

Abstract
Mammalian pyruvate kinase catalyzes the final step of glycolysis, and its M2 isoform (PKM2) is widely expressed in proliferative tissues. Mutations in PKM2 are found in some human cancers, however, the effects of these on enzyme activity and regulation are unknown. Here, we characterized five cancer-associated PKM2 mutations, occurring at various locations on the enzyme, with respect to substrate kinetics and activation by the allosteric activator fructose-1,6-bisphosphate (FBP). The mutants exhibit reduced maximal velocity, reduced substrate affinity, and/or altered activation by FBP. The kinetic parameters of five additional PKM2 mutants that have been used to study enzyme function or regulation also demonstrate the deleterious effects of mutations on PKM2 function. Our findings indicate that PKM2 is sensitive to many amino acid changes and support the hypothesis that decreased PKM2 activity is selected for in rapidly proliferating cells.
INTRODUCTION
Mammalian pyruvate kinase isoforms function as homotetramers to catalyze the final step of glycolysis, which is the transfer of phosphate from phosphoenolpyruvate (PEP) to ADP to produce pyruvate and ATP. There are four mammalian pyruvate kinase isoforms, and the activity of each isoform is regulated in a manner that suits its physiological role [1–3]. The muscle (PKM1) isoform is found in non-proliferating and highly catabolic tissues such as the heart, brain, and skeletal muscle, where it functions as a highly active, constitutive tetramer with few regulatory inputs [1, 2, 4–7]. The liver (PKL) and red blood cell (PKR) isoforms are subject to allosteric feed-forward activation by an upstream glycolytic intermediate, fructose-1,6-bisphosphate (FBP) [8, 9]. Both of these enzymes are also inhibited by phosphorylation, with the liver isoform regulated to minimize futile cycles during gluconeogenesis [10]. The M2 isoform of pyruvate kinase (PKM2) is expressed in most other tissue types, including the developing embryo and virtually all cancers and proliferative tissues studied to date [1, 2, 4, 11]. In proliferating tissues, PKM2 catalytic activity is tightly regulated to balance the catabolic and anabolic needs of proliferating cells [12, 13].
PKM2 activity is modulated by a host of regulatory inputs. Like PKL and PKR, fructose-1,6-bisphosphate (FBP) is a major allosteric activator of PKM2 [14, 15], and its activity is also affected by other allosteric effectors, including thyroid hormone T3, serine, phenylalanine, and select other amino acids [7, 16–19]. FBP binding increases affinity of PKM2 for one of its substrates, PEP, and stabilizes the PKM2 tetramer in a fully active conformation [15, 20]. The PKM2 tetramer assembles as a dimer of dimers, and the enzyme exists in a tetramer-dimer-monomer equilibrium with the less-active, FBP-free tetramer conformation prone to dissociation to inactive dimers and monomers. Tetramer dissociation significantly reduces enzyme activity, while the binding of FBP favors tetramer assembly and results in an increase in Vmax due to an apparent increase in concentration of functional enzyme [7, 16]. This association-dissociation phenomenon is also observed in vivo [20–22].
Release of FBP, and subsequent down-regulation of enzyme activity, is stimulated by interaction of the enzyme FBP-binding pocket with tyrosine phosphorylated proteins generated by growth signaling [23]. Additional mechanisms can also reduce pyruvate kinase activity in proliferating cells including lysine acetylation, cysteine oxidation, and degradation of the enzyme in nutrient-replete conditions [24, 25]. Reduction of PKM2 activity via intracellular signaling may facilitate biosynthesis [26], and genetic or pharmacologic activation of pyruvate kinase disrupts proliferative metabolism and is detrimental to tumor growth [20, 27, 28]. Decreased pyruvate kinase activity seems particularly important for nucleobase synthesis [28], and genetic experiments using mouse cancer models suggest that loss of pyruvate kinase activity can be selected for in tumors [11, 29].
We reported heterozygous missense mutations in PKM2 that were found in human cancers [29]. Here, we consider mutations in PKM2 reported in The Cancer Genome Atlas (TCGA) affecting 23 amino acids throughout the tertiary structure of the enzyme (Supplemental Figure 1). Each PKM2 subunit contains three main domains: the A domain, a TIM barrel that hosts the active site; the B domain, which closes down on the active site during substrate binding and catalysis; and the C domain, which comprises most of the dimer-dimer interface and contains the FBP binding site [30, 31] (Supplemental Figure 1C). Pyruvate kinase is highly conserved, and PKM2 function appears to be sensitive to amino acid substitutions, even in solvent exposed residues [32], suggesting that the PKM2 mutations occurring in human tumors may have an effect on enzyme activity. Given that a reduction in intracellular pyruvate kinase activity supports proliferative metabolism [2, 20, 24, 28, 29], we hypothesized that these cancer-associated mutations may reduce or abolish PKM2 pyruvate kinase activity.
Non-glycolytic signaling functions of PKM2 have also been proposed as an alternative explanation for PKM2 selection in cancer [12, 33, 34]. In this context, amino acid substitutions in PKM2 have been used to experimentally separate the proposed non-glycolytic functions from the role of the enzyme in glycolysis [35, 36], or to alter the response of the enzyme to other allosteric inputs [17, 23]. While some pyruvate kinase enzyme activity is retained in these mutants, the effects of these mutations on enzyme kinetics have not been well characterized. To understand how these mutations affect PKM2 function in proliferating cells, kinetic characterization is necessary, particularly in light of controversy surrounding the proposed functions of PKM2 [3, 37].
EXPERIMENTAL PROCEDURES
PCR Mutagenesis.
PCR mutagenesis was used to generate mutations in the coding sequence of the human PKM2 (hPKM2) cDNA cloned into pET-28a with an in-frame N-terminal 6x-His tag [23]. The conditions for mutagenesis PCR were 90 s at 98°C, followed by 16 cycles of 10 s at 98°C, 30 s at 55°C, and 10 min at 72°C, with an additional 10 min final elongation step at 72°C. The forward primers used were as follows (5’−3’, underlined bases indicate mutations):
S37A: CGCCTGGACATTGATTACCCACCCATCACAGCC;
P117L: CTAGACACTAAAGGACTTGAGATCCGAACTGGG;
R246S: TTTGCGTCATTCATCAGCAAGGCATCTGATGTC;
K367M: TCTGGAGAAACAGCCATGGGGGACTATCCTCTG;
R399E: TTATTTGAGGAACTCGAACGCCTGGCGCCCATT;
G415R: GAAGCCACCGCCGTGCGTGCCGTGGAGGCCTCC;
K433E: ATAATCGTCCTCACCGAGTCTGGCAGGTCTGCT;
R455Q: ATCATTGCTGTGACCCAGAATCCCCAGACAGCT;
H464A: ACAGCTCGTCAGGCCGCCCTGTACCGTGGCATC;
R516C: GTGCTGACCGGATGGTGCCCTGGCTCCGGCTTC.
The reverse primer sequences are the reverse complement of the forward primers. Successful mutagenesis was verified by sequencing the coding region of the plasmid using T7 promoter and terminator primers.
Native Protein Expression.
Wild-type and mutant PKM2 6x-His tagged proteins were expressed in E. coli BL21(DE3) and the soluble protein was purified from the cell lysates. A 50 mL starter culture was inoculated with a colony of freshly transformed E. coli and grown at 37°C overnight in LB broth containing 50 μg/mL of kanamycin (LB-kan). The starter culture was diluted 1/40 into 1 L LB-kan containing 2 mM MgCl2. The expression culture was grown at 37°C to OD600 of 0.7 and induced for 6 hours at 25°C with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Following centrifugation, the cell pellet was snap frozen using liquid nitrogen and stored at −80° C prior to subsequent purification.
Protein Preparation 1: One-Column PKM2 Purification.
E. coli BL21(DE3) cells were transformed, induced, pelleted, and frozen as described in “Native Protein Expression” above. All protein purification steps were performed on ice or at 4°C. The cell pellet was resuspended in Buffer C (50 mM Tris, pH 8.5; 10 mM MgCl2; 300 mM NaCl; 10% glycerol; 5mM imidazole), lysed by sonication and clarified prior to supernatant collection and addition of β-mercaptoethanol (BME) to 0.1% (v/v) final concentration. Recombinant protein was batch bound to Ni-NTA (nickel-nitriloacetic acid) agarose beads (Qiagen 30210, 4 mL bead volume per 1 L of culture). Beads were batch-washed 4 times with 30 mL Buffer D (50 mM Tris, pH 8.5, 10 mM MgCl2, 300 mM NaCl, 10% glycerol, 30 mM imidazole) and packed into a gravity flow column at 4°C. Protein was eluted with Buffer E (50 mM Tris pH 8.5, 10 mM MgCl2, 250 mM NaCl, 10% glycerol, 250 mM imidazole) and collected in 1 mL fractions. The three fractions containing the peak of eluted protein were identified using the Bradford assay, pooled, and dialyzed against 1 L Buffer F (50 mM Tris pH 7.5, 10 mM MgCl2, 25 mM NaCl, 20% glycerol, 21 mM β-mercaptoethanol) for a total 24 hours at 4° C, with one change of buffer after 12 hours.
Protein Preparation 2: Inclusion Body Production and On-Column Refolding.
