

Abstract
The deubiquitinase (DUB) Ubiquitin C-terminal Hydrolase L1 (UCHL1) is expressed primarily in the central nervous system under normal physiological conditions. However, UCHL1 is overexpressed in various aggressive forms of cancer with strong evidence supporting UCHL1 as an oncogene in lung, glioma, and blood cancers. In particular, the level of UCHL1 expression in these cancers correlates with increased invasiveness and metastatic behavior, as well as poor patient prognosis. Although UCHL1 is considered an oncogene with potential as a therapeutic target, there remains a significant lack of useful small-molecule probes to pharmacologically validate in vivo targeting of the enzyme. Herein, we describe the characterization of a new covalent cyanopyrrolidine-based UCHL1 inhibitory scaffold in biochemical and cellular studies to better understand the utility of this inhibitor in elucidating the role of UCHL1 in cancer biology.
Keywords: Covalent inhibitor, deubiquitinase, enzyme inhibition, hydrolase, structural biology
Graphical Abstract
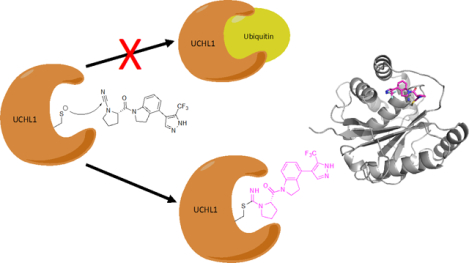
We present biochemical, structural and cellular characterization of a newly reported class of cyanopyrrolidine covalent inhibitors of the important oncotarget UCHL1. The insight into mechanism of action and structural analysis learned from this work provides information for the future design of potent and selective UCHL1 covalent inhibitors.
Introduction
The addition of ubiquitin (Ub) to proteins is a post-translational modification important for regulating many aspects of eukaryotic biology.[1] Ubiquitin is added to Lys residues of target proteins through a covalent bond between the ε-amino group of the Lys residue and the last carboxylate group of the terminal Gly76 of Ub through an isopeptide bond. This covalent addition is catalyzed by the sequential action of three enzymes known as the E1 Ub activating enzyme,[2] E2 Ub conjugating enzyme,[3] and E3 Ub ligases in an ATP-driven process.[4] This usually results in attachment of several Ub groups as a polyubiquitin-chain modification of the target proteins; wherein, successive Ub groups are added to the first Ub via one of seven Lys residues on Ub or its Met1 group in isopeptide (or peptide) links that connect Ub monomers in the polyubiquitin chain. Eight distinct types of polyubiquitin chains resulting from linkage through each distinct amino group of Ub, along with branched architectures, can result in complex biological signals that form the basis of the Ub signaling system in eukaryotes.[5] Ub can be released from proteins (and other Ub groups in polyubiquitin chains) through hydrolysis by means of one of several enzymes that are collectively known as deubiquitinases (DUBs).[6] Humans have close to 100 DUBs, most of which are cysteine proteases, with one separate enzymatic class being the so-called JAMM proteases, which require Zn dependent catalysis.[7] The cysteine protease DUBs are grouped into six types: the Ubiquitin Specific Proteases (USP), Ubiquitin C-terminal Hydrolase (UCH), Machado-Josephin Domain, Ovarian Tumor Proteases, and the more recently discovered Mindy and ZUFSP groups.[7–8] Regulation of the ubiquitination and deubiquitination processes has implications on various disease states including neurodegenerative diseases and cancer.[9] Many cellular processes are subject to precise regulation though a balance of ubiquitination and its removal by DUBs; accordingly, DUBs have emerged as key players in cellular homeostasis and, when deregulated have impact on various disease states. Thus, DUBs are novel targets for therapeutic intervention in cancer, auto-immune, and neurodegenerative diseases.[9g]
The DUB known as Ubiquitin C-terminal Hydrolase L1 (UCHL1), is a 25 kDa protein that is expressed almost exclusively in the central and peripheral nervous system under normal physiological conditions.[10] While UCHL1 has been studied for nearly 20 years, its biological role under normal and pathophysiological conditions has yet to be elucidated. As one of the most abundant soluble proteins in the brain, it has been linked to the progression of Alzheimer’s and Parkinson’s disease.[11] There is also strong evidence supporting UCHL1 as an oncogene in various cancers including glioblastoma, small-cell lung cancer, and blood cancers, among others.[9j, 12] In particular, increased UCHL1 levels in these cancers correlates with increased invasiveness and metastatic behavior as well as poor patient prognosis.[13] In many cases reducing UCHL1 activity either by performing genetic depletion experiments or expression of catalytically inactive mutants results in a significant reduction in the metastatic behavior of certain cancer cell lines.[13b, 14]
Although UCHL1 is considered an oncogene with potential as a therapeutic target, there is a significant lack of small-molecule inhibitor development in the field. LDN-57444[9f] and LDN-91946[15] (Figure 1) were identified in the early 2000’s. LDN-57444, the more potent of the two (IC50 value of 0.88 ± 0.14 μM), serves as the de facto chemical probe for UCHL1 (Figure 1). However, this molecule has liabilities including off-target toxicity and chemical instability.[13d] A peptide-based covalent inhibitor VAEFMK (Figure 1) has been identified and characterized structurally with UCHL1, though no cellular or inhibitory data have been reported.[16] Recently, a new series of cyanopyrrolidine-based inhibitors have been identified in patent literature (Figure 1, compound 1),[17] however, there was little inhibitory information and no on-target cellular validation has been reported. Given the relative paucity of UCHL1 inhibitors in the literature, we set out to characterize this new class of inhibitor in biochemical and cellular assays, as well as determine its mechanism of action. Successfully determining the mechanism of action and validating on-target cellular activity could provide new insights into future UCHL1 inhibitor design.
Figure 1.
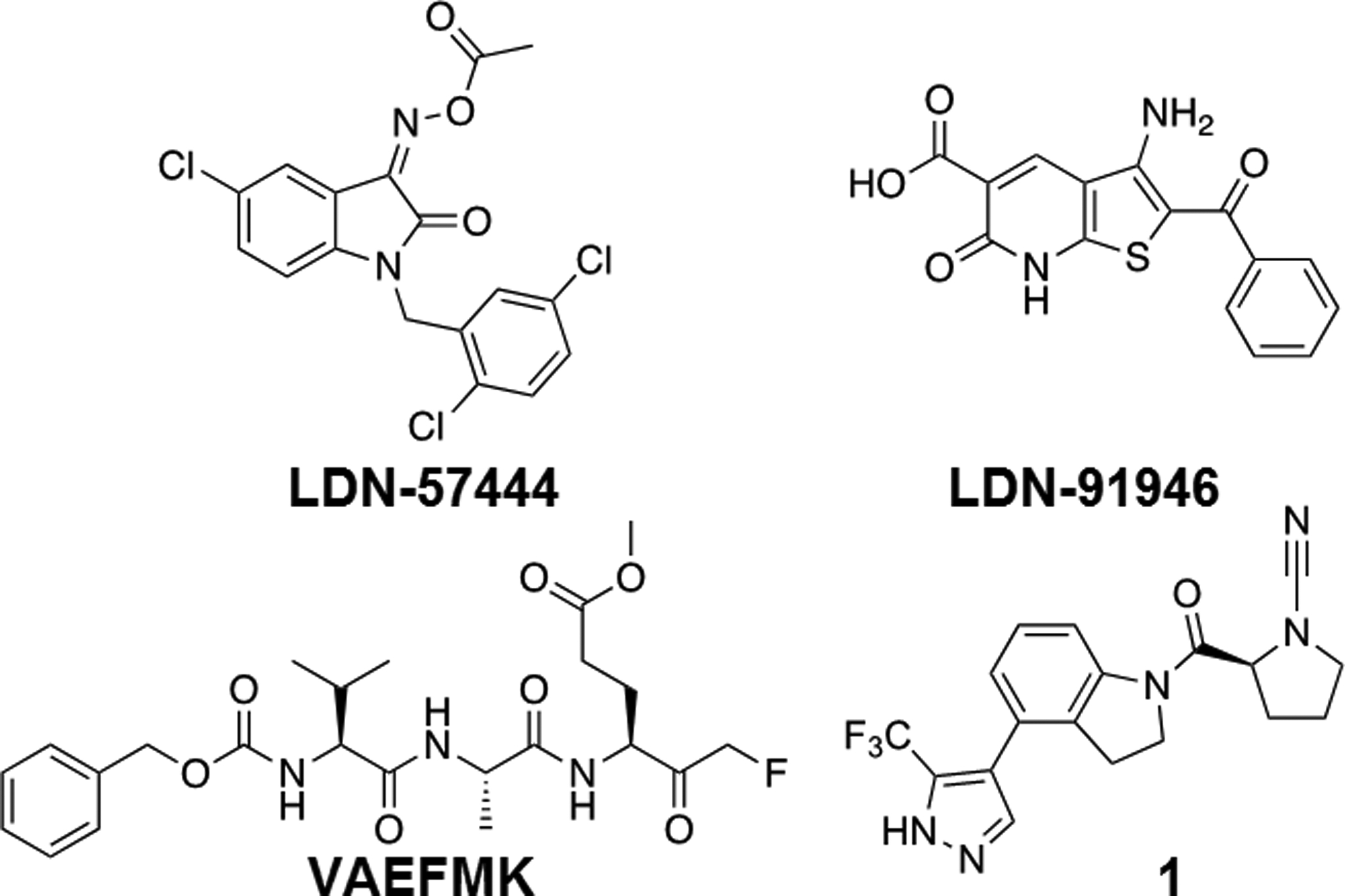
Reported UCHL1 inhibitors.
Cyanopyrrolidines have been previously utilized as cysteine reactive electrophilic warheads to target cathepsins[18] where the active site cysteine undergoes nucleophilic attack on the electrophilic center at the carbon of the nitrile moiety. Given that UCHL1 is also a cysteine protease, we hypothesized that this molecule may be acting via covalent modification of the active site cysteine (Figure 2). We selected a potent analog from the patent report to carry out the characterization.[17a] The results of these studies and cellular on-target validation as well as toxicity assays are presented herein.
Figure 2.
