

Abstract
Background and Purpose
Intrahepatic cholestasis is mainly caused by dysfunction of bile secretion and has limited effective treatment. Rosiglitazone is a synthetic agonist of PPARγ, whose endogenous agonist is 15‐deoxy‐Δ12,14‐PGJ2 (15d‐PGJ2). Reticulon 4B (Nogo‐B) is the detectable Nogo protein family member in the liver and secreted into circulation. Here, we determined if rosiglitazone can alleviate intrahepatic cholestasis in mice.
Experimental Approach
Wild‐type, hepatocyte‐specific PPARγ or Nogo‐B knockout mice received intragastric administration of α‐naphthylisothiocyanate (ANIT) and/or rosiglitazone, followed by determination of intrahepatic cholestasis and the involved mechanisms. Serum samples from primary biliary cholangitis (PBC) patients and non‐PBC controls were analysed for cholestasis‐related parameters.
Key Results
Rosiglitazone prevented wild type, but not hepatocyte‐specific PPARγ deficient mice from developing ANIT‐induced intrahepatic cholestasis by increasing expression of bile homeostatic proteins, reducing hepatic necrosis, and correcting abnormal serum parameters and enterohepatic circulation of bile. Nogo‐B knockout provided protection similar to that of rosiglitazone treatment. ANIT‐induced intrahepatic cholestasis decreased 15d‐PGJ2 but increased Nogo‐B in serum, and both were corrected by rosiglitazone. Nogo‐B deficiency in the liver increased 15d‐PGJ2 production, thereby activating expression of PPARγ and bile homeostatic proteins. Rosiglitazone and Nogo‐B deficiency also alleviated cholestasis‐associated dyslipidemia. In addition, rosiglitazone reduced symptoms of established intrahepatic cholestasis in mice. In serum from PBC patients, the decreased 15d‐PGJ2 and increased Nogo‐B levels were significantly correlated with classical cholestatic markers.
Conclusions and Implications
Levels of 15d‐PGJ2 and Nogo are important biomarkers for intrahepatic cholestasis. Synthetic agonists of PPARγ could be used for treatment of intrahepatic cholestasis and cholestasis‐associated dyslipidemia.
Abbreviations
- 15d‐PGJ2
15‐deoxy‐Δ12,14‐PGJ2
- ALP
alkaline phosphatase
- ALT
alanine transaminase
- ANIT
α‐naphthylisothiocyanate
- AST
aspartate transaminase
- BSEP
bile salt export pump
- CYP7A1
cholesterol 7α‐hydroxylase
- γ‐GT
γ‐glutamyltransferase
- H/LPGDS
haematopoietic or lipocalin PGD2 synthase
- HDL‐C
HDL cholesterol
- LDL‐C
LDL cholesterol
- PBC
primary biliary cholangitis
- p‐PPARγ
phosphorylated PPARγ
- RTN‐4/Nogo
reticulon‐4
- RTN‐4B/Nogo‐B
reticulon 4B
- SHP
small heterodimer partner
- TBA
total bile acids
- TBIL
total bilirubin
- T‐CHO
total cholesterol
- TG
triglycerides
- UDCA
ursodeoxycholic acid
What is already known
Defects in proteins involved in bile secretion cause intrahepatic cholestasis
There are few effective treatments for intrahepatic cholestasis, at present
What this study adds
Rosiglitazone protected mice against α‐naphthylisothiocyanate‐induced intrahepatic cholestasis, by activating PPARγ.
Deletion of the hepatic protein Nogo‐B was also protective in this model of intrahepatic cholestasis
What is the clinical significance
15d‐PGJ2 and Nogo are important biomarkers for intrahepatic cholestasis.
Synthetic agonists of PPARγ could be used to treat primary biliary cholangitis.
1. INTRODUCTION
Intrahepatic cholestasis includes primary biliary cholangitis (PBC) and primary sclerosing cholangitis. It can cause fibrosis, cirrhosis, and eventually liver failure. Currently, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7104) is used to treat PBC patients at the early stages while having little effect on primary sclerosing cholangitis or other types of cholestasis (Beuers, Trauner, Jansen, & Poupon, 2015). Recently, another treatment, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435, has been approved and can be used in combination with UDCA or as a monotherapy for PBC patients with poor response or intolerance to UDCA, although adverse effects of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435, including dyslipidemia, have been reported (Kowdley et al., 2018; Neuschwander‐Tetri et al., 2015; Nevens et al., 2016). Liver transplantation might be the only and last option for cholestatic patients at the advanced stages.
Defects in bile acid synthesis and secretion, mainly secretion, result in intrahepatic cholestasis. Accumulated bile acids and bile salts in the liver are toxic to hepatocytes and cause liver damages (Yerushalmi, Dahl, Devereaux, Gumpricht, & Sokol, 2001). Production of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4351 from cholesterol by action of cholesterol 7α‐hydroxylase (CYP7A1) is the rate‐limiting step for bile acid synthesis and subsequent secretion of bile from hepatocytes is mediated by a number of ATP‐binding cassette (ABC) transporters. ABCB4 is essential for secretion of phospholipids (Morita et al., 2007). Its mutations are associated with a wide spectrum of cholestatic conditions including the progressive familial intrahepatic cholestasis type 3, adult cholestatic liver disorders, https://www.guidetopharmacology.org/GRAC/DiseaseDisplayForward?diseaseId=508, low phospholipid‐associated cholelithiasis, and PBC (Ghonem, Ananthanarayanan, Soroka, & Boyer, 2014). The ABCG5/ABCG8 heterodimer is also a pre‐requisite for cholesterol and oxysterols secretion (Yu, Hammer, et al., 2002; Yu, Li‐Hawkins, et al., 2002). Disruption of ABCG5 or ABCG8 reduces, while overexpression of both enhances, biliary cholesterol and oxysterol secretion in mice (Langheim et al., 2005). The bile salt export pump (BSEP) is responsible for secretion of bile salts and deficiency of this transporter causes progressive familial intrahepatic cholestasis type 2 (Strautnieks et al., 1998).
The https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=603) is a nuclear transcription factor and it is activated by agonists such as endogenous bile acids or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435 (Forman et al., 1995). FXR induces BSEP and ABCB4 expression to enhance secretion of bile salts and phospholipids (Ijssennagger et al., 2016). Meanwhile, FXR inhibits bile acid synthesis by inactivating CYP7A1 in an indirect manner via https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=636) and FGF 15/19 (Goodwin et al., 2000; Inagaki et al., 2005). https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 is another ligand‐activated transcription factor which is activated either by its endogenous agonist, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=refs&ligandId=1877) or synthetic ligands such as the thiazolidinediones. Two thiazolidinediones, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1056 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2694, are currently used to treat patients with type 2 diabetes.
https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2838B (RTN‐4B or Nogo‐B) is a member of the Nogo protein family with a molecular weight of 45 kDa in mouse or 55 kDa in human. Nogo‐B has been found to participate in several biological functions in the liver, such as wound healing, hepatocyte proliferation, liver fibrosis, and regeneration (Cantalupo et al., 2015; Gao et al., 2013; Zhang et al., 2011). Nogo‐B appears to be the only member of the Nogo family which is detected in the liver (Zhang et al., 2011). This protein is also secreted into the circulation and an increase in serum Nogo‐B levels has been found in patients with a wide variety of diseases, such as pulmonary arterial hypertension, hepatic cirrhosis, and https://www.guidetopharmacology.org/GRAC/DiseaseDisplayForward?diseaseId=255 (Gao et al., 2015; Men et al., 2015; Sutendra et al., 2011). Recently, pioglitazone, a synthetic PPARγ agonist, was shown to regress advanced liver fibrosis in patients with non‐alcoholic steatohepatitis (Musso, Cassader, Paschetta, & Gambino, 2017). Intrahepatic cholestasis is one cause of liver fibrosis, so the regression of liver fibrosis by pioglitazone could be attributed to anti‐intrahepatic cholestasis effects of PPARγ. However, whether PPARγ agonists can prevent or treat intrahepatic cholestasis remains unknown.
In this study, we have determined the effects of pretreatment or post‐treatment with rosiglitazone (another synthetic agonist of PPARγ) on intrahepatic cholestasis induced by intragastric administration of α‐naphthylisothiocyanate (ANIT) in mice. We also assessed the relevance of circulating levels of 15d‐PGJ2 or Nogo to our results in this model.
2. METHODS
2.1. Construction and transfection of PPARγ2 expression vectors
The wild‐type PPARγ2 was generated by reverse transcription with total cellular RNA isolated from HepG2 cells and an oligo (dT)18 primer followed by PCR with forward and backward primers: 5′‐TCTCGAGCTCAATGGGTGAAACTCTGGGAG‐3′ and 5′‐CCGCGGTACCCTAATACAAGTCCTTGTAGATCTCCT‐3′. After the sequence was confirmed, the PCR product was digested with SacI and KpnI and then subcloned into a pEGFP‐C2 expression vector, and this wild‐type PPARγ2 expression vector was named as C2‐PPARγ2. The non‐p‐PPARγ2 (Ser112 is mutated to Ala) and constitutively p‐PPARγ2 (Ser112 is mutated to Asp) expression vectors were constructed using the Phusion site‐directed mutagenesis kit from New England Biolabs, C2‐PPARγ plasmid DNA and primers with the corresponding mutated sequences, and they were named as C2‐PPARγ2‐S112A (SA) and C2‐PPARγ2‐S112D (SD) respectively. HepG2 cells cultured in six‐well plates were transfected with wild‐type or mutant PPARγ2 expression vector with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
2.2. Cell culture and construction of Nogo‐B deficient HepG2 cells
HepG2 cells, a human hepatic cell line, were purchased from ATCC (Manassas, VA, USA, RRID:CVCL_0027) and cultured in DMEM medium containing 10% FBS, 50 mg·ml−1 penicillin/streptomycin and 2 mmol·L−1 glutamine. Cells (~90% confluence) received treatment in serum‐free medium.
LentiCRISPR was used to construct Nogo knockout (KO) plasmid (Cong & Zhang, 2015). The target sequence is 5′‐CGTTCAAGTACCAGTTCGTG‐3′, which locates in the second exon 63 bp downstream of the start codon. After ligation, HepG2 cells were transfected with the plasmid DNA for 24 hr. Cells were then treated with puromycin (3 μg·ml−1) for 4 days for selection. The limiting dilution assay was employed to obtain several monoclones. After culture expansion, cells were harvested to confirm Nogo deficiency by Western blot and sequence analysis. Based on the fact that Nogo‐B is the major member of Nogo family detected in HepG2 cells by Western blot (Figure S1A, left panel), the Nogo deficiency in HepG2 cells was simply referred to as Nogo‐B deficiency and these cells were named Nogo‐B‐cas9 cells. The corresponding control cells were named as Ctrl‐cas9 cells. All the experiments with cells were conducted in a blinded and random fashion.
2.3. Collection of serum samples from PBC patients and non‐PBC controls for biochemical assay
With a protocol approved by the Clinical Ethics Committee of the First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China) and adhering strictly to the Declaration of Helsinki Principles (2008), we recruited seven female patients with an age range of 45–65 years old who had been diagnosed with PBC and were undergoing UDCA treatment. We also recruited nine non‐PBC female controls with matched age range but no symptoms of PBC, or other diseases. At the time of recruitment, all the participants were fully informed and signed their consents.
After collection, blood samples were used to isolate serum by technicians who were blinded to the samples, in the subsequent determination of albumin, total bilirubin (TBIL), alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), γ‐glutamyltransferase (γ‐GT), total cholesterol (T‐CHO), HDL cholesterol (HDL‐C), and LDL cholesterol (LDL‐C) using an automatic biochemical analyser (Model #3100, Hitachi High‐Technologies Corporation, Japan). Levels of 15d‐PGJ2 and Nogo were measured using the corresponding ELISA assay kits for human samples. Unequal group sizes resulted from the limitation of recruitment of PBC patients or from the exclusion of outliers.