E. coli strain BL21(DE3) transformed with pET28a-hPKM2 was grown in 1 L of LB-kan containing 5% (w/v) sucrose at 37°C. The culture was induced at OD600 of 0.4 with 1 mM IPTG and shaking continued for 3 hours at 37°C. Following centrifugation, the cell pellet was resuspended in Buffer G (50 mM Tris pH 7.5, 100 mM KCl, 20% glycerol; 60 mL per 1 L culture) and lysed by sonication. Inclusion bodies were isolated by centrifugation at 15,000xg for 15 minutes, washed twice with Inclusion Body Wash Buffer 1 (50 mM Tris pH 7.5, 10 mM EDTA, 2% Triton X-100, 500 mM NaCl, 5 mM DTT), once with Inclusion Body Wash Buffer 2 (50 mM Tris pH 7.5), and collected in a pre-weighed tube. The inclusion body pellet was resolubilized overnight at 4°C in 1 mL of Resuspension Buffer (50 mM Tris pH 8.0, 6 M guanidinium, 40 mM imidazole, 5 mM DTT) per 30 mg wet pellet weight. A gravity flow column was packed with Ni-NTA beads (2 mL) at 22°C and washed with 4 column volumes (CV) of Resuspension Buffer. Resolubilized protein was applied to the column and the column was washed with 15 CV of Column Wash Buffer (50 mM Tris pH 8.0, 4 M urea, 40 mM imidazole, 1 mM DTT). To initiate refolding, the column was washed with 4 CV of Refolding Buffer (50 mM Bis-Tris Propane pH 8.0, 100 mM KCl, 20% glycerol, 2 mM MgCl2). The column outlet was stopped and refolding continued at 22° C for 1 h. Protein was eluted using Buffer H (50 mM Bis-Tris Propane pH 8.0, 100 mM KCl, 20% glycerol, 2 mM MgCl2, 250 mM imidazole, 1 mM DTT). Fractions (1 mL) containing protein were pooled and DTT was added to a final concentration of 25 mM.
Protein Preparation 3: Two-Column Purification of Native Proteins.
E. coli BL21(DE3) cells were transformed, induced, pelleted, and frozen as described in “Native Protein Expression” above. All protein purification steps were performed on ice or at 4°C. The cell pellet from 1 L of culture was resuspended in a total of 60 mL Buffer A (50 mM Tris, pH 7.5, 10 mM MgCl2, 300 mM KCl, 10% glycerol, 5 mM imidazole) with protease inhibitors (Roche, 11836170001). Resuspended cells were lysed by sonication and the lysate was clarified by centrifugation at 20,000xg for 45 minutes. The supernatant was filtered using a 0.22 μm filter and applied to a Ni2+-charged IMAC column (GE Healthcare HisTrap 1mL) using an Äkta FPLC system. The column was washed with 15 column volumes of Wash Buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 300 mM KCl, 20 mM imidazole), then with 20 column volumes of Wash Buffer containing 20% Elution Buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 250 mM KCl, 250 mM imidazole), and the protein was eluted with a 6 column volume linear gradient from 20% to 100% Elution Buffer. Fractions containing the protein of interest were pooled and concentrated using Amicon Ultra centrifugal filters (Millipore, UFC903024) and further purified by size exclusion chromatography (GE Healthcare HiPrep 16/60, Sephacryl S-200) with Buffer B (25 mM Bis-tris-propane, pH 7.5, 10 mM MgCl2, 25 mM KCl) as the mobile phase. Fractions of interest were again pooled and spin-concentrated. Glycerol was added to the concentrated protein to 20% v/v, concentration of purified PKM2 determined by absorbance at 280 nm using an extinction coefficient of 29,910 M−1cm−1, and stored at −80° C.
SDS-PAGE.
Purified proteins were analyzed by SDS-PAGE using 8% polyacrylamide gels and visualized by Coomassie blue staining. Images of stained gels were obtained using a LI-COR Biosciences Odyssey imager.
Kinetic Assays.
A lactate-dehydrogenase-linked spectrophotometric assay was used to determine pyruvate kinase activity by measuring the oxidation of NADH via absorbance at 340 nm. The reaction buffer consisted of 50 mM Bis-Tris Propane pH 7.5, 200 mM KCl, 15 mM MgCl2, 100 units/mL lactate dehydrogenase (LDH), 2 mM ADP (or PEP), 180 μM NADH and PEP (or ADP) concentrations from 0 to 2 mM as indicated. Enzymes were pre-incubated with or without 50 mM FBP prior to the start of the assay, and the reaction was initiated by adding reaction buffer to enzyme to a final volume of 100 μL in a well of a 96-well plate. Final FBP concentration was 5 mM when present in the assay. Final pyruvate kinase concentrations were chosen to allow measurement of initial rates; for example, wild-type enzymes were assayed at concentrations near 1 μg/mL. The reaction was monitored using a Tecan Infinite M200 Pro plate reader. Km and Vmax values were calculated by fitting the initial rate data using GraphPad Prism software. One unit is defined as the amount of pyruvate kinase activity causing oxidation of one μmole of NADH per minute at 25° C in the LDH-linked assay.
FBP Activation Assay.
Enzymes were pre-incubated in concentrations of FBP as indicated and the reaction initiated by addition of reaction buffer containing the same FBP concentration. PEP concentrations were chosen to be insufficient to induce cooperativity. Apparent half maximal effective concentration (EC50) values for FBP binding were determined by fitting to data to a sigmoid dose response curve with variable slope using GraphPad Prism software.
Size Exclusion Chromatography.
5 μg of wild-type and mutant PKM2 proteins in a volume of 10 μL were subjected to HPLC separation on a Yarra SEC-3000 size exclusion column (Phenomenex) with an aqueous mobile phase containing 25 mM bis-tris-propane-HCl (pH 7.1), 25 mM KCl, and 10 mM MgCl2 at a flow rate of 1 mL/min. The proteins used in this assay were prepared using Protein Preparation 3 and were assayed in the absence of FBP.
RESULTS
Kinetic Characterization of PKM2 and PKM1
Including a 6x-histidine-tag on proteins facilitates purification using Ni-affinity chromatography, and the kinetic parameters of PKM2 are not altered by a 6x-histidine tag [15]. To determine whether single step Ni-affinity could be used to prepare recombinant PKM2 proteins for kinetic analysis, 6x-His-PKM2 expressed in E. coli was isolated from bacterial lysates via binding to Ni-NTA beads, single step elution with imidazole, followed by dialysis as reported previously [20, 29, 38]. 6x-His-PKM2 prepared using this one-step protocol (Protein Preparation 1 in Experimental Methods; hereafter “Prep 1”) was active and exhibited hyperbolic kinetics with respect to PEP substrate; however, addition of the allosteric activator FBP to the reaction did not substantially increase affinity for PEP or increase the maximal velocity of the enzyme (Figure 1A). These results suggested that most of the enzyme was already bound to FBP from the bacteria despite purification as previously reported [19, 23, 39]. Independent preparations of PKM2 using this approach occasionally produced enzyme that showed some FBP activation; however, this variability in FBP activation complicated detailed kinetic analysis. Ammonium sulfate precipitation has been previously used to remove allosteric effectors from liver pyruvate kinase preparations [40, 41], as sulfate ions appear to interfere with the interaction between FBP and the liver enzyme [41]. Including 300 mM ammonium sulfate in the kinetic assay allowed FBP activation to be observed when PKM2 prepared in this manner was studied (Figure 1B). In the presence of ammonium sulfate, FBP increased the affinity of the enzyme for PEP without affecting Vmax, suggesting that the PKM2 tetramer is incompletely saturated with bacterially-derived FBP under these conditions. In order to analyze PKM2 that was not bound to bacterial FBP, unfolded 6x-histidine-tagged PKM2 was isolated in the form of bacterial inclusion bodies, washed extensively in a denatured state incapable of binding FBP, and then refolded in the absence of FBP using an on-column refolding protocol (Protein Preparation 2 in Experimental Methods; hereafter “Prep 2”). PKM2 prepared using Prep 2 exhibited activation by FBP, as evidenced by a decrease in apparent Km for PEP and an increase in Vmax (Figure 1C). The observed increase in Vmax is consistent with an FBP-induced assembly of fully-active PKM2 tetramers and is consistent with previous studies [7, 16]. An activity-based determination of FBP activation of refolded PKM2 protein yielded an AC50 value of ~2 μM (Figure 1D), which is also comparable to previously reported values for PKM2 activation [7] and binding of FBP to non-phosphorylated PKL [41], although somewhat higher than a more recently reported AC50 of 118 nM [19]. Despite the kinetic behavior of the refolded PKM2 in response to FBP being more consistent with past studies, Prep 2 was laborious and we sought an alternative preparation of native, bacterially-expressed PKM2 that is largely free of bacterial FBP.
Figure 1. Steady-State Kinetics and FBP Activation of 6x-His-PKM2 prepared using different approaches.
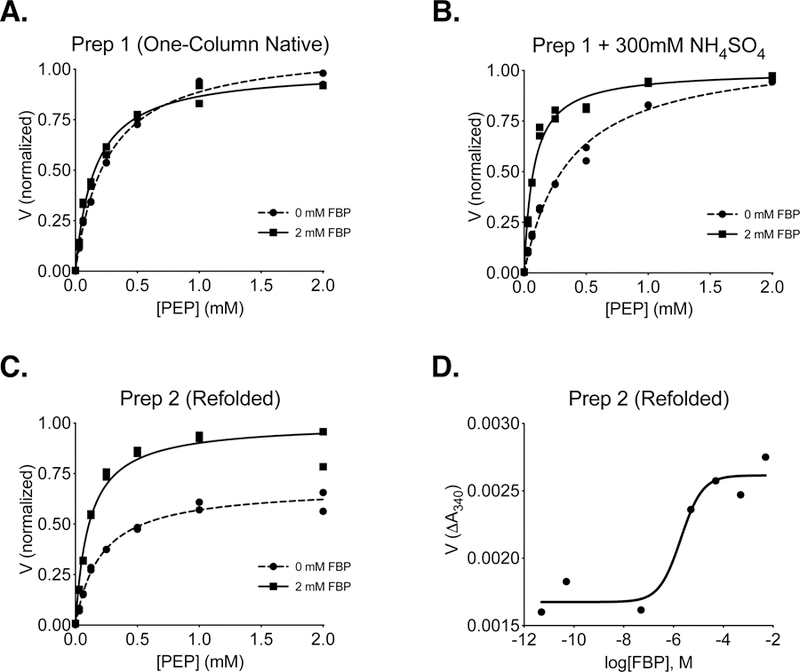
Individual data points are plotted, and assays were conducted using a saturating ADP concentration (5 mM) and the indicated concentration of FBP. (A) PKM2 reaction rate (V) with respect to PEP concentration for enzyme purified using one-step Ni-NTA affinity chromatography (Prep 1). (B) Reaction rate with respect to PEP concentration for Prep 1 PKM2 in the presence of 300 mM ammonium sulfate. (C) Reaction rate (V) with respect to PEP concentration for PKM2 isolated from inclusion bodies and refolded in the absence of FBP (Prep 2). (D) Activation of refolded Prep 2 PKM2 by FBP in the presence of subsaturating (0.125 mM) PEP and saturating (5 mM) ADP.
