

Abstract
The extracellular matrix (ECM) has force-responsive (i.e., mechanochemical) properties that enable adaptation to mechanical loading through changes in fibrous network structure and inter-fiber bonding. Imparting such properties into synthetic fibrous materials would allow re-enforcement under mechanical load, the potential for material self-adhesion, and the general mimicking of ECM. Here, we develop multi-fiber hydrogel networks through the electrospinning of multiple fibrous hydrogel populations, where fibers contain complementary chemical moieties (e.g., aldehyde groups, hydrazide groups) that form covalent bonds within minutes when brought into contact under mechanical load. These fiber interactions lead to microscale anisotropy, as well as increased material stiffness and plastic deformation. Macroscale structures (e.g., tubes, layered scaffolds) are fabricated from these materials through inter-fiber bonding and adhesion when placed into contact while maintaining a microscale fibrous architecture. The design principles for engineering plasticity described here can be applied to numerous material systems to introduce unique properties, from textiles to biomedical applications.
Keywords: responsive materials, electrospinning, self-adhesive hydrogels, mechanochemical, biomimetic materials
Graphical Abstract
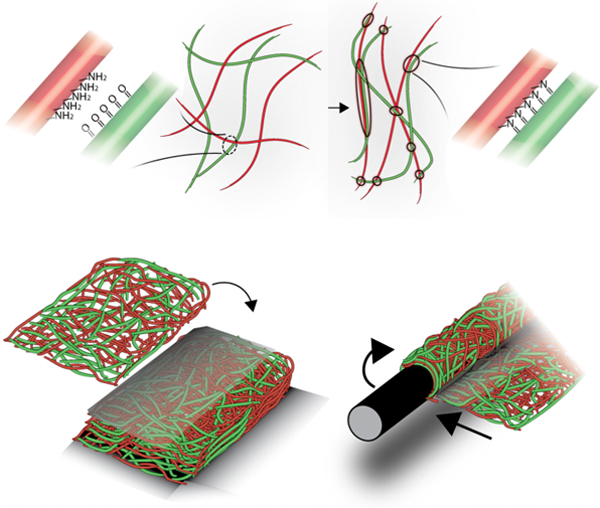
Force-induced self-adhesion within and between fibrous hydrogels is induced through chemical reactions via the incorporation of complementary chemical groups into electrospun multi-fiber hydrogel networks. Adhesive fibers adapt to tensile forces by increasing fiber alignment, stiffness, and length, while layers can be bonded through compression to fabricate thick constructs and structures such as helices and lumens.
Natural biopolymers, such as those that comprise the fibrillar extracellular matrix (ECM), have force-responsive (i.e., mechanochemical) properties that allow tissues to adapt to mechanical load.[1] For example, in response to strain, fibrillar proteins (e.g., collagen, fibrin) undergo acute non-linear changes in stiffness (e.g., strain-stiffening)[2] and inter-fiber bonding,[3,4] that result in plastic deformation and changes in material structure[5,6] and mechanics.[7] The incorporation of these mechanochemical features of natural fibrous proteins into synthetic materials provides a route to design force-responsive and self-adhesive materials.