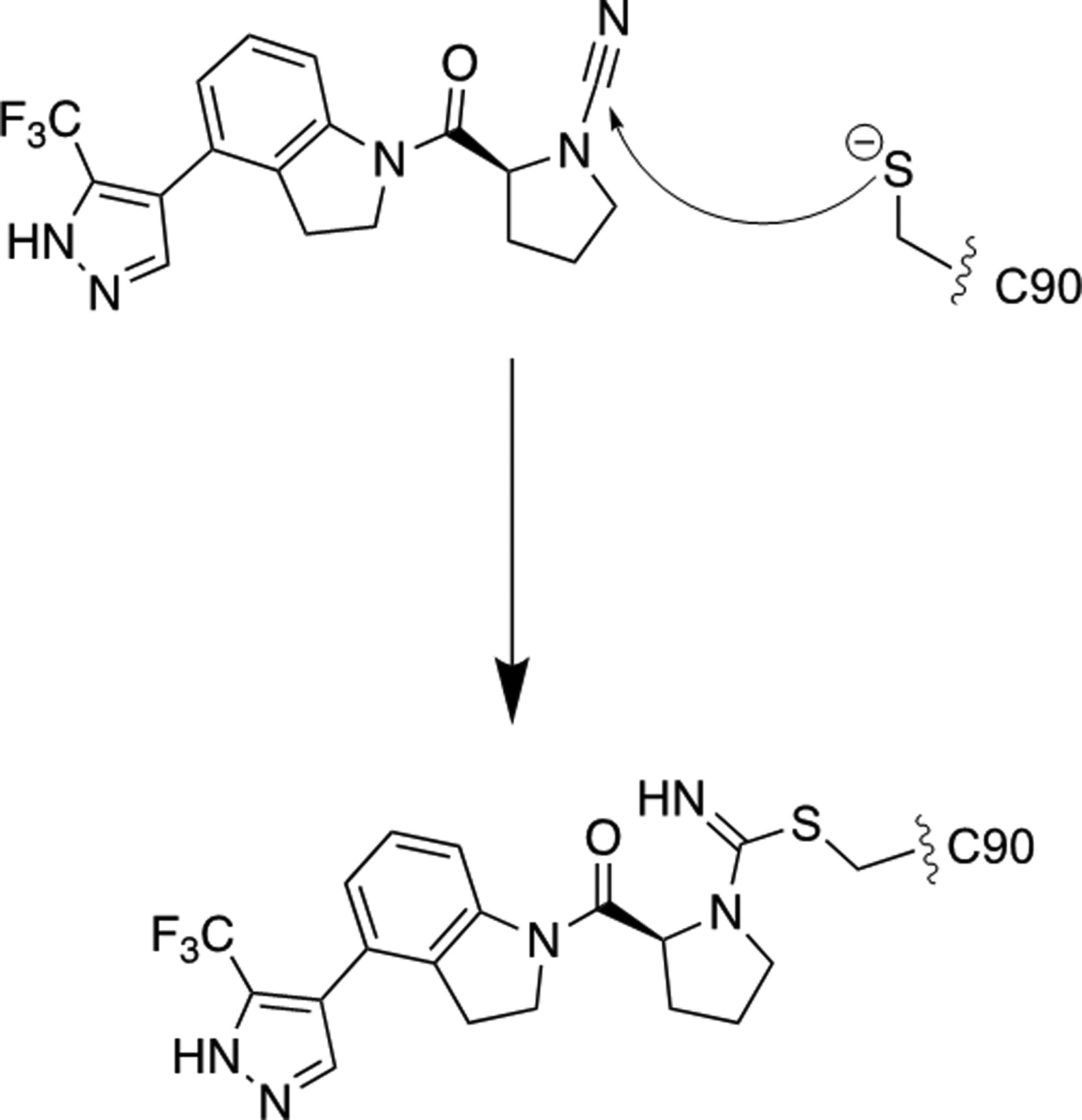
Proposed mechanism for inhibition of UCHL1 by 1.
Results and Discussion
Chemistry
Compound 1 was synthesized according to a modified procedure from the original report (Scheme 1).[17a] In brief, Boc-L-proline was reacted with 4-bromoindoline via amide bond coupling using HATU to form intermediate 3. Intermediate 3 was converted to 4 using a microwave assisted Suzuki-Miyaura reaction. This was followed by Boc-deprotection and subsequent cyanamide formation using cyanogen bromide to provide the final compound 1.
Scheme 1.
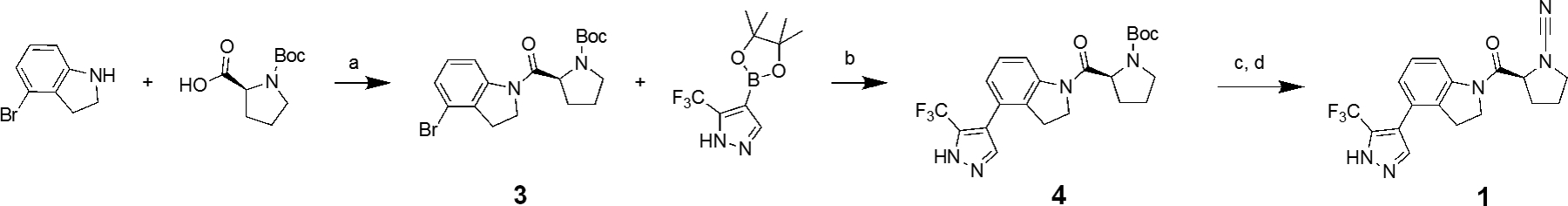
Reagents and Conditions: a) HBTU (1.25 eq), DIPEA (1.5 eq), THF, 25 °C, 16 hr; b) NaCO3H (2 eq), Pd(dppf)Cl2 (0.1 eq), 9:1 DMF:H2O, 110 °C, 1.5 hr; c) TFA (20 eq), DCM, 25 °C, 2 hr; d) K2CO3 (2.2 eq), BrCN (1.0 eq), THF, 0 °C, 30 minutes.
Biochemical Characterization of Cyanopyrrolidine Inhibition
The in vitro inhibition of UCHL1, and the closely related UCHL3, by 1 was determined by monitoring cleavage of rhodamine110 from ubiquitin substrate (Ub-Rho).[19] Both UCHL1 and UCHL3 were pre-incubated with compound 1 for 30 minutes before the addition of Ub-Rho. Compound 1 inhibited both UCHL1 (Figure 3A, black line) and UCHL3 (Figure 3A, red line) with IC50 ± standard error values of 0.67 ± 1.0 μM and 6.4 ± 1.1 μM, respectively when pre-incubated for 30 minutes. While this represents a slight improvement in potency compared to the current best-in-class UCHL1 inhibitor (Figure 1, LDN-57444, IC50 value of 0.88 ± 0.14 μM), it also has decreased selectivity for UCHL1 over UCHL3 compared to LDN-57444.
Figure 3.
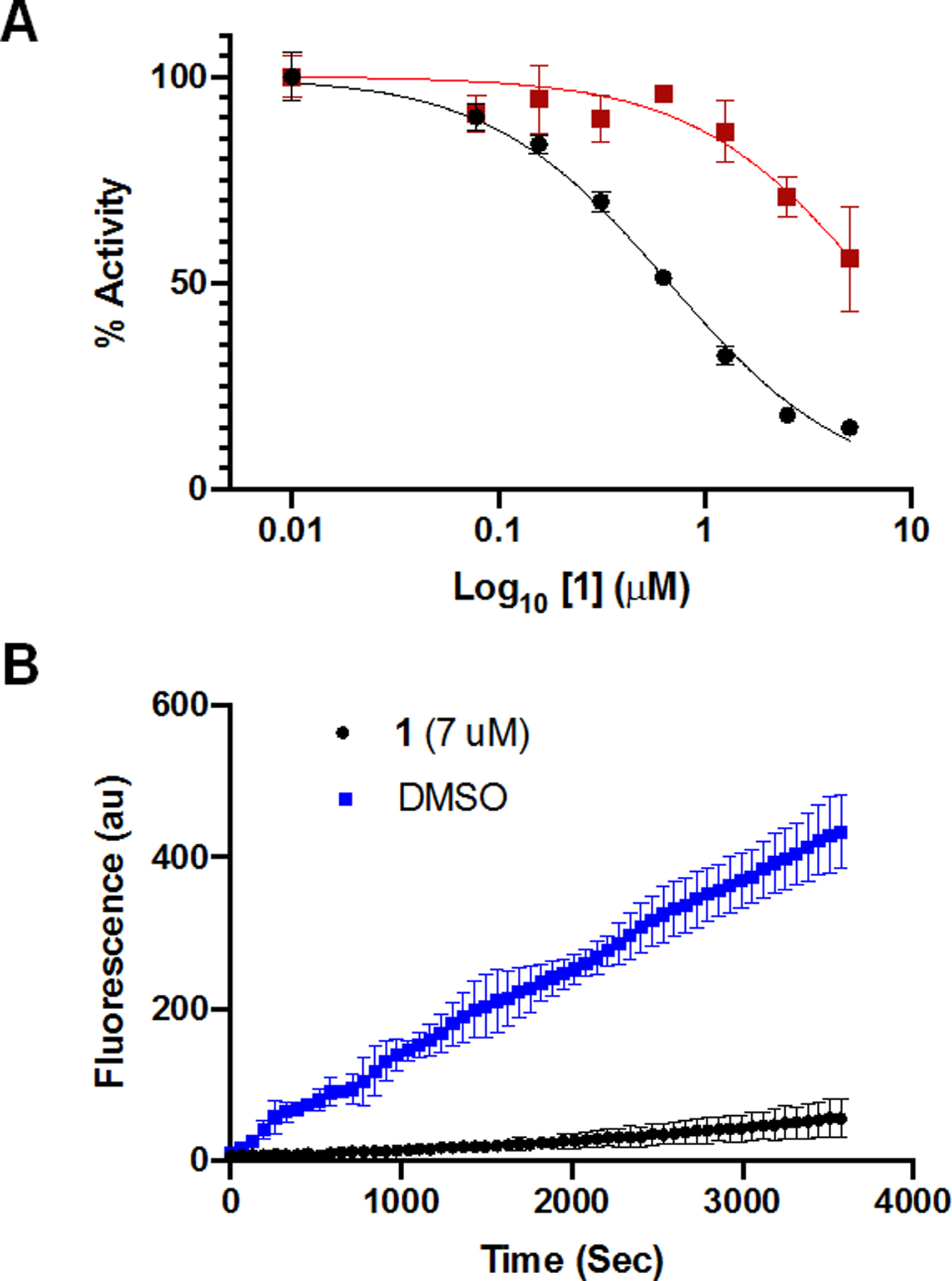
A) IC50 curves for compound 1 against UCHL1 (black) and UCHL3 (red) after 30 minutes of preincubation. B) Progress curves for UCHL1 treated with DMSO (blue) or compound 1 (black) following dilution into substrate.
Nitrile electrophiles are common warheads for other proteases, forming an isothiourea as the result of the nucleophilic attack by the cysteine (Figure 2).[18a] The reactive free energies of various nitriles have been calculated computationally, and the reaction energy of a cyanamide such as in molecule 1 is higher than a typical nitrile, though such an electrophile is known to be reversible.[20] To confirm that compound 1 is indeed a covalent modifier of UCHL1 and assess the rate of reversibility, we performed a jump-dilution experiment[21] in which UCHL1 was pre-incubated with 1 at 7 μM, approximately 10-fold the IC50 value, for 30 minutes. The enzyme-inhibitor complex was then rapidly diluted 100-fold into a solution containing Ub-Rho. Upon dilution into a substrate-containing solution, enzymes treated with non-covalent or quickly reversible covalent inhibitors are expected to regain activity and display progress curves similar to DMSO treated controls. Very slowly reversible or irreversible covalent inhibitors will not regain activity and their progress curves are expected to look similar to fully inhibited enzyme in a non-jump-dilution experiment. The resulting progress curve for compound 1 matched that of a very slowly reversible or irreversible inhibitor (Figure 3B).[22] Additionally, His-tagged UCHL1 Wild-type (WT) and catalytically inactive mutant His-UCHL1C90A were incubated with either compound 1 or DMSO for 30 minutes and subjected to analysis by electrospray ionization-mass spectrometry. Detected mass-to-charge ratios were deconvoluted using the analysis software (DataExpress version 6.0.11.3, Advion). The sample of His-UCHL1WT treated with compound 1 displayed an average mass shift of molecular weight approximately 372, which corresponds to the addition of one molecule of 1 (molecular weight 375 g/mol), while the DMSO treated sample of His-UCHL1WT matched the mass of apo UCHL1. His-UCHL1C90A did not display a change in mass corresponding to the addition of 1 (Table S1).
The rate of reversibility was then determined by observing the change in enzymatic activity over time when preincubated with 1. Compound 1 was most potent within 30 minutes, with a steady decrease in potency as time progressed out to 24 hours (Figure S1). Irreversible inhibitors display a steady increase in potency until all of the molecule has formed a covalent bond with the target protein and should result in a linear change in IC50.[23] The slow recovery of enzymatic activity over time confirmed the very slow reversibility of the inhibitor, likely into a non-reactive product.
To confirm that the molecule engages UCHL1 in lysates a ubiquitin activity-based probe gel shift assay was employed.[24] Using hemagglutinin-tagged Ub-vinylmethylester (HA-Ub-VME) compound 1 activity in HEK293T cell lysate was assessed. Blotting for UCHL1 showed the expected molecular weight shift of the newly formed HA-Ub-UCHL1 adduct (Figure 4, top panel) compared to UCHL1 alone. Compound 1 inhibited UCHL1 activity in these lysates in a dose-dependent manner as evidenced by the decreased intensity of the band corresponding to HA-Ub-UCHL1 with increasing dose of molecule. Subsequently blotting for the HA-tag showed compound 1 exhibited dose-dependent inhibition of another DUB in these lysates along with UCHL1. The identity of this off-target DUB remains to be determined (Figure 4, bottom panel).