Ni-affinity chromatography followed by size exclusion chromatography of soluble 6x-His tagged PKM2 allowed the isolation of FBP-responsive PKM2 with high specific activity (Supplemental Figure 2). PKM2 protein prepared via this method (Protein Preparation 3 in Experimental Methods; hereafter “Prep 3”) retained the ability to be activated by FBP, and similar to refolded FBP-free PKM2 generated using Prep 2, FBP caused an increase in Vmax and a reduction in Km with respect to PEP (Figure 2A). FBP had little effect on Km with respect to ADP for protein produced in this manner (Figure 2B). The qualitative similarity between kinetics of refolded PKM2 (Prep 2) and PKM2 that was prepared using two-column purification (Prep 3) suggests that the addition of size exclusion chromatography is sufficient to separate PKM2 from the FBP present in the bacterial lysate. We also observed little variation in enzyme qualities when PKM2 was independently purified using the two-column Prep 3 (Supplemental Figure 3). As expected, PKM1 protein prepared using Prep 3 exhibited hyperbolic kinetics with respect to PEP and was not activated by FBP (Figure 2C, D). These results are summarized in Table 1, and the kinetic behavior of PKM2 and PKM1 are consistent with those reported previously, including a requirement for FBP to improve PKM2 catalytic function [7, 15]. Of note, the slightly lower Vmax observed for PKM1 compared to FBP-activated PKM2 should not be interpreted as PKM1 being an inferior enzyme. These results also establish Prep 3 as a method for preparing mutant enzymes to study how mutations affect the kinetic parameters of PKM2.
Figure 2. Steady-State Kinetics of 6x-His-PKM2 and 6x-His-PKM1 Prepared Using Two-Column Purification (Prep 3).
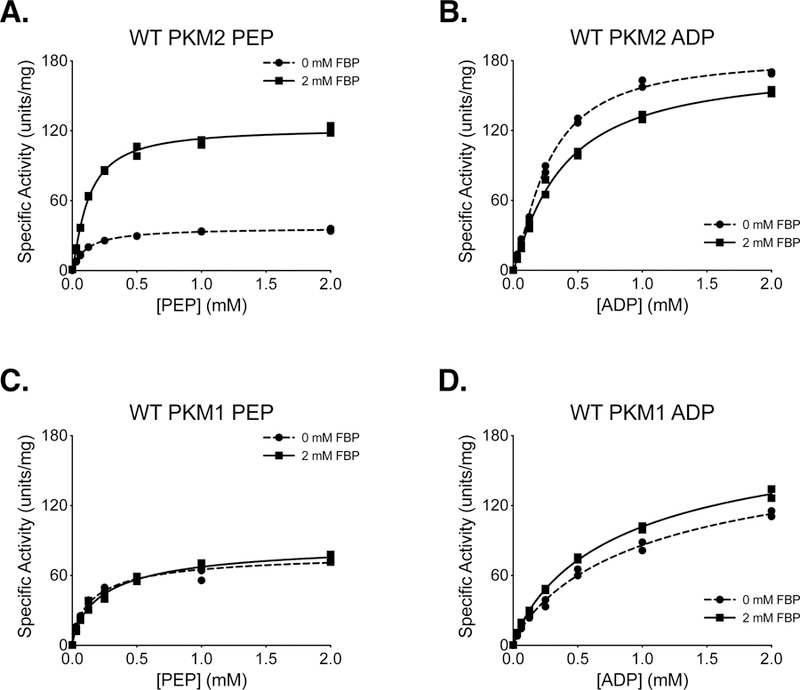
All conditions were performed in duplicate using proteins prepared with Prep 3, and individual data points are shown. (A) PKM2 activity with varying concentrations of PEP and saturating (5 mM) ADP. (B) PKM2 activity with varying concentrations of ADP and saturating (5 mM) PEP. (C) PKM1 activity with varying concentrations of PEP and saturating (5 mM) ADP. (D) PKM1 activity with varying concentrations of ADP and saturating (5 mM) PEP.
Table 1.
Kinetic Parameters for PEP in the Presence and Absence of 2 mM FBP for Wild-Type and Mutant PKM2.
| Enzyme | FBP | Km (μM) | Vmax (U/mg) | Vmax Normalized | nH | Fit†/R2 | FBP AC50 (95% CI) |
|---|---|---|---|---|---|---|---|
| PKM2 Prep 1 | − | 254.5 ± 18.02 | 111%* | (1.0) | M/0.9934 | ||
| + | 155.8 ± 12.80 | 100%* | (1.0) | M/0.9908 | |||
| PKM2 Prep 1 + 300mM (NH4)2 SO4 | − | 360.2 ± 33.56 | 110%* | (1.0) | M/0.9904 | ||
| + | 74.08 ± 6.798 | 100%* | (1.0) | M/0.9862 | |||
| PKM2 Prep 2 | − | 202.7 ± 17.49 | 94.82 ± 2.466 | 68.5%* | (1.0) | M/0.9898 | |
| + | 109.5 ± 14.38 | 138.4 ± 4.694 | 100%* | (1.0) | M/0.9739 | 1.98 μM (0.207−18.9) | |
| PKM1 Prep 3 | − | 126.6 ± 13.76 | 73.45 ± 2.134 | 90.8%* | (1.0) | M/0.9813 | |
| + | 191.2 ± 19.46 | 80.74 ± 2.441 | 100%* | (1.0) | M/0.9849 | ||
| PKM2 Prep 3 | − | 109.1 ± 3.787 | 36.92 ± 0.3305 | (1.0) | M/0.9981 | ||
| + | 141.4 ± 9.801 | 129.5 ± 2.472 | (1.0) | M/0.9929 | 48.7 nM (8.46–281) | ||
| PKM2 P117L | − | 171.0 ± 18.34 | 30.07 ± 1.260 | 1.4 ± 0.56 | H/0.9837 | ||
| + | 173.2 ± 21.34 | 74.84 ± 3.275 | 0.97 ± 0.11 | H/0.9898 | 2.37 μM (0.567–9.77) | ||
| PKM2 R246S | − | 517.8 ± 90.46 | 57.13 ± 6.216 | 2.1 ± 0.56 | H/0.9514 | ||
| + | 194.2 ± 33.39 | 94.74 ± 5.651 | 0.97 ± 0.11 | H/0.9858 | 788 nM (102–6111) | ||
| PKM2 G415R | − | 1124 ± 140.3 | 82.68 ± 6.323 | 1.4 ± 0.10 | H/0.9977 | ||
| + | 3170 ± 2219 | 234.8 ± 94.16 | 0.99 ± 0.12 | H/0.9915 | |||
| PKM2 R455Q | − | 404.6 ± 75.60 | 172.7 ± 11.91 | 0.93 ± 0.08 | H/0.9927 | ||
| + | 129.3 ± 4.126 | 161.3 ± 1.837 | 1.2 ± 00.4 | H/0.9988 | 693 nM (398–1208) | ||
| PKM2 R516C | − | 128.7 ± 18.62 | 10.51 ± 0.4431 | 0.72 ± 0.06 | H/0.9953 | ||
| + | 177.5 ± 17.98 | 10.91 ± 0.3963 | 1.1 ± 0.09 | H/0.9928 | |||
| PKM2 S37A | − | 853.1 ± 377.6 | 148.4 ± 21.71 | 0.71 ± 0.08 | H/0.9917 | ||
| + | 144.6 ± 9.667 | 159.4 ± 3.964 | 1.3 ± 0.09 | H/0.9940 | |||
| PKM2 K270M | − | 101.5 ± 37.25 | 5.144 ± 0.6810 | 1.5 ± 0.70 | H/0.7595 | ||
| + | 68.01 ± 17.26 | 6.253 ± 0.5325 | 1.2 ± 0.40 | H/0.8954 | |||
| PKM2 K367M | − | 299.2 ±71.17 | 43.72 ± 3.717 | 0.94 ± 0.12 | H/0.9832 | ||
| + | 85.89 ± 6.838 | 33.03 ± 0.8520 | 1.4 ± 0.15 | H/0.9893 | |||
| PKM2 R399E | − | 503.8 ± 97.94 | 84.90 ± 6.284 | 0.93 ± 0.08 | H/0.9936 | ||
| + | 158.8 ± 13.71 | 115.8 ± 3.784 | 1.3 ± 0.12 | H/0.9905 | |||
| PKM2 K433E | − | 739.5 ± 338.8 | 94.12 ± 14.63 | 0.75 ± 0.09 | H/0.9881 | ||
| + | 137.1 ± 9.522 | 85.26 ± 2.071 | 1.1 ± 0.07 | H/0.9958 | |||
| PKM2 H464A | − | 804.8 ± 69.91 | 139.2 ± 8.135 | 1.9 ± 0.17 | H/0.9947 | ||
| + | 88.53 ± 4.996 | 113.8 ±2.243 | 1.3 ± 0.10 | H/0.9937 | |||
Substrate-velocity data were fit using Michaelis-Menten (M) or Hill (H) models.
Maximum velocities relative to the FBP condition.
Mutations in PKM2 found in human cancer
Missense mutations throughout the coding sequence of PKM2 were found from sequencing of primary human cancers [29]. To determine whether cancer-associated mutations found in the TCGA occurred in conserved residues, the protein sequences of pyruvate kinase isoforms from various species were aligned and the locations of all 23 mutations identified in human cancers were compared to residues found in other pyruvate kinase proteins (Figure 3). More than half (13/23) of the mutations were found in residues that were identical across all pyruvate kinase protein considered, suggesting that many of these residues may be important for enzyme function.
Figure 3. Multiple Sequence Alignment of Pyruvate Kinase Isoforms from Different Species.

The locations of mutations found in human tumours [29] are marked with blue or red boxes. The red boxes indicate the mutations characterized in this study. Green boxes mark residues mutated in past studies of PKM2 function [17, 23, 35, 36, 43] that were characterized in this study.