2.4. In vivo studies with animals
All animal care and experimental protocols for in vivo studies complied with the Guide for the Care and Use of Laboratory Animals published by the NIH (NIH publication no. 85‐23, revised 1996) and were approved by the Ethics Committee of Nankai University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. The mice were housed in SPF units of the Animal Center at the College of Life Sciences, Nankai University animal units (with a 12‐hr light cycle from 8 a.m. to 8 p.m., 23 ± 1°C, 60–70% humidity) and maintained on a standard rodent diet with free access to water in plastic bottles. Mice were allowed to acclimatize to their housing environment for at least 7 days before experiments. Up to five mice were kept per plastic cage with corn cob bedding material.
To generate specific hepatocyte‐specific PPARγ deficient mice, the PPARγfl/fl mice were crossbred with the Alb‐Cre transgenic mice (purchased from Jackson Laboratory, USA). Thus, the control mice are PPARγfl/fl/Cre−/− and hepatocyte‐specific PPARγ deficient mice are PPARγfl/fl/Cre+/−, and they were named as PPARγfl/fl and hepatocyte‐specific PPARγ KO (RRID:IMSR_RMRC13187) mice respectively. The sex‐ and age‐matched PPARγfl/fl and hepatocyte‐specific PPARγ KO mice were used in this study.
Generation of Nogo deficient mice (C57BL/6J background, RRID:IMSR_JAX:000664) was completed by Nanjing Biomedical Research Institute of Nanjing University (Nanjing, Jiangsu, China) based on the CRISPR/Cas9 system. Briefly, a pair of sgRNAs were designated to target exon 2 and 8, respectively, which can delete ~37 Kb in the genome and corresponding 2,824 bp of the transcript. The sequence of exon 2–8 was also present in mRNA of all other members of Nogo family. Therefore, such deletion also impaired expression of other Nogo family members. The offspring were genotyped by PCR method as follows: the genomic DNA was isolated from a small piece of mouse tail using the DNA isolation kit purchased from Vazyme Biotech Co., Ltd (Nanjing, Jiangsu, China) followed by PCR with the primers 2260Rtn4‐gtF1: 5′‐GGGTCATCTTTCCTGTTATGC‐3′, 2260Rtn4‐gtR1: 5′‐ATGAGATCCATACACAGCAG‐3′, 2260Rtn4‐delgtF1: 5′‐CAAGAGCGTTAAGGATGCCA‐3′, and 2260Rtn4‐gtR2: 5′‐TCACCAAGTGCCTCATCTGG‐3′. The products of PCR were determined by DNA electrophoresis in a 1.0% agarose gel.
In this study, we mainly focused on the effect of Nogo deficiency on intrahepatic cholestasis and expression of molecules involved in bile homeostasis. Nogo‐B is the member of the Nogo family, detectable in the liver. Therefore, the Nogo deficient mice were also referred to as Nogo‐B KO mice. The validated Nogo‐B KO and the littermate control mice were bred in the Animal Center of Nankai University (Tianjin, China).
We decided sample size for animal studies based on a survey of data from published research or preliminary studies. In this study, the number of mice was not predetermined by a statistical method, and no mice were excluded for statistical analysis. We conducted treatment to mice in a blinded fashion. The drugs used for treating animals were prepared by researchers who did not carry out the treatments. In addition, all animals were randomized before they received treatment. The animals were daily checked for food intake, water drinking, and bodyweight gain during the treatment.
Intrahepatic cholestasis in mice was induced by treating animals with ANIT, which can generate cholestatic symptoms, similar to those observed in humans (Kossor et al., 1995; Zhang et al., 2018). Adult male C57BL/6J mice (18 ± 1 g, 8 ± 0.5 weeks) were purchased from Vital River Inc. (Beijing, China). Totally, 22 C57BL/6J, 24 PPARγfl/fl, and 22 hepatocyte‐specific PPARγ deficient (PPARγ−/−) mice (all are males, 18 ± 1 g, 8 ± 0.5 weeks) were randomly divided into four groups (≥5 mice per group) and received the following treatments by intragastric administration (Figure 1a). (a) Vehicle group: corn oil daily for five days (n = 6 for C57BL/6J; n = 5 for PPARγfl/fl; and hepatocyte‐specific PPARγ−/−); (b) rosiglitazone group (Rosi): rosiglitazone (30 mg·kg−1) dissolved in corn oil daily for five days (n = 6 for C57BL/6J; n = 5 for PPARγfl/fl and hepatocyte‐specific PPARγ−/−); (c) ANIT group: corn oil on Days 1–3 and 5 for four days, and ANIT (80 mg·kg−1) on Day 4 once (n = 7); and (d) rosiglitazone plus ANIT (Rosi + ANIT) group: rosiglitazone daily for five days and ANIT on Day 4 once (n = 7 for C57BL/6J and PPARγfl/fl; n = 5 for hepatocyte‐specific PPARγ−/−). Unequal group size resulted from efforts to match age of experimental cohorts arising from in‐house breeding or from exclusion of outliers. At 48 hr after ANIT administration, all mice were weighed and then killed. To determine the effect of rosiglitazone on the established intrahepatic cholestasis, a total of 15 C57BL/6J mice (18 ± 1 g, 8 ± 0.5 weeks) were randomly divided into three groups (n = 5) and received treatment as scheduled in Figure 7a. After killing at the end of experiment, blood, liver, and intestine samples were collected gently and individually from all the mice.
Figure 1.
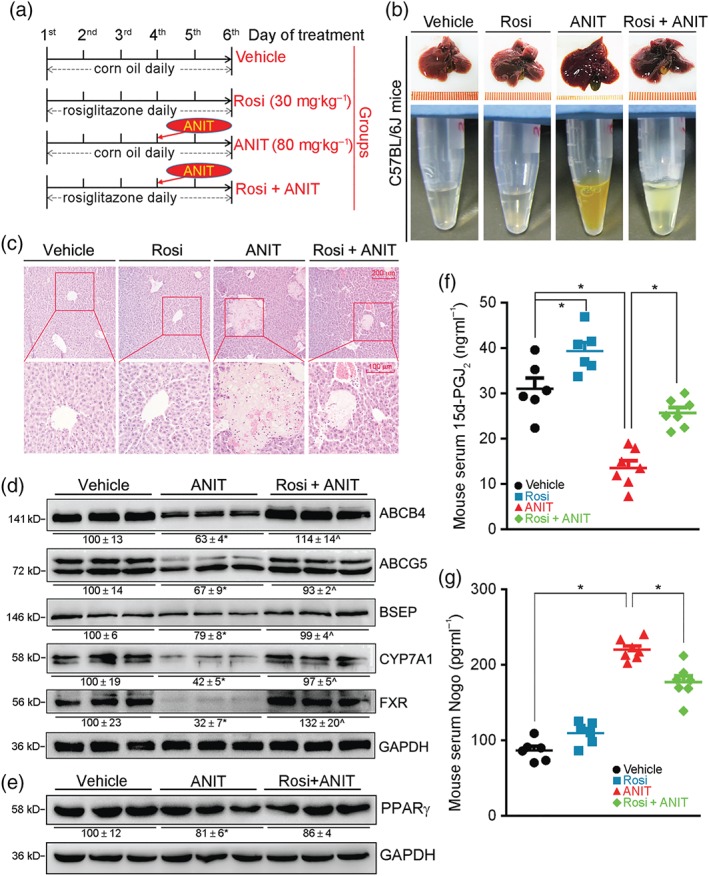
Rosiglitazone protects C57BL/6J mice from ANIT‐induced intrahepatic cholestasis by activating bile homeostatic proteins and production of 15d‐PGJ2, while reducing Nogo. (a) (The schedule of treatment): C57BL/6J mice were randomly divided into four groups (eight per group) and received intragastric administration of corn oil (Vehicle), rosiglitazone (Rosi, 30 mg·day−1·kg−1), ANIT (80 mg·kg−1) once on Day 4, or ANIT plus Rosi as indicated. Two days after ANIT treatment, mouse tissue samples were collected. (b) Representative photographs of liver and serum samples from each group. (c) Liver paraffin sections were stained with H&E. (d) Expression of ABCB4, ABCG5, BSEP, CYP7A1, FXR was determined as total protein extracted from liver. (e) PPARγ protein was determined by western blot with quantification of band density. * ^ P < .05, significantly different from Vehicle group, n = 6. (f, g) Levels of 15d‐PGJ2 and Nogo were determined in serum samples using ELISA assay kits. n = 6 for Vehicle and Rosi groups, n = 7 for ANIT and ANIT + Rosi groups. * P < .05, significantly different as indicated
Figure 7.
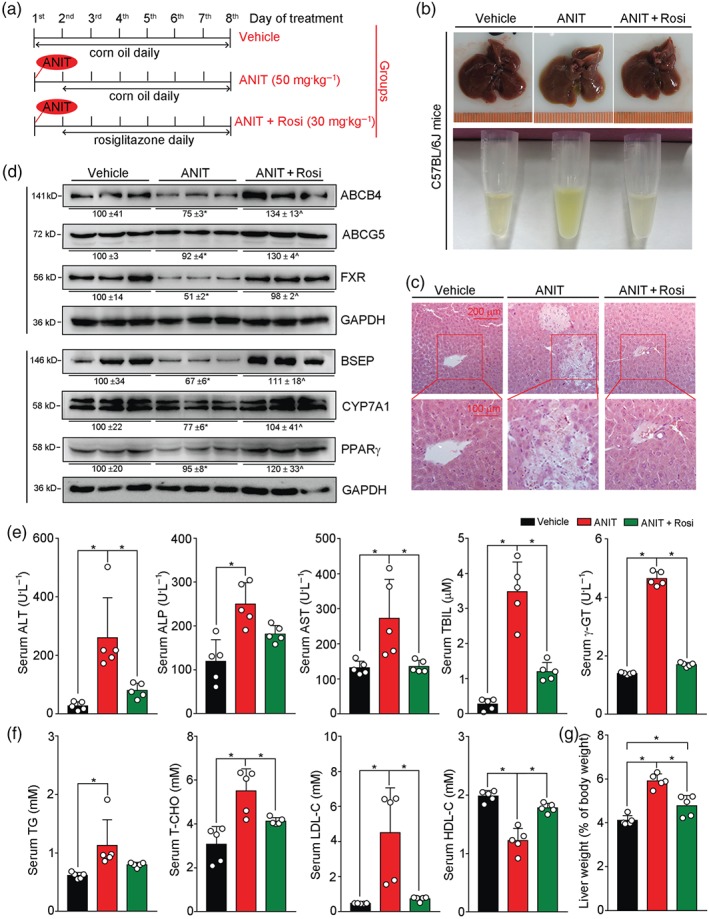
Rosiglitazone ameliorates intrahepatic cholestasis induced by ANIT. (a) (The schedule of treatment): 8 ± 0.5‐week‐old male C57BL/6J mice were randomly divided into three groups (five per group) and received the following treatments, by intragastric administration, (1) Vehicle group: corn oil daily for seven days; (2) ANIT group: ANIT (50 mg·kg−1 ) on Day 1, corn oil daily for six days from Days 2 to 7; (3) ANIT plus Rosi group, ANIT (50 mg·kg−1) on Day 1 and rosiglitazone (30 mg·kg−1) daily for six days from Days 2 to 7. (b) Representative photographs of liver and serum samples from each group; (c) Liver paraffin sections were stained with H&E. (d) Expression of proteins (ABCB4, ABCG5, BSEP, CYP7A1, FXR, and PPARγ) extracted from the livers. (e,f) Serum ALT, ALP, AST, TBIL, γ‐GT, TG, T‐CHO, LDL‐C, and HDL‐C levels were determined. (g) Ratio of liver weight to bodyweight in each mouse was calculated. For (e, f, g), * P < .05, significantly different as indicated; n = 5
Serum was prepared from blood and used to determine ALP, AST, ALT, γ‐GT, TBIL, T‐CHO, HDL‐C, LDL‐C, and triglycerides (TG) using an automated biochemical analyser if the volume as large enough. For 15d‐PGJ2, Nogo, and γ‐GT we used the corresponding ELISA assay kits for mouse samples. The liver was completely removed and weighed, followed by calculation of the ratio of liver weight to bodyweight (%). A piece of liver was then taken and fixed in 4% paraformaldehyde/PBS, followed by preparation of liver sections for H&E staining.