Several strategies have been used to recapitulate such mechanochemical behavior in materials. For instance, strain-stiffening synthetic hydrogels have been engineered through the synthesis of polymers with semi-flexible backbones, which organize into a mesh-like architecture and mechanical loading induces polymer extension and packing.[8,9] Additionally, mechanochemical polymers have been designed with latent mechanophores that are activated under destructive shear forces and undergo crosslinking to increase bond density to higher levels than before bond breakage (i.e., strengthening)[10] or create mechano-radicals in response to bond scission, that promote growth of polymer networks and increased crosslinking (i.e., toughening).[11] Polymers containing cryptic reactive groups that are chemically exposed with mechanical loading have also been engineered to undergo force-responsive changes in stiffness.[12] Lastly, many self-healing and self-adhesive materials have been engineered, where dynamic bonds are utilized to facilitate rapid recovery of mechanical properties and adhesion between polymer chains and bulk hydrogels after mechanical load.[14,15] While these material systems capture some of the responsive properties of the ECM, they are inherently non-fibrous – importantly, it is the fibrous network structure and fiber stretching and buckling under mechanical load that allows network densification and inter-fiber interactions within the ECM.[6,7,13]
In the present work, we introduce a generalizable strategy to generate fibrous materials with mechanochemical and self-adhesive properties by combining dynamic covalent chemistry with multi-fiber electrospinning techniques. Synthetic fibrous materials can be engineered through techniques such as electrospinning, where material structure, biochemical composition,[14,15] and heterogeneity[16] can be tuned to mimic the ECM. Electrospun hydrogel fibers can be tailored to have single fiber stiffnesses within the range of ECM fibers and to undergo deformation under cellular tractions.[17,18] In our approach, multi-fiber electrospinning is used to fabricate fibrous hydrogel networks, where commonly used complementary chemistries (e.g., aldehydes and hydrazides) are introduced into distinct fiber populations that are mixed within the same network of fibers (Figure 1a). These fiber populations are separated from each other after fabrication; however, under mechanical load, the fibers are brought into close enough proximity (e.g., physical contact between fibers) so that a chemical reaction can occur (e.g., the formation of hydrazone bonds from the interaction of aldehydes and hydrazides) (Figure 1b). This “adhesive” interaction alters the local fiber microstructure and can be used to bind adjacent materials together. Conventional, “non-adhesive” fibrous hydrogel networks of uniform composition or those that lack either of the reactive groups involved in these mechanochemical reactions do not exhibit these properties (Figure 1c).
Figure 1.

Mechanochemical interactions in multi-fiber hydrogel networks through the reaction of complementary chemical groups. (a) Schematic of the electrospinning of two fiber populations from hyaluronic acid modified with norbornenes (NorHA), as well as either hydrazides (NorHA-Hyd, red) or aldehydes (NorHA-Ald, green). Norbornenes are used for intra-fiber and inter-fiber crosslinking through a photoinitiated thiol-ene reaction to stabilize the fiber, while the hydrazides and aldehyde groups permit additional inter-fiber reactions when fibers are brought into contact. Inset shows hydrazide and aldehyde groups exposed on fiber surface after electrospinning, stabilization, and hydration. (b) Schematic (top) and fluorescent images (bottom) of mechanical strain-induced interactions between hydrazide (colored red or containing red fluorophore) and aldehyde (colored green or containing green fluorophore) containing NorHA fibers (i) before, (ii) during, and (iii) after applying strain for 40 minutes. After removal of strain, permanent deformations of “adhesive” fibers are observed through fluorescent microscopy (yellow indicating coinciding red and green fluorophores). (c) Schematic (top) and fluorescent images (bottom) indicating a lack of mechanical strain-induced interactions in “non-adhesive” fiber control under the same loading conditions. The multi-fiber hydrogel control is fabricated from mixed fibers of unmodified NorHA and NorHA-Ald fiber populations, where no chemical reaction is expected when the fibers contact each other. Scale bar is 100 µm.
Electrospun fibers were fabricated from hyaluronic acid (HA) due to its biocompatibility and potential for chemical modification for hydrogel formation[19]. HA was first modified with norbornenes (NorHA) through an anhydrous esterification reaction to couple norbornene carboxylic acid to HA.[20] NorHA was then separately modified with either hydrazides (NorHA-Hyd) through an amidation reaction between adipic acid dihydrazide and carboxylic acids of HA or aldehydes (NorHA-Ald) through oxidation of NorHA with sodium periodate (Figure S1 and S2).[21] NorHA, NorHA-Hyd, and NorHA-Ald could then be electrospun into swollen multi-fibrous materials (fibers of NorHA-Hyd and NorHA-Ald to form “adhesive” fibrous networks or fibers of NorHA and NorHA-Ald to form “non-adhesive” fibrous networks) via electrospinning, stabilization through photocrosslinking, and hydration (Figure S3). Hydrazides and aldehydes were chosen as they undergo rapid Schiff Base reactions to form hydrazone bonds under physiological conditions and have been widely used to form dynamic hydrogels for biomedical applications[22–24]. However, this approach could be used across a wide selection of chemistries, where combination induces chemical reactions (e.g., methacrylates and thiols via Michael Addition reactions, amines and aldehydes to form imine derivatives), where the selection of the chemical pairs will control the kinetics and strength of fiber interactions.