The P117L, R246S, G415R, R455Q, and R516C cancer mutants were selected for study because they were examples of mutations from a variety of enzyme locations (Supplemental Figure 4). These mutations were among the first identified by the TCGA in the human PKM gene, and they occurred at low frequency in a variety of tumours, including uterine, kidney, and lung adenocarcinoma (Supplemental Table 1). These PKM2 mutants were prepared using the two-column purification (Prep 3), and enzyme kinetics were determined with respect to PEP and ADP (Figure 4, 5). A functional FBP activation assay was also used to determine FBP AC50 values for WT, P117L, R246S, G415R, and R455Q (Figure 6A–E). Given the relatively low enzyme concentrations required for activity-based assays, the response to FBP during AC50 determination is likely a factor of both FBP binding affinity and the tetramer-monomer equilibrium in solution [18], either of which may be affected by mutations. The location of the mutations and the effects on PKM2 function are described below.
Figure 4. Steady-State Kinetics of PKM2 Cancer Mutants With Respect to PEP Concentration.
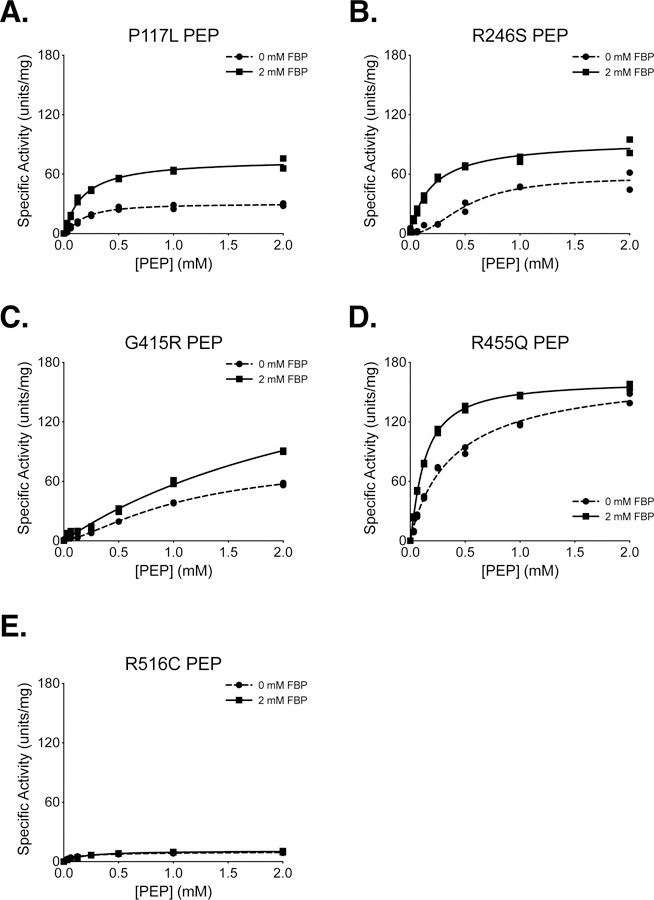
All proteins were prepared using Prep 3. For each indicated mutant, the ADP concentration was held constant at 2 mM. Assays were performed in duplicate and all data points are shown with allosteric sigmoidal (Hill) fit lines.
Figure 5. Steady-State Kinetics of PKM2 Cancer Mutants With Respect to ADP Concentration.
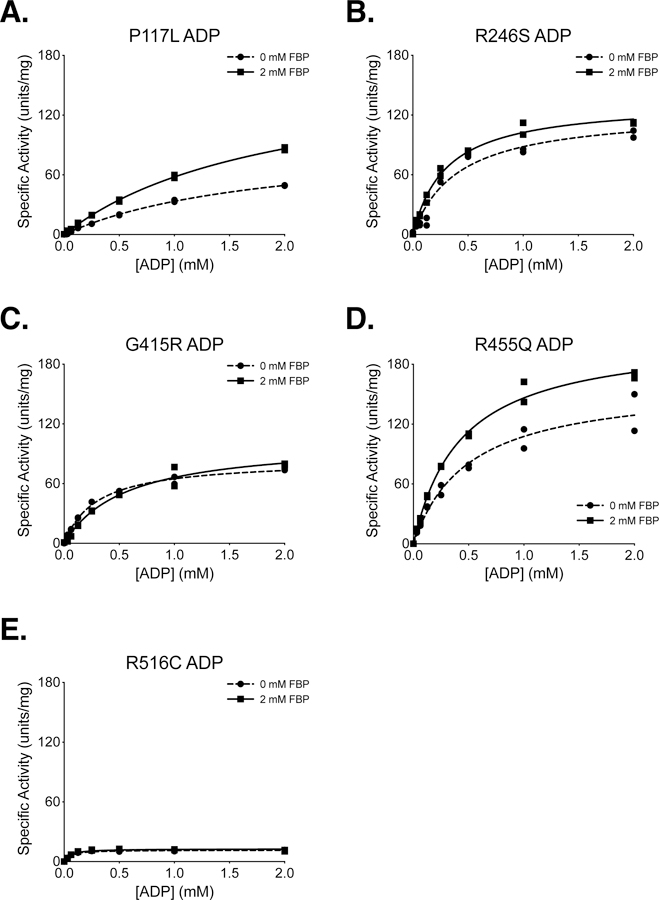
All proteins were prepared using Prep 3. For each indicated mutant, the PEP concentration was held constant at 2 mM. Assays were performed in duplicate and all data points are shown with Michaelis-Menten fit lines.
Figure 6. Activation of PKM2 and PKM2 Cancer Mutants by FBP.
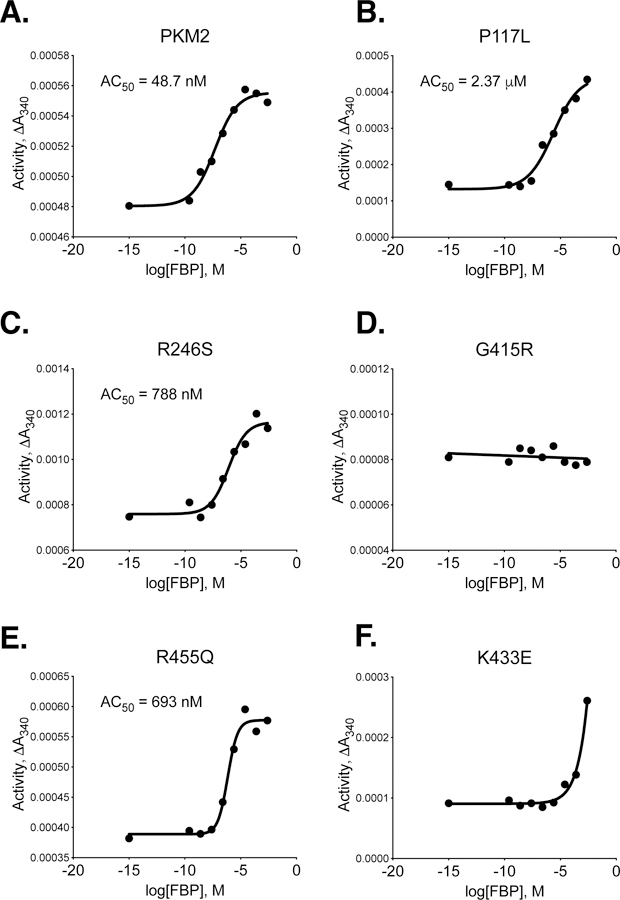
All proteins were prepared using Prep 3. For each indicated mutant, activity was assessed with 2 mM ADP and 0.125 mM PEP and data points are means of duplicate measurements. Data were fit to a sigmoid dose response curve with variable slope, except that G415R was fit with linear regression.
P117 is found in the “hinge” region of the protein between the A and B domains that allows closure of the active site upon substrate binding (Supplemental Figure 5); P117 is conserved from humans to E. coli. Despite a reduction in overall velocity, the P117L mutant retains near-wild-type affinity for PEP in the presence and absence of FBP, and the enzyme appears capable of forming stable tetramers in response to FBP binding as addition of FBP increases enzyme Vmax (Figures 4A,B). The main effect of the P117L substitution is to greatly reduce affinity of the enzyme for ADP (Figure 5A). The Km with respect to ADP is increased by a factor of 4 to 5, to 1.84 mM in the absence of FBP, and 1.93 mM in the presence of FBP (Table 2). These data suggest that FBP acts on this PKM2 mutant to stabilize a tetrameric form of the enzyme despite its reduced ADP affinity. The P117L mutation has some effect on apparent FBP activation, as the AC50 for FBP is increased relative to wild-type PKM2 (Figure 6A–B). Interestingly, a large selective reduction of ADP binding affinity has been observed in the R119C mutant of PKM1 [42]. R119 also lies near the hinge region and the active site cleft; however, the positive side chain of R119 is exposed in the active site and may play a more direct role in ADP binding than does P117.
Table 2.
