after renal IRI. We found that TLR2 and NOD1 and 2 signaling induces neutrophil, macrophage and dendritic cell migration in vitro, however their blockade only decreases neutrophil infiltration into ischemic kidneys. The results of this study suggest that future therapies targeted to innate immune blockade should consider that either TLR2 or NOD1/2 blockade could decrease neutrophil inflammation following an ischemic insult to the kidney, however blockade of these PRRs would not likely impact infiltration of dendritic cells or macrophages. Developing rational approaches that target innate immunity in IRI-induced acute kidney injury requires an understanding of the relative role of PRRs in directing inflammation in the kidney.
Keywords: Ischemia/reperfusion injury, Acute kidney injury, TLR2, NOD1, NOD2
1. Introduction
Renal ischemia/reperfusion injury (IRI) often leads to acute kidney injury and subsequent chronic kidney disease [1]. There are two phases of IRI. The first is the ischemic phase during which low oxygen delivery leads to apoptotic and necrotic forms of cell death and release of molecules (danger-activated molecular patterns – DAMPs) that stimulate receptors constitutively expressed on parenchymal and immune cells (pattern recognition receptors – PRRs). The second phase occurs during the restoration of blood flow and is marked by innate and adaptive inflammatory cell infiltrates that respond to DAMPs and proinflammatory molecules released during the ischemic phase [2–5]. Both phases of renal IRI involve activation of innate and adaptive inflammatory mediators [6]; and PRR blockade can protect the kidney experimentally from IRI [7,8].
Recent data show that TLRs and NLRs cooperate to induce robust inflammatory responses in several experimental models [9–11]. In some cases, activation of both TLRs and NLRs is needed to mount an inflammatory response [12], and yet in other cases their activation can compensate for one another [13]. Since crosstalk has been described between TLRs and NLRs (and since TLR2 and NODs 1 and 2 play key roles in renal IRI [7,8]), this study assessed the relative roles of TLR2 and NOD signaling on activation of inflammatory cells that are known to contribute to renal inflammation after IRI. Developing rational approaches to IRI-induced acute kidney injury requires an understanding of the relative role of PRRs in directing inflammatory responses.
2. Methods and materials
2.1. Mice
All mice were housed in either the UCSD or Scripps Research vivariums and were approved for use by the Institutional Animal Care and Use Committee of Scripps Research and UCSD Animal Research Centers. All animals were handled according to the recommendations of the Humanities and Sciences and the standards of the Association for Assessment and Accreditation of Laboratory Animal Care. BALB/cByJ and C57BL/6 J mice were obtained from Jackson Laboratories, Bar Harbor MN. The NOD1 × 2−/− mice were obtained from J. Matheson at the Scripps Research Institute, La Jolla, CA.
2.2. Myeloid cell isolation
Neutrophils were isolated from bone marrow of either WT or NOD1x2−/− mice using Histopaque 1077 and 1119 reagents (Sigma-Aldrich, St. Louis, MO), and purity confirmed by Geimsa-Wright stain. Neutrophil purity was > 95%. Macrophages and dendritic cells were isolated from bone marrow of WT and NOD1x2−/− mice. Briefly the bone marrow cells were cultured with 5% GM-CSF for 5 days with addition of fresh media every 48 h, Macrophages were isolated by positive magnetic selection using F4/80 antibody (BioLegend, San Diego CA) and dendritic cells using CD11c + antibody (BioLegend, San Diego CA). Purity verified by flow cytometry was > 90% for each cell type. There were no significant differences in purity of the cells between the WT and NOD1x2−/− mice.
2.3. Stimulation of myeloid cells
Individual myeloid cell types were stimulated with ultrapure synthetic agonists; TLR2 agonist, PAM3CSK4 (PAM) (Enzo Life Sciences, Farmingdale, NY); NOD1 agonist, L-alanyl-γ-D-glutamyl-meso-diami-nopimelic acid (T-DAP) plus NOD2 agonist, muramyl dipeptide (MDP) (Enzo Life Sciences, Farmingdale, NY) for the times/doses indicated in the figure legends.
2.4. Cell surface activation markers
Activation of immune cells was analyzed by staining with fluorescent antibodies (anti-CD62L, -CD11b, -MHCII, -CD80, and -CD86]) from Biolegend (San Diego, CA) against their respective activation markers followed by flow cytometric analysis on a LSRII flow cytometer (BD bioscience, Franklin Lakes, NJ).
2.5. Cytokine assays
Supernatants of proliferating cells were collected and used to analyze the indicated cytokines: CXCL1 [chemokine (C-X-C motif) ligand 1, also called KC]; IL-6 (R&D Systems, Minneapolis, MN) by ELISA following the manufacturer’s methods.
2.6. Myeloid cell migration
To test for migration, the different myeloid cell types were seeded at a density of 2 × 105 cells/well on a polycarbonate filter with 5-μm pore size in 24-well transwell chambers (Corning Costar, Cambridge, MA) coated with FBS for 30 min at room temperature. The lower chambers contained 600 μL 10 K RPMI media alone or media supplemented with either CXCL8 for neutrophil migration or CCL19 for macrophage or DC migration (R&D Systems, Minneapolis, MN). Myeloid cells were stimulated for 4 h with PAM3 or T-DAP + MDP or were unstimulated, and were added to the upper chamber of the transwells at a density of 2 × 105 cells/well in a total volume of 100 μL and incubated for 4 h at 37 °C. Migrated cells were counted using a cell counter (Cellometer, Nexcelom, Lawrence, MA).