To examine their mechanochemical properties, the “adhesive” fibrous hydrogel networks were electrospun as suspended networks and the fibers were compressed using a micromanipulator and micropipette (Figure S4). The micropipette compresses suspended fibers together to create points of contact, while the microscope permits simultaneous visualization of fluorescently-labeled fibers to evaluate either transient (i.e., reversible) or plastic (i.e., irreversible) deformation when the compressive forces are removed. When examined, plastic deformation was observed for the adhesive fibers within 1 minute and further increased after loading for up to 5 minutes (Figure S4) and even up to 40 minutes (Figure 1b, Video S1). These kinetics are consistent with the reaction behavior of aldehydes and hydrazides to form hydrazone bonds as previously reported in hydrogel formation.[22,23] Importantly, this behavior was not observed in the non-adhesive fibers that did not possess the complementary bonds for reaction (Figure 1c, Video S1) or in the adhesive fibers that were treated with an aldehyde capping molecule (i.e., tert-butyl carbazate, TBC) to deactivate the aldehydes and prevent hydrazone bond formation (Figure S5, Video S1). To provide further evidence of hydrazone bond formation, plastic deformation was rapidly disrupted by lowering the pH with acetate buffer (pH 4.3), which disrupts the pH sensitive hydrazone bond formed between fibers (Figure S6, Video S2). Thus, these findings indicate that mechanical load-induced interactions between fiber populations within multi-fiber hydrogel networks induces a chemical reaction to permanently change the fibrous architecture of the material.
Fibrous ECMs[25] and tissues[26] adapt to loads by aligning in the direction of strain, which leads to anisotropic properties and topographic cues for cells.[27] Strain-induced alignment likely occurs due to the formation of bonds between ECM fibers; however, the nature of these bonds in the ECM is unclear.[6] Similar to ECM, we hypothesized that the developed adhesive fibers would adapt to tensile strains by forming new hydrazone bonds in the strained state, leading to residual alignment when the strain is removed. Using a custom straining device, fibrous hydrogel networks were strained 50% (150% of their original length), held for 1 hour to allow inter-fiber bonds to form, and then the strain released with the sample brought back to the original length (Figure 2a). Confocal imaging of adhesive fibers showed significant differences in fiber alignment before and after the application of strain (p< 0.001), with fibers aligning along the axis of strain (Figure 2b). However, non-adhesive fibers returned to their isotropic orientation after strain removal, likely due to the lack of inter-fiber bond formation during strain (Figure S7). Additionally, adhesive fibrous hydrogel network porosity significantly decreased after strain, while non-adhesive fibrous hydrogel network porosity did not, indicating a change in pore structure with fiber adhesion (Figure S8).
Figure 2.

Strain-induced changes in fiber orientation in multi-fiber hydrogel networks. (a) Schematic of the experimental device to strain fibrous hydrogel networks, where clamped samples were strained 50%, held for 1 hour, and the strain removed. (b) Left: representative confocal images of adhesive fibers (i) before straining, (ii) while strained, and (iii) after strain removal. Right: normalized frequency of fiber orientations before and after straining (flat line indicates no orientation, whereas the presence of a peak indicates orientation at that angle). (c) Left: fiber network model of adhesive fibers (i) before straining, (ii) while strained, and (iii) after strain removal. Right: normalized frequency of fiber orientations before and after straining. Scale bar is 50 µm, ***p≤0.001.
As another measure of fiber anisotropy, human mesenchymal stromal cells (hMSCs) were seeded onto fibrous networks, containing either adhesive or non-adhesive fibers and modified with the adhesive peptide RGD (Figure S9). Without the addition of strain (i.e., stretching), cell orientation remained unorganized, likely due to the random orientation of the fibers (Figure S10). Alternatively, the fibrous networks were strained after the seeding of hMSCs for one hour to introduce fiber alignment, released, and the cell behavior assessed again 24 hours later. Consistent with fiber orientation, hMSCs on the adhesive fibers aligned in the direction of strain, whereas there was no cell alignment observed on non-adhesive fibers. This is an additional measure of the plastic deformation of fibers to introduce alignment, as fiber alignment has been previously shown to guide cell orientation. [28]
To support these experimental findings, we developed a discrete fiber network model (described within Supporting Information) where various fiber parameters (e.g., porosity, mixtures of fiber populations, inter-fiber bonding) within random networks can be controlled.[6,29] Networks contained initial crosslinks at fiber junctions that constrained the relative motion and rotation of connected fibers and individual fibers were modeled as elastic beams with circular cross-sections. Uniaxial displacement was introduced with free network contractions in the transverse direction to mimic the uniaxial mechanical experiments. The finite element model was updated to account for the new adhesions, which after unloading caused plasticity at the scale of the network. Importantly, the fiber network model displayed residual alignment, similar to the experimental observations, when comparing fiber orientation before and after uniaxial tension (Figure 2c).