Figure 4.
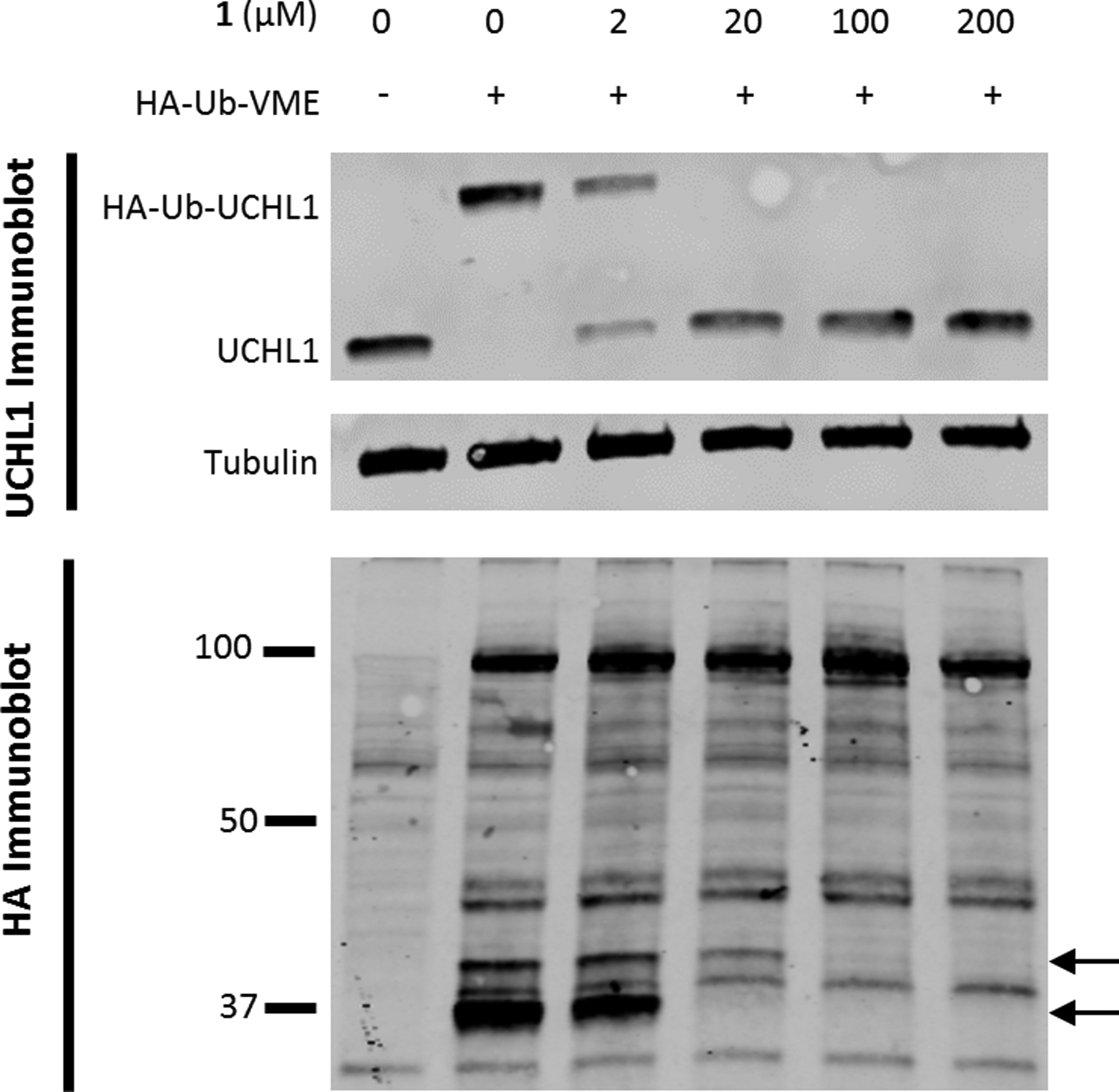
HEK293T cell lysate was incubated with 1 for 10 minutes at room temperature before addition of HA-Ub-VME. This was incubated for 10 additional minutes then quenched by the addition of 4x laemlli buffer and heated at 90 °C for 5 minutes. UCHL1 immunoblot shows a reduction of HA-Ub-UCHL1 formation with increased concentration of 1. HA immunoblot shows two bands decrease as the concentration of 1 is increased, denoted by black arrows.
Structural Characterization and Molecular Modelling of Cyanopyrrolidine Inhibition
While multiple crystal structures of UCHL1 have been deposited into the Protein Data Bank (PDB), only one is bound to a small-molecule inhibitor (PDB ID 4DM9). To better understand the ligand binding mode with UCHL1 for this new class of inhibitor we turned to high-resolution nuclear magnetic resonance (NMR) experiments. 15N-heteronuclear single quantum coherence (HSQC) chemical shift assignments known for the unligated UCHL1 were kindly provided by Shang-Te Hsu (National Taiwan University – Taipei), and side chain 13C and 1H resonance assignments were from the Biological Magnetic Resonance bank (BMRB) (ID 17260).
Information about the site of interaction between 1 and UCHL1 was obtained from ligand-induced changes in the 15N-HSQC spectrum of UCHL1. Upon addition of 1, a number of peaks shifted in frequency (Figure S2). The difference in chemical shift in the absence and presence of 1 was quantified for well-resolved resonances, and the residues with the changes in chemical shift >0.15 ppm were mapped to the structure in Figure 5A (blue). Most of the mapped residues are localized to a region near the cross-over loop (residues 149–160) and Cys 90, which indicates 1 binds near the active site pocket.
Figure 5.
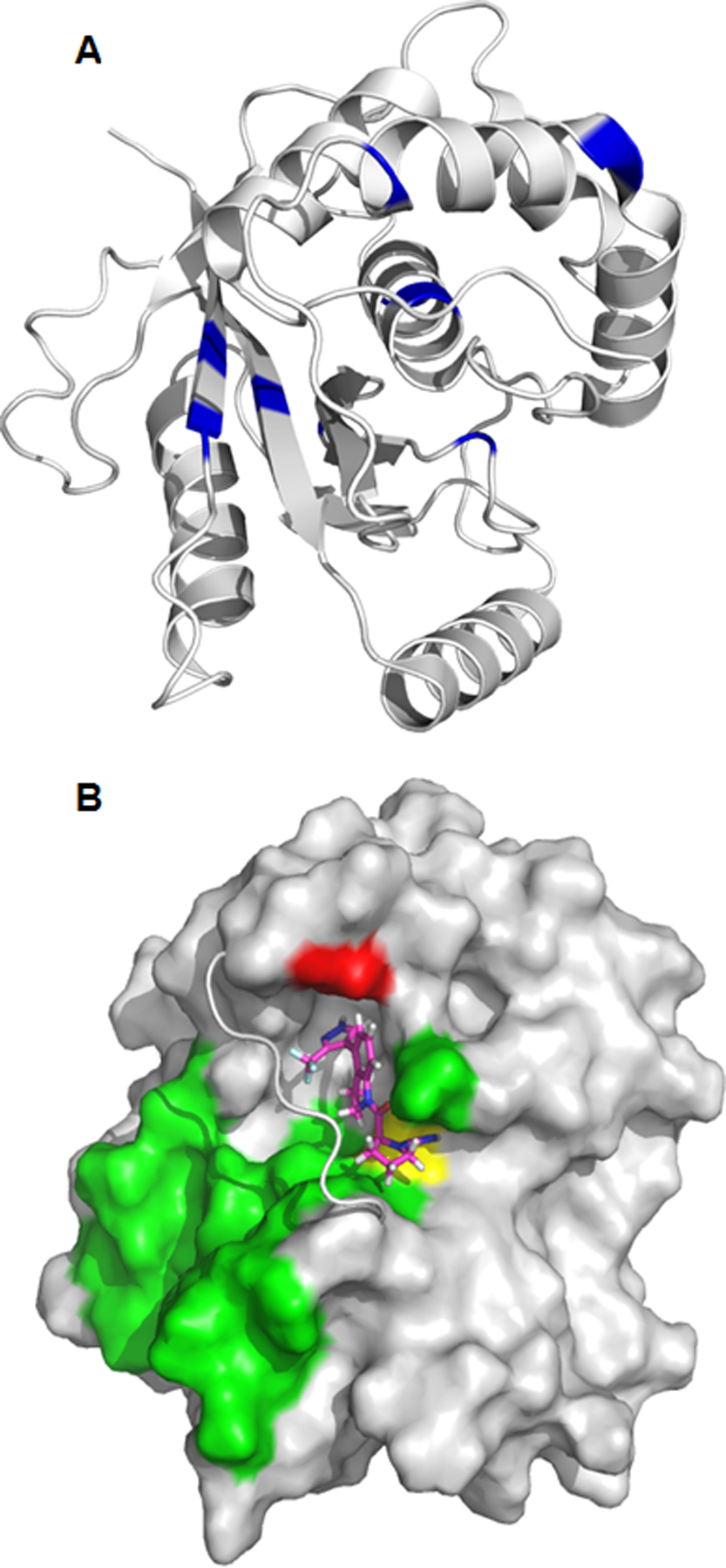
A) Chemical shift changes () greater than 0.15 ppm are mapped to the structure of UCHL1 (PDB ID: 2ETL). The difference in 15N-HSQC chemical shifts between unligated UCHL1 and after addition of 1 were measured at a molar ratio [1:1]. The orientation of UCHL1 is approximately the same as Figure 5B. B) Predicted binding pose of compound 1 (magenta sticks) with UCHL1 (grey), supported by 3D NOE NMR experiments. The crossover loop of UCHL1 is shown as a cartoon for clarity. Compound 1 binds to UCHL1 on the same side of the crossover loop as the ubiquitin binding interface of UCHL1 (green surface). Compound 1 forms a covalent bond with cysteine 90 (yellow surface) and has an experimentally determined NOE with alanine 147 (red surface).
Intermolecular Nuclear Overhauser Effects (NOE’s) were measured[25] with 15N-13C labeled UCHL1 to further define the interaction with 1. A number of isotope-filtered NOE crosspeaks were observed between ligand protons and protein protons bonded to 13C-labeled carbons. The intermolecular distance information from the NOE crosspeaks was used in an interactive, self-consistent approach involving potential crosspeak assignments and computationally modeling the complex with NOE-distance restraints.
For the modeling studies we used the crystal structure of UCHL1 (PDB ID 2ETL)[26] and prepared the protein coordinates for computational studies using Maestro (Schrödinger, LLC). Using CovDock (Schrödinger, LLC) a docking site was defined around the active site of UCHL1 as indicated by the chemical shift changes (Figure 5A), a NOE constraint of 1 to 6 angstroms was set as a requirement between aromatic indoline proton 3 of the ligand and CβH3 side chain of Ala147 (Figure 5, red surface and Figure 6), and Cys90 (Figure 5, yellow surface and Figure 6) was set as the nucleophilic residue.
Figure 6.