2.7. Induction of in vivo ischemia/reperfusion injury
All experimental mice were age-matched (8–12 weeks), and only male mice were used. As previously published [7,8,14], the following methods were used to induce IR injury. The mice were anesthetized with isofluorane and injected intraperitoneally with ketamine (100 mg/kg)/xylazine (8 mg/kg) in saline. Core body temperatures were maintained between 36 °C and 37.0 °C during surgery by continuous monitoring with a rectal thermometer and automatic heating blanket. Both kidneys were exposed with bilateral flank incisions, and ischemia was induced by clamping both renal arteries with nontraumatic microvessel clamps (S&T, Neuhausen, Switzerland) for 25 min. Renal veins remained unoccluded. Cessation of blood flow was documented by visual inspection. After 25 min of ischemia, the clamps were released, and reflow was verified by visual inspection of the kidneys. All mice received 200 μL of saline dripped over the open flanks during surgery to keep the tissue moist, and 30 μL of saline per gram body weight was injected subcutaneously after surgery to replenish fluid loss. Measurement of renal function was conducted 24 h after reperfusion. The mice were anesthetized and blood was collected from the inferior vena cava before sacrifice into a syringe preloaded with 3.8% sodium citrate. Plasma was isolated by centrifugation at 4000 xg for 10 min at 4 °C. Renal function was assessed using the Pointe Scientific creatinine kit (Canton, MI). All samples were run in duplicate and measurements repeated three times.
2.8. Measurement of creatinine
Measurement of renal function by creatinine was conducted 24 h after reperfusion. The mice were anesthetized and blood was collected from the inferior vena cava, before sacrifice, into a syringe preloaded with 3.8% sodium citrate. Plasma was isolated by centrifugation at 4000 xg for 10 min at 4 °C. Renal function was assessed using the Pointe Scientific creatinine kit (Canton, MI). All samples were run in duplicate and measurements repeated three times.
2.9. Immune cell infiltration in IR injured kidneys
After 24 h of reperfusion, the animals were euthanized and the kidneys were harvested and individually prepared to a single cell suspension slurry and then the cells were stained with fluorescent detection antibodies for neutrophils (GR-1), macrophages (F4/80), and dendritic cells (CD11c), purchased from BioLegend, San Diego, CA, and analyzed on a LSRII flow cytometer (BD bioscience, Franklin Lakes, NJ). The groups were normalized by counting 20,000 leukocytes from each sample to obtain equivalent cell numbers measured between the samples. The percentage difference between the individual phenotype markers was analyzed on equivalently gated events.
2.10. Statistical analysis
Groups were compared using nonparametric t-test or ANOVA (GraphPad Prism Software, GraphPad, La Jolla, CA). All values in graphs represent mean ± standard deviation (SD). A value of P < .05 was considered statistically significant.
3. Results
3.1. Effect of TLR2 and NLR signals on neutrophil responses
Neutrophils rapidly infiltrate the kidney during the reperfusion phase of renal IRI [15–17]. Both TLR2 and NODs 1 and 2 are expressed on neutrophils and are thought to direct inflammatory responses to the ischemic tissue [18]. The relative contribution of each of these PRR types to neutrophil responses is not yet known. Furthermore, it is not known whether there is crosstalk between TLR2 and NODs1 and 2.
In Fig. 1, we asked whether neutrophils were activated by TLR2 and/or NODs 1 and 2. We used NOD1x2−/− mice to simultaneously block NOD signaling because compensatory signaling between NOD1 and NOD2 is known to occur [19]. In Fig. 1 Panel A, we found that both WT and NOD1x2−/− neutrophils treated with the TLR2 agonist (PAM) expressed the characteristic neutrophil activation signature; upregulation of CD11b and downregulation of CD62 (WT- treated, PAM; NOD1x2−/− treated, PAM)). Expression of these activation molecules was not, however, affected by the NOD1/2 agonists (T-DAP + MDP) or by the absence of NOD1 and NOD2 (NOD1x2−/−), demonstrating that cell surface expression of neutrophil activation markers was dependent on TLR2 and did not involve NOD 1/2 signals.
Fig. 1.