In response to mechanical manipulation, fibrous ECM changes in geometry (e.g., lengthening) and mechanical properties (e.g., stiffening), which are thought to prevent tissues from prematurely failing.[25] To explore strain-induced changes in adhesive fibrous hydrogel network geometry, we applied tensile strain (50%) to samples via an electromechanical testing machine (Instron) and imaged the samples before, during (holding for 10 minutes), and after strain removal (Figure 3a). Upon removing strain, the length of the adhesive fiber mat increased as shown by the bowed appearance in the side view of the strained sample (Figure 3a, Video S3), whereas non-adhesive fibers returned to their original length (Figure S11, Video S3). This macroscale lengthening is likely a consequence of the plasticity of the network with mechanical loading, which changes the microscale fiber alignment. Supporting this, cyclic loading tests showed Mullins type softening behavior in adhesive fibers, which is associated with rearrangements of ECM fibers in tissues,[30] while non-adhesive fibers had cycle independent stress-strain behavior (Figure S12).
Figure 3.

Strain-induced changes in adhesive fiber properties. (a) Adhesive fibers were strained to 50%, held for 10 minutes, and then strain removed. Images of the sample from the side before straining (left) and after strain removal (right). (b) Representative plot of the experimental tangent modulus (Et) (light blue circles) from adhesive fibers strained with holding at 50% strain (left) or without holding (right). Plots also include results of Et determined through a continuum model simulation of these loading conditions (black, diamonds). Scale bar is 15 mm.
Since adhesive fibers align and form new inter-fiber bonds under strain, we hypothesized that the mechanical properties of these materials would change under load, such as under tensile testing until failure. When the adhesive and non-adhesive fibrous hydrogels were tensile tested without any holding to permit inter-fiber crosslinking, we observed that the adhesive material exhibited a greater modulus and increased stress at failure when compared to the non-adhesive materials, whereas the non-adhesive materials exhibited greater elongation prior to failure (Figure S13). We suspect that this behavior is due to the initially enhanced inter-fiber crosslinking at junction points due to hydrazone bond formation during material fabrication (Figure S14). To further assess changes in material properties under tensile strain, materials were strained to various extents and held to allow new inter-fiber hydrazone bond formation and then loaded to failure. The material mechanical properties are reported as instantaneous changes in the tangent modulus (Et) to measure the modulus above the linearly elastic regime (i.e. high strains). Adhesive fibers held at 50% strain showed a step-change in Et (1.7 fold increase in Et compared to before holding, p < 0.0001) upon re-application of loading (Figure 3b), whereas adhesive fibers loaded without holding or at strains ≤ 30% did not show this step-change in Et (Figure S15). Additionally, non-adhesive fibers did not show a change in Et in response to straining and holding (Figure S11). We believe that microscale alignment and crosslinking between fibers that occurs when adhesive fibers are held at 50% strain contributes to stiffening. Supporting this, aligned adhesive fibrous hydrogels have significantly higher moduli than non-aligned materials (Figure S13). Additionally, extended creep testing showed that adhesive fibers are stabilized under load, likely due to hydrazone bond formation, while non-adhesive fibers display higher rates of creep (Figure S16).
To model the changes in Et at the scale of the entire networks, we developed a reactive continuum fiber model (Table S1, details in Supporting Information). This model is based on our previous models of the plastic deformation of collagen networks.[6] The model accounts for the strain-stiffening of the networks due to fiber realignment, the Poisson effect, and the subsequent formation of crosslinks at high strains. When strained in tension and held, the reactive continuum model fits the adhesive fiber experimental data well and accurately predicted the step-change in modulus (Figure 3b). This rapid increase in the modulus was dependent on sustained bond formation time (i.e., length of time the strain was held), since the same model strained without holding did not show a step-change in Et. Along with recapitulating the behavior of the material system developed, the reactive continuum model can also be used as a framework for predicting how other fibrous material formulations will behave under mechanical manipulation and highlights bond formation time as a critical parameter for mechanical changes.