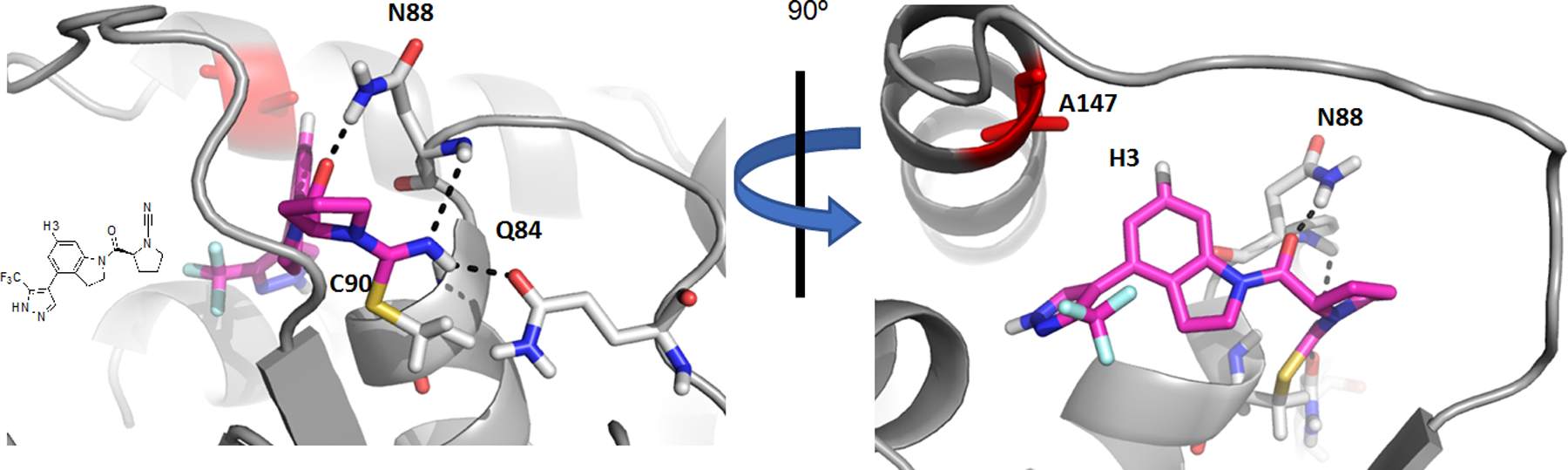
Predicted binding pose of compound 1 (magenta) with UCHL1 (grey), supported by 3D intermolecular-NOE NMR experiments. Constraints provided when docking included formation of a covalent bond with cysteine 90 (yellow), and a distance between 1 and 6 angstroms between ligand proton 3 (H3) and alanine 147 (red sticks). The resulting pose shows hydrogen bonds (black dashed lines) between compound 1 and the oxyanion hole (Q84 and N88) of UCHL1.
In the crystal structure, the first nine N-terminal residues (Met1 – Asn9) lie below the cross-over loop in a groove between the two amino acids used as constraints, Ala147 and Cys90. Given the experimental NOE observed between the ligand and Ala147 and the inherent flexibility of terminal residues, it was hypothesized that the N-terminal portion of UCHL1 may adopt an alternative position upon ligand binding. To account for this possibility, docking was performed both with the full-length crystallographic structure and with the N-terminal residues removed. The pose accepted from iterative docking with Cys90 as the nucleophilic residue and iterative NOE-derived distance restraints is shown in Figure 5 and Figure 6. This pose predicts the molecule threads into a cavity under the cross-over loop of UCHL1 and that the isothiourea resulting from the nucleophilic attack of Cys90 on the cyanamide carbon of 1 interacts with the putative oxyanion hole stabilizing residues Gln84 and Asn88,[26] consistent with previous observations between other cyanamide inhibitors and their corresponding cysteine protease targets.[18a] The inhibitor is also stabilized by an interaction between the amide carbonyl of 1 and the sidechain of Asn88. While further studies are necessary to validate this binding pose it is consistent with an experimentally observed intermolecular NOE assignment to Ala147 Hβ and ligand H3, and the cyanamide electrophile reacting with Cys90.
Cellular Characterization
UCHL1 levels of expression and enzymatic activity are correlated with increased metastatic behavior in various cancers, including small-cell lung cancer, myeloma, and lymphoma.[9h, 12b-d, 13d, 27] Three cell lines were selected to evaluate the efficacy of compound 1 based on their sensitivity to genetic depletion of UCHL1: SW1271 (small-cell lung cancer line), KMS11, and KMS12 (myeloma cell lines). SW1271 and KMS11 cells have high levels of UCHL1 expression and are sensitive to shRNA depletion of UCHL1, as determined by the project Achilles score. [12b, 28] In contrast, KMS12 cells have low levels of UCHL1 and are not dependent on the DUB for proliferation (Figure 7A).[12b] Thus, we sought to evaluate differential cell growth in these cell lines upon pharmacological inhibition of UCHL1. As predicted, treatment of SW1271 cells with compound 1 displayed a dose-dependent response with a CC50 = 139 nM as monitored using a CellTiter-Glo® assay (Figure 7B). Compound 1 activity was also assessed in KMS11 and KMS12 cells by tracking percent proliferation over time at various concentrations of 1. Consistent with their differential expression of UCHL1, compound 1 resulted in cell toxicity in the KMS11 cells while only reducing proliferation in the KMS12 cells upon treatment with the same concentration compound (Figure 7C). The enhanced inhibition of proliferation in the KMS11 cells was observed at several concentrations of compound 1 (Figure S3). Taken together, these data suggest on target inhibition of UCHL1, but the observed growth inhibition induced by 1 in the KMS12 cells is likely due to off-target interactions.
Figure 7.
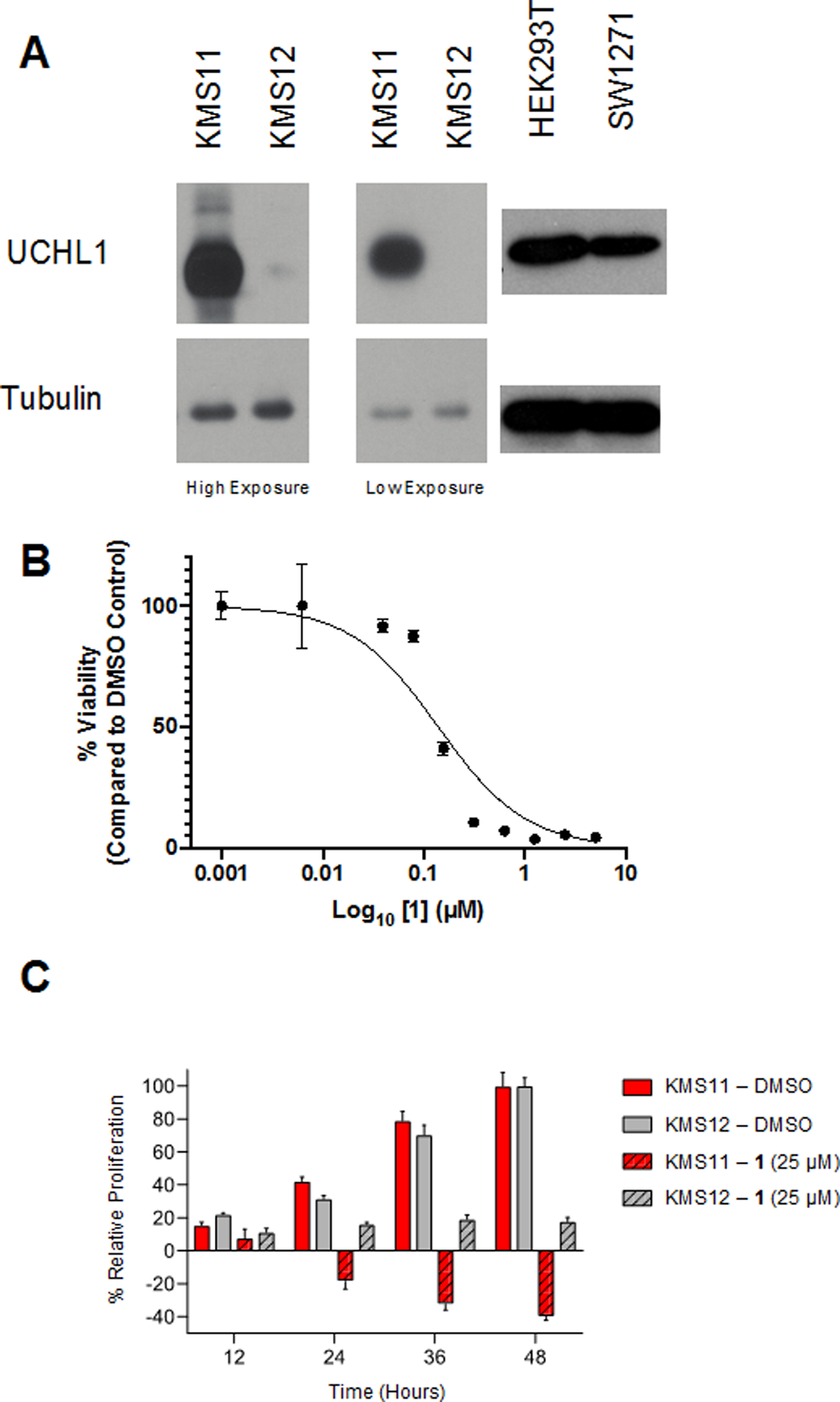
A) Immunoblots of KMS11, KMS12, HEK293, and SW1271 showing UCHL1 expression. B) Compound 1 displays a dose-dependent effect on the viability of SW1271 cells (CC50 = 138.9 nM). C) Compound 1 decreases the rate of proliferation for both KMS11 and KMS12 cells at 25 μM.
To further investigate if the inhibitor engages UCHL1 in intact cells and to identify potential off-targets for compound 1 an alkyne containing analog was synthesized. Intermediate 4 was N-alkylated to append an alkyne moiety on the pyrazole ring (Scheme 2). Subsequent deprotection of the Boc group using TFA and addition of the cyanamide via cyanogen bromide provided final compound 2. Compound 2 retained UCHL1 activity albeit with a 10-fold reduction in potency (IC50 value of 6.4 ± 1.2 μM, Figure S4). This loss of activity is consistent with the predicted binding pose as the alkylated nitrogen of the pyrazole points towards the protein surface, likely perturbing the binding of the inhibitor. The IC50 for UCHL3 did not significantly change (5.5 ± 1.1 μM) effectively making 2 equipotent against each enzyme.
Scheme 2.
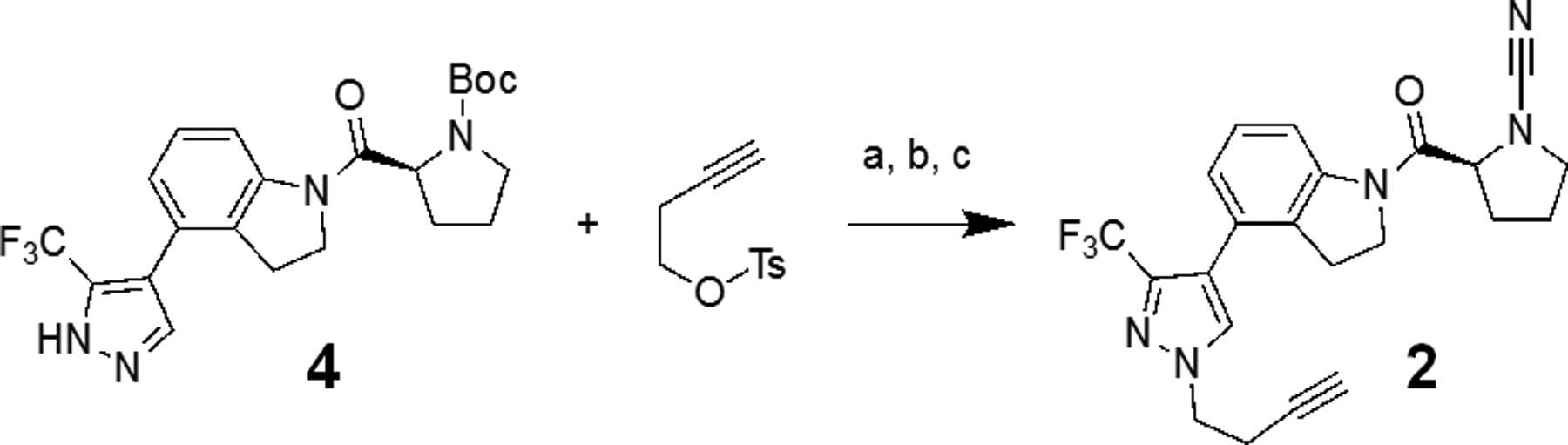
Reagents and Conditions: a) K2CO3 (2 eq), ACN, 110 °C, 1.5 hr; b) TFA (20 eq), DCM, 25 °C, 2 hr; c) K2CO3 (2.2 eq), BrCN (1.0 eq), THF, 0 °C, 30 minutes.