A total of 21 Nogo‐B KO and 21 corresponding littermate control mice (all are males, 18 ± 1 g, 8 ± 0.5 weeks) were randomly divided into three groups (seven mice/group) and received the following treatments: Vehicle, ANIT, and ANIT plus rosiglitazone, with the same schedule and assays as in wild‐type mice, described above.
2.5. Determination of ABCB4, ABCG5, CYP7A1, FXR, Nogo‐B, and PPARγ protein expression by Western blot
The antibody‐based procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). Cells cultured in six‐well plates or ~20‐mg liver were lysed or homogenized in the lysis buffer containing 50 mmol·L−1 Tris, pH 7.5, 150 mmol·L−1 NaCl, 1% Triton X‐100, 1% sodium deoxycholate, 1 mmol·L−1 PMSF, 50 mmol·L−1 sodium fluoride, 1 mmol·L−1 sodium orthovanadate, 50 μg·ml−1 aprotinin/leupeptin and 1× PMSF (Enzo, #ALX‐270‐184‐G025) and cocktail (Millipore, #539131‐10VL) to extract total proteins. The cellular lysate or tissue homogenates were transferred into 1.5‐ml tube and centrifuged for 10 min at 17,000 g and 4°C. The supernatant was collected as total cellular or tissue protein and used immediately or stored at −20°C for less than 1 week. Samples were frozen and thawed not more than three times.
The same amount (~50 μg) of total proteins from each sample was loaded on a 10% or 12% SDS‐PAGE. After ~100‐min electrophoresis, proteins were transferred onto a nitrocellulose membrane (PALL, Mexico). The membrane was then blocked with PBS containing 5% fresh non‐fat dry milk (Solarbio, Beijing, China) for 1 hr at room temperature, followed by incubation with 1:1,000 diluted fresh primary antibody or anti‐GAPDH antibody (1:5,000) in PBS containing 1% fresh dry fat‐free milk overnight at 4°C. Membranes were washed for 3 × 10 min with PBS containing 0.05% Tween 20 (PBST) and then re‐blocked and incubated with 1:5,000 diluted fresh HRP‐conjugated donkey anti‐rabbit or donkey anti‐goat IgG in PBS containing 1% fresh dry fat‐free milk for 1 hr at room temperature. After washing for 3 × 10 min with PBST, the membrane was incubated in Western blotting chemiluminescence reagents 1 and 2 (Millipore, Billerica, MA, USA), followed by exposure to a film or scanning with Clinx ChemiCapture (CLINX, Shanghai, China). The images were analysed with Image J software Version 1.43u.
2.6. Isolation of total RNA and determination of mRNA expression by quantitative real‐time PCR
After treatment, total RNA was extracted using the Trizol reagent (Invitrogen) which contains DNase to eliminate genomic DNA according to the classic protocol. The lysate or homogenate was well mixed with chloroform and centrifuged at 17,000 g for 15 min at 4°C. The upper aqueous phase, which contains RNA, was collected and mixed with isopropanol and stored at −20°C overnight to precipitate the total RNA. The next day, RNA was centrifuged and treated with 75% and 100% ethanol specifically. Total RNA was dissolved in water finally. RNA was assessed by OD 260/280 between 1.8−2.0 and OD 260/230 > 2.0. The cDNA was then synthesized with 1‐μg total RNA using a reverse transcription kit purchased from New England Biolab (Ipswich, MA, USA). The reaction mixture for reverse transcription consisted of 1‐μg RNA, 2‐μl 5× qRT Super Mix and nuclease‐free water up to total 10 μl. The programme is as follows: 25°C for 5 min, 50°C for 30 min, and 75°C for 5 min. The synthesized cDNA was stored at −20°C until analysis. The primer sequences listed in Table 1 were designed by NCBI and spanned introns. The primers were synthesized and validated by GENEWIZ (South Plainfield, NJ, RRID:SCR_003177). qPCR reactions contained 2‐μl cDNA, 1 μl each primer (10 mM), 10‐μl SYBR Green PCR Master Mix (Roche, USA), and 6‐μl nuclease‐free water (total volume is 20 μl). qPCR was carried out at 95°C for 5 min for pre‐degeneration, 50 cycles of 95°C for 15 s, 55°C for 15 s, 72°C for 30 s, and read melting curve for 20 min. Expression of mRNA for ABCB4, ABCG5, BSEP, CYP7A1, FXR, haematopoietic PGD2 synthase (HPGDS), and lipocalin PGD2 synthase (LPGDS) was determined with the primers listed in Table 1 and normalized by GAPDH mRNA in the corresponding samples.
Table 1.
Sequences of primers for qRT‐PCR
| Genes | ID | Primer | Sequence | Product (bp) |
|---|---|---|---|---|
| hABCB4 | 5244 | Forward | CAGTACTGGTGCACTTTCTACAAGACTT | 85 |
| Reverse | TGCAATTAAAGCCAACCTGGTT | |||
| hABCG5 | 64240 | Forward | TCGGAGAGCTGATTTTCTGTG | 147 |
| Reverse | CTATTTCCCGTTCCTTGCTTTG | |||
| hBSEP | 8647 | Forward | TTCCAGGAAAAGCATGTGTG | 110 |
| Reverse | CATTTCGCTCTCGATGTTCA | |||
| hCYP7A1 | 1581 | Forward | CAGAACTGAATGACCTGCCA | 108 |
| Reverse | GGTGCAAAGTGAAATCCTCC | |||
| hFXR | 9971 | Forward | CACAGCGTTTTTGGTAATGC | 98 |
| Reverse | TTGTTTGTGGAGACAGAGCCT | |||
| hGAPDH | 2597 | Forward | AAGGTGAAGGTCGGAGTCAA | 108 |
| Reverse | AATGAAGGGGTCATTGATGG | |||
| mHPGDS | 54486 | Forward | AATGTCAAGCTGATGCAGTG |
120 |
| Reverse | GTGCTTGATGTGTGAGCAAT | |||
| mLPGDS | 19215 | Forward | ATGACGAGTA CGCTCTGCTA |
110 |
| Reverse | CTCCTTCAGCTCGTCCTTCA | |||
| mSHP | 23957 | Forward | AGGAGTATGCGTACCTGAAG | 203 |
| Reverse | AAGGGTGCCTGGAATGTTCTT | |||
| mLRH‐1 | 26424 | Forward | TCATGCTGCCCAAAGTGGAGA | 232 |
| Reverse | TGGTTTTGGACAGTTCGCTT | |||
| mGAPDH | 14433 | Forward | TGTGTCCGTCGTGGATCTGA | 234 |
| Reverse | TTGCTGTTGAAGTCGCAGGAG |
Abbreviations: h, human; m, mouse.
2.7. Determination of mouse serum γ‐GT
Mouse serum γ‐GT was determined using the corresponding assay kit purchased from BioSino Bio‐Technology and Science Inc. (Beijing, China; Shaw, Stromme, London, & Theodorsen, 1983). Before the assay, serum samples in the group of mice treated with ANIT or ANIT plus rosiglitazone were diluted five times with saline (0.9% NaCl). All samples and the working buffer were warmed to 37°C. Either 10‐μl saline (as a blank) or 10‐μl serum sample was added into a 96‐well plate followed by addition of 100‐μl working buffer. The absorbance at 405 nm was read using a Microplate reader (BioTek, Winooski, VT, USA) at 1, 2, and 3 min after addition of the working solution. The variation rate per minute was calculated to obtain the average change rate (△), which was used to calculate γ‐GT activity as γ‐GT (U·L−1) = △ × 1,158.
2.8. Extraction and determination of total bile acids in mouse liver and intestine
Total bile acid (TBA) was extracted from liver and intestine and determined using an assay kit (Li et al., 2011). Briefly, after treatment, a piece of liver (~100 mg) or whole intestine were added to 1‐ or 5‐ml 95% ethanol respectively. Samples were incubated for 3 hr in a 60°C water bath. Samples were then centrifuged for 10 min at 6,150 g, and the supernatant was transferred into a new tube. The remaining pellet was re‐suspended in 80% ethanol and incubated for another 3 hr in a 60°C water bath. Samples were centrifuged for 10 min at 6,150 g again and the supernatant was collected. The pellet was re‐suspended in a mixture of chloroform and methanol (2:1, v/v; 0.5 ml for liver and 5 ml for intestine) and incubated for 1 hr at room temperature. After centrifugation for 10 min at 6,150 g, the supernatant was collected. The supernatant collected from the three steps above was mixed and centrifuged for 10 min at 6,150 g and room temperature to remove debris.
TBA in the supernatant (there was 2.5 ml and 15 ml for liver and intestine samples respectively) above were determined using the TBA assay kit purchased from Diazyme (Poway, CA, USA). Briefly, the supernatant of intestine sample was pre‐diluted ~20–100 times with 80% ethanol. The reagent R1 and R2 supplied in the assay kit were pre‐warmed to 37°C. Five μl of sample (the supernatant of liver or the diluted supernatant of intestine sample) or 80% ethanol (as the blank) was added into a 96‐well plate followed by addition of 135‐μl reagent R1 and incubated for 5 min at 37°C. Then, 45‐μl reagent R2 was added to each sample followed by determination of absorbance at 405 nm immediately which was considered as the value at 0 min. The absorbance of samples was then measured every min for 3–5 min. The △405 nm was calculated as subtraction of the absorbance read at 3 min by the absorbance read at 2 min. The results of TBA in each sample was calculated as TBA (μmol·ml−1) = × standard (50 μmol·ml−1) × diluted factors. The TBA (μmol) in tissue samples was calculated as follows: in the liver, TBAliver (μmol) = TBA (μmol·ml−1) × volume (2.5 ml) × LW (g)/0.1 g (LW: total liver weight) and in the intestine, TBAintestine (μmol) = TBA (μmol·ml−1) × volume (15 ml).
2.9. Determination of 15d‐PGJ2 levels in serum, cellular lysate, and cell‐conditioned medium
To determine 15d‐PGJ2 levels in cells and conditioned medium, cells at the same density and number were switched into serum‐free medium and cultured for 24 hr. The 24‐hr conditioned medium was collected and centrifuged for 20 min at 1,0000 x g to remove cell debris. Cells remaining in dishes were washed twice with cold PBS and then lifted by addition of trypsin. The collected cells were centrifuged for 5 min at 1,000 x g. Cells were washed once with PBS and then re‐suspended in cold PBS (~1 × 106 cell/150 μl PBS) and homogenized. A portion of homogenates was saved to determine protein content, and the rest of homogenates was centrifuged for 10 min at 1,500 x g. The supernatant was saved. Samples of human or mouse serum, conditioned medium, and supernatant of cellular lysate were used to determine 15d‐PGJ2 levels using the corresponding ELISA kits. The cellular 15d‐PGJ2 levels were normalized by cellular protein content and expressed as ng·μg−1 protein.