Having demonstrated that new inter-fiber bonds form within adhesive fibers under mechanical loading, we next sought to explore how this property could be exploited for interfacial bonding between fibrous materials. We first performed interfacial over-lap adhesion tests between layers of fibers to assess adhesive strengths (Figure 4a). A non-hydrated fiber mat was cut, overlaid, compressed, and hydrated with compression for 20 minutes to permit inter-fiber bond formation for adhesion testing (Figure S17). Notably, adhesive fibers had a 14-fold higher adhesive strength than non-adhesive fibers (p < 0.0001) (Figure 4b). Owing to the dynamic nature of hydrazone bonds, we also found that fiber mats subjected to interfacial adhesion testing with failure at the interface could also be re-adhered with a similar adhesive strength, an important property if repeated adhesion is desired. Since the concentration of reactive groups between fibers can be tuned, we fabricated fibrous hydrogels with lower concentrations of hydrazide groups (20% of the original hydrazide formulation), and observed a ~50% decrease in adhesive strength with a reduction in the hydrazide group concentration (Figure 4c). To increase the adhesive strength, we increased the density of fibers and consequently the density of reactive groups by fabricating aligned fibrous hydrogels, which display a 2-fold increase in adhesive strength when compared to non-aligned adhesive fibers (Figure 4d).
Figure 4.

Interfacial bonding with adhesive fibers. (a) Schematic of interfacial bonding between layers of adhesive fibers assessed with tensile loading. (b) Representative stress strain curves during interfacial adhesion testing (top) and average adhesive strength (bottom) of non-adhesive (black), adhesive (blue), and adhesive fibers that have been re-adhered after one cycle of adhering and breaking adhesion (pink). (c) Representative stress strain curves during interfacial adhesion testing (top) and average adhesive strength (bottom) of adhesive fibers with low hydrazide concentration (light blue, 20% of original hydrazide) and adhesive fibers (blue) from panel (b) for comparison. (d) Representative stress strain curves during interfacial adhesion testing (top) and average adhesive strength (bottom) of aligned adhesive fibers (green), and adhesive fibers (blue) from panel (b) for comparison. One-way ANOVA and Tukey post hoc test across all conditions was used to test for significance. *,**,**** represent p≤0.05, p≤0.01, p≤0.0001.
For many textile and biomedical applications, the self-adhesive properties of such adhesive fibers can be explored to fabricate interesting and unique structures without the need for external glues or additional chemicals We first assessed if helical structures could be fabricated by twisting a layer of fibers to induce new bonds to stabilize the twisted structure (Figure 5a). The adhesive fibers maintained their helical structures under vigorous mechanical agitation, whereas non-adhesive fibers unraveled with agitation (Figure S18). Next, we investigated the formation of laminated structures through the layering of adhesive fibers, motivated by the limited thicknesses possible with electrospinning and previously explored harsh processing conditions or use of other materials to glue scaffolds together.[31] To overcome this limitation, laminated constructs of 20 adhesive fiber mats were fabricated by layering and hydrating adhesive fibers under compression, which led to bonding between layers and their stability with mechanical agitation (Figure 5b, Video S4). Although not shown here, this layering process could also be utilized to fabricate constructs where fiber orientation changes with depth, mimicking the complex fibrous structures in tissues such as cardiac tissue and meniscal tissue.[32,33]
Figure 5.

Macroscale structure fabrication with inter-fiber and interfacial bonding in adhesive fibers. (a) Adhesive fibers were twisted into helical structures, hydrated and mechanically agitated to show maintenance of the folded helical structure at (i) low magnification (scale bar is 10 mm) and (ii) high magnification (scale bar is 2 mm). (b) Layered adhesive fibrous networks (shown with 20 layers) laminated into a thick construct, and visualized (i) after hydration and mechanical agitation (scale bar is 3 mm) and (ii) with fluorescence microscopy to examine interlamellar layers (scale bar is 0.5 mm). (c) Luminal structures fabricated with adhesive fibers by wrapping around a needle and visualized (i) during removal from the support needle and (ii) while extruding 70 kDa rhodamine labeled dextran (red) (scale bar is 20 mm). Fibers were illuminated with ultraviolet light to enhance visualization (a (i), and c (i, ii)).