To determine the off-target interactions between 1 and proteins other than UCHL1, we utilized copper catalyzed azide-alkyne cycloaddition (CuAAC) to ligate either biotin or a sulfo-Cyanine5 (Cy5-Azied) fluorophore to the protein-ligand complex (Figure S5).[29] First it was confirmed that the CuACC ligation could proceed while 2 was bound to recombinant UCHL1. UCHL1 was treated with 2 in a dose-dependent manner, followed by CuAAC ligation with Cy5-Azide. This sample was then subjected to gel electrophoresis and imaged. The results confirmed successful CuACC addition of the fluorophore to compound 2 while bound to UCHL1 as evidenced by increased fluorescence band intensity as the concentration of 2 increased (Figure S6). UCHL1 contains five surface exposed cysteines in addition to the catalytic Cys90. It is possible that the fluorescent labeling of UCHL1 with 2 could be due to a covalent adduct formed with any one of the surface exposed cysteines. To rule this possibility out the same experiment was performed by treating both wild-type UCHL1 and a catalytically inactive Cys90Ala UCHL1 mutant (UCHL1C90A) in parallel. Samples were subjected to gel electrophoresis, which revealed that WT-UCHL1 was fluorescently labeled while UCHL1C90A exhibited no detectable fluorescence confirming the probe covalently targets the active site cysteine (Figure S7). This data, combined with the ESI-MS data described above confirms the molecule preferentially modifies the Cys90.
We next utilized the alkyne probe to assess off-targets in both KMS11 and SW1271 cells. These cells were incubated with 2 in dose-response for 4 hours before being washed 3 times with PBS, collected, and lysed. The normalized cell lysate was then subjected to CuAAC reaction with Cy5-azide to label all proteins with which 2 was able to interact. In both cell lines, a dose-dependent labelling of proteins was observed (Figure S8). Additionally, to confirm that 2 was specifically labelling UCHL1 in a dose-dependent manner, an out-competition experiment was performed by treating cells simultaneously with 2 (10 μM) in the presence of increasing concentrations of 1 (0 – 20 μM). Here, it was observed that 1 was able to out-compete 2 for UCHL1 in both SW1271 and KMS11 cell lines in a dose-dependent manner (Figure 8, Loading controls in Figure S9). However, the alkyne probe maintained binding to higher molecular weight proteins and the dose-dependent decrease in fluorophore intensity was not as robust as with UCHL1. Given the loss of UCHL1 selectivity resulting from the addition of the alkyne moiety, the apparent non-specific interactions observed here could feasibly be a result of the reduced selectivity from the alkyne addition. However, we believe there is some degree of off-target binding as the bands at approximately 37 kDa and 50 kDa have slight reduction of the fluorescence signal from the alkyne probe 2 at the highest concentration of the non-alkyne containing competitor 1.
Figure 8.
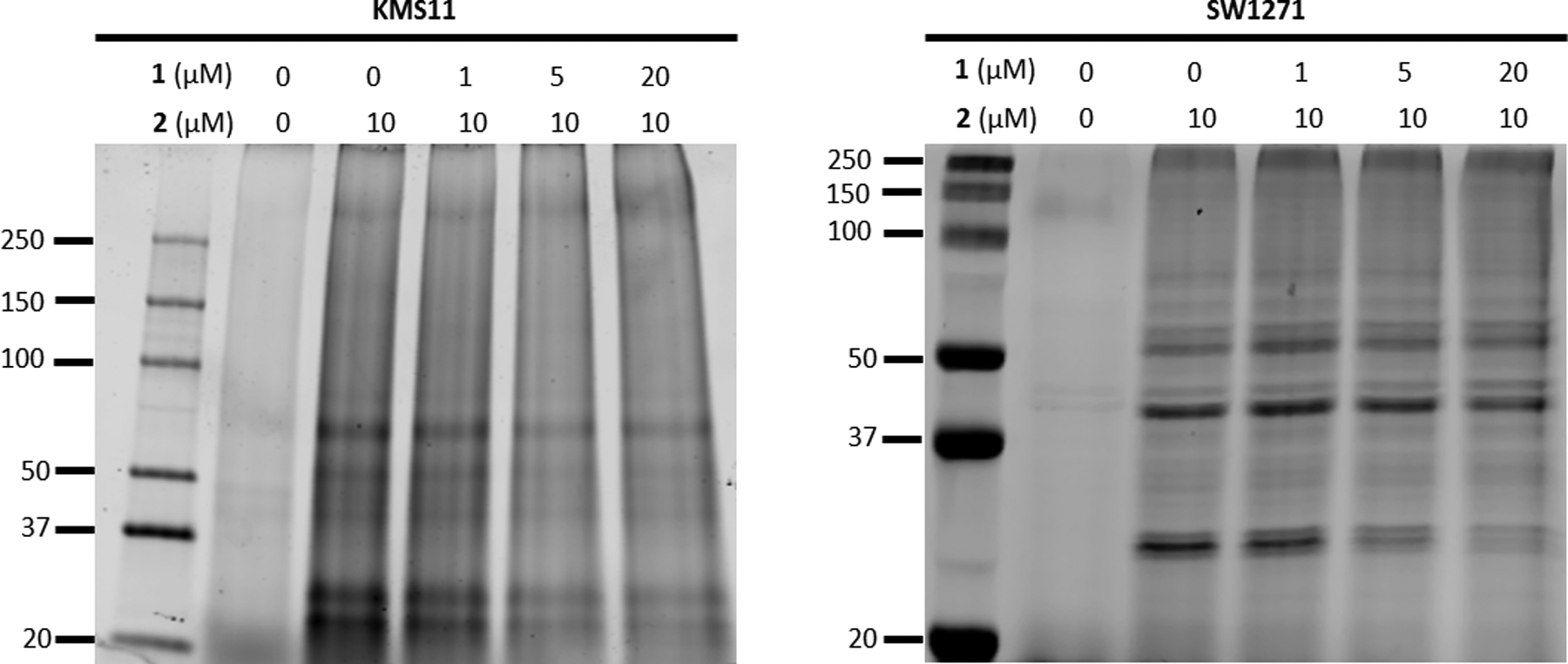
Fluorescence imaged gels from KMS11 and SW1271 cells treated simultaneously with 10 μM of compound 2 and increasing concentrations of compound 1 for 4 hrs then subjected to CuAAC labeling with Cy5 dye show a dose-dependent decrease in intensity for a band near 25 kDa.
In an attempt to identify these non-specific binding and off-targets for 2, a biotin-streptavidin pull-down experiment was performed. UCHL1 expressing KMS11 cells were treated with either 1 or 2 for 4 hours before being washed, collected, and lysed. A CuAAC reaction was performed using biotin-azide, biotinylating all proteins with which 2 interacted in the cells. This sample was subjected to pulldown using streptavidin-bound magnetic beads. The beads, washed of all non-specific proteins, were eluted and analyzed by gel electrophoresis and immunoblotting for UCHL1 (Figure S10). Tryptic digestion followed by mass spectrometry confirmed the presence of UCHL1 in the streptavidin-enriched sample, matching a peptide sequence from recombinantly expressed UCHL1 (Figure S11). However, no other DUBs were present in a sufficient quantity at the level of our detection compared to DMSO control to be considered off-target interactors. Therefore, even though it is observed that 2 does covalently modify proteins other than UCHL1 in intact cells using the fluorophore-linked 2, we were not able to definitively identify these proteins in this round of pull-down/mass spectrometry experiments. A tabular list of the highest confidence putative off-target interactions is provided in the supplemental information (Table S2) and will be validated in future work.
Conclusions
In summary, we have presented the characterization of a new class of UCHL1 inhibitor. The cyanopyrrolidine scaffold was confirmed to bind to UCHL1 via covalent modification of the active site Cys90 with very slow reversibility. The molecule was shown to inhibit UCHL1, along with at least one other DUB, in HEK293 cell lysate by using the HA-Ub-VME activity-based probe. NMR and docking studies suggest that the inhibitor lies in a region overlapping the ubiquitin contact surface of the UCHL1 active site groove directly underneath the active site cross-over loop (Figure 5), a feature of UCHL1 believed to play a key role in size-based selection of ubiquitinated substrates.[26] In addition to the isothiourea linkage produced from the covalent attack of the catalytic cysteine, the molecule may engage in some prominent interactions with the active site pocket, specifically with the oxyanion hole via hydrogen bonding with Gln84 and an adjacent residue Asn88. The molecule exhibits efficacy at sub-micromolar concentrations against cancer cell lines KMS11 and SW1271 that are known to be sensitive UCHL1 knockdown while also displaying moderate growth inhibition against KMS12 myeloma cells that are not sensitive to UCHL1 depletion. These data suggest potential off-target interactions may contribute to the inhibition of cell growth. Using an alkyne-tagged probe analog the molecule was confirmed to inhibit UCHL1 in cells while also binding to other proteins in a dose-dependent manner; however, further work is necessary to definitively identify these proteins and define the nature of their interaction with the probe molecule. While the cyanopyrrolidine scaffold of compound 1 was likely developed for therapeutic purposes to target UCHL1 it may not serve as a suitable chemical probe due to the lack of selectivity and off-target toxicity. However, the compound may prove useful for validating on target engagement of future UCHL1 inhibitors in intact cells and provide insight into effective inhibitory strategies for the UCHL1.
Experimental Section
General:
1H and 13C NMR spectra were recorded on Bruker DRX500 spectrometer (operating at 500 and 126 MHz) in DMSO-d6 or CDCL3 with or without the internal standard of TMS at 0.05% v/v. The chemical shifts (d) reported as parts per million (ppm) and the coupling constants are reported as s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet. The purity of all final compounds was >95 % purity as assessed by HPLC according to current American Chemical Society guidelines for publication. Final compounds were analyzed on an Agilent 1200 series chromatograph. The chromatographic method utilized as ThermoScientific Hypersil GOLD C-18 or silica column. UV detection wavelength = 220/254 nm; flow-rate = 1.0 mL/min; solvent = acetonitrile/water for reverse phase. Both organic and aqueous mobile phases contain 0.1% v/v formic acid. The mass spectrometer used is an Advion CMS-L Compact Mass Spectrometer with an ESI or an APCI source. Samples are submitted for analysis using the atmospheric solids analysis probe (ASAP). Compounds were prepared according to following protocols and are detailed below.