Michaelis-Menten Kinetic Parameters for ADP in the Presence and Absence of 2 mM FBP for Wild-Type and Mutant PKM2.
| Enzyme | FBP | Km (μM) | Vmax (U/mg) | R2 |
|---|---|---|---|---|
| PKM2 (Prep 3) | − | 364.6 ± 33.20 | 209.6 ± 6.740 | 0.9909 |
| + | 453.6 ± 29.40 | 189.8 ± 4.630 | 0.9957 | |
| PKM1 | − | 744.4 ± 62.40 | 153.1 ± 5.628 | 0.9940 |
| + | 623.7 ± 37.24 | 168.4 ± 4.168 | 0.9967 | |
| PKM2 P117L | − | 1836 ± 132.7 | 94.74 ± 4.013 | 0.9978 |
| + | 1932 ± 109.0 | 169.6 ± 5.699 | 0.9987 | |
| PKM2 R246S | − | 406.4 ± 90.97 | 124.0 ± 10.12 | 0.9511 |
| + | 302.9 ± 26.98 | 133.5 ± 3.987 | 0.9905 | |
| PKM2 G415R | − | 294.1 ± 20.84 | 83.96 ± 1.891 | 0.9949 |
| + | 558.6 ± 81.54 | 103.2 ± 5.989 | 0.9820 | |
| PKM2 R455Q | − | 495.7 ± 87.61 | 161.0 ± 10.99 | 0.9683 |
| + | 426.7 ± 32.36 | 208.7 ± 5.848 | 0.9938 | |
| PKM2 R516C | − | 45.75 ± 6.187 | 11.64 ± 0.3176 | 0.9718 |
| + | 50.34 ± 9.253 | 12.82 ± 0.4903 | 0.9492 | |
| PKM2 S37A | − | 338.0 ± 36.89 | 87.31 ± 3.294 | 0.9858 |
| + | 459.4 ± 61.19 | 157.1 ± 7.904 | 0.9816 | |
| PKM2 K270M | − | 275.7 ± 47.84 | 6.063 ± 0.3438 | 0.9644 |
| + | 185.7 ± 72.53 | 5.909 ± 0.7172 | 0.8012 | |
| PKM2 K367M | − | 274.7 ± 30.90 | 45.47 ± 1.670 | 0.9844 |
| + | 297.4 ± 24.85 | 28.31 ± 0.7892 | 0.9911 | |
| PKM2 R399E | − | 513.1 ± 34.49 | 172.5 ± 4.515 | 0.9956 |
| + | 657.1 ± 71.43 | 166.4 ± 8.336 | 0.9903 | |
| PKM2 K433E | − | 444.0 ± 80.40 | 113.8 ± 7.702 | 0.9656 |
| + | 713.5 ± 31.26 | 191.6 ± 3.632 | 0.9984 | |
| PKM2 H464A | − | 456.9 ± 36.59 | 138.6 ± 4.185 | 0.9933 |
| + | 612.2 ± 42.74 | 213.1 ± 6.132 | 0.9957 | |
R246 is partially solvent-exposed and is part of the TIM barrel of the enzyme A domain, and is located within 14 angstroms of the active site (Supplemental Figure 6). In the wild-type protein, the R246 side chain appears to hydrogen bond with the backbone oxygen of F244 and form an electrostatic interaction with the negatively-charged side chain of D250. The R246S mutant displays a reduction in overall specific activity and a decrease in affinity for PEP relative to wildtype (Figure 4B). The disruption of catalytic efficiency of the enzyme is likely due to changes in active site conformation caused by disruption of the tertiary structure through loss of structural interactions in the A domain. This mutation had little effect on ADP and apparent FBP activation (Figures 5B, 6C).
G415 is located at the dimer-dimer interface of the tetramer. This glycine is located on an α-helix at the subunit interface and positioned such that the G415 of one subunit is in contact with the G415 of the subunit on the opposite side of the interface (Supplemental Figure 7). The lack of a bulky side chain allows tight packing of the α-helices at the interface. The G415R substitution introduces a large, charged residue at this interface and appears to abolish FBP activation of the enzyme (Figures 4C, 5C, 6D). PKM2 G415R exhibits cooperativity with respect to PEP binding in the absence of FBP (nH = 1.4), and this cooperativity is reduced in the presence of FBP (nH = 0.99). The reduction of cooperativity and Vmax increase caused by FBP suggest that FBP does indeed bind to the enzyme, but the G415R mutation blunts FBP activation to levels that may not be physiologically meaningful.
R455 is located near the FBP binding pocket and forms intra-chain electrostatic contacts with D476 and D485 (Supplemental Figure 8). Loss of charge due to the R455Q substitution might be expected to disrupt tertiary structure and affect FBP binding. This mutation appears to slightly decrease affinity for ADP and increase the apparent FBP AC50 to 693 nM, but the R455Q mutant otherwise retains near-wild-type activity (Figures 4D, 5D, 6E).
R516 is located on the FBP binding loop of the enzyme, but this residue is solvent-exposed and does not make contacts with other parts of the protein or bound FBP (Supplemental Figure 9). Interestingly, the R516C mutation greatly reduces the catalytic efficiency of the enzyme for unclear reasons (Figures 4E, 5E).
A summary of kinetic parameters is provided in Tables 1 and 2. Overall, the PKM2 mutants identified in human cancers that were evaluated exhibited either lowered maximal activity or reduced affinity for substrates and/or FBP when compared to wild-type PKM2.
Mutations in PKM2 Generated to Study Enzyme Functions
Several studies have introduced mutations into PKM2 in an attempt to abolish non-glycolytic aspects of protein function, or to alter the response of the enzyme to allosteric effectors [17, 23, 35, 36]. The effects of these mutations on the kinetic parameters of PKM2 as a glycolytic enzyme have not been extensively characterized. The substitutions considered here include S37A, K367M, K270M, R399E, K433E, and H464A [17, 23, 35, 36, 43, 44]. These mutant enzymes were prepared using two-column purification (Prep 3), and all enzymes were assayed for kinetics with respect to PEP and ADP in the absence and presence of FBP (Figure 7A–F, 8A–F).
Figure 7. Steady-State Kinetics of PKM2 “Literature” Mutants with Respect to PEP Concentration.
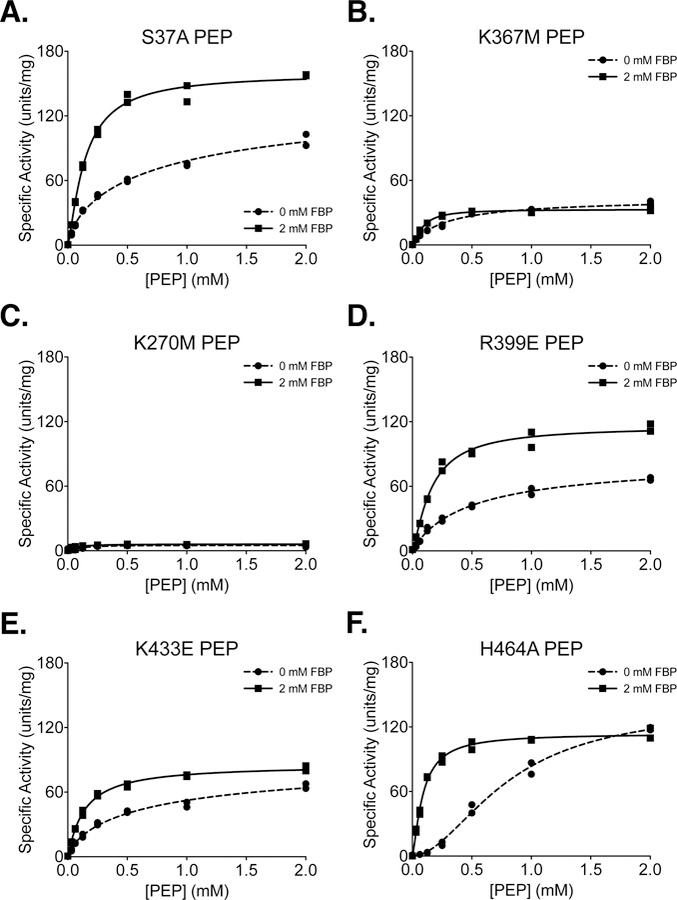
All proteins were prepared using Prep 3. For each indicated mutant, activity was assessed with ADP concentration held constant at 2 mM. Assays were performed in duplicate and all data points are shown with allosteric sigmoidal (Hill) fit lines.
Figure 8. Steady-State Kinetics of PKM2 “Literature” Mutants with Respect to ADP Concentration.
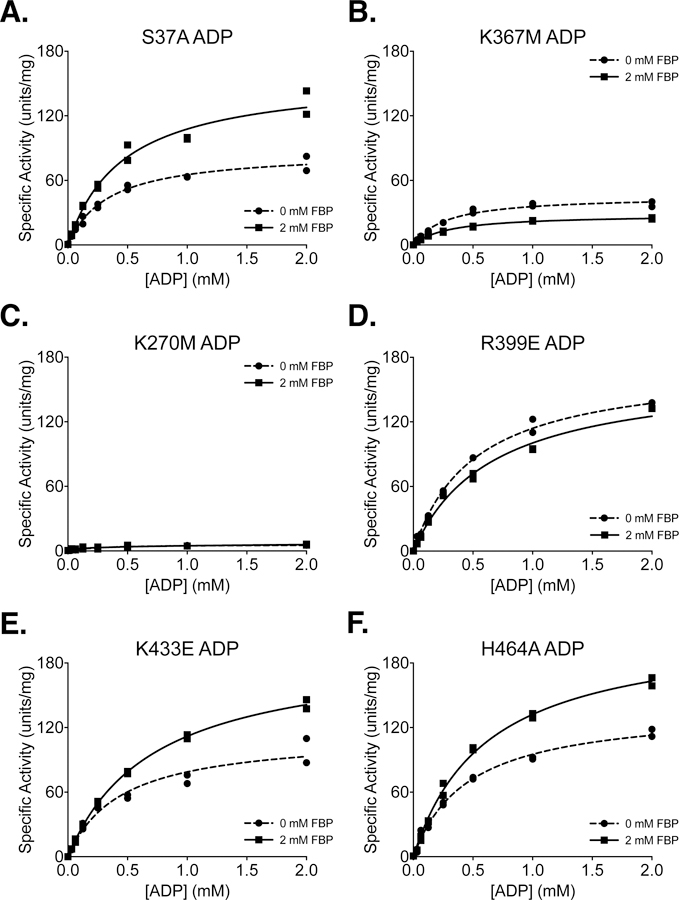
All proteins were prepared using Prep 3. For each indicated mutant, activity was assessed with PEP concentration held constant at 2 mM. Assays were performed in duplicate and all data points are shown with Michaelis-Menten fit lines.
Phosphorylation of PKM2 on S37 is reported to facilitate translocation of PKM2 to the nucleus for a role in oncogenic signaling [36]. The S37A mutation eliminates phosphorylation at S37 and reduces orthotopic xenograft growth of glioblastoma cells in mice. In the absence of added FBP, PKM2 S37A exhibited an increase in the Km for PEP (Table 1) and a reduction in Vmax with respect to ADP (Table 2) when compared to wild-type PKM2, indicating that the S37A mutation may affect activity of the enzyme despite being solvent exposed and relatively far from ligand binding sites (Supplemental Figure 10).