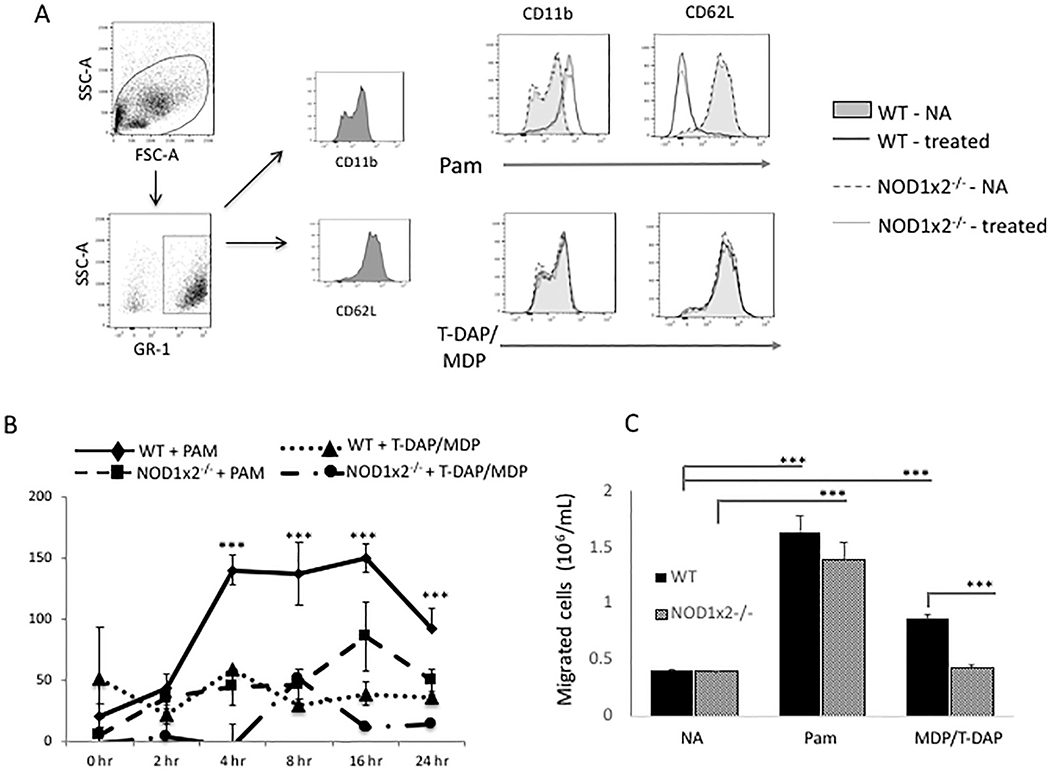
Effect of TLR2 and NOD1/2 agonists (PAM and T-DAP/MDP) on neutrophil activation. Panel A. Expression of CD11b and CD62L on cell surface of neutrophils from WT or NOD1x2−/− mice stimulated with either the TLR2 agonist PAM (1 μg/mL) or combined NOD1 and NOD2 agonists T-DAP/MDP (10 μg/mL). Panel B. Secretion of CXCL1 in supernatants over time from cultures of WT or NOD1x2−/− neutrophils stimulated with PAM (1 μg/mL) or T-DAP/MDP (10 μg/mL). Error bars represent SDs of 3 samples/gp. Panel C. Neutrophil migration detected in a transwell assay comparing WT vs NOD1x2−/− neutrophils. Neutrophils were first cultured for 24 h with either the TLR2 agonist PAM (1 μg/mL) or the combined NOD1/2 agonists T-DAP/MDP (10 μg/mL). Migration to CXCL8 (250 ng/mL) in the lower chamber was assessed at 4 h. Error bars represent SD of three samples. Each of the graphs represent one of three identical experiments. *** p < .001.
Neutrophils produce chemokines that recruit other neutrophils and inflammatory cells to the injured kidney. Fig. 1 Panel B shows that TLR2 stimulation causes WT neutrophils to secrete the chemotactic chemokine CXCL1 [keratinocyte chemoattractant (KC)] (WT + PAM). In the absence of NODs 1 and 2 however, TLR2 was not able to induce CXCL1 secretion from neutrophils (NOD1x2−/− + PAM). WT neutrophils treated with the NOD1/2 agonists alone did not produce CXCL1 (WT + T-DAP/MDP). These data show that TLR2 and NOD1/2 signals synergize for CXCL1 production from neutrophils.
Activated neutrophils migrate into kidneys during the reperfusion phase of IRI, and they accumulate in the interstitium of the kidney [20]. To determine whether TLR2 and/or NOD1/2 stimulation could induce neutrophil migration in vitro, WT neutrophils were treated with TLR2 or NOD1/2 agonists (PAM and T-DAP/MDP respectively) and then tested for migration across transwell chambers. In Fig. 1, Panel C, it is shown that both TLR2 and NOD1/2 stimulation (PAM and T-DAP/MDP) caused neutrophil migration, however TLR2 stimulation resulted in more neutrophils migrating across the transwells. Therefore, neutrophil migration can occur under the direction of either TLR2 or NOD1s1/2, however TLR2 appears to be a more potent stimulus in vitro.
3.2. Effect of TLR and NLR signals on macrophage function
DAMPs activate macrophages and potently induce proinflammatory cytokine/chemokine secretion and proliferation [21,22]. TLR2 agonists upregulate both NOD1 and NOD2 mRNA in macrophages [23], and crosstalk between TLR and NOD signaling in macrophages has previously been reported [24,25].
The relative contribution of TLR2 and NOD1/2 signaling to macrophage activation is shown in Fig. 2. Shown in Fig. 2 Panel A, stimulation of WT macrophages with either the TLR2 or NOD1/2 agonists upregulated MHC class II, CD86 and CD80 cell surface expression on macrophages in WT and NOD1x2−/− macrophages (WT-treated, NOD1x2−/− treated; PAM). Stimulation with the NOD1/2 agonist only had a minimal effect on MHC class II and CD86 expression (WT treated, T-DAP/MDP). This is different than in neutrophils, where only TLR2 stimulation regulates cell surface activation markers.
Fig. 2.