Chemical Synthesis:
tert-butyl (S)-2-(4-bromoindoline-1-carbonyl)pyrrolidine-1-carboxylate (3)
To a solution of boc-L-proline (1.2 g, 4.9 mmol, 1.2 eq) in THF was added HATU (2.3 g, 6.1 mmol, 1.5 eq) and DIPEA (1.4 mL, 8.1 mmol, 2.0 eq) at 25 °C. The reaction mixture was stirred for 1 hour. 4-Bromoindoine (0.8 mg, 4.0 mmol, 1.0 eq) was added to the reaction mixture. The reaction mixture was stirred at 25 °C for 15 hours. The resulting mixture was poured into saturated NaHCO3 (10 mL). The organic phase was collected, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (5–50% Ethyl Acetate in Hexanes). The material was washed with hexanes and dried yielding tert-butyl (S)-2-(4-bromoindoline-1-carbonyl)pyrrolidine-1-carboxylate (3) (1.4 g, 86%). 1H NMR (300 MHz, DMSO-d6) δ 8.16 – 7.93 (m, 1H), 7.30 – 7.03 (m, 2H), 4.64 – 4.42 (m, 1H), 4.33 – 4.05 (m, 2H), 3.56 – 3.35 (m, 2H), 3.15 (t, J = 9.0 Hz, 2H), 2.36 – 2.12 (m, 1H), 1.95 – 1.75 (m, 3H), 1.43 – 1.12 (m, 9H). MS APCI+: 295.0 [M-Boc+H].
tert-butyl (S)-2-(4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carboxylate (4)
To a solution of intermediate 3 (856 mg, 2.2 mmol, 1.0 eq) in DMF:Water (9:1, 1.5 mL) was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trifluoromethyl-1H-pyrazol (567 mg, 2.2 mmol, 1 eq) and NaHCO3 (364 mg, 4.30 mmol, 2.0 eq). The reaction mixture was stirred at 25 °C in a microwave tube and degassed for 15 minutes. Pd(dppf)Cl2 (158 mg, 0.22 mmol, 0.1 eq) was added and the reaction tube was sealed and heated at 110 °C for 1.5 hours in a microwave. The reaction mixture was poured into water (20 mL) and extracted with Ethyl Acetate (3 × 50 mL). The combined organic phase was washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (5% Methanol in DCM). The material obtained from chromatographic purification was azeotropically distilled with a mixture of hexane:diethyl ether (1:1, 50 mL) yielding tert-butyl (S)-2-(4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carboxylate (4) (636.4 mg, 65.2%). 1H NMR (300 MHz, DMSO-d6) δ 13.81 (s, 1H), 8.23 – 8.01 (m, 2H), 7.31 – 7.12 (m, 1H), 6.93 (d, J = 7.6 Hz, 1H), 4.61 – 4.43 (m, 1H), 4.34 – 3.98 (m, 2H), 3.50 – 3.36 (m, 2H), 3.06 (t, J = 8.6 Hz, 2H), 2.37 – 2.12 (m, 1H), 2.07 – 1.64 (m, 3H), 1.42 – 1.18 (m, 9H). APCI-MS: m/s 351.2 [M-Boc+H]+.
(S)-2-(4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carbonitrile (1)
Step 1.
To a solution of intermediate 4 (100 mg, 0.22 mmol, 1 eq) in DCM was added TFA (0.5 mL) at 25 °C. The reactiom mixture was stirred for 2 hours, and then concentrated under reduced pressure. The obtained residue was triturated with diethyl ether (1 mL) yielding (S)-1-prolyl-5-(5-carbonly)pyrrolidine-1-carboxylate TFA salt (70.9 mg, 68.8% yield).APCI-MS: m/z 351.2 [M + H]+. This material was used direclty for the next step without further purification.
Step 2.
To a solution of (S)-1-prolyl-5-(5-carbonly)pyrrolidine-1-carboxylate TFA salt (25.0 mg, 54 μM, 1 eq) was in THF was added K2CO3 (17.0 mg, 120 μM, 2.2 eq) at 0 °C. Cyanogen bromide (6.0 mg, 54 uM, 1.0 eq) was added to the reaction mixture at 0 °C. The reaction mixture was stirred at 25 °C for 30 minutes and then poured onto water (50 mL). The resulting mixture was extracted with Ethyl Acetate (3 × 50 mL), combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (5% Methanol in Ethyl Acetate) yielding title compound S)-2-(4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carbonitrile (1) (14 mg, 69%). 1H NMR (500 MHz, Chloroform-d) δ 8.27 (d, J = 8.1 Hz, 1H), 7.67 (s, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.03 (d, J = 7.7 Hz, 1H), 4.63 – 4.42 (m, 1H), 4.21 (d, J = 9.7 Hz, 1H), 4.04 (d, 1H), 3.65 (m, 2H), 3.29 – 2.95 (m, 2H), 2.40 – 2.21 (m, 1H), 2.20 – 2.07 (m, 2H), 2.07 – 1.92 (m, 2H).13C NMR (126 MHz, CDCl3) δ 167.8, 142.5, 130.4, 129.6, 127.8, 127.1, 126.3, 120.4, 118.5, 117.1, 116., 62.3, 51.5, 47.6, 30.0, 29.6, 27.7, 24.3. APCI-MS: m/z 376.1 [M + H]+. HPLC retention time: 11.251 min. HPLC Purity: 98.17%.
(S)-2-(4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carbonitrile (2)
Step 1.
To a solution of tert-butyl (2S)-2-(4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-2,3-dihydro-1H-indene-1-carbonyl)pyrrolidine-1-carboxylate (50 mg, 1 eq, 0.11 mmol) in acetonitrile. K2CO3 (31 mg, 2 eq, 0.22 mmol) was added, followed by but-3-yn-1-yl 4-methylbenzenesulfonate (75 mg, 3.3 eq, 0.33 mmol) was added to the reaction mixture. The reaction was heated to 110 oC in a microwave for 1 hour. The resulting reaction mixture was quenched with water (50 mL)and brine (50 mL), extracted with ethyl acetate (3×50 ml), rinsed with saturated NaHCO3 (50 mL), and rinsed with brine (50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting oil was purified using flash chromatography (0->50% EtOAc over 10 min, hold 5 min, 50->80% EtOAc over 10 min, hold 5 min.). The resulting residue was azeotroped with dichloromethane and dried using under reduced pressure yielding tert-butyl (S)-2-(4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carboxylate. 1H NMR (300 MHz, Chloroform-d) δ 8.26 (t, J = 8.6 Hz, 1H), 7.53 (d, J = 8.6, 1.1 Hz, 1H), 7.24 – 7.13 (m, 1H), 7.03 – 6.91 (m, 1H), 4.64 – 4.42 (m, 1H), 4.33 (td, 2H), 4.24 – 3.96 (m, 2H), 3.78 – 3.39 (m, 2H), 3.04 (q, J = 7.2, 6.5 Hz, 2H), 2.80 (tt, J = 6.6, 2.6 Hz, 2H), 2.33 – 2.09 (m, 2H), 2.07 (t, J = 2.7 Hz, 1H), 2.01 – 1.79 (m, 2H), 1.49 – 1.27 (m, 9H). APCI-MS: m/z 403.1 [M-Boc+H]+.
Step 2.
To a solution of tert-butyl (S)-2-(4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carboxylate (38.9 mg, 0.077 mmol, 1 eq) in DCM was added TFA (0.5 mL) at 25 °C. The reactiom mixture was stirred for 2 hours, and then concentrated under reduced pressure. The obtained residue was triturated with diethyl ether (1 mL) yielding (S)-4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-1-prolylindoline, 2,2,2-trifluoroacetate salt. MS APCI+: 403.2 [M+H]. This material was used direclty for the next step.
Step 3.
To a solution of S)-4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-1-prolylindoline, 2,2,2-trifluoroacetate salt (40 mg, 0.08 mmol, 1 eq) in THF was added K2CO3 (24 mg, 0.17 mmol, 2.2 eq) at 0 °C. Cyanogen bromide (9.53 mg, 0.09 mmol, 1.2 eq) was added to the reaction mixture at 0 °C. The reaction mixture was stirred at 25 °C for 30 minutes and then poured onto water (50 mL). The resulting mixture was extracted with Ethyl Acetate (3 × 50 mL), combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (5% Methanol in Ethyl Acetate) yielding title compound (S)-2-(4-(1-(but-3-yn-1-yl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)indoline-1-carbonyl)pyrrolidine-1-carbonitrile (2) (24 mg, 72%). 1H NMR (300 MHz, Chloroform-d) δ 8.25 (d, J = 8.1 Hz, 1H), 7.55 (s, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.01 (d, J = 7.7 Hz, 1H), 4.47 (dd, J = 8.1, 3.4 Hz, 1H), 4.34 (t, J = 6.5 Hz, 2H), 4.27 – 4.14 (m, 1H), 4.08 – 3.94 (m, 1H), 3.82 – 3.44 (m, 2H), 3.20 – 2.95 (m, 2H), 2.80 (td, J = 6.5, 2.6 Hz, 2H), 2.38 – 2.21 (m, 1H), 2.19 – 2.09 (m, 2H), 2.07 (t, J = 2.6 Hz, 2H), 2.04 – 1.89 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 167.9, 142.6, 131.6, 131.0, 130.5, 127.9, 127.3, 126.3, 123.1, 119.5, 118.8, 117.2, 116.3, 79.8, 71.8, 71.0, 62.4, 62.1, 51.5, 30.1, 27.8, 24.4, 20.5. APCI-MS: m/z 428.2 [M+H]+. HPLC retention time: 12.453 min. HPLC Purity: 99.0%.
Biochemical and Cellular Assays
Protein Expression and Purification:
UCHL1 for NMR experiments (Plasmid provided by Chittaranjan Das – Purdue University) was grown in M9 minimal media supplemented with 2 g/L 13C Glucose and 1g/L 15NH4Cl (Cambridge Isotopes) as the sole carbon and nitrogen sources. Bacterial cultures were grown at 37 °C to an optical density of 0.6–0.8. After 0.1 mM IPTG induction at 17 °C for 18 hours, the bacteria were lysed and pelleted at 15,000 × g, and clarified lysate was purified via glutathione-Sepharose (GE Life Sciences) column according to manufacturer’s instructions. The protein was purified further by size exclusion chromatography on a Superdex S75 column (Amersham Pharmacia).
UCHL1, UCHL1C90A, and UCHL3 (Expressed in pET15b by GenScript) for biochemical assays were grown in LB growth medium at 37 °C to an optical density of 0.6–0.8. After 0.1 mM IPTG induction at 17 °C for 18 hours the bacteria were lysed and pelleted at 15,000 × g, and clarified lysate was purified via HisPur Ni-NTA resin (Thermo Scientific) according to manufacturer’s instructions.
ESI-MS for Intact UCHL1 and UCHL1 - Compound 1 Complex:
Recombinantly expressed His-UCHL1WT or His-UCHL1C90A were diluted into assay buffer (50 mM Tris pH 7.6, 0.5 mM EDTA, 1 mM DTT) to a concentration of 5 mg/mL in the presence of DMSO (1% v/v) or compound 1 (200 μM) and incubated at room temperature for 30 minutes. This was then diluted 8-fold into mass spectrometry buffer (75% acetonitrile, 25% water, 0.1% formic acid, 0.1% ammonium formate). 40 μL of a buffer-containing blank, a DMSO treated samples, or compound 1 treated samples were injected into an Advion CMS-L Compact Mass Spectrometer outfitted with an ESI ionization source. After subtracting the blank from each sample, the data was manually deconvoluted and analyzed using DataExpress (Advion, version 6.0.11.3).
Fluorescence-based Deubiquitinase Activity Assay:
Reactions were performed in black 384 well plates (Fisher 12566624) in a final volume of 50 μL. DUBs were diluted in reaction buffer (50 mM Tris pH 7.6, 0.5 mM EDTA, 5 mM DTT, 0.1% (w/v) BSA) to a concentration of 2.5 nM or 0.25 nM for UCHL1 and UCHL3 respectively (Final concentration in well 1 nM or 0.1 nM for UCHL1 and UCHL3 respectively). To each well was added 20 μL of DUB containing solution and 10 μL of 5X inhibitor dissolved in reaction buffer (final concentration 5, 2.5, 1.5, 0.625, 0.3125, 0.15625, 0.078125, 0 μM) and this was allowed to incubate for 30 minutes at room temperature. Reactions were initiated by the addition of 20 μL 450 nM Ub-Rho (Boston Biochem U-555, Final concentration 180 nM). Reactions were incubated at room temperature and read immediately (Excitation = 485 nm, Emission = 535 nm) for 20 minutes. Readings were performed on a Synergy Neo2. Biochemical IC50 were calculated using GraphPad Prism 8 (GraphPad Software, San Diego, California USA, www.graphpad.com) and the standard deviation was determined over three independent experiments.