Electrospun scaffolds have also been used extensively for fabricating luminal structures for vascular tissue engineering and tissue conduits[34,35]; however, these structures are generally preformed or require annealing or suturing to hold a luminal shape, which increases the complexity of fabrication. Here, adhesive fibers were wrapped around an 18G needle, hydrated, and removed (Figure S19) and maintained a stable luminal structure, even after vigorous mechanical agitation (Figure 5c, Video S5). 70 kDa rhodamine-dextran could be injected through lumens without noticeable leakage through the laminated area of the tube, which suggested the inter-lamellar bonds formed a tight seal (Video S6). Luminal structures were stable in cell culture medium for at least 13 days at 37°C (Figure S20), whereas non-adhesive materials lost their luminal structure soon after removal from the needle (Figure S18). With such a technique, damaged tissues (e.g., vessels, peripheral nerves) could be mechanically supported by adhesive fibers that are wrapped around the tissue and secured simply through the rapid interfacial bonding when the material is placed in contact with itself. Further, the mild processing techniques (e.g., water as a solvent) to fabricate adhesive fibrous hydrogel networks allow for the incorporation of therapeutics such as biologics, where their release can be further controlled through fiber engineering.[36]
Natural ECM has mechanochemical properties that allow the ECM to adapt to loading[3,5,7,25]. Although efforts have been made to model these properties in materials,[10,11] [12] none have done so without the addition of external inputs (e.g., additional monomer and chemical treatments) and while maintaining the important fibrous network architecture observed in the ECM.[1] Here, we combined multi-fiber electrospinning with complementary dynamic chemistry (i.e. hydrazone bonds) to fabricate adhesive fibrous hydrogel networks where mechanical loading induces fibers to come into contact for the reaction of chemical groups on the various fiber populations. These materials have force responsive self-adhesive properties, which can be utilized to impart changes is stiffness, geometry, and microscale anisotropy with mechanical loading, as well for the fabrication of complex structures via the adhesion of adjacent fibrous materials. Although a specific formulation of self-adhesive fibrous materials is described here, this approach can be easily extended to other fibrillar materials and reactive groups. Furthermore, the fiber network and continuum models developed can be expanded to inform future responsive material fabrication. In the rapidly growing field of fibrous material production, including as biomaterials, such self-adhesive fiber systems may facilitate the production of stimuli-responsive materials whose outputs can be tuned based on material composition, stimulus, and structure.
Experimental Section
Polymer Synthesis and Multifiber Electrospinning Parameters:
Detailed procedures for NorHA, NorHA-Ald and NorHA-Hyd synthesis, and electrospinning parameters are provided within the Supporting Information.
Fiber Manipulation, Straining, and Imaging:
For microscale compression studies, suspended fibrous hydrogel networks were created as previously described.[37] Briefly, fibers were electrospun onto thiolated PDMS molds with 2mm diameter wells and crosslinked (10mW/cm2 2 hrs) in an inert environment (N2). Suspended fibers were then hydrated with DPBS or DPBS containing 50mM tert-butyl carbazate to inhibit hydrazone bond formation (Sigma-Aldrich), and placed on an inverted spinning disk microscope (Nikon) with a micromanipulator setup (Eppendorf). Micropipette tips, fabricated using a pipette puller (Sutter Instrument Co), were coated with 1% BSA (Sigma-Aldrich) for 15 minutes, and then inserted into suspended fibrous hydrogel networks for compression studies. Suspended fibers were simultaneously imaged while compressing by translating the micropipette tip 300µm in the x-y plane at a rate of ~5µm/sec. Inter-fiber hydrazone bond kinetics were determined by holding suspended fibers in a strained position for increasing amounts of time and then removing strain to assess plastic deformation indicative of fiber bonding. To disrupt hydrazone bonds that form during compression, plastically compressed fibers were hydrated with acetate buffer (pH 4.3), which causes hydrazone bonds to break (Figure S6). To assess residual fiber alignment after straining, a previously described custom straining device,[38] was used to strain fibers and image before, during and after straining on a confocal microscope (Leica).