2.10. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Based on our previous studies and/or preliminary experiments, we calculated the group size for in vitro study. Data are presented as mean ± SEM and were generated from at least five independent experiments. For Western blot assay, after images were obtained, the density of each band was quantified by a technician (blinded to the treatments) with Image J software (National Institutes of Health, Bethesda, MD, USA, RRID:SCR_003070). The density of target band was normalized to GAPDH in the corresponding sample to reduce variance. For quantitative real‐time PCR, target gene mRNA expression was normalized by GAPDH mRNA in the corresponding samples. The data of quantitative real‐time PCR or quantification of Western blot were expressed as folds of the control group's mean value which was defined as one plus its own SD. GraphPad Prism software (version 7.0, GraphPad Software, San Diego, CA, RRID:SCR_002798) was used for one‐way or two‐way ANOVA or unpaired t‐test (two‐tailed) or unpaired t‐test (two‐tailed) with Welch's correction. For ANOVA, Tukey's post hoc test was performed for data with F at P < .05 and no significant variance inhomogeneity. Correlation coefficients were calculated using the Spearman correlation test, and one‐tailed P value was calculated. The differences between group means were considered significant when P < .05 (n ≥ 5). The declared group size is the number of independent values and the statistical analysis was carried out using these independent values.
2.11. Materials
Rosiglitazone was purchased from Alexis Biochemicals (San Diego, CA, USA). ANIT was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Rabbit polyclonal antibodies recognizing both human and mouse molecules were purchased from the following sources: anti‐ABCB4 (#NBP2‐30887), ABCG5 (#NBP1‐59803, RRID:AB_11017123), and Nogo (#NB100‐56681, RRID:AB_838641) from Novus Biologicals (Littleton, CO, USA); anti‐BSEP (#A14694, RRID:AB_2761569) from Abclonal (Wuhan, China); anti‐CYP7A1 (#sc‐25536, RRID:AB_2088578) and GAPDH (#sc‐25778, RRID:AB_10167668) from Santa Cruz Biotechnology (Dallas, Texas, USA); anti‐PPARγ (#16643‐1‐AP, RRID:AB_10596794) from Proteintech Group (Chicago, IL, USA); anti‐FXR (#ab28480, RRID:AB_726991) and phosphorylated PPARγ (p‐PPARγ, #ab60953, RRID:AB_946311) from Abcam (Cambridge, MA, USA). The ELISA kits for human Nogo (#E‐EL‐0087c) and mouse Nogo (#CSB‐EL020572HU) were purchased from Elabscience (Wuhan, Hubei, China) and CUSABIO Technology LLC (Houston, TX, USA) respectively. Due to the high homology among Nogo family members and the limited skills and knowledge, the cross reactivity among Nogo family members or analogues cannot be excluded by the current ELISA assay kits. The 15d‐PGJ2 ELISA kits used for mouse sample (#E‐EL‐0087c) and human samples (#JL41064‐48 T) were purchased from Elabscience and Jianglaibio (Shanghai, China) respectively.
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski et al., 2019; Alexander, Fabbro et al., 2019).
3. RESULTS
3.1. Rosiglitazone protects mice from ANIT‐induced intrahepatic cholestasis
To determine if rosiglitazone can prevent intrahepatic cholestasis, C57BL/6J mice were pretreated with rosiglitazone for 3 days followed by ANIT treatment for 48 hr (Figure 1a). In ANIT‐treated mice, dark coloured bile, livers with macroscopic signs of damages and blurry serum of a light brown colour were observed, effects not observed in mice treated with vehicle. Treatment with rosiglitazone protected mice from ANIT‐induced damage (Figure 1b). Histological analysis of liver sections showed multiple large areas of necrosis with infiltration of inflammatory cells in ANIT‐treated mouse liver. However, such necrosis was substantially reduced by rosiglitazone (Figure 1c).
Besides hepatomegaly (the ratio of liver weight to bodyweight was increased by about 37%), the biochemical assays showed that ANIT markedly increased serum ALT, AST, ALP, γ‐GT, and TBIL (Table 2). Liver TBA was increased while intestine TBA was decreased correspondingly, indicating abnormal enterohepatic circulation of bile acids. The sum of TBA in liver and intestine was also reduced by ANIT treatment, suggesting the secretion of TBA into the circulation was increased substantially which is consistent with changes of serum colour (Figure 1b). Rosiglitazone alone had mild effects on these biomarkers (Table 2). However, it clearly corrected ANIT‐caused abnormal serum biomarkers. Compared with ANIT alone, serum ALT, AST, ALP, γ‐GT, and TBIL were markedly reduced by rosiglitazone. Rosiglitazone reduced liver TBA but increased intestine TBA and the sum of liver and intestine TBA (Table 2), thereby correcting the impaired enterohepatic circulation of bile acids.
Table 2.
Rosiglitazone alleviates intrahepatic cholestasis: Biochemical assay
|
Parameters |
Treatment | ||||
|---|---|---|---|---|---|
| Vehicle | Rosi | ANIT | ANIT + Rosi | ||
| LW/BW (%) | 4.30 ± 0.11 | 4.92 ± 0.06 | 5.91 ± 0.18* | 4.72 ± 0.32# | |
| BW (g) | 18.27 ± 0.51 | 18.20 ± 1.04 | 16.51 ± 1.36* | 16.72 ± 0.66 | |
| Serum enzyme activity, total bilirubin, and lipids | ALT | 27.2 ± 2.0 | 22.9 ± 0.68 | 1,749 ± 182* | 347 ± 81.8# |
| AST | 123.8 ± 2.5 | 117.8 ± 6.3 | 5,416 ± 626* | 956 ± 341.0# | |
| ALP | 129.3 ± 3.7 | 109.8 ± 3.5 | 383 ± 24.7* | 247.5 ± 6.4# | |
| γ‐GT | 1.27 ± 0.22 | 1.27 ± 0.12 | 21.54 ± 2.68* | 9.81 ± 1.40# | |
| TBIL | 0.26 ± 0.02 | 0.16 ± 0.01 | 22.16 ± 3.92* | 2.2 ± 0.63# | |
| T‐CHO | 2.66 ± 0.31 | 3.46 ± 0.13 | 7.10 ± 1.03* | 5.51 ± 0.21 | |
| LDL‐C | 0.44 ± 0.05 | 0.49 ± 0.02 | 2.09 ± 0.15* | 0.5 ± 0.10# | |
| HDL‐C | 1.95 ± 0.22 | 2.44 ± 0.80 | 0.98 ± 0.15* | 3.22 ± 0.23# | |
| TG | 0.58 ± 0.01 | 0.68 ± 0.04 | 3.43 ± 0.60* | 0.87 ± 0.07# | |
| Liver TBA | 0.73 ± 0.05 | 0.55 ± 0.09 | 2.52 ± 0.22* | 1.67 ± 0.34# | |
| Intestine TBA | 12.24 ± 0.81 | 17.85 ± 1.43 | 6.14 ± 0.94* | 19.34 ± 1.33# | |
Note. C57BL/6J mice received vehicle (corn oil), rosiglitazone (Rosi), α‐naphthylisothiocyanate (ANIT), or rosiglitazone plus α‐naphthylisothiocyanate (Rosi + ANIT) treatment as indicated in Figure 1a. After treatment, mouse bodyweight (BW) and liver weight (LW) were determined to calculate the ratio of LW/BW (%). Enzyme activity of ALT, AST, ALP, and γ‐GT (as U·L−1) and concentration of TBIL (μmol·L−1), T‐CHO, LDL‐C, HDL‐C, and TG (mmol·L−1) were measured in serum samples. Total bile acids (TBA, μmol) were determined in samples of liver and intestine.
P < .05, significantly different from Ctrl (vehicle) # P < .05, significantly different from ANIT (n = 5).
In addition to reduction of HDL‐C, ANIT increased serum T‐CHO, LDL‐C and TG, indicating it caused severe cholestasis‐associated hypercholesterolemia or dyslipidemia in mice (Table 2). However, this ANIT‐induced cholestasis‐associated dyslipidemia was potently ameliorated by rosiglitazone (Table 2).
The effect of ANIT and/or rosiglitazone on expression of proteins involved in bile homeostasis was determined. Figure 1d shows that ANIT inhibited expression of molecules for bile synthesis (CYP7A1), secretion (ABCB4, ABCG5, and BSEP), and the transcription factor (FXR) in mouse liver. Rosiglitazone alone activated expression of these proteins (Figure S2). More importantly, rosiglitazone reversed ANIT‐inhibited expression of these proteins.
The results above implied that PPARγ activity was regulated either by PPARγ expression or by production of endogenous agonist. As shown in Figure 1e, ANIT slightly reduced PPARγ expression which was not affected by rosiglitazone. Therefore, PPARγ activity was not regulated by alterations in PPARγ expression. The potent endogenous PPARγ agonist 15d‐PGJ2 (Nagy, Tontonoz, Alvarez, Chen, & Evans, 1998) binds to PPARγ to trigger its transcriptional activity and induce PPARγ expression. We therefore determined the effect of ANIT or rosiglitazone on 15d‐PGJ2 production in vivo. As shown in Figure 1f, rosiglitazone alone increased serum 15d‐PGJ2 levels but more importantly, it potently blocked the ANIT‐induced decrease in circulating 15d‐PGJ2 (Figure 1f).
Increased circulation Nogo‐B levels in patients with hepatic cirrhosis (Men et al., 2015) implies that circulating Nogo‐B might also be involved in ANIT‐induced intrahepatic cholestasis and rosiglitazone prevention. We initially observed that Nogo‐B was the only Nogo family member detected in mouse serum by Western blot (Figure S1B). However, due to the lower sensitivity of Western blot than ELISA and the limited information on secretion of Nogo family members, we have referred to the ELISA results as Nogo in this study. Interestingly, associated with ANIT‐induced intrahepatic cholestasis, serum Nogo levels were significantly increased, and this increase was substantially reduced by rosiglitazone treatment (Figure 1g).
3.2. PPARγ expression is essential for the prevention, by rosiglitazone, of intrahepatic cholestasis
To further investigate the anti‐cholestatic functions and mechanisms of rosiglitazone, we conducted in vitro studies with HepG2 cells (a human hepatic cell line). As shown in Figure 2a, rosiglitazone induced expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR protein in a concentration‐ or time‐dependent manner.
Figure 2.