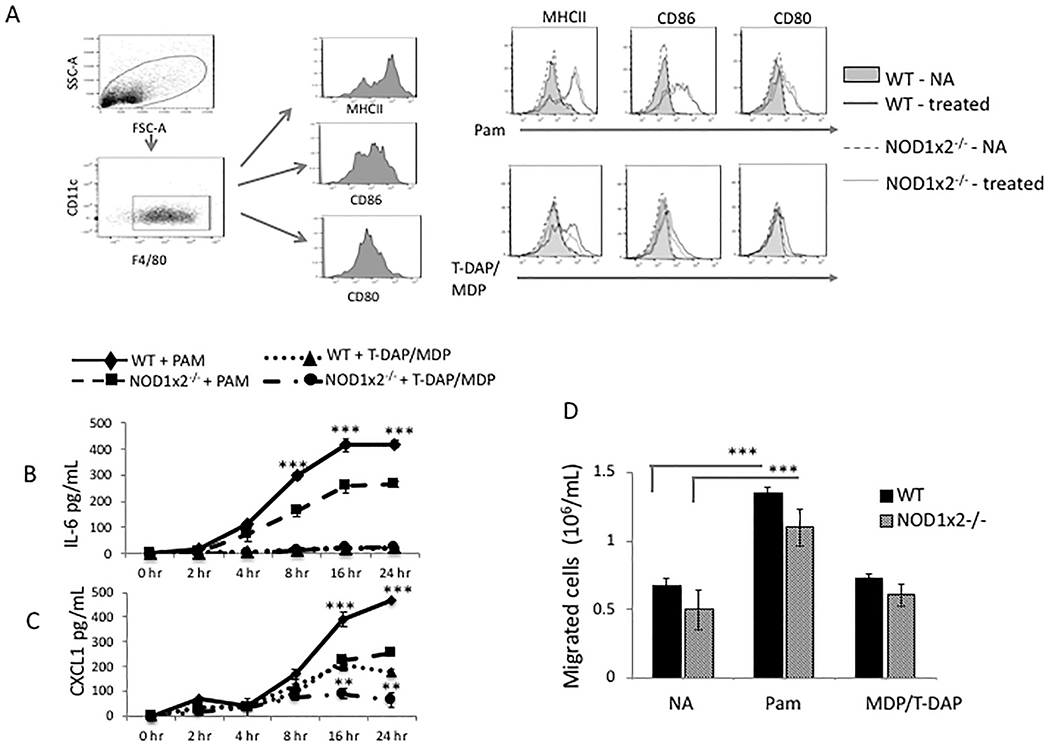
Effect of TLR2 and NOD1/2 agonists on macrophage activation. Panel A. Expression of MHC class II, CD86 and CD80 on cell surface of macrophages derived from bone marrows of WT or NOD1x2−/− mice stimulated overnight with either the TLR2 agonist PAM (1 μg/mL) or the combined NOD1/2 agonists T-DAP + MDP (10 μg/mL) and analyzed by FACS. Panels B and C. IL-6 and CXCL1 (respectively) detected in supernatants over time of cultures of WT vs. NOD1x2−/− macrophages stimulated with PAM (1 μg/mL). Error bars represent SD of triplicates. Panel D. Macrophage migration detected in a transwell assay comparing WT vs NOD1x2−/− macrophages. Macrophages were cultured for 24 h with either the TLR2 agonist PAM (1 μg/mL) or the combined NOD1 and NOD2 agonists T-DAP/MDP (10 μg/mL). Migration to CXCL8 (250 ng/mL) in the lower chamber was assessed at 4 h. Error bars represent SD of three samples. In each of the experiments error bars represent SD of triplicates. Each graph represents one of three identical experiments. In Panel B **p < .01, ***p < .001 (between WT and NOD1x2−/−, PAM treated). In Panel C ** p < .01 and ***p < .001 (between WT and NOD1x2−/−, PAM treated – top two lines of graph) and ** p < .01 and ***p < .001 (between WT and NOD1x2−/−.T-DAP/MDP treated – bottom two lines of graph).
Stimulation of TLR2 has been shown in experimental models to potently induce macrophage secretion of cytokines such as IL-6 and CXCL1 [26]. Fig. 2, Panels B and C show that TLR2-induced IL-6 and CXCL1 secretion (respectively) was significantly decreased in macrophages from NOD1x2−/− mice compared to WT macrophages (WT + PAM vs NOD1x2−/− + PAM, in both panels). Treatment of WT macrophages with the NOD1/2 agonists also increased CXCL1 secretion (WT + T-DAP/MDP, panel C). These data showed that, like in neutrophils, there was crosstalk TLR2 and NODs1/2 signaling for CXCL1 induction.
Fig. 2, Panel D, compares in vitro migration of WT macrophages stimulated with either TLR2 or NOD1/2 agonists. The TLR2 agonist (PAM) increased macrophage migration across transwells in both WT and NOD1x2−/− macrophages, but the NOD1/2 agonist (T-DAP/MDP) had little effect on macrophage migration, suggesting that macrophage migration was affected by TLR2 stimulation, but not by NOD1/2 stimulation, which was different that we observed for neutrophil migration, which was affected by both TLR2 and NOD1x2 signals.