Jump-Dilution Experiment:
Reactions were performed in black 384 well plates (Fisher 12566624) in a final volume of 50 μL. UCHL1 was diluted in reaction buffer (50 mM Tris pH 7.6, 0.5 mM EDTA, 5 mM DTT, 0.1% (w/v) BSA) to a concentration of 100 nM (Final concentration in well 1 nM). To this was added 7 μM compound 1 or DMSO, and the mixture was incubated for 30 minutes at room temperature. This was then diluted 100-fold into Ub-Rho containing buffer (stock concentration 181.2 nM, final concentration 180 nM in well). Reactions were incubated at room temperature and read immediately (Excitation = 485 nm, Emission = 535 nm) for 20 minutes. Readings were performed on a Synergy Neo2.
Time-Dependent Activity Assay:
Reactions were performed in black 384 well plates (Fisher 12566624) in a final volume of 50 μL. UCHL1 was diluted in reaction buffer (50 mM Tris pH 7.6, 0.5 mM EDTA, 5 mM DTT, 0.1% (w/v) BSA) to a concentration of 2.5 nM (Final concentration in well 1 nM). To each well was added 20 μL of DUB containing solution and 10 μL of 5X inhibitor dissolved in reaction buffer (final concentration 5, 2.5, 1.5, 0.63, 0.31, 0.16, 0.078, 0 μM) and this was allowed to incubate for 12, 42, 72, 102, 132, 162, 1095, and 1410 minutes at room temperature. Reactions were initiated by the addition of 20 μL 450 nM Ub-Rho (Boston Biochem U-555, Final concentration 180 nM). Reactions were incubated at room temperature and read immediately (Excitation = 485 nm, Emission = 535 nm) for 20 minutes. Readings were performed on a Synergy Neo2. Biochemical IC50 were calculated using GraphPad Prism 8 (GraphPad Software, San Diego, California USA, www.graphpad.com).
Probe Labelling Assays:
HEK293T cells (2.8×106 cell/mL * 8 mL) were lysed in 400 uL of lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 5 mM DTT, 2 mM ATP, 0.5% NP-40, 10% glycerol) and incubated on ice for 30 min. Cells were lysed on ice using sonication, and centrifuged at 17,000 × g for 10 min to remove cell debris. Concentration of cell lysate was measured using a Bradford assay. To microcentrifuge tubes was added 245 μg cell lysate and 10 μL of DMSO or 2, and the mixture was allowed to incubate at room temperature for 10 minutes. HA-Ub-VME was added to a final concentration of 0.49 μM, and the mixture was incubated at room temperature for 10 minutes. The reaction was quenched using 4x laemelli buffer and heated to 90 °C for 5 minutes before being analyzed by immunoblot.
Cell Proliferation Assay:
The proliferation of KMS11 and KMS12 cells was monitored using an IncuCyte devise (Sartorius; www.essenbioscience.com). Cells (3 × 104 per well; 200μl) were seeded in triplicate in 96 well plates the night prior to the addition of compounds. The IncuCyte was set to collect images from five fields per well at four hour intervals using the nuclear dye Nuclight Red (Sartorius; www.essenbioscience.com). At each timepoint, the number of cells per well was determined by taking the mean of the five locations in each well. The relative proliferation at each timepoint was normalized, with 100% set as the number of cells observed for incubated with DMSO (0.1%) controls at 72 hours.
Cell Toxicity Assay:
SW1271 cells were purchased from the ATCC and cultured in DMEM containing 10% FBS and 1% Penicillin/Streptomycin. For growth inhibition assays, five thousand cells were planted per well in a 96-well white wall plate in the presence of the indicated doses of compound 1. Cells were grown for a period of 144 hours at which point cell viability was quantified using the Cell Titer Glo Assay (Promega #G7572). Changes in luminescence units were normalized to vehicle control wells and plotted using a log(inhibitor) vs. response curve fit analysis.
CuAAC Ligation and in-gel Fluorescence – Dose-Response with Recombinant Protein:
1 mg/mL UCHL1 (49 μL) was incubated with 2 (2, 1, 0.5, 0.25, 0.125, 0.0625 μM) in PBS blocking buffer (ThermoScientific # 37538) for 30 minutes at room temperature. To each reaction sample is added: 90 μL of 1X PBS, 20 μL of 2.5 mM Cy5 Azide (Click Chemistry Tools #AZ118), 10 μL of 100 mM THPTA pre-mixed with 10 μL 20 mM CuSO4, and 10 μL of 300 mM Sodium Ascorbate. This mixture mixed by gently vortexing and allowed to incubate for 2 hours protected from light at room temperature. Protein was precipitated by sequential addition of MeOH (600 μL), CHCl3 (150 μL) and water (400 μL). Protein was pelleted by centrifugation at 17,000 × g for 5 min and then further washed in methanol (450 μL × 2) and re-pelleted. Pellets were air dried protected from light for one hour and stored at −20 °C until ready for use. Samples were resuspended in 1x PBS with 2% w/v SDS before adding 4x laemelli buffer and heated to 95 °C for 5 minutes prior to SDS page. Samples were separated by SDS page and the gels were scanned using a Licor Odyssey at 600 nM. Gels were subsequently Coomassie stained to determine loading.
CuAAC Ligation and in-gel Fluorescence – UCHL1WT and UCHL1C90A:
1 mg/mL His-UCHL1WT or His-UCHL1C90A was incubated with 1 μM 2 (2% v/v DMSO final) for 30 minutes at room temperature. To each reaction sample is added: μL of 1X PBS, 20 μL of 2.5 mM Cy5 Azide (Click Chemistry Tools #AZ118), 10 μL of 100 mM THPTA pre-mixed with 10 μL 20 mM CuSO4, and 10 μL of 300 mM Sodium Ascorbate. This mixture mixed by gently vortexing and allowed to incubate for 2 hours protected from light at room temperature. Protein was precipitated by sequential addition of MeOH (600 μL), CHCl3 (150 μL) and Water (400 μL). Protein was pelleted by centrifugation at 17,000 × g for 5 min and then further washed in methanol (450 μL × 2) and re-pelleted. Pellets were air dried protected from light for one hour and stored at −20 °C until ready for use. Samples were resuspended in 1x PBS with 2% w/v SDS before adding 4x laemelli buffer and heated to 95 °C for 5 minutes prior to SDS page. Samples were separated by SDS page and the gels were scanned using a Licor Odyssey at 600 nM. Gels were subsequently Coomassie stained to determine loading.
CuAAC Ligation and in-gel Fluorescence – Treated Cells:
Cells were treated with 0, 1, 5, or 20 ¨μM 1 with concurrently with 10 μM 2 for 4 hours before being washed, scraped, and collected. Cells were lysed in 200 μL lysis buffer (1x PBS, 0.1% w/v SDS, 1% v/v Triton X-100, 1X HALT protease inhibitor) on ice for 30 minutes with vigorous vortexing every 10 minutes. Cells were centrifuged at 17,000 × g for 10 minutes, and the supernatant was collected. The concentration was measured by BCA, and cell lysate was normalized to 2 mg/mL and used immediately. The remainder of the lysate was stored at −80oC until use. 50 μL of each sample (at 2 mg/mL) was added to a new tube. To each tube was added 6 μL of a freshly prepared click cocktail: 3 μL 1.7 mM THPTA in 1:4 DMSO:tButOH (100 μM final concentration), 1 μL 50 mM CuSO4 in water (1 mM final concentration), 1 μL 1.25 mM Cy5-N3 (25 uM final concentration), 1 μL 50 mM TCEP (1 mM final concentration – prepared directly before use). The samples were vortexed briefly and let to react for 2 hours at room temperature, protected from light. The reactions were quenched by the addition of 17 μL 4x laemelli buffer. 15 μL was added per land, and bands were separated by SDS-PAGE. Gels were subsequently Coomassie stained to determine loading.
Sample Preparation for Proteomic Analysis:
Cells were lysed in 100 μL lysis buffer (1x PBS, 0.1% w/v SDS, 1% v/v Triton X-100, 1X HALT protease inhibitor) on ice for 30 minutes with vigorous vortexing every 10 minutes. Cells were centrifuged at 17,000 × g for 10 minutes, and the supernatant was collected. The concentration was measured by BCA, and cell lysate was normalized to 6 mg/mL and stored at −80oC until use. 50 μL of each sample (at 6 mg/mL) was added to a new tube. To each tube was added: 90 μL 1X PBS, 20 μL of 2.5 mM Biotin-PEG3-Azide (Click Chemistry Tools Product # AZ104–25), 20 μL of pre-mixed (100 mM THPTA - 10 μL and 20 mM CuSO4 – 10 μL), and 10 μL of 300 mM Sodium Ascorbate. The mixture was gently vortexed with each addition, and then allowed to react at room temperature for 2 hours protected from light. Samples were precipitated by adding 600 μL MeOH, 150 μL Chloroform, and 400 uL mq water to each tube and vortexing. Samples were centrifuged at 17,000 × g for 5 minutes, and the aqueous layer removed. The precipitate was washed with 450 μL MeOH centrifuged at 17,000 g, and decanted (2X) before being air dried for 1 hour while protected from light. Samples were then stored at −20 °C overnight. To each sample was added 300 μL Binding Buffer (1X TBS pH 7.6, 0.1% Tween-20). Samples were vortexed, sonicated, gently heated, and centrifuged at 17,000 × g for 5 minutes. This sample was added to 100 μL of pre-washed Pierce Streptavidin Magnetic Beads (Product # 88816). Samples were incubated at room temperature on a rocker for 1.5 hours before separating liquid from beads using a magnetic stand. Beads were washed with 300 μL of Wash Buffer (1X TBS pH 7.6 + 2M Urea) (3X). Beads were eluted sequentially using 100 μL of: 1) 1X TBS + 5 mM Biotin, 2) 1X TBS + 2% w/v SDS and, 3) 4X Laemelli Buffer (Biorad Product #1610747). 5 μL of 4X Laemelli Buffer was added to 15 μL of each sample, and 10 μL of this was loaded onto a 12% SDS PAGE gel before analysis by immunoblot.
NMR Experiments:
All NMR data were collected at 300 K on a Bruker Avance-III-800 spectrometer equipped with a QCI cryoprobe. Samples of 15N,13C labelled UCHL1 (0.6–0.8 mM in 50 mM d-Tris, pH 7.4, 50 mM NaCl, 1 mM DTT) alone or in complex with unlabelled 1 (molar ratio 1:1) were prepared as aforementioned.
2D 15N-HSQC experiments (pulse program HSQCETF3GPSI2) were performed with 1024 and 128 complex points, spectral width 14 and 32 ppm, and carrier frequency on water and 118.5 ppm for the proton and nitrogen dimension, respectively. 2D 13C-HSQC experiments (pulse program HSQCETGPSISP2.2) were performed with 1024 and 256 complex points, spectral width 16 and 72 ppm, and carrier frequency on water and 36 ppm for the proton and carbon dimension, respectively. Intermolecular NOEs were measured by a 3D filtered NOE experiment (pulse program NOESYHSQCGPWGX13D) with 1024, 64, and 32 complex points and spectral width 14, 70, and 12 ppm in the protein proton, aliphatic carbon, and ligand proton dimension, respectively. The carrier frequencies were set on water for proton dimensions and 34.5 ppm for carbon dimension. Spectra were collected with 150 ms mixing time.
The chemical shift perturbation analysis for the apo and bound UCHL1 is described in a previous publication (Cite Feng et al PROTEIN SCIENCE 2018 VOL 27:1780–1796 DOI: 10.1002/pro.3489). Details for analysing the Intermolecular NOE data are described in Supporting Information.