Fiber Network and Continuum Models:
Detailed procedures for fiber network and continuum model development and parameters are contained within the Supplemental Information.
Mechanical Testing:
Fibrous mats (~60–130µm thick) were electrospun and cut into dogbone shapes using a surgical blade. Samples were annealed to PDMS molds for clamping using non-cured PDMS. Samples were clamped using custom grips, hydrated with saline in a custom bath, pre-tension was applied (0.002 N), and samples were strained at a rate of 1% strain/per second (Instron, 5848, 5.0 N load cell). For different tests, samples were either held at indicated strains for set amounts of time and then loaded to failure at the same rate or unloaded back to the original length. Tangent modulus (Et) was calculated using the stress-strain curve. For adhesion tests, samples were sectioned mid-substance, overlaid, compressed and hydrated under compression for 20 minutes (Figure S10). Adhered samples were clamped, loaded onto the Instron within a saline bath, and loaded to failure. For extended creep testing, short (6mm) rectangular samples were fabricated and tested in the hydrated state using dynamic mechanical analysis (DMA, Texas Instruments).
Statistical Analysis:
Statistical significance between groups was determined using a t-test, or one-way ANOVA with post hoc Tukey honestly significant difference test between groups, and Watson’s two-sample test of homogeneity was used to determine significant differences between angular data with an alpha value of 0.05.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr. Edward Bonnevie, Dr. Kwang Hoon Song, Dr. Katrina Wisdom, and Jonathan Galarraga for assistance with NMR and helpful discussions on material synthesis and characterization. This study was supported by the National Institutes of Health (F32 DK117568, R01 AR056624) the National Science Foundation MRSEC (DMR-1720530) and Center for Engineering MechanoBiology STC (CMMI: 15–48571). This work was carried out in part at the Singh Center for Nanotechnology, part of the National Nanotechnology Coordinated Infrastructure Program, which is supported by the National Science Foundation grant NNCI-1542153. M. D’Este received support from the Swiss National Science Foundation grant number IZSEZ0_177045.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Matthew D. Davidson, Department of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA
Ehsan Ban, Center for Engineering Mechanobiology and Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA.
Anna C. M. Schoonen, Department of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA
Mu-Huan Lee, Department of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA.
Matteo D’Este, AO Research Institute Davos, Davos, Switzerland.
Vivek B. Shenoy, Center for Engineering Mechanobiology and Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
Jason A. Burdick, Department of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA
References:
- [1].Burla F, Mulla Y, Vos BE, Aufderhorst-Roberts A, Koenderink GH, Nat. Rev. Phys 2019, 1, 249. [Google Scholar]
- [2].Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA, Nature 2005, 435, 191. [DOI] [PubMed] [Google Scholar]
- [3].Kurniawan NA, Vos BE, Biebricher A, Wuite GJL, Peterman EJG, Koenderink GH, Biophys. J 2016, 111, 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nam S, Hu KH, Butte MJ, Chaudhuri O, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cheema U, Chuo CB, Sarathchandra P, Nazhat SN, Brown RA, Adv. Funct. Mater 2007, 17, 2426. [Google Scholar]
- [6].Ban E, Franklin JM, Nam S, Smith LR, Wang H, Wells RG, Chaudhuri O, Liphardt JT, Shenoy VB, Biophys. J 2018, 114, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vos BE, Liebrand LC, Vahabi M, Biebricher A, Wuite GJL, Peterman EJG, Kurniawan NA, MacKintosh FC, Koenderink GH, Soft Matter 2017, 13, 8886. [DOI] [PubMed] [Google Scholar]