The K367M mutation was generated in an effort to disrupt the predicted ADP binding site of PKM2 and generate a kinase-inactive mutant [43]; however, the functional effect of this amino substitution was never assessed for the human enzyme. Inspection of the ADP-bound PKM2 crystal structure shows that K367 is indeed near the active site (Supplemental Figure 11), but the side chain does not make direct contact with the bound substrate (Supplemental Figure 12). While the K367M mutation does decrease specific activity and affinity for both PEP and ADP, the mutant still retains some activity with Vmax about 30% that of the wild-type enzyme (Figures 7B, 8B). These findings are similar to those reported for this mutation at a single set of substrate conditions [45]. Because the K367M substitution does not fully abolish PKM2 enzymatic activity, caution is advised in interpreting studies that rely on this mutation as completely disrupting PKM2 catalytic function for the pyruvate kinase reaction [43, 45–48].
An alternative catalytically-dead mutant of PKM2 can be generated by introducing a K270M mutation [44]. K270 is a catalytic lysine in the enzyme active site (Supplemental Figure 11) that serves to stabilize the pentacoordinate transition state that forms during the transfer of phosphate between reactant and product [15]. Substitution of the analogous lysine in Saccharomyces cerevisiae (K240) and Bacillus stearothermophilus (K221) with methionine results in properly-folded enzymes with severely reduced catalytic activity [49, 50]. The activity of the yeast K240M mutant is reduced by approximately 1000-fold when using Mg2+ as a cofactor [49]. Testing the effect of introducing this mutation into the human enzyme demonstrates that a K270M mutation reduces the overall activity of human PKM2 to less than 5% of the wild-type enzyme (Figures 7C, 8C). Both the K270M and K367M mutants can be produced via Prep 3 with similar yield and purity when compared with wild-type PKM2 (Supplemental Figure 13).
The R399E mutation results in loss of inter-subunit contacts across the dimer-dimer interface (Supplemental Figure 14), and has been reported to be a constitutive dimer with enhanced protein kinase activity [35], although a later study suggested that the R399E mutant can indeed form tetramers [51]. PKM2 R399E exhibits increased cooperativity with respect to PEP (nH=1.3) in the absence of FBP when compared to wild-type PKM2 (Table 1). FBP increases affinity of the R399E mutant enzyme for PEP but not ADP, as evidenced by a large decrease in the Km for PEP (Tables 1 and 2; Figures 7D, 8D). In addition to the effect of FBP on PEP binding affinity, Vmax for PEP increases somewhat during FBP activation (Figure 7D, Table 1), suggesting tetramer assembly or stabilization in the active state.
K433 is located on a loop that forms part of the FBP binding pocket (Supplemental Figure 15). This positively-charged residue is reported to interact with phosphotyrosine residues on other proteins during intracellular signaling and facilitate the inactivation of PKM2 by causing release of the allosteric activator FBP [23]. The K433E substitution has been reported to interfere with phosphotyrosine-based FBP release, but not with FBP activation of the protein. PKM2 K433E was responsive to activation by FBP (Figures 6F, 7E, 8E). FBP increased affinity for PEP as evidenced by a decrease in Km with respect to PEP; however, the mutant enzyme shows a somewhat reduced Vmax with FBP as compared to the wild-type enzyme (Table 1). Additionally, the affinity of the enzyme for FBP appears to be reduced relative to wild-type PKM2, as shown by the effect of FBP titration on enzyme activity (Figure 6F).
Serine acts as an allosteric activator of PKM2 with a reported AC50 of 1.3 mM [17], and H464 is found in the serine/alanine/phenylalanine binding pocket (Supplemental Figure 16) [7, 17, 52]. Substitution of H464 with alanine is reported to abolish serine binding without affecting FBP activation [17]. Our results confirm that PKM2 H464A is activated by FBP (Figures 7F, 8F). When compared to wild-type enzyme, the H464A mutation reduces Vmax in the FBP-bound state and increases the cooperativity of PEP binding in the absence of FBP.
A summary of kinetic parameters for the engineered mutants studied is also provided in Tables 1 and 2. While each cancer mutation and engineered mutant has a unique effect on enzyme kinetics, one theme that emerges is that these amino acid substitutions often reduce the apparent affinity of PKM2 for PEP compared to wild-type enzyme, especially in the absence of FBP. This pattern is evident in Table 1, as 7 of 11 mutants exhibit a two-fold or greater increase in the Km for PEP compared to wild type when FBP is absent.
Effect of Mutations on Tetramer-Monomer Equilibrium
We sought to determine the effect of mutations on the ability of PKM2 mutants to form stable tetramers when prepared via Prep 3 and assayed without exogenous FBP. HPLC-based size exclusion chromatography showed that wild-type PKM2 formed stable tetramers under our separation conditions (Figure 9). This HPLC separation method allows injection of enzyme at a relatively high concentration of 0.5 mg/ml, which is approximately 500-fold more concentrated than enzyme concentrations used for the kinetic assays. This provides an equivalent concentration of monomers (~86 μM) that is almost two orders of magnitude greater than the recently-reported tetramer-dimer and tetramer-monomer dissociation constants of around 1 μM for wild-type PKM2 [18, 39]. The non-dilutive HPLC assay conditions should thus favor PKM2 tetramer formation and provide a more direct measure of tetramer stability than the functional AC50 determinations. Interestingly, most mutants separated on size exclusion chromatography as tetramers using this assay, with the exception of G415R and R399E. The G415R mutation is expected to disrupt the packing of α-helices at the dimer-dimer interface of the tetramer (Supplemental Figure 7), and this mutant protein eluted at a time consistent with it being predominantly a monomer. The R399E mutant enzyme mostly eluted as a tetramer; however, some protein eluted at a volume consistent with a dimer, consistent with the loss of inter-subunit contacts across the dimer-dimer interface in this mutant (Supplemental Figure 14) only partially disrupting tetramer formation in these relatively concentrated conditions. Taken together, these results imply that most PKM2 mutations studied may have less effect on the tetramer-monomer equilibrium in vivo than suggested by the steady-state kinetics or FBP AC50 values.
Figure 9. HPLC Size Exclusion Chromatography of PKM2 and PKM2 Mutants.
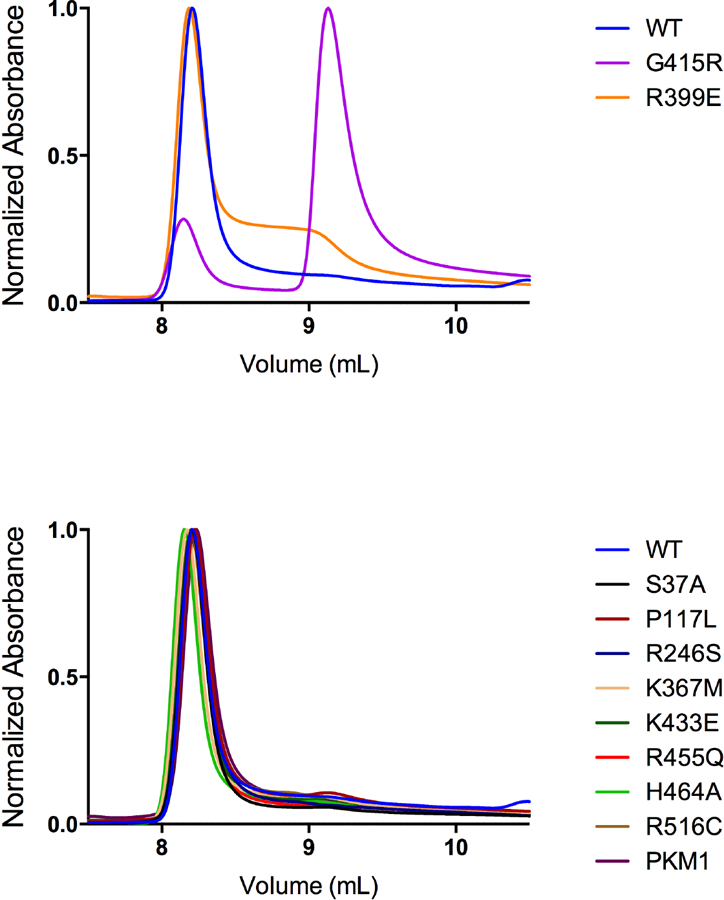
PKM2 proteins were prepared using Prep 3, then separated by HPLC size exclusion in the absence of FBP to determine the effect of mutations on tetramer-monomer equilibrium. This separation method accommodates a high concentration of enzyme (0.5 mg/ml) relative to the kinetic assays. An elution volume of ~8.25 mL is consistent with tetramers while an elution volume of ~9.25 mL is consistent with monomer. Only the G415R and R399E mutants showed significant amounts of protein eluting at a volume consistent with the enzyme being a dimer or monomer.
DISCUSSION
Single amino acid substitutions affecting PKM2 are found in human cancers, and the mutations considered in this study tended to lower affinity of the enzyme for substrate, reduce its maximal activity, or alter activation by FBP. That these mutations have functional effects is not surprising for two reasons. First, these mutations affect residues that are highly conserved, and many of the cancer mutations considered here are located near functional sites on the enzyme. Second, PKM2 activity was found to be sensitive to amino acid substitution when SNPs in the human population that lead to PKM2 missense mutations were studied and found to result in reduced enzyme activity, reduced thermal stability, or altered allosteric regulation [32]. Characterization of missense PKM2 mutations in patients with Bloom Syndrome, the underlying cause of which is defective DNA damage repair, revealed that the mutations studied also negatively affect PKM2 function [53–55].