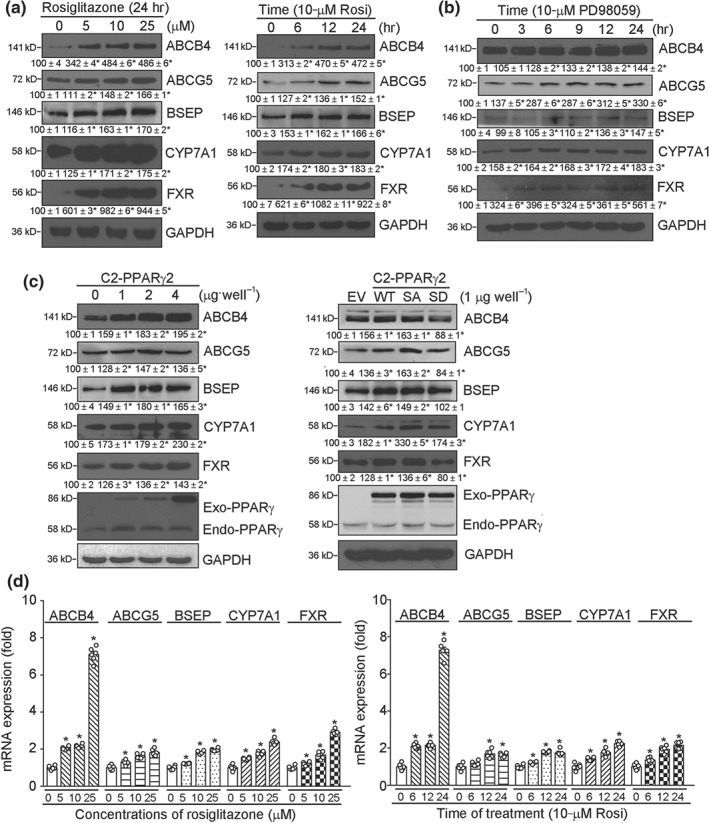
PPARγ activates expression of bile homeostatic proteins in HepG2 cells. (a, b) HepG2 cells were treated with rosiglitazone (Rosi) at the indicated concentrations for 24 hr or at 10 μmol·L−1 for the indicated times (a); PD98059 at 10 μmol·L−1 for the indicated times (b); (c) HepG2 cells in six‐well plates were transfected with wild‐type PPARγ expression vector (C2‐PPARγ) at the indicated concentrations (left panel); or expression vector of wild‐type PPARγ (WT), non‐phosphorylated PPARγ mutant (C2‐PPARγ2‐S112A or SA) or constitutively phosphorylated PPARγ mutant (C2‐PPARγ2‐S112D or SD; right panel) for 6 hr. Cells were switched into complete medium and cultured for another 24 hr, followed by assy for expression of ABCB4, ABCG5, BSEP, CYP7A1, FXR, exogenous PPARγ (Exo‐PPARγ), and endogenous PPARγ (Endo‐PPARγ) protein. * P < .05, significantly different from control, n = 6; (d) HepG2 cells in serum‐free medium were treated with rosiglitazone at the indicated concentrations for 16 hr (left panel) or with 10 μmol·L−1 rosiglitazone (Rosi) for the indicated times (right panel). Expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR mRNA was determined by qRT‐PCR, n = 6. * P < .05, significantly different from corresponding control group
Dephosphorylation of Ser112 in PPARγ protein by MEK1/2 inhibition, using https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5241, can also enhance its transcriptional activity (Han et al., 2000). In our model, PD98059 induced expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR protein in a time‐dependent manner (Figure 2b). In addition, high expressing normal PPARγ (C2‐PPARγ) and non‐p‐PPARγ (γS112A or SA) mutant, but not constitutive p‐PPARγ (γS112D or SD) mutant, increased expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR (Figure 2c). Associated with increased protein levels, expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR mRNA was activated by rosiglitazone in a concentration‐ or time‐dependent manner (Figure 2d).
In vivo, PPARγfl/fl and hepatocyte PPARγ KO mice were treated with ANIT, rosiglitazone, or rosiglitazone plus ANIT using the same schedule as in Figure 1a. Similar to wild‐type mice, ANIT induced severe intrahepatic cholestasis in PPARγfl/fl mice, such as blurry, brown coloured, saturated serum and necrosis in the liver. Rosiglitazone protected PPARγfl/fl mice from the ANIT‐induced damage (upper half, Figure 3a,b). In contrast, lack of PPARγ expression in hepatocytes blocked the protection by rosiglitazone (lower half, Figure 3a,b).
Figure 3.
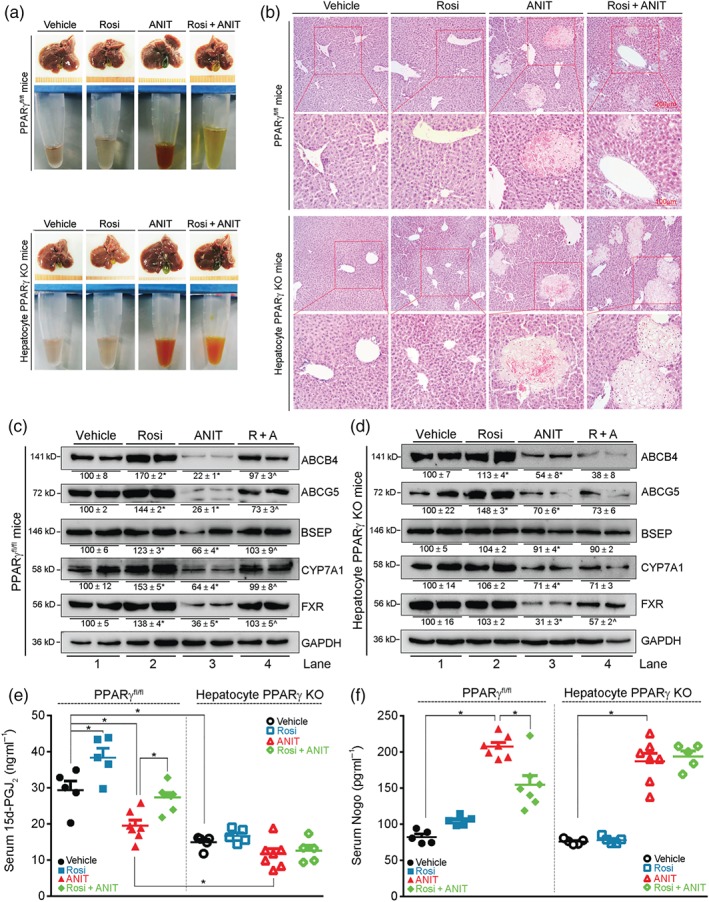
PPARγ expression is essential for rosiglitazone to protect against intrahepatic cholestasis. (a, b) Floxed PPARγ (PPARγfl/fl) and hepatocyte‐specific PPARγ knockout (hepatocyte PPARγ KO) mice received the same treatment for C57BL/6J mice, as indicated in Figure 1a. Liver and serum samples from each group were photographed (a) and (b) liver paraffin sections were stained with H&E. (c, d) expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR protein was determined in total proteins extracted from PPARγfl/fl or hepatocyte PPARγ KO mouse liver. * P < .05: Lane 2 or 3 significantly different from Lane 1; ^ P < .05: Lane 4 significantly different from Lane 3, n = 6. (e, f) Levels of 15d‐PGJ2 (e) and Nogo (f) were determined in serum samples using ELISA assay kits. n = 5 for Vehicle and Rosi groups, n = 7 for ANIT groups and ANIT + Rosi in PPARγfl/fl mice, n = 5 for ANIT + Rosi in hepatocyte PPARγ KO mice. * P < .05, significantly different as indicated
At the molecular level, ANIT decreased expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR in PPARγfl/fl mouse liver. Rosiglitazone alone activated expression of these proteins and substantially reversed the ANIT‐inhibited expression (Figure 3c). In hepatocyte PPARγ KO mice, rosiglitazone had little effect on ANIT‐inhibited expression of bile homeostatic molecules (Figure 3d).
As observed in wild‐type mice, rosiglitazone increased but ANIT reduced serum 15d‐PGJ2 in PPARγfl/fl mice, while the ANIT‐induced reduction of 15d‐PGJ2 was blocked by rosiglitazone (left half, Figure 3e). In contrast, lack of hepatocyte PPARγ expression not only reduced basal 15d‐PGJ2 levels but also blocked the effect of rosiglitazone on recovery of 15d‐PGJ2 levels after ANIT (right half, Figure 3e). ANIT increased circulating Nogo in both PPARγfl/fl and hepatocyte PPARγ KO mice with a greater effect in PPARγfl/fl mice (Figure 3f). Expression of Nogo‐B was induced by ANIT equally in PPARγfl/fl and hepatocyte PPARγ KO mouse liver (Figure S3). Although rosiglitazone alone had little effect on serum Nogo levels in both PPARγfl/fl and hepatocyte PPARγ KO mice, it substantially reduced the induction of serum Nogo by ANIT in PPARγfl/fl, but not in hepatocyte PPARγ KO mice (Figure 3f).
3.3. Lack of Nogo‐B expression enhances expression of bile homeostatic molecules
The association between circulating Nogo and intrahepatic cholestasis implies inhibition of Nogo‐B in the liver may reduce the disease by activating expression of proteins involved in bile homeostasis. In vitro, lack of Nogo‐B expression in HepG2 cells activated expression of ABCB4, ABCG5, BSEP, CYP7A1, and FXR (Figure 4a,b [Lane 4 vs. 1], c [Lane 5 vs. 1]). In vivo, Nogo‐B deficiency increased expression of these proteins in mouse liver (Figure 4d). In addition, PPARγ expression was increased in both Nogo‐B KO cells and mouse liver (Figure S4).
Figure 4.
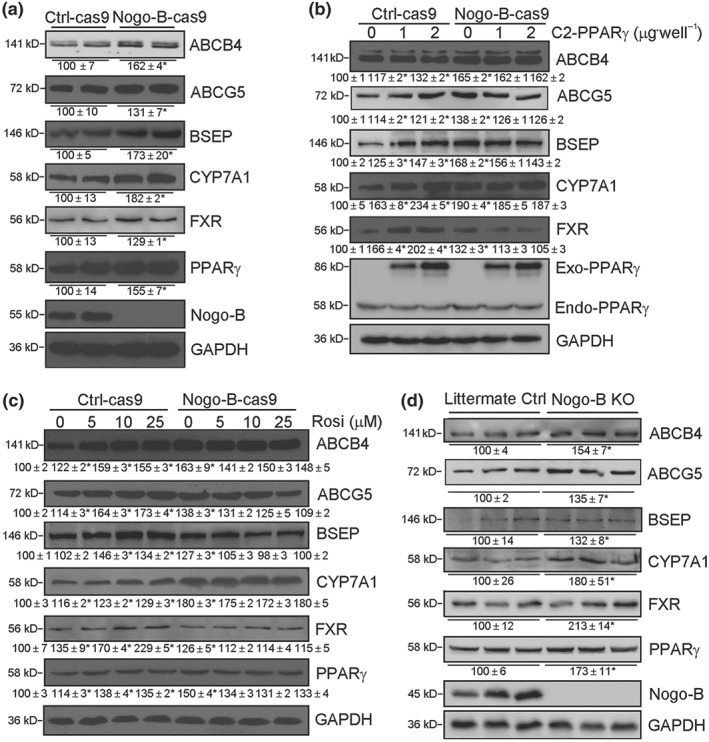
Nogo‐B deficiency activates expression of bile homeostatic proteins and PPARγ. (a) Nogo‐B knockout (Nogo‐B‐cas9) and the corresponding control (Ctrl‐cas9) HepG2 cells; (b, c) Ctrl‐cas9 and Nogo‐B‐cas9 cells in six‐well plates were transfected with PPARγ expression vector at the indicated concentrations for 24 hr (b) or treated with rosiglitazone (c) at the indicated concentrations for 16 hr; (d) littermate control or Nogo‐B KO mouse liver. Total proteins extracted from above samples were used to determine expression of ABCB4, ABCG5, BSEP, CYP7A1, FXR, Nogo‐B, and endogenous (Endo)/exogenous (Exo) PPARγ protein. * P < .05, significantly different from control, n = 6
High expression of PPARγ or rosiglitazone treatment induced expression of bile homeostatic proteins in Ctrl‐cas 9 cells (left half, Figure 4b,c) and normal HepG2 cells (Figure 2), but neither of these conditions were able to induce expression of these proteins in Nogo‐B deficient cells (right half, Figure 4b,c). Our data show that Nogo‐B deficiency increased expression of bile homeostatic proteins at basal levels while blocking the induction of these proteins by PPARγ activation.
3.4. Nogo‐B deficient mice are resistant to ANIT‐induced intrahepatic cholestasis
Next, we tested the effects of Nogo‐B deficiency on the resistance of mice to ANIT‐induced intrahepatic cholestasis. Both littermate and Nogo‐B mice were treated with ANIT or ANIT plus rosiglitazone. As shown in Figure 5a, ANIT induced intrahepatic cholestasis in littermate control mice, which was characterized by severe damage to liver and obvious changes in serum colour. Co‐treatment of mice with rosiglitazone protected littermate control mice from ANIT‐induced damage (left panel, Figure 5a). In contrast, ANIT alone caused much less damage to liver and little change in serum colour, in Nogo‐B KO mice. Meanwhile, the protective effects of rosiglitazone Against ANIT induced damage were much less, in Nogo‐B KO mice (right panel, Figure 5a).