3D NOE Determination:
Computational Docking:
UCHL1 was prepared using Protein PrepWizard from PDB ID 2ETL using OPLS3 force fields within the Maestro suite version 2016–3 (Schrödinger LLC). Compound 1 was prepared within LigPrep generating possible states at pH 7.0 ± 2.0, retaining specified chiralities using an OPLS3 forcefield. Docking of 1 within UCHL1’s active site was performed using CovDock. The receptor was centered around cysteine 90, cysteine 90 was defined as the nucleophilic residue, and a nucleophilic addition to a triple bond was selected as the reaction type. A NOE constraint was defined, requiring the placement of ligand proton H3 to be between 1 and 6 angstroms from Cβ of alanine 147. No torsional constraints were defined. Resulting poses were analysed using Pymol.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Chad Hewitt for providing UCHL3 and UCHL1C90A and Dr. Huaping Mo for assistance in NMR data collection. This work was funded by NIH P30CA023168 and R01GM039478. The NMR and Mass Spectrometry data were acquired by the PINMRF and Purdue Proteomics Facility supported by P30CA023168 as part of the Purdue Center for Cancer Research shared resources funds. Support from the Purdue University Center for Cancer Research Small Grants Program, in particular the Phase 1 Concept Award, is also gratefully acknowledged. Finally, the authors also would like to acknowledge the Purdue University College of Pharmacy for additional support.
References
- [1].Komander D, Rape M, Annu. Rev. Biochem 2012, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- [2].Schulman BA, Harper JW, Nat. Rev. Mol. Cell Biol 2009, 10(5), 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ye Y, Rape M, Nat. Rev. Mol. Cell Biol 2009, 10(11), 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Deshaies RJ, Joazeiro CAP, Annu. Rev. Biochem 2009, 78. [DOI] [PubMed] [Google Scholar]
- [5].a) Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J, Cell 2009, 137(1), 133–145; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP, Nat. Biotechnol 2003, 21(8), 921. [DOI] [PubMed] [Google Scholar]
- [6].Komander D, Clague MJ, Urbé S, Nat. Rev. Mol. Cell Biol 2009, 10(8), 550. [DOI] [PubMed] [Google Scholar]
- [7].Komander D, Biochem. Soc. Trans 2009, 37(5), 937–953. [DOI] [PubMed] [Google Scholar]
- [8].a) Harrigan JA, Jacq X, Martin NM, Jackson SP, Nat. Rev. Drug Discovery 2018, 17(1), 57; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hewings DS, Heideker J, Ma TP, AhYoung AP, Oualid F. El, Amore A, Costakes GT, Kirchhofer D, Brasher B, Pillow T, Nat. Commun 2018, 9(1), 1162; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rehman SAA, Kristariyanto YA, Choi S-Y, Nkosi PJ, Weidlich S, Labib K, Hofmann K, Kulathu Y, Mol. Cell 2016, 63(1), 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Setsuie R, Wada K, Neurochem. Int 2007, 51(2–4), 105–111; [DOI] [PubMed] [Google Scholar]; b) Graham SH, Liu H, Ageing Res. Rev 2017, 34, 30–38; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD, The Journal of Pathology 1990, 161(2), 153–160; [DOI] [PubMed] [Google Scholar]; d) Atkin G, Paulson H, Front. Mol. Neurosci 2014, 7, 63; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lim K-H, Baek K-H, Curr. Pharm. Des 2013, 19(22), 4039–4052; [DOI] [PubMed] [Google Scholar]; f) Liu Y, Lashuel HA, Choi S, Xing X, Case A, Ni J, Yeh L-A, Cuny GD, Stein RL, Lansbury PT Jr, Chemistry & Biology 2003, 10(9), 837–846; [DOI] [PubMed] [Google Scholar]; g) Hussain S, Zhang Y, Galardy P, Cell Cycle 2009, 8(11), 1688–1697; [DOI] [PubMed] [Google Scholar]; h) Hibi K, Westra WH, Borges M, Goodman S, Sidransky D, Jen J, Am. J. Pathol 1999, 155(3), 711–715; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kim HJ, Magesh V, Lee J-J, Kim S, Knaus UG, Lee K-J, Oncotarget 2015, 6(18), 16287; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Sanchez-Diaz PC, Chang JC, Moses ES, Dao T, Chen Y, Hung JY, PLoS ONE 2017, 12(5), e0176879; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Jang MJ, Baek SH, Kim JH, Cancer Lett. 2011, 302(2), 128–135. [DOI] [PubMed] [Google Scholar]
- [10].Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J, Science 1989, 246(4930), 670–673. [DOI] [PubMed] [Google Scholar]
- [11].a) Xue S, Jia J, Brain Res. 2006, 1087(1), 28–32; [DOI] [PubMed] [Google Scholar]; b) Öhrfelt A, Johansson P, Wallin A, Andreasson U, Zetterberg H, Blennow K, Svensson J, Dementia and Geriatric Cognitive Disorders Extra 2016, 6(2), 283–294; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gong B, Radulovic M, Figueiredo-Pereira ME, Cardozo C, Front. Mol. Neurosci 2016, 9, 4; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kabuta T, Furuta A, Aoki S, Furuta K, Wada K, J. Biol. Chem 2008, 283(35), 23731–23738; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kabuta T, Setsuie R, Mitsui T, Kinugawa A, Sakurai M, Aoki S, Uchida K, Wada K, Hum. Mol. Genet 2008, 17(10), 1482–1496; [DOI] [PubMed] [Google Scholar]; f) Nishikawa K, Li H, Kawamura R, Osaka H, Wang Y-L, Hara Y, Hirokawa T, Manago Y, Amano T, Noda M, Biochem. Biophys. Res. Commun 2003, 304(1), 176–183; [DOI] [PubMed] [Google Scholar]; g) Setsuie R, Wang Y-L, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun Y-J, Neurochem. Int 2007, 50(1), 119–129; [DOI] [PubMed] [Google Scholar]; h) Andersson FI, Werrell EF, McMorran L, Crone WJK, Das C, Hsu S-TD, Jackson SE, J. Mol. Biol 2011, 407(2), 261–272. [DOI] [PubMed] [Google Scholar]
- [12].a) Sanchez Diaz PC, Chang JC, Dao T, Chen Y, Hung JY, The FASEB Journal 2016, 30, 1108–1104; [Google Scholar]; b) Bedekovics T, Hussain S, Galardy PJ, Catalytically Active UCH-L1 Is Required for MYC Driven B-Cell Lymphomagenesis, Am Soc Hematology, 2017; [Google Scholar]; c) Hussain S, Foreman O, Perkins SL, Witzig TE, Miles RR, Van Deursen J, Galardy PJ, Leukemia 2010, 24(9), 1641; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bedekovics T, Hussain S, Feldman AL, Galardy PJ, Blood 2016, 127(12), 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Seliger B, Fedorushchenko A, Brenner W, Ackermann A, Atkins D, Hanash S, Lichtenfels R, Clin. Cancer Res 2007, 13(1), 27–37; [DOI] [PubMed] [Google Scholar]; b) Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, Kobayashi M, Hirota K, Itasaka S, Yoshimura M, Nat. Commun 2015, 6, 6153; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ma Y, Zhao M, Zhong J, Shi L, Luo Q, Liu J, Wang J, Yuan X, Huang C, J. Cell. Biochem 2010, 110(6), 1512–1519; [DOI] [PubMed] [Google Scholar]; d) Hussain S, Bedekovics T, Chesi M, Bergsagel PL, Galardy PJ, Oncotarget 2015, 6(38), 40704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Kim HJ, Kim YM, Lim S, Nam YK, Jeong J, Lee KJ, Oncogene 2009, 28(1), 117; [DOI] [PubMed] [Google Scholar]; b) Lien H-C, Wang C-C, Lin C-H, Lu Y-S, Huang C-S, Hsiao L-P, Yao Y-T, Hum. Pathol 2013, 44(9), 1838–1848. [DOI] [PubMed] [Google Scholar]
- [15].Mermerian AH, Case A, Stein RL, Cuny GD, Bioorg. Med. Chem. Lett 2007, 17(13), 3729–3732. [DOI] [PubMed] [Google Scholar]
- [16].Davies CW, Chaney J, Korbel G, Ringe D, Petsko GA, Ploegh H, Das C, Bioorg. Med. Chem. Lett 2012, 22(12), 3900–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Kemp M, Stockley M, Jones A, U.S. Patent Application No. 15/738,900, 2018; [Google Scholar]; b) Kemp MI, Woodrow MD, U.S. Patent Application No. 16/060,299, 2018. [Google Scholar]
- [18].a) Deaton DN, Hassell AM, McFadyen RB, Miller AB, Miller LR, Shewchuk LM, Tavares FX, Willard DH, Wright LL, Bioorg. Med. Chem. Lett 2005, 15(7), 1815–1819; [DOI] [PubMed] [Google Scholar]; b) Yadav MR, Shinde AK, Chouhan BS, Giridhar R, Menard R, J. Enzyme Inhib. Med. Chem 2008, 23(2), 190–197. [DOI] [PubMed] [Google Scholar]
- [19].Hassiepen U, Eidhoff U, Meder G, Bulber J-F, Hein A, Bodendorf U, Lorthiois E, Martoglio B, Anal. Biochem 2007, 371(2), 201–207. [DOI] [PubMed] [Google Scholar]
- [20].De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N, Eur. J. Med. Chem 2017, 138, 96–114. [DOI] [PubMed] [Google Scholar]
- [21].Mons E, Jansen IDC, Loboda J, van Doodewaerd BR, Hermans J, Verdoes M, van Boeckel CAA, van Veelen PA, Turk B, Turk D, J. Am. Chem. Soc 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Strelow J, Dewe W, Iversen PW, Brooks HB, Radding JA, McGee J, Weidner J, Mechanism of action assays for enzymes In Assay Guidance Manual [Internet]. Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2012. [Google Scholar]
- [23].Strelow JM, SLAS DISCOVERY: Advancing Life Sciences R&D 2017, 22(1), 3–20. [DOI] [PubMed] [Google Scholar]
- [24].Ekkebus R, Flierman D, Geurink PP, Ovaa H, Curr. Opin. Chem. Biol 2014, 23, 63–70; [DOI] [PMC free article] [PubMed] [Google Scholar]; b McLellan L, Forder C, Cranston A, Harrigan J, Jacq X, in Proteostasis, Springer, 2016, pp. 411–419. [DOI] [PubMed] [Google Scholar]
- [25].a) Breeze AL, Prog. Nucl. Magn. Reson. Spectrsoc 2000, 36(4), 323–372; [Google Scholar]; b) Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE, J. Am. Chem. Soc 1997, 119(29), 6711–6721; [Google Scholar]; c) Ogura K, Terasawa H, Inagaki F, J. Biomol. NMR 1996, 8(4), 492–498; [DOI] [PubMed] [Google Scholar]; d) Iwahara J, Wojciak JM, Clubb RT, J. Biomol. NMR 2001, 19(3), 231–241. [DOI] [PubMed] [Google Scholar]
- [26].Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, Petsko GA, Proc. Natl. Acad. Sci 2006, 103(12), 4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Chen G, Gharib TG, Huang C-C, Thomas DG, Shedden KA, Taylor JMG, Kardia SLR, Misek DE, Giordano TJ, Iannettoni MD, Clin. Cancer Res 2002, 8(7), 2298–2305; [PubMed] [Google Scholar]; b) Otsuki T, Yata K, Takata‐Tomokuni A, Hyodoh F, Miura Y, Sakaguchi H, Hatayama T, Hatada S, Tsujioka T, Sato Y, Br. J. Haematol 2004, 127(3), 292–298; [DOI] [PubMed] [Google Scholar]; c) Hussain S, Bedekovics T, Liu Q, Hu W, Jeon H, Johnson SH, Vasmatzis G, May DG, Roux KJ, Galardy PJ, Blood 2018, 132(24), 2564–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WFJ, Pantel SE, Sci. Data 2014, 1, 140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Kolb HC, Finn MG, Sharpless KB, Angew. Chem., Int. Ed 2001, 40(11), 2004–2021; [DOI] [PubMed] [Google Scholar]; b) Ward JA, McLellan L, Stockley M, Gibson KR, Whitlock GA, Knights C, Harrigan JA, Jacq X, Tate EW, ACS Chem. Biol 2016, 11(12), 3268–3272; [DOI] [PubMed] [Google Scholar]; c) Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Nat. Chem. Biol 2015, 11(8), 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.