The high affinity of PKM2 for FBP is shared by other mammalian pyruvate kinase isoforms such as PKL, but not by the pyruvate kinase isoforms of unicellular organisms. The values reported elsewhere for half-maximal binding or half-maximal activation of mammalian pyruvate kinases are in the sub- to low-micromolar range (0.34–7.5 μM) [7, 39, 41], with the most recent work reporting KD = 25.5 nM for direct binding of FBP to PKM2 (after correction for co-purified FBP) and AC50 = 118 nM for FBP activation of wild-type PKM2 [19]. While the FBP AC50 value reported in the present study for WT PKM2 (48.7 nM, Figure 6A and Table 1) compares favorably to those values, the FBP AC50 we observe for refolded PKM2 is higher (1.98 μM), although still within the range of previously reported results. While this discrepancy may be an artefact of the preparation methods, there is greater imprecision in the AC50 parameter for refolded PKM2. A reported value for the intracellular FBP concentration in mammalian cells is 80 μM [56], which is at least one order of magnitude greater than the concentration required for half-maximal activation of PKM2. Thus, in the absence of other inputs, FBP binding to mammalian pyruvate kinases may be saturated in many cellular conditions in vivo [19]. In contrast, the K0.5 values for FBP activation are around 45 μM for S. cerevisiae pyruvate kinase [57], and 70 μM for E.coli pyruvate kinase [58], making the yeast and E.coli pyruvate kinase isoforms better suited to respond to changes in intracellular FBP occurring in the physiological concentration range. Estimates of FBP concentration in E.coli and yeast can be higher than those found in mammalian cells [59, 60], arguing that FBP concentrations can be high enough to saturate the microbial enzymes under some conditions. The mammalian isoforms appear to have traded some allosteric control based on FBP concentration for regulation by intracellular signaling to control metabolism in different specialized tissue contexts. Inactivation of PKL by phosphorylation during gluconeogenesis in the liver is one example, and inactivation of PKM2 due to FBP release caused by growth signaling in proliferating cells is another.
Most wild-type and mutant PKM2 protein eluted at a volume consistent with tetramers during HPLC size exclusion chromatography, despite the observation that maximal enzyme activity (Vmax) from the same protein preparations could be increased by FBP, consistent with formation of active tetramers in a tetramer-dimer or tetramer-monomer equilibrium. These seemingly contradictory observations may be explained by the enzyme concentrations used in the respective assays. HPLC size exclusion chromatography evaluates relatively concentrated samples (0.5 mg/ml) that are a better approximation of cellular conditions than is the kinetic assay, which requires significant dilution of this highly active enzyme (to approximately 1 μg/mL or 17 nM) to allow determination of initial rates. The in vivo concentration of pyruvate kinase has been reported as 172 uM or 10 mg/mL [56], a value well above the reported KD of 1 μM for tetramer-monomer and tetramer-dimer equilibria [18, 39]. The HPLC size exclusion results were thus obtained from samples at concentrations above the KD for tetramer dissociation, while the kinetic assays can only be conducted with samples at concentrations below the KD which favors tetramer dissociation. The kinetic assays employed by us and others for determining AC50 values for FBP activation of PKM2 are thus limited in that they measure the enzyme in an artificially dilute state, absent of the molecular crowding present in the cytosol. Our size exclusion results differ from a recent report [18] in that we observe almost exclusively the tetramer form of PKM2 in the absence of FBP; this may be due to a number of factors in the current study, including a 5-fold greater concentration of protein, high flow rates allowed by HPLC that limit time-dependent dissociation of PKM2 tetramers, and the presence of Mg2+ available during separation to bind the active site and stabilize the protein. These data suggest that the majority of PKM2 in cells may exist in a tetrameric state rather than as mixture of inactive monomers/dimers and active tetramers.
The mutations that have been used to study non-canonical PKM2 functions were found to alter some aspect of PKM2 function as a glycolytic enzyme. The effect exhibited by these disparate mutations on enzyme kinetics suggests that generation of PKM2 mutations that abolish or alter specific protein functions is challenging. Therefore, interpretation of data relying on PKM2 mutants to study aspects of cell biology linked to cell proliferation also requires consideration of the metabolic consequences of those mutants.
The determination of kinetic parameters in this study was restricted to the analysis of PKM2 homomultimers. The cancer mutations reported are all heterozygous, suggesting that the mutated form of the enzyme is co-expressed with wild-type enzyme in the affected cancer cells. A similar situation has been found in individuals with Bloom syndrome, where heterozygous missense mutations occur in PKM2 [53]. Like the mutations studied here, the single amino acid substitutions found in Bloom Syndrome patients alter enzyme kinetics [54], despite the mutant subunits being capable of heterotetramerization with wild-type PKM2 [55]. PKM2 has been observed to form heterotetramers with PKM1 and PKL [20, 61], and heterotetramerization is likely to occur between wild-type PKM2 and PKM2 mutants, as even chicken PKM1 and bovine PKL can form functional heterotetramers [62]. The kinetic parameters of heterotetramers containing wild-type and cancer mutation subunits remain to be determined, but they are likely to be intermediate between the kinetics of homotetramers of the two isoforms [63, 64]. Because PKM2 and PKM1 differ by only one exon, they share significant sequence identity and four of the five cancer mutations studied here (P117L, R246S, R455Q, and R516C) would also occur in the M1 isoform if expressed in the cancer cell.
The presence of PKM2 mutations in human cancers demonstrates that some tumor cells tolerate, and perhaps select for, reduced PKM2 activity. PKM2 is not required for growth of many cancers [11, 29, 65–70], and loss-of-function mutations may be advantageous for cancer cells since decreased pyruvate kinase activity promotes a proliferative metabolic program [11, 20, 28, 29]. Retention of one wild-type copy of PKM2 may provide metabolic flexibility by allowing upregulation of PKM2 activity to promote cancer cell survival under nutrient stress conditions [29]. Mutations in PKM2 are therefore not oncogenic, but may play a part in creating a metabolic state in the cancer cell that is permissive for proliferation. Regardless, this analysis demonstrates that the kinetic properties of PKM2 are sensitive to amino acid substitution and informs our understanding of how PKM2 expression impacts cancer biology.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Katherine R. Mattaini and Katherine “TK” Allsop for technical assistance. V.M.L. and A.J.H. were supported by the MIT Undergraduate Research Opportunities Program. A.M.H. was supported by an HHMI graduate student fellowship and was a Vertex Scholar. Z.L. and W.J.I. were supported in part by T32GM007287. W.J.I. also acknowledges support from the Sara and Frank McKnight Fund for Biochemical Research, and NIH DP5OD021365. M.V.H. acknowledges support from NIH R01CA168653, P30CA1405141, the Burroughs Wellcome Fund, the Ludwig Center at MIT, SU2C, the Lustgarten Foundation, the MIT Center for Precision Cancer Medicine, and a Faculty Scholars award from HHMI.
Footnotes
DISCLOSURES
M.G.V.H. is a consultant and scientific advisory board member of Agios Pharmaceuticals, Aeglea Biotherapeutics, and Auron Therapeutics.
REFERENCES
- 1.Mazurek S (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells, The international journal of biochemistry & cell biology. 43, 969–80. [DOI] [PubMed] [Google Scholar]
- 2.Dayton TL, Jacks T & Vander Heiden MG (2016) PKM2, cancer metabolism, and the road ahead, EMBO Rep. 17, 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris RA & Fenton AW (2019) A critical review of the role of M2PYK in the Warburg effect, Biochim Biophys Acta Rev Cancer. 1871, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imamura K & Tanaka T (1972) Multimolecular forms of pyruvate kinase from rat and other mammalian tissues. I. Electrophoretic studies, Journal of biochemistry. 71, 1043–51. [DOI] [PubMed] [Google Scholar]
- 5.David CJ, Chen M, Assanah M, Canoll P & Manley JL (2010) HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer, Nature. 463, 364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG & Krainer AR (2010) The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism, Proceedings of the National Academy of Sciences of the United States of America. 107, 1894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan HP, O’Reilly FJ, Wear MA, O’Neill JR, Fothergill-Gilmore LA, Hupp T & Walkinshaw MD (2013) M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation, Proceedings of the National Academy of Sciences of the United States of America. 110, 5881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor CB & Bailey E (1967) Activation of liver pyruvate kinase by fructose 1,6-diphosphate, The Biochemical journal. 102, 32C–33C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koler RD & Vanbellinghen P (1968) The mechanism of precursor modulation of human pyruvate kinase I by fructose diphosphate, Advances in enzyme regulation. 6, 127–42. [DOI] [PubMed] [Google Scholar]
- 10.Kahn A & Marie J (1982) Pyruvate kinases from human erythrocytes and liver, Methods Enzymol. 90 Pt E, 131–40. [DOI] [PubMed] [Google Scholar]
- 11.Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T & Vander Heiden MG (2016) Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma, Genes Dev. 30, 1020–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaneton B & Gottlieb E (2012) Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer, Trends in biochemical sciences. 37, 309–16. [DOI] [PubMed] [Google Scholar]
- 13.Gui DY, Lewis CA & Vander Heiden MG (2013) Allosteric regulation of PKM2 allows cellular adaptation to different physiological states, Science signaling. 6, pe7. [DOI] [PubMed] [Google Scholar]
- 14.Imamura K, Taniuchi K & Tanaka T (1972) Multimolecular forms of pyruvate kinase. II. Purification of M 2 -type pyruvate kinase from Yoshida ascites hepatoma 130 cells and comparative studies on the enzymological and immunological properties of the three types of pyruvate kinases, L, M 1, and M 2, J Biochem. 72, 1001–15. [DOI] [PubMed] [Google Scholar]
- 15.Dombrauckas JD, Santarsiero BD & Mesecar AD (2005) Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis, Biochemistry. 44, 9417–29. [DOI] [PubMed] [Google Scholar]
- 16.Ashizawa K, McPhie P, Lin KH & Cheng SY (1991) An in vitro novel mechanism of regulating the activity of pyruvate kinase M2 by thyroid hormone and fructose 1, 6-bisphosphate, Biochemistry. 30, 7105–11. [DOI] [PubMed] [Google Scholar]