Figure 5.
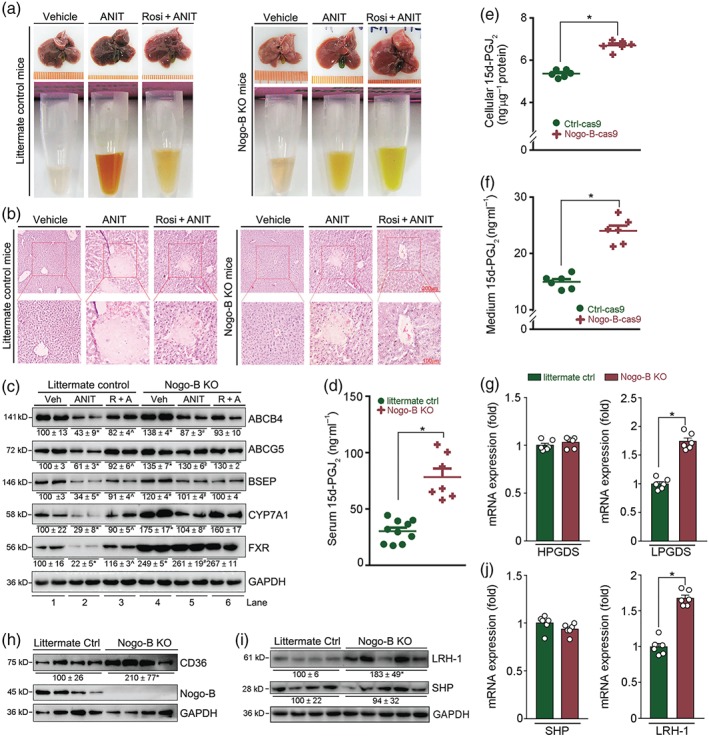
Nogo‐B deficiency protects mice against ANIT‐induced intrahepatic cholestasis by activating 15d‐PGJ2 production. (a, b) Nogo‐B KO or littermate control mice were randomly divided into three groups and given corn oil (Vehicle) by intragastric administration, ANIT (80 mg·kg−1) or ANIT plus rosiglitazone (Rosi, 30 mg·day−1·kg−1), as shown in Figure 1a. After 48 hr of ANIT treatment, mouse tissue samples were collected. Liver and serum samples were photographed (a), and (b) liver paraffin sections stained with H&E. (c) Expression of proteins (ABCB4, ABCG5, BSEP, CYP7A1, FXR, and PPARγ) extracted from littermate and Nogo‐B KO mouse liver. * P < .05, Lane 2 or 4 * P < .05, significantly different from Lane 1; ^,# P < .05, Lane 3 or 5 significantly different from Lane 2. (d) Levels of 15d‐PGJ2 in serum samples from littermate controls and Nogo‐B KO mice; all are males, 18 ± 1 g, 8 ± 0.5 weeks, n = 10 for control mice, n = 7 for Nogo‐B KO mice. * P < .05, significantly different as indicated. (e, f) Both cellular lysate and 24‐hr serum‐free conditioned medium were collected from Ctrl‐cas9 and Nogo‐B‐cas9 cells at the same density (~80% confluence). Levels of 15d‐PGJ2 in cellular lysate (e), and conditioned medium (f) were determined using an ELISA assay kit. Cellular protein content was determined and used to normalize cellular 15d‐PGJ2 content (ng·μg−1 protein, f). * P < .05, significantly different as indicated; (n = 6). (g–j) Total cellular proteins or RNA were extracted from liver samples of littermate control and Nogo‐B KO mice. Expression of haematopoietic PGDS (HPGDS) and lipocalin PGDS (LPGDS) mRNA (g), CD36 protein (h), LRH‐1, and SHP protein (i) or mRNA (j) was determined by Western blot or qRT‐PCR. * P < .05, significantly different as indicated; (n = 6)
The results of H&E staining confirmed the protective effects of Nogo‐B KO against ANIT‐induced intrahepatic cholestasis. ANIT caused severe necrosis in littermate control mouse liver, which was substantially reduced by rosiglitazone (left panel, Figure 5b). In contrast, necrosis caused by ANIT was significantly reduced, while the effect of rosiglitazone on liver necrosis was attenuated in Nogo‐B KO mice (right panel, Figure 5b).
Similar to wild‐type mice, Table 3 shows ANIT increased ALT, AST, ALP, γ‐GT, and TBIL in serum, and liver TBA, but decreased intestinal TBA or the sum of liver and intestine TBA, in littermate control mice. Dyslipidemia was also observed in littermate control mice after ANIT treatment. However, rosiglitazone clearly ameliorated these effects of intrahepatic cholestasis. For instance, TBA levels in both liver and intestine were substantially restored to normal.
Table 3.
Nogo‐B deficiency protects mice against intrahepatic cholestasis: Biochemical assay
| Parameters | Control mice | Nogo‐B KO mice | |||||
|---|---|---|---|---|---|---|---|
| Vehicle | ANIT | ANIT + Rosi | Vehicle | ANIT | ANIT + Rosi | ||
| LW/BW (%) | 4.62 ± 0.04 | 5.89 ± 0.04¶ | 4.47 ± 0.05§ | 4.60 ± 0.09 | 5.44 ± 0.20 | 5.32 ± 0.13 | |
| Serum enzyme activity, total bilirubin, and lipids | ALT | 38.6 ± 4.6 | 1,930 ± 177¶ | 304 ± 39§ | 26.0 ± 2.7 | 837 ± 122§ | 566 ± 103 |
| AST | 143 ± 8 | 5,815 ± 390¶ | 951 ± 188§ | 90 ± 8.4 | 661 ± 84§ | 415 ± 81 | |
| ALP | 136 ± 7 | 361 ± 26¶ | 243 ± 16§ | 172 ± 13 | 245 ± 24§ | 287 ± 20 | |
| γ‐GT | 1.02 ± 0.07 | 17.91 ± 1.89¶ | 7.78 ± 1.20§ | 2.55 ± 0.34 | 6.00 ± 0.88§ | 5.76 ± 1.07 | |
| TBIL | 0.28 ± 0.04 | 18.13 ± 1.66¶ | 3.58 ± 0.36§ | 0.16 ± 0.02 | 9.84 ± 2.23§ | 9.06 ± 1.04 | |
| T‐CHO | 2.68 ± 0.17 | 6.11 ± 0.1 0¶ | 4.71 ± 0.15§ | 2.80 ± 0.04 | 2.60 ± 0.21§ | 2.61 ± 0.21 | |
| LDL‐C | 0.43 ± 0.04 | 2.08 ± 0.06¶ | 0.50 ± 0.04§ | 0.29 ± 0.03 | 0.38 ± 0.09§ | 0.26 ± 0.03 | |
| HDL‐C | 1.99 ± 0.11 | 1.46 ± 0.07¶ | 2.70 ± 0.08§ | 1.83 ± 0.03 | 1.85 ± 0.18 | 1.65 ± 0.07 | |
| TG | 0.65 ± 0.04 | 2.96 ± 0.06¶ | 0.95 ± 0.05§ | 0.77 ± 0.05 | 1.04 ± 0.17§ | 1.09 ± 0.03 | |
| Liver TBA | 0.92 ± 0.07 | 2.16 ± 0.18¶ | 1.30 ± 0.11§ | 0.66 ± 0.05 | 0.71 ± 0.11§ | 0.94 ± 0.02 | |
| Intestine TBA | 12.07 ± 0.81 | 7.94 ± 0.35¶ | 13.13 ± 0.73§ | 11.83 ± 0.48 | 13.47 ± 1.05§ | 11.38 ± 0.88 | |
Note. Littermate control or Nogo‐B KO mice received vehicle (corn oil), α‐naphthylisothiocyanate (ANIT), or α‐naphthylisothiocyanate plus rosiglitazone (ANIT + Rosi) treatment. After treatment, mouse serum, liver, and intestine were used to complete the same assays as described in Table 2.
P < .05, significantly different from Ctrl (Vehicle).
P < .05, significantly different from ANIT‐treated control mice (n = 6).
Interestingly, compared with littermate control mice, ANIT increased serum intrahepatic cholestatic parameters to a much lower extent in Nogo‐B KO mice (Table 3). For instance, γ‐GT was increased ~18‐fold in littermate control but ~2.4‐fold in Nogo‐B KO mice. Liver or intestine TBA and the sum of liver and intestine TBA were not affected by ANIT in Nogo‐B KO mice and serum cholesterol levels were not changed either. In addition, rosiglitazone had no effect on serum parameters in ANIT‐treated Nogo‐B KO mice (Table 3).
As shown in Figure 5c, expression of bile homeostatic proteins in littermate control mouse liver was inhibited by ANIT (Lane 2 vs. 1), and the inhibition was reversed by rosiglitazone (Lane 3 vs. 2). Compared with littermate control mice, basal expression of ABCB4, ABCG5, BSEP, and FXR in Nogo‐B KO mouse liver was clearly increased (Lane 4 vs. 1). More importantly, the effects of ANIT on expression of these proteins were much weaker than those in littermate control mice (Lane 5 vs. 2). For example, FXR expression in Nogo‐B KO mouse liver was not affected by ANIT. Meanwhile, the effects of rosiglitazone on expression of these proteins in Nogo‐B KO mice were mild, except for CYP7A1. Our data would suggest that Nogo‐B deficiency enhances resistance of mice to ANIT‐induced intrahepatic cholestasis and cholestasis‐associated dyslipidemia.
3.5. Nogo‐B deficiency activates 15d‐PGJ2 production
Our results above suggested 15d‐PGJ2 might be an endogenous inhibitor of intrahepatic cholestasis and that its production could be increased by Nogo‐B deficiency. Indeed, serum 15d‐PGJ2 levels in Nogo‐B KO mice were much higher than in littermate control mice (Figure 5d), even to a level much higher than that induced by rosiglitazone in wild‐type mice (Figures 1f and 3e). Furthermore, both 15d‐PGJ2 production and secretion were significantly increased by Nogo‐B deficiency in HepG2 cells (Figure 5e,f). Haematopoietic and/or lipocalin PGD2 synthases (HPGDS and LPGDS) play an important role in cellular 15d‐PGJ2 production (Seo & Oh, 2017). At the mRNA levels, Figure 5g shows that Nogo‐B deficiency activated LPGDS expression in mouse liver, while having little effect on expression of HPGDS. Accompanying the enhanced 15d‐PGJ2 production, expression of CD36, a well‐known PPARγ target (Nagy et al., 1998), was also higher in Nogo‐B KO mouse liver (Figure 5h), which further supports PPARγ activation by Nogo‐B deficiency.
3.6. Reduces 15d‐PGJ2 and increased Nogo in serum are associated with PBC in patients
PBC is a major type of intrahepatic cholestatic liver disease that occurs more frequently in women (Kaplan & Gershwin, 2005). To assess the relevance of our experimental results to clinical disease, we collected serum samples from female PBC patients and corresponding non‐PBC controls. As observed in our animal model, serum 15d‐PGJ2 in PBC patients was reduced by about 40% (Figure 6a). Meanwhile, circulating Nogo was more than doubled (Figure 6b). The classical biomarkers for PBC diagnosis, γ‐GT (ALP and TBIL] were higher in PBC patients than non‐PBC controls (Figure 6c). The biomarkers for general liver injury, ALT and AST were increased while albumin was reduced in PBC patients (Figure 6d). HDL‐C was substantially decreased in PBC patients, indicating the disordered cholesterol metabolism that is associated with the disease (Figure 6e).
Figure 6.