3.3. Effect of TLR and NLR signals on dendritic cell function
TLR2 and NODs 1 and 2 are highly expressed in dendritic cells (DCs) and crosstalk between these PRRs has also been reported [27]. To determine the effect of TLR2 and NOD1/2 stimulation on DCs, DCs from WT and NOD1x2−/− mice were stimulated with the TLR2 agonist PAM and assessed for expression of activation markers, cytokine/chemokine production and migration in vitro. Shown in Fig. 3 Panel A, there were no differences in TLR2-induced cell surface expression of MHC class II, CD86 or CD80 between WT and NOD1x2−/− DCs treated with the TLR2 agonist (WT treated or NOD1x2−/− treated, PAM). The NOD1/2 agonist had no effect on MHC II, CD86 or CD80 expression in either WT or NOD1x2−/− dendritic cells (WT treated or NOD1x2−/− treated, T-DAP/MDP).
Fig. 3.
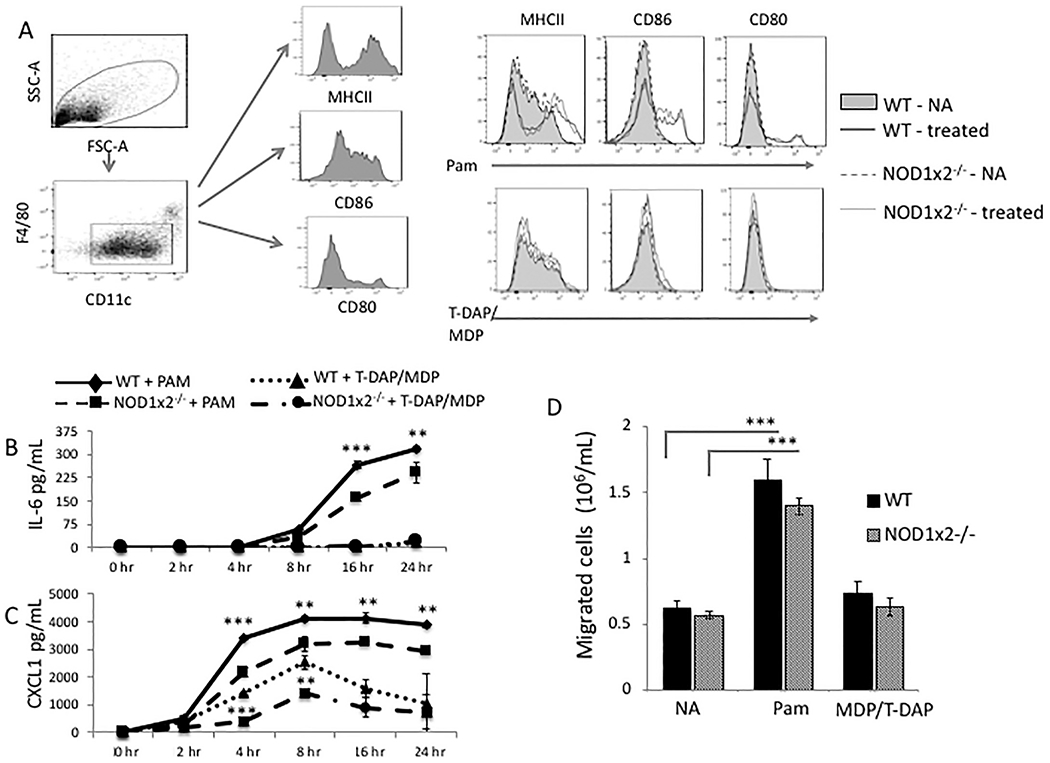
Effect of TLR2 and NOD1 x NOD2 agonists on dendritic cell activation. Panel A. Expression of MHC Class II, CD86, and CD80 on cell surface of dendritic cells derived from spleens of WT or NOD1x2−/− mice stimulated overnight with either the TLR2 agonist PAM (1 μg/mL) or combined NOD1/2 agonists T-DAP + MDP (10 μg/mL) and analyzed by FACS. Panel B. Detection of IL-6 or CXCL1 (Panel C) over time in culture supernatants of WT vs. NOD1x2−/− dendritic cells stimulated overnight with PAM (1 μg/mL). Panel D. Dendritic cell migration detected in a transwell assay comparing WT vs NOD1x2−/− dendritic cells. Dendritic cells were cultured for 24 h with either the TLR2 agonist PAM (1 μg/mL) or the combined NOD1/2 agonists T-DAP + MDP (10 μg/mL). Migration to CXCL8 (250 ng/mL) in the lower chamber was assessed at 4 h. Error bars represent SD of three samples. Each graph represents one of three identical experiments. **p < .01, ***p < .001.
In Fig. 3, Panels B and C, WT or NOD1x2−/− DCs were treated with the TLR2 agonist, PAM, and supernatants assayed for IL-6 (Panel B) and CXCL1 (Panel C). TLR2 stimulation induced the proinflammatory molecules from both WT and NOD1x2−/− DCs, however in the absence of NODs 1 and 2, the secretion of IL-6 and CXCL1 was significantly decreased (WT + PAM vs NOD1x2−/− + PAM). As noted for macrophages, treatment of WT dendritic cells with the NOD1/2 agonist induced CXCL1 secretion (WT + T-DAP/MDP vs NOD1x2−/− + T-DAP/MDP).
Fig. 3, Panel D, compares migration in vitro of WT DCs stimulated with either the TLR2 or NOD1/2 agonists (PAM or T-DAP/MDP). The TLR2 agonist (PAM) significantly increased DC migration across the transwells in both WT and NOD1x2−/− groups, but the NOD1/2 agonist (T-DAP/MDP) had no effect on DC migration. These data show, as was the case with macrophages, that DC migration in vitro was affected by TLR2 stimulation, but not by NOD1/2 stimulation.