- 17.Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O’Reilly M & Gottlieb E (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2, Nature. 491, 458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan M, McNae IW, Chen Y, Blackburn EA, Wear MA, Michels PAM, Fothergill-Gilmore LA, Hupp T & Walkinshaw MD (2018) An allostatic mechanism for M2 pyruvate kinase as an amino-acid sensor, The Biochemical journal. 475, 1821–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macpherson JA, Theisen A, Masino L, Fets L, Driscoll PC, Encheva V, Snijders AP, Martin SR, Kleinjung J, Barran PE, Fraternali F & Anastasiou D (2019) Functional cross-talk between allosteric effects of activating and inhibiting ligands underlies PKM2 regulation, Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ & Vander Heiden MG (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis, Nat Chem Biol. 8, 839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashizawa K, Willingham MC, Liang CM & Cheng SY (1991) In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate, J Biol Chem. 266, 16842–6. [PubMed] [Google Scholar]
- 22.Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA & Satin LS (2013) Direct measurements of oscillatory glycolysis in pancreatic islet beta-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 activity, J Biol Chem. 288, 33312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christofk HR, Vander Heiden MG, Wu N, Asara JM & Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein, Nature. 452, 181–6. [DOI] [PubMed] [Google Scholar]
- 24.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL & Lei QY (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth, Molecular cell. 42, 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG & Cantley LC (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses, Science. 334, 1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eigenbrodt E, Reinacher M, Scheefers-Borchel U, Scheefers H & Friis R (1992) Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells, Crit Rev Oncog. 3, 91–115. [PubMed] [Google Scholar]
- 27.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL & Cantley LC (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth, Nature. 452, 230–3. [DOI] [PubMed] [Google Scholar]
- 28.Lunt SY, Muralidhar V, Hosios AM, Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo PN, Hollern DP, Bellinger G, Dayton TL, Christen S, Elia I, Dinh AT, Stephanopoulos G, Manalis SR, Yaffe MB, Andrechek ER, Fendt SM & Vander Heiden MG (2015) Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation, Molecular cell. 57, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC & Vander Heiden MG (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells, Cell. 155, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stammers DK & Muirhead H (1977) Three-dimensional structure of cat muscle pyruvate kinase at 3–1 A resolution, Journal of molecular biology. 112, 309–16. [DOI] [PubMed] [Google Scholar]
- 31.Muirhead H, Clayden DA, Barford D, Lorimer CG, Fothergill-Gilmore LA, Schiltz E & Schmitt W (1986) The structure of cat muscle pyruvate kinase, The EMBO journal. 5, 475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allali-Hassani A, Wasney GA, Chau I, Hong BS, Senisterra G, Loppnau P, Shi Z, Moult J, Edwards AM, Arrowsmith CH, Park HW, Schapira M & Vedadi M (2009) A survey of proteins encoded by non-synonymous single nucleotide polymorphisms reveals a significant fraction with altered stability and activity, The Biochemical journal. 424, 15–26. [DOI] [PubMed] [Google Scholar]
- 33.Lu Z & Hunter T (2018) Metabolic Kinases Moonlighting as Protein Kinases, Trends Biochem Sci. 43, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amin S, Yang P & Li Z (2019) Pyruvate kinase M2: A multifarious enzyme in non-canonical localization to promote cancer progression, Biochim Biophys Acta Rev Cancer. 1871, 331–341. [DOI] [PubMed] [Google Scholar]
- 35.Gao X, Wang H, Yang JJ, Liu X & Liu ZR (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase, Molecular cell. 45, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC & Lu Z (2012) ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect, Nature cell biology. 14, 1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hosios AM, Fiske BP, Gui DY & Vander Heiden MG (2015) Lack of Evidence for PKM2 Protein Kinase Activity, Molecular cell. 59, 850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J & Chen J (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth, Science signaling. 2, ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gavriilidou AFM, Holding FP, Mayer D, Coyle JE, Veprintsev DB & Zenobi R (2018) Native Mass Spectrometry Gives Insight into the Allosteric Binding Mechanism of M2 Pyruvate Kinase to Fructose-1,6-Bisphosphate, Biochemistry. 57, 1685–1689. [DOI] [PubMed] [Google Scholar]
- 40.Blair JB & Walker RG (1984) Rat liver pyruvate kinase: influence of ligands on activity and fructose 1,6-bisphosphate binding, Archives of biochemistry and biophysics. 232, 202–13. [DOI] [PubMed] [Google Scholar]
- 41.El-Maghrabi MR, Claus TH, McGrane MM & Pilkis SJ (1982) Influence of phosphorylation on the interaction of effectors with rat liver pyruvate kinase, J Biol Chem. 257, 233–40. [PubMed] [Google Scholar]
- 42.Cheng X, Friesen RH & Lee JC (1996) Effects of conserved residues on the regulation of rabbit muscle pyruvate kinase, J Biol Chem. 271, 6313–21. [DOI] [PubMed] [Google Scholar]
- 43.Le Mellay V, Houben R, Troppmair J, Hagemann C, Mazurek S, Frey U, Beigel J, Weber C, Benz R, Eigenbrodt E & Rapp UR (2002) Regulation of glycolysis by Raf protein serine/threonine kinases, Advances in enzyme regulation. 42, 317–32. [DOI] [PubMed] [Google Scholar]
- 44.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A & Semenza GL (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1, Cell. 145, 732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang P, Li Z, Fu R, Wu H & Li Z (2014) Pyruvate kinase M2 facilitates colon cancer cell migration via the modulation of STAT3 signalling, Cell Signal. 26, 1853–62. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, Zhou Q, Majumder S, Bi E, Liu DX, Huang S & Lu Z (2014) PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells, Nature communications. 5, 5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K & Lu Z (2011) Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation, Nature. 480, 118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK & Lu Z (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis, Cell. 150, 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bollenbach TJ, Mesecar AD & Nowak T (1999) Role of lysine 240 in the mechanism of yeast pyruvate kinase catalysis, Biochemistry. 38, 9137–45. [DOI] [PubMed] [Google Scholar]
- 50.Sakai H (2005) Mutagenesis of the active site lysine 221 of the pyruvate kinase from Bacillus stearothermophilus, Journal of biochemistry. 137, 141–5. [DOI] [PubMed] [Google Scholar]
- 51.Wang P, Sun C, Zhu T & Xu Y (2015) Structural insight into mechanisms for dynamic regulation of PKM2, Protein Cell. 6, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams R, Holyoak T, McDonald G, Gui C & Fenton AW (2006) Differentiating a ligand’s chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase, Biochemistry. 45, 5421–9. [DOI] [PubMed] [Google Scholar]
- 53.Anitha M, Kaur G, Baquer NZ & Bamezai R (2004) Dominant negative effect of novel mutations in pyruvate kinase-M2, DNA Cell Biol. 23, 442–9. [DOI] [PubMed] [Google Scholar]
- 54.Akhtar K, Gupta V, Koul A, Alam N, Bhat R & Bamezai RN (2009) Differential behavior of missense mutations in the intersubunit contact domain of the human pyruvate kinase M2 isozyme, J Biol Chem. 284, 11971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta V, Kalaiarasan P, Faheem M, Singh N, Iqbal MA & Bamezai RN (2010) Dominant negative mutations affect oligomerization of human pyruvate kinase M2 isozyme and promote cellular growth and polyploidy, J Biol Chem. 285, 16864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srivastava DK & Bernhard SA (1986) Metabolite transfer via enzyme-enzyme complexes, Science. 234, 1081–6. [DOI] [PubMed] [Google Scholar]
- 57.Murcott TH, Gutfreund H & Muirhead H (1992) The cooperative binding of fructose-1,6-bisphosphate to yeast pyruvate kinase, The EMBO journal. 11, 3811–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waygood EB, Mort JS & Sanwal BD (1976) The control of pyruvate kinase of Escherichia coli. Binding of substrate and allosteric effectors to the enzyme activated by fructose 1,6-bisphosphate, Biochemistry. 15, 277–82. [DOI] [PubMed] [Google Scholar]
- 59.Xu YF, Zhao X, Glass DS, Absalan F, Perlman DH, Broach JR & Rabinowitz JD (2012) Regulation of yeast pyruvate kinase by ultrasensitive allostery independent of phosphorylation, Molecular cell. 48, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ & Rabinowitz JD (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli, Nature chemical biology. 5, 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardenas JM & Dyson RD (1978) Mammalian pyruvate kinase hybrid isozymes: tissue distribution and physiological significance, J Exp Zool. 204, 361–7. [DOI] [PubMed] [Google Scholar]
- 62.Cardenas JM, Blachly EG, Ceccotti PL & Dyson RD (1975) Properties of chicken skeletal muscle pyruvate kinase and a proposal for its evolutionary relationship to the other avian and mammalian isozymes, Biochemistry. 14, 2247–52. [DOI] [PubMed] [Google Scholar]
- 63.Cardenas JM & Dyson RD (1973) Bovine pyruvate kinases. II. Purification of the liver isozyme and its hybridization with skeletal muscle pyruvate kinase, J Biol Chem. 248, 6938–44. [PubMed] [Google Scholar]
- 64.Hubbard DR & Cardenas JM (1975) Kinetic properties of pyruvate kinase hybrids formed with native type L and inactivated type M subunits, J Biol Chem. 250, 4931–6. [PubMed] [Google Scholar]
- 65.Cortes-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, Muller A, Haberkorn A, Chene P, Sellers WR & Hofmann F (2013) M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth, Proceedings of the National Academy of Sciences of the United States of America. 110, 489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang YH, Israelsen WJ, Lee D, Yu VW, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG & Scadden DT (2014) Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis, Cell. 158, 1309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tech K, Tikunov AP, Farooq H, Morrissy AS, Meidinger J, Fish T, Green SC, Liu H, Li Y, Mungall AJ, Moore RA, Ma Y, Jones SJM, Marra MA, Vander Heiden MG, Taylor MD, Macdonald JM & Gershon TR (2017) Pyruvate Kinase Inhibits Proliferation during Postnatal Cerebellar Neurogenesis and Suppresses Medulloblastoma Formation, Cancer Res. 77, 3217–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lau AN, Israelsen WJ, Roper J, Sinnamon MJ, Georgeon L, Dayton TL, Hillis AL, Yilmaz OH, Di Vizio D, Hung KE & Vander Heiden MG (2017) PKM2 is not required for colon cancer initiated by APC loss, Cancer Metab. 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hillis AL, Lau AN, Devoe CX, Dayton TL, Danai LV, Di Vizio D & Vander Heiden MG (2018) PKM2 is not required for pancreatic ductal adenocarcinoma, Cancer Metab. 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dayton TL, Gocheva V, Miller KM, Bhutkar A, Lewis CA, Bronson RT, Vander Heiden MG & Jacks T (2018) Isoform-specific deletion of PKM2 constrains tumor initiation in a mouse model of soft tissue sarcoma, Cancer Metab. 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.