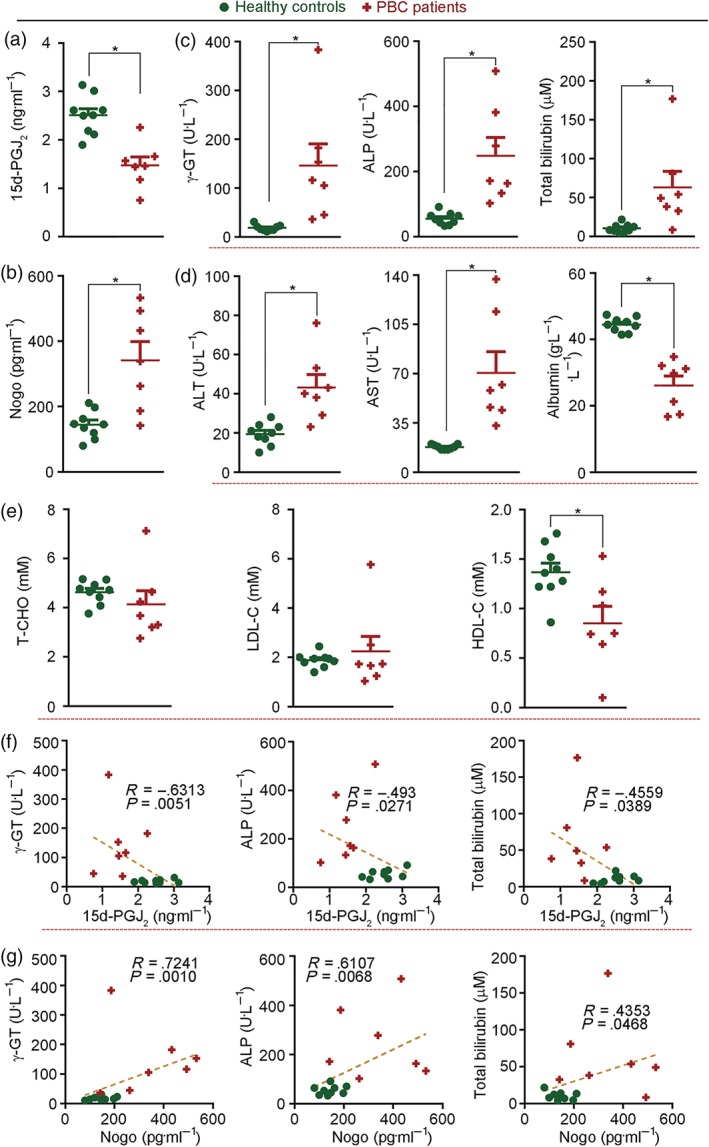
Decreased levels of 15d‐PGJ2 and increased levels of Nogo in serum are associated with PBC in patients. Serum samples were collected from PBC patients and non‐PBC controls and the following components were determined: (a) 15d‐PGJ2; (b) Nogo; (c) classical PBC biomarkers of γ‐GT, ALP, and TBIL; (d) General biomarkers of liver injury (ALT, AST, and albumin). (e) Cholesterol levels. n = 9 for healthy controls, n = 7 for PBC patients; (f, g) The correlation coefficient (R) between classical PBC biomarkers (γ‐GT, ALP, and TBIL) and 15d‐PGJ2 or Nogo was analysed using Spearman correlation test with 95% confidence interval; all correlations were significant, P < .05
We further analysed the correlation between 15d‐PGJ2 or Nogo and classical cholestatic biomarker, γ‐GT, ALP, or TBIL. Figure 6f shows that 15d‐PGJ2 was negatively correlated to γ‐GT, ALP, and TBIL. Meanwhile, Nogo was positively correlated to γ‐GT, ALP, and TBIL (Figure 6g). These data suggest reduced 15d‐PGJ2 and decreased Nogo in serum were also present in PBC patients.
3.7. Rosiglitazone reduces established intrahepatic cholestasis in mice
To determine if rosiglitazone can reduce established intrahepatic cholestasis, C57BL/6J mice were pretreated with ANIT for 1 day to induce the condition and then treated with rosiglitazone for 6 days (Figure 7a). As observed in our prevention study, the ANIT‐induced blurry serum with a light‐yellow colour was corrected by rosiglitazone treatment (Figure 7b). Also the ANIT‐caused necrosis in the liver was clearly blocked by rosiglitazone (Figure 7c).
Similarly, ANIT inhibited expression of the bile homeostatic proteins (CYP7A1, ABCB4, ABCG5, BSEP, and FXR) in mouse liver, and this inhibition was reversed by rosiglitazone (Figure 7d). Furthermore, the ANIT‐induced abnormal serum biomarkers, such as ALT, AST, γ‐GT, and TBIL were significantly corrected by rosiglitazone treatment (Figure 7e). Rosiglitazone also corrected ANIT‐induced cholestasis‐associated dyslipidemia (Figure 7f) and restored mouse liver size to normal (Figure 7g). Taken together, our data showed that rosiglitazone was able to prevent and also to reverse intrahepatic cholestasis induced by ANIT.
4. DISCUSSION
Until the recent approval of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435, UDCA was the only approved treatment for patients with intrahepatic cholestatic liver disease (Lindor et al., 2009; Poupon, Poupon, & Balkau, 1994). UDCA has multiple anti‐cholestatic effects, such as neutralization of toxic bile acids, activation of bile secretion, and anti‐apoptosis/inflammation (Beuers et al., 2015). Agonists of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=593, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7186, reduce serum ALP and γ‐GT and enhance ABCB4 expression and biliary phosphatidylcholine secretion (Ghonem et al., 2014; Ghonem, Assis, Boyer, 2015). Fenofibrate has been used as an adjuvant medicine to UDCA for intrahepatic cholestasis treatment (Kita et al., 2006). However, several adverse effects of PPARγ ligands on the patients including increased creatinine kinase, nausea, and heartburn have been observed (Ghonem, Assis, Boyer, 2015).
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2693, a synthetic PPARγ ligand used to treat type 2 diabetes, was withdrawn from the market because of severe side‐effects, particularly inducing intrahepatic cholestasis by increasing serum bile salt concentrations and inhibiting BSEP expression in rat liver (Funk, Pantze, et al., 2001; Funk, Ponelle, Scheuermann, & Pantze, 2001). The cholestatic potential of troglitazone may have decreased enthusiasm for investigating other PPARγ ligands as treatments for intrahepatic cholestasis. However, in this study, we have shown that rosiglitazone can potently alleviate intrahepatic cholestasis. Either pretreatment or post‐treatment of mice with rosiglitazone reduced ANIT‐induced liver damage and corrected ANIT‐induced abnormal serum biochemical parameters and enterohepatic circulation of bile (Figures 1b,c and 7c–g; Table 2). The hepatic expression of proteins involved in bile homeostasis, inhibited by ANTI, was substantially restored by rosiglitazone (Figures 1d and 7d). We further demonstrated the essential role of PPARγ in the alleviation of intrahepatic cholestasis by rosiglitazone‐ (Figure 3).
Nogo‐B deficiency in mice increased basal expression of bile homeostatic proteins in liver (Figure 4d) and increased resistance to ANIT‐induced intrahepatic cholestasis (Figure 5a,b; Table 3), which was mainly attributed to potent activation of 15d‐PGJ2 production (Figure 5d). Activation of 15d‐PGJ2 production by rosiglitazone and Nogo‐B deficiency was linked to PPARγ activation, suggesting that the enzyme(s) responsible for 15d‐PGJ2 production could be a PPARγ target. Indeed, we conducted a sequence alignment assay and found three putative PPARγ responsive elements (PPREs) in the proximal region of the LPGDS promoter. Therefore, LPGDS expression may be activated at a transcriptional level. Our findings also suggested that endogenous 15d‐PGJ2 might be an endogenous inhibitor of intrahepatic cholestasis. Consistent with this possibility, we found decreased serum levels of 15d‐PGJ2 in PBC patients (Figure 6a) and cholestatic mice. Furthermore, the reduction of ANIT‐induced intrahepatic cholestasis by rosiglitazone‐ was associated with corresponding changes in serum levels of 15d‐PGJ2 (Figures 1f and 3e).
Hypercholesterolemia is commonly found in cholestatic patients (Dann et al., 2006; Gylling, Farkkila, Vuoristo, & Miettinen, 1995) and treatment with UDCA has little effect on this disorder while https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435 exacerbates hypercholesterolemia (Neuschwander‐Tetri et al., 2015; Nevens et al., 2016). Although it is unclear if this cholestasis‐associated hypercholesterolemia can increase risk of coronary heart disease (Allocca et al., 2006; Crippin et al., 1992; Longo et al., 2002; Van Dam & Gips, 1997), a combined therapy of UDCA and statin may have to be applied, as clinical trials have demonstrated that statins do not affect liver function in patients (Ritzel et al., 2002; Stojakovic et al., 2007). In this study, we found hypercholesterolemia in both PBC patients (Figure 6e) and the mouse model (Tables 2 and 3, Figure 7f). However, rosiglitazone treatment potently ameliorated lipid profiles (Tables 2 and 3, Figure 7), while Nogo‐B deficiency blocked cholestasis‐associated dyslipidemia (Table 3). Activation of CYP7A1, the critical protein for bile acid synthesis and cholesterol metabolism, by PPARγ or Nogo‐B deficiency (Figures 1d, 2, 3c, 4, and 5c) should be an important mechanism for alleviation of cholestasis‐associated dyslipidemia. It also suggests that PPARγ ligands can be more effective than UDCA or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3435 for intrahepatic cholestasis treatment, as neither of these compounds ameliorates cholestasis‐associated hypercholesterolemia.
FXR inhibits CYP7A1 expression to reduce bile acid synthesis. FXR initially induces SHP expression and then reduces https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=633, a transcription factor activating CYP7A1 expression (Goodwin et al., 2000; Lu et al., 2000). We found rosiglitazone or Nogo‐B deficiency induced FXR and CYP7A1 expression simultaneously (Figures 1d, 2, 3c, 4, 5c, and 7d), indicating the minor role of FXR in the alleviation of intrahepatic cholestasis in this study. We further observed that Nogo‐B deficiency slightly affected SHP expression but potently activated liver receptor homolog‐1 expression (Figure 5i,j).
Increased circulating Nogo‐B has been reported in several different disease states (Gao et al., 2015; Men et al., 2015; Sutendra et al., 2011), whereas Nogo‐B deficiency is known to ameliorate liver fibrosis and portal hypertension by inactivating the TGFβ signalling pathway (Tashiro, Satoh, Utsumi, Chung, & Iwakiri, 2013; Zhang et al., 2011) and to reduce alcoholic liver disease by regulating Kupffer cell polarization (Park et al., 2017). In this study, we found that Nogo‐B might facilitate intrahepatic cholestasis. Furthermore, our study suggests that both 15d‐PGJ2 and Nogo‐B are important biomarkers for intrahepatic cholestasis. Synthetic PPARγ ligands might provide a novel approach to the treatment of intrahepatic cholestasis and cholestasis‐associated hypercholesterolemia (Figure S5).
AUTHOR CONTRIBUTIONS
S.Z., M.Y., F.G., X.Y., Y.C., and C.M. performed in vivo experiments. Q.L., Z.W., X.L., Q.R.M., Y. Zhu, and W.H. completed molecular biological experiments and biochemical assays. H.W., H.H., Y. Zhang, and D.K. completed collection of human serum samples and determination of biochemical parameters in human samples. J.H. and Y.D. designed experiments, interpreted results, and wrote the manuscript. D.P.H. edited the manuscript.