3.4. Effect of TLR2 and NOD1/2 on inflammatory infiltrates after renal IRI
The in vitro studies showed us that TLR2 signaling cooperated with NOD1/2 signaling to boost IL-6 and CXCL1 production; and they also had a significant impact on neutrophil migration in vitro. Since CXCL1 plays a pivotal role in vivo in neutrophil recruitment [28], we tested for the collaboration of these PRRs in neutrophil, macrophage and dendritic cell recruitment in vivo using a well-characterized model of renal IRI, and mice deficient in TLR2 and NOD1s1/2. Fig. 4, Panel A, shows that the absence of either TLR2 (TLR2−/−) or NOD1/2 (NOD1x2−/−) significantly decreased plasma creatinine by 24 h after bilateral renal artery clamping, compared to WT mice (WT).
Fig. 4.
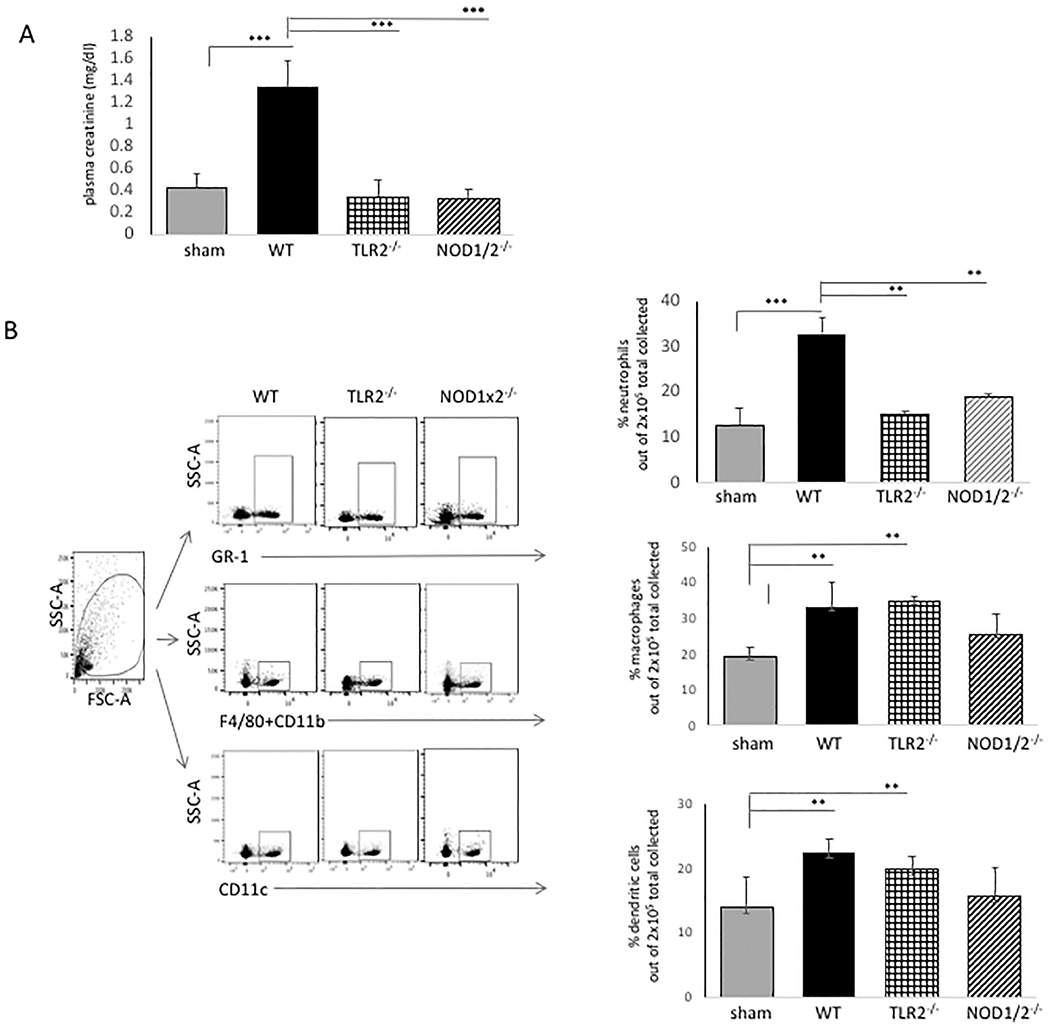
Effect of TLR2 and NOD1x2 deficiency in renal IRI. WT vs. TLR2−/− or NOD1x2−/− mice were treated with 25 min bilateral renal artery clamping followed by 24 h reperfusion. At end of reperfusion, plasma creatinine was detected and the kidneys were harvested and single cell suspension of each tissue prepared. The suspension was stained for inflammatory infiltrates and analyzed by FACS using florescence antibodies to GR-1 (neutrophils); F4/80 and CD11b (macrophages); CD11c (dendritic cells). Error bars represent standard deviations of triplicate samples. **p < .01, *** p < .001.