- [8].Kouwer PHJ, Koepf M, Le Sage VAA, Jaspers M, van Buul AM, Eksteen-Akeroyd ZH, Woltinge T, Schwartz E, Kitto HJ, Hoogenboom R, Picken SJ, Nolte RJM, Mendes E, Rowan AE, Nature 2013, 493, 651. [DOI] [PubMed] [Google Scholar]
- [9].Romera M. Fernandez-Castano, Lafleur RPM, Guibert C, Voets IK, Storm C, Sijbesma RP, Angew. Chemie Int. Ed 2017, 56, 8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ramirez ALB, Kean ZS, Orlicki JA, Champhekar M, Elsakr SM, Krause WE, Craig SL, Nat. Chem 2013, 5, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matsuda T, Kawakami R, Namba R, Nakajima T, Gong JP, Science (80-. ) 2019, 363, 504. [DOI] [PubMed] [Google Scholar]
- [12].Tran YH, Rasmuson MJ, Emrick T, Klier J, Peyton SR, Soft Matter 2017, 13, 9007. [DOI] [PubMed] [Google Scholar]
- [13].Britton S, Kim O, Pancaldi F, Xu Z, Litvinov RI, Weisel JW, Alber M, Acta Biomater. 2019, 94, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wade RJ, Bassin EJ, Gramlich WM, Burdick JA, Adv. Mater 2015, 27, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wade RJ, Bassin EJ, Rodell CB, Burdick J. a., Nat. Commun 2015, 6, 6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qu F, Holloway JL, Esterhai JL, Burdick JA, Mauck RL, Nat. Commun 2017, 8, 1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim IL, Khetan S, Baker BM, Chen CS, Burdick JA, Biomaterials 2013, 34, 5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, Shenoy VB, Burdick JA, Chen CS, Nat. Mater 2015, 14, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Highley CB, Prestwich GD, Burdick JA, Curr. Opin. Biotechnol 2016, 40, 35. [DOI] [PubMed] [Google Scholar]
- [20].Gramlich WM, Kim IL, Burdick JA, Biomaterials 2013, 34, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang LL, Highley CB, Yeh YC, Galarraga JH, Uman S, Burdick JA, J. Biomed. Mater. Res. - Part A 2018, 106, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McKinnon DD, Domaille DW, Cha JN, Anseth KS, Adv. Mater 2014, 26, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA, Freels PD, Gorman JH, Gorman RC, Spinale FG, Burdick JA, Nat. Mater 2014, 13, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lou J, Liu F, Lindsay CD, Chaudhuri O, Heilshorn SC, Xia Y, Adv. Mater 2018, 30, DOI 10.1002/adma.201705215. [DOI] [PubMed]
- [25].Susilo ME, Paten JA, Sander EA, Nguyen TD, Ruberti JW, Interface Focus 2016, 6, 20150088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tower TT, Neidert MR, Tranquillo RT, Ann. Biomed. Eng 2002, 30, 1221. [DOI] [PubMed] [Google Scholar]
- [27].Carey SP, Goldblatt ZE, Martin KE, Romero B, Williams RM, Reinhart-King CA, Integr. Biol. (United Kingdom) 2016, 8, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mitchell GR, Tojeira A, in Procedia Eng, Elsevier, 2013, pp. 117–125. [Google Scholar]
- [29].Abhilash AS, Baker BM, Trappmann B, Chen CS, Shenoy VB, Biophys. J 2014, 107, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quinn KP, Winkelstein BA, J. Biomech. Eng 2011, 133, 064506. [DOI] [PubMed] [Google Scholar]
- [31].Yang G, Li X, He Y, Ma J, Ni G, Zhou S, Prog. Polym. Sci 2018, 81, 80. [Google Scholar]
- [32].Peters NS, Wit AL, Circulation 1998, 97, 1746. [DOI] [PubMed] [Google Scholar]
- [33].Li Q, Qu F, Han B, Wang C, Li H, Mauck RL, Han L, Acta Biomater. 2017, 54, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang X, Ding B, Li B, Mater. Today 2013, 16, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Greco Song H-H, Rumma RT, Ozaki CK, Edelman ER, C. S. Chen, 2018, DOI 10.1016/j.stem.2018.02.009. [DOI] [PMC free article] [PubMed]
- [36].Kim IL, Pfeifer CG, Fisher MB, Saxena V, Meloni GR, Kwon MY, Kim M, Steinberg DR, Mauck RL, Burdick JA, Tissue Eng. Part A 2015, 21, 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Davidson MD, Song KH, Lee M-H, Llewellyn J, Du Y, Baker BM, Wells RG, Burdick JA, ACS Biomater. Sci. Eng 2019, 5, 3899. [DOI] [PubMed] [Google Scholar]
- [38].Bonnevie ED, Gullbrand SE, Ashinsky BG, Tsinman TK, Elliott DM, Chao PG, Smith HE, Mauck RL, Nat. Biomed. Eng 2019, DOI 10.1038/s41551-019-0458-4. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.