CONFLICT OF INTEREST
All the authors state no conflict of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://doi.org/10.1111/bph.14207, https://doi.org/10.1111/bph.14208, and https://doi.org/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Expression of Nogo family members in HepG2 cells, human and mouse serum A: total cellular proteins were extracted from Ctrl‐cas9 and Nogo‐B‐cas9 HepG2 cells. Expression of Nogo family members in HepG2 cells and human serum was determined by Western blot; B: serum samples were colleted from littermate control and Nogo‐B KO mice, followed by determination of Nogo family member protein expression by Western blot. Both Nogo‐B2 and Nogo‐B can be determined in HepG2 cells, while only Nogo‐B was detectable in both human and mouse serum.
Figure S2. Rosiglitazone induces expression of molecules for bile homeostasis in mouse liver.
Total proteins were extracted from a piece of mouse liver after treatment as indicated in Figure 1A. Expression of ABCB4, ABCG5, BSEP, CYP7A1 and FXR protein was determined by Western blot with quantification of band density.*p<0.05 vs. Vehicle, n=6.
Figure S3. ANIT induces Nogo‐B expression in PPARγfl/fl and hepatocyte PPARγ KO mouse liver.
Total proteins were extracted from a piece of liver of PPARγfl/fl and hepatocyte PPARγ KO mice after ANIT treatment as indicated in Figure 3A. Expression of Nogo‐B in the liver was determined by Western blot with quantification of band density.*,^p<0.05 vs. Vehicle in corresponding type of mice, n=5 for Vehicle groups, n=7 for ANIT groups.
Figure S4. Nogo‐B deficiency induces PPARγ protein expression in HepG2 cells and mouse liver.
Total proteins were extracted from Ctrl‐cas9 andNogo‐B‐cas9 HepG2 cells, ora piece of liver of littermate control and Nogo‐B KO mice, followed by determination of PPARγ protein expression by Western blot with quantification of band density.*p<0.05 vs. Ctrl‐cas9 cells or littermate control mice, n=6.
Figure S5. The model depicts anti‐intrahepatic cholestasis properties of rosiglitazone and Nogo‐B deficiency.
In wildtype mice, ANIT induces intrahepatic cholestasis by inhibiting expression of molecules for bile homeostasis, which is associated with reduction of 15d‐PGJ2and increase of Nogo in circulation. Treatment of cholestatic mice with rosiglitazone activates PPARγ to correct ANIT‐inhibited hepatic expression of molecules for bile homeostasis and circulating 15d‐PGJ2and Nogo, thereby reducing intrahepatic cholestasis. In Nogo KO mice, lack of Nogo‐B expression in the liver enhances 15d‐PGJ2production to activate PPARγ, therefore, the basal expression of molecules for bile homeostasis is activated which results in that Nogo KO mice are resistant to intrahepatic cholestasis naturally.
ACKNOWLEDGEMENTS
Dr Miao Yu and Miss Shuang Zhang received the American Society of Biochemistry and Molecular Biology Graduate/Postdoctoral Travel Awards for presenting the different parts of this work at the “Experimental Biology 2016” and the “Experimental Biology 2018” in San Diego, California, April 2016 and April 2018 respectively. They made equal contributions to this work. The homozygous floxed (+/+) PPARγ mice for this study were kindly provided by Dr Shengzhong Duan from Shanghai Jiaotong University (Shanghai, China).
This work was supported by the International Science and Technology Cooperation Programs of China Grant 2017YFE0110100 to J.H., Y.D., Y.C., and X.Y.; the NSFC Grants 81722046 to Y.D.; 81773727 and 81973316 to J.H.; 31770863 to Y.C.; and 81803517 to X.Y.; and the Fundamental Research Funds for the Central Universities of China to Y.D., X.Y., and Y.C.
Zhang S, Yu M, Guo F, et al. Rosiglitazone alleviates intrahepatic cholestasis induced by α‐naphthylisothiocyanate in mice: The role of circulating 15‐deoxy‐Δ12,14‐PGJ2 and Nogo. Br J Pharmacol. 2020;177:1041–1060. 10.1111/bph.14886
Contributor Information
Jihong Han, Email: jihonghan2008@nankai.edu.cn.
Yajun Duan, Email: yduan@hfut.edu.cn.
REFERENCES
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear hormone receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocca, M. , Crosignani, A. , Gritti, A. , Ghilardi, G. , Gobatti, D. , Caruso, D. , … Battezzati, P. M. (2006). Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut, 55(12), 1795–1800. 10.1136/gut.2005.079814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuers, U. , Trauner, M. , Jansen, P. , & Poupon, R. (2015). New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. Journal of Hepatology, 62(1 Suppl), S25–S37. [DOI] [PubMed] [Google Scholar]
- Cantalupo, A. , Zhang, Y. , Kothiya, M. , Galvani, S. , Obinata, H. , Bucci, M. , … di Lorenzo, A. (2015). Nogo‐B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nature Medicine, 21(9), 1028–1037. 10.1038/nm.3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , & Zhang, F. (2015). Genome engineering using CRISPR‐Cas9 system. Methods in Molecular Biology, 1239, 197–217. [DOI] [PubMed] [Google Scholar]
- Crippin, J. S. , Lindor, K. D. , Jorgensen, R. , Kottke, B. A. , Harrison, J. M. , Murtaugh, P. A. , & Dickson, E. R. (1992). Hypercholesterolemia and atherosclerosis in primary biliary cirrhosis: What is the risk? Hepatology, 15(5), 858–862. 10.1002/hep.1840150518 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann, Kenyon, A. P. , Wierzbicki, A. S. , Seed, P. T. , Shennan, A. H. , & Tribe, R. M. (2006). Plasma lipid profiles of women with intrahepatic cholestasis of pregnancy. Obstetrics and Gynecology, 107(1), 106–114. [DOI] [PubMed] [Google Scholar]
- Forman, B. M. , Goode, E. , Chen, J. , Oro, A. E. , Bradley, D. J. , Perlmann, T. , … Weinberger, C. (1995). Identification of a nuclear receptor that is activated by farnesol metabolites. Cell, 81(5), 687–693. 10.1016/0092-8674(95)90530-8 [DOI] [PubMed] [Google Scholar]
- Funk, C. , Pantze, M. , Jehle, L. , Ponelle, C. , Scheuermann, G. , Lazendic, M. , & Gasser, R. (2001). Troglitazone‐induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology, 167(1), 83–98. 10.1016/s0300-483x(01)00460-7 [DOI] [PubMed] [Google Scholar]
- Funk, C. , Ponelle, C. , Scheuermann, G. , & Pantze, M. (2001). Cholestatic potential of troglitazone as a possible factor contributing to troglitazone‐induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Molecular Pharmacology, 59(3), 627–635. [PubMed] [Google Scholar]
- Gao, B. , Xu, Y. , Leng, J. , Wang, K. , Xia, B. , & Huang, J. (2015). Clinical implications of increased Nogo‐B levels in patients with acute coronary syndromes and stable angina pectoris. International Heart Journal, 56(3), 341–344. [DOI] [PubMed] [Google Scholar]
- Gao, L. , Utsumi, T. , Tashiro, K. , Liu, B. , Zhang, D. , Swenson, E. S. , & Iwakiri, Y. (2013). Reticulon 4B (Nogo‐B) facilitates hepatocyte proliferation and liver regeneration in mice. Hepatology, 57(5), 1992–2003. 10.1002/hep.26235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonem, N. S. , Ananthanarayanan, M. , Soroka, C. J. , & Boyer, J. L. (2014). Peroxisome proliferator‐activated receptor α activates human multidrug resistance transporter 3/ATP‐binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatology, 59(3), 1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghonem, N. S. , Assis, D. N. , & Boyer, J. L. (2015). Fibrates and cholestasis. Hepatology, 62(2), 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, B. , Jones, S. A. , Price, R. R. , Watson, M. A. , McKee, D. D. , Moore, L. B. , … Kliewer, S. A. (2000). A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Molecular Cell, 6(3), 517–526. 10.1016/S1097-2765(00)00051-4 [DOI] [PubMed] [Google Scholar]
- Gylling, H. , Farkkila, M. , Vuoristo, M. , & Miettinen, T. A. (1995). Metabolism of cholesterol and low‐ and high‐density lipoproteins in primary biliary cirrhosis: Cholesterol absorption and synthesis related to lipoprotein levels and their kinetics. Hepatology, 21(1), 89–95. [PubMed] [Google Scholar]
- Han, J. , Hajjar, D. P. , Tauras, J. M. , Feng, J. , Gotto, A. M. Jr. , & Nicholson, A. C. (2000). Transforming growth factor‐β1 (TGF‐β1) and TGF‐β2 decrease expression of CD36, the type B scavenger receptor, through mitogen‐activated protein kinase phosphorylation of peroxisome proliferator‐activated receptor‐γ. The Journal of Biological Chemistry, 275(2), 1241–1246. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijssennagger, N. , Janssen, A. W. F. , Milona, A. , Ramos Pittol, J. M. , Hollman, D. A. A. , Mokry, M. , … Kersten, S. (2016). Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. Journal of Hepatology, 64(5), 1158–1166. 10.1016/j.jhep.2016.01.016 [DOI] [PubMed] [Google Scholar]
- Inagaki, T. , Choi, M. , Moschetta, A. , Peng, L. , Cummins, C. L. , McDonald, J. G. , … Kliewer, S. A. (2005). Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metabolism, 2(4), 217–225. 10.1016/j.cmet.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Kaplan, M. M. , & Gershwin, M. E. (2005). Primary biliary cirrhosis. The New England Journal of Medicine, 353(12), 1261–1273. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita, R. , Takamatsu, S. , Kimura, T. , Kokuryu, H. , Osaki, Y. , & Tomono, N. (2006). Bezafibrate may attenuate biliary damage associated with chronic liver diseases accompanied by high serum biliary enzyme levels. Journal of Gastroenterology, 41(7), 686–692. [DOI] [PubMed] [Google Scholar]
- Kossor, D. C. , Goldstein, R. S. , Ngo, W. , DeNicola, D. B. , Leonard, T. B. , Dulik, D. M. , & Meunier, P. C. (1995). Biliary epithelial cell proliferation following α‐naphthylisothiocyanate (ANIT) treatment: Relationship to bile duct obstruction. Fundamental and Applied Toxicology, 26(1), 51–62. 10.1006/faat.1995.1074 [DOI] [PubMed] [Google Scholar]
- Kowdley, K. V. , Luketic, V. , Chapman, R. , Hirschfield, G. M. , Poupon, R. , Schramm, C. , … for the Obeticholic Acid PBC Monotherapy Study Group (2018). A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology, 67, 1890–1902. 10.1002/hep.29569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langheim, S. , Yu, L. , von Bergmann, K. , Lutjohann, D. , Xu, F. , Hobbs, H. H. , & Cohen, J. C. (2005). ABCG5 and ABCG8 require MDR2 for secretion of cholesterol into bile. Journal of Lipid Research, 46(8), 1732–1738. 10.1194/jlr.M500115-JLR200 [DOI] [PubMed] [Google Scholar]
- Li, T. , Matozel, M. , Boehme, S. , Kong, B. , Nilsson, L. M. , Guo, G. , … Chiang, J. Y. L. (2011). Overexpression of cholesterol 7α‐hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology, 53(3), 996–1006. 10.1002/hep.24107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor, K. D. , Gershwin, M. E. , Poupon, R. , Kaplan, M. , Bergasa, N. V. , Heathcote, E. J. , & American Association for Study of Liver Diseases (2009). Primary biliary cirrhosis. Hepatology, 50(1), 291–308. 10.1002/hep.22906 [DOI] [PubMed] [Google Scholar]
- Longo, M. , Crosignani, A. , Battezzati, P. M. , Squarcia Giussani, C. , Invernizzi, P. , Zuin, M. , & Podda, M. (2002). Hyperlipidaemic state and cardiovascular risk in primary