In Fig. 4, Panel B, it can be seen that neutrophils, macrophages and dendritic cells infiltrated the kidneys within 24 h after the ischemic injury in WT mice (Fig. 4, Panel B, WT). The absence of either TLR2 or NOD1/2 significantly decreased the neutrophil infiltration compared to WT mice (Panel B, WT vs TLR2−/− or NOD1x2−/−, top panel). Despite apparent protection from renal injury, as noted in Fig. 4, Panel A, however, the absence of TLR2 or NOD1/2 did not decrease macrophage or dendritic cell infiltration after the ischemic renal injury compared to WT mice (Fig. 4, Panel B, middle and lower panels WT vs TLR2−/− or NOD1x2−/−).
4. Discussion
This study evaluated the relative roles of TLR2 and NOD1/2 signals on myeloid cells that are known to participate in renal IRI. Several recent reports have shown TLRs and NLRs can have redundant roles in production of proinflammatory cytokines and chemokines, and have highlighted the fact that there can be crosstalk between TLRs and NLRs in inflammatory responses to tissue injury. Appreciating this crosstalk is highly relevant for clinical strategies directed at dampening inflammation in response to ischemic tissue injury, as it is likely that targeting both cell membrane and cytoplasmic PRRs is necessary in order to abrogate inflammatory responses to IRI.
This study shows that TLR2 and NOD1/2 signals synergize to produce the chemokine CXCL1 in neutrophils, macrophages and dendritic cells. However, both TLR2 and NOD1/2 stimulation could independently signal neutrophil migration in vitro and in vivo. TLR and NLR signaling has been linked in several cell types, suggesting there might be integrated responses due to cooperative signaling between these PRRs [29]. It has already been shown that TLRs and NODs can participate in production of proinflammatory molecules that enhance immune responses [11,30,31]. For instance, TLR2 and NOD2 have been shown to synergize for cytokine production in response to Mycobacterium tuberculosis, however they signal cytokine production through independent pathways [32]. Reports have also linked TLR and NOD pathways by negative crosstalk. For instance, Watanabe et al. showed that NOD2 negatively regulated TLR2-mediated Th1 responses [33]. However, this negative crosstalk was not confirmed in other models [10]. Chen and Park have shown TLR and NLR crosstalk at the level of MAPK and RIP2 activation [34,35], while other investigators suggest that synergism is likely to be specific to the stimuli and to the specific cell type [29].
Many studies have shown that ischemic kidney injury leads to renal inflammation [1,36], and we too found a significant increase in neutrophil, macrophage and dendritic cell infiltration into the WT kidneys at 24 h after reperfusion. The absence of TLR2 or NOD1/2 only had an effect on neutrophil infiltration, but not on macrophage or dendritic cell infiltrates, despite the apparent lack of injury in the TLR2−/− and NOD1x2−/− kidneys (as noted by plasma creatinine). Since creatinine might not accuratley reflect renal injury, and in fact is a late marker of disease [37], a range of injury and reperfusion kinetics will be needed to further assess the role of macrophage and DC infiltration and the relation to TLR2 and NODs1 and 2 signals. In addition, future studies will need to focus on potential cross-talk between different inflammatory cells in the ischemic mileu.
The results of this study suggest that future therapies targeted to innate immune blockade should consider the possibility that crosstalk between TLRs and NLRs occurs in inflammatory cells (particularly neutrophils) that participate in renal inflammation following an ischemic insult to the kidney.
Acknowledgments
Funding
This work was funded by grants to DBM awarded by the National Institutes of Health, R01DK113162 and R01 DK091136 and by the Scripps Clinic Renal Research Collaborative through the Scripps Clinic Foundation, La Jolla, CA.
Footnotes
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- [1].Zuk A, Bonventre JV, Acute kidney injury, Annu. Rev. Med 67 (2016) 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Boros P, Bromberg JS, New cellular and molecular immune pathways in ischemia/reperfusion injury, Am. J. Transplant 6 (2006) 652–658. [DOI] [PubMed] [Google Scholar]
- [3].Devarajan P, Cellular and molecular derangements in acute tubular necrosis, Curr. Opin. Pediatr 17 (2005) 193–199. [DOI] [PubMed] [Google Scholar]
- [4].Dagher PC, Herget-Rosenthal S, Ruehm SG, et al. , Newly developed techniques to study and diagnose acute renal failure, J. Am. Soc. Nephrol 14 (2003) 2188–2198. [DOI] [PubMed] [Google Scholar]
- [5].Li C, Jackson RM, Reactive species mechanisms of cellular hypoxia-reoxygenation injury, Am J Physiol Cell Physiol 282 (2002) C227–C241. [DOI] [PubMed] [Google Scholar]
- [6].Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H, The local and systemic inflammatory transcriptome after acute kidney injury, J. Am. Soc. Nephrol 19 (2008) 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shigeoka AA, Holscher TD, King AJ, et al. , TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways, J. Immunol 178 (2007) 6252–6258. [DOI] [PubMed] [Google Scholar]
- [8].Shigeoka AA, Kambo A, Mathison JC, et al. , NOD1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury, J. Immunol 184 (2010) 2297–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Uehara A, Fujimoto Y, Fukase K, Takada H, Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines, Mol. Immunol 44 (2007) 3100–3111. [DOI] [PubMed] [Google Scholar]
- [10].Kobayashi KS, Chamaillard M, Ogura Y, et al. , Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract, Science 307 (2005) 731–734. [DOI] [PubMed] [Google Scholar]
- [11].van Heel DA, Ghosh S, Butler M, et al. , Synergistic enhancement of Toll-like receptor responses by NOD1 activation, Eur. J. Immunol 35 (2005) 2471–2476. [DOI] [PubMed] [Google Scholar]
- [12].Netea MG, Ferwerda G, de Jong DJ, et al. , The frameshift mutation in Nod2 results in unresponsiveness not only to Nod2- but also Nod1-activating pepti-doglycan agonists, J. Biol. Chem 280 (2005) 35859–35867. [DOI] [PubMed] [Google Scholar]
- [13].Kawai T, Akira S, Toll-like receptors and their crosstalk with other innate receptors in infection and immunity, Immunity 34 (2011) 637–650. [DOI] [PubMed] [Google Scholar]
- [14].Shigeoka AA, Mueller JL, Kambo A, et al. , An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury, J. Immunol 185 (2010) 6277–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Awad AS, Rouse M, Huang L, et al. , Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury, Kidney Int 75 (2009) 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F, Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction, J. Biol. Chem 274 (1999) 10689–10692. [DOI] [PubMed] [Google Scholar]
- [17].Perera PY, Mayadas TN, Takeuchi O, et al. , CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-in-ducible gene expression, J. Immunol 166 (2001) 574–581. [DOI] [PubMed] [Google Scholar]
- [18].Zhou H, Coveney AP, Wu M, et al. , Activation of both TLR and NOD signaling confers host innate immunity-mediated protection against microbial infection, Front. Immunol 9 (2018) 3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moreira LO, Zamboni DS, NOD1 and NOD2 signaling in infection and inflammation, Front. Immunol 3 (2012) 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shvedova AA, Kisin ER, Mercer R, et al. , Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice, Am J Physiol Lung Cell Mol Physiol 289 (2005) L698–L708. [DOI] [PubMed] [Google Scholar]
- [21].Mantovani A, Sica A, Sozzani S, Alla vena P, Vecchi A, Locati M, The chemokine system in diverse forms of macrophage activation and polarization, Trends Immunol 25 (2004) 677–686. [DOI] [PubMed] [Google Scholar]
- [22].Williams TM, Little MH, Ricardo SD, Macrophages in renal development, injury, and repair, Semin. Nephrol 30 (2010) 255–267. [DOI] [PubMed] [Google Scholar]
- [23].Takahashi Y, Isuzugawa K, Murase Y, et al. , Up-regulation of NOD1 and NOD2 through TLR4 and TNF-alpha in LPS-treated murine macrophages, J. Vet. Med. Sci 68 (2006) 471–478. [DOI] [PubMed] [Google Scholar]
- [24].Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G, The cytosolic sensors NOD1 and Nod2 are critical for bacterial recognition and host defense after exposure to toll-like receptor ligands, Immunity 28 (2008) 246–257. [DOI] [PubMed] [Google Scholar]
- [25].Kapetanovic R, Nahori MA, Balloy V, et al. , Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages, Infect. Immun 75 (2007) 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De Filippo K, Henderson RB, Laschinger M, Hogg N, Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways, J. Immunol 180 (2008) 4308–4315. [DOI] [PubMed] [Google Scholar]
- [27].Tada H, Aiba S, Shibata K, Ohteki T, Takada H, Synergistic effect of NOD1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells, Infect. Immun 73 (2005) 7967–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sawant KV, Poluri KM, Dutta AK, et al. , Chemokine CXCL1 mediated neutrophil recruitment: role of glycosaminoglycan interactions, Sci. Rep 6 (2016) 33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Becker CE, O’Neill LA, Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs, Semin. Immunopathol 29 (2007) 239–248. [DOI] [PubMed] [Google Scholar]
- [30].Fritz JH, Girardin SE, Fitting C, et al. , Synergistic stimulation of human monocytes and dendritic cells by toll-like receptor 4 and NOD1-and NOD2-activating agonists, Eur. J. Immunol 35 (2005) 2459–2470. [DOI] [PubMed] [Google Scholar]
- [31].Li J, Moran T, Swanson E, et al. , Regulation of IL-8 and IL-lbeta expression in Crohn’s disease associated NOD2/CARD15 mutations, Hum. Mol. Genet 13 (2004) 1715–1725. [DOI] [PubMed] [Google Scholar]
- [32].Ferwerda G, Girardin SE, Kullberg BJ, et al. , NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis, PLoS Pathog 1 (2005) 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A, Innate immune response in Th1- and Th2-dominant mouse strains, Shock 22 (2004) 460–466. [DOI] [PubMed] [Google Scholar]
- [34].Chen CM, Gong Y, Zhang M, Chen JJ, Reciprocal cross-talk between Nod2 and TAK1 signaling pathways, J. Biol. Chem 279 (2004) 25876–25882. [DOI] [PubMed] [Google Scholar]
- [35].Park JH, Kim YG, McDonald C, et al. , RICK/RIP2 mediates innate immune responses induced through NOD1 and Nod2 but not TLRs, J. Immunol 178 (2007) 2380–2386. [DOI] [PubMed] [Google Scholar]
- [36].Kinsey GR, Li L, Okusa MD, Inflammation in acute kidney injury, Nephron Exp Nephrol 109 (2008) el 02–el 07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Slocum JL, Heung M, Pennathur S, Marking renal injury: can we move beyond serum creatinine? Transl. Res 159 (2012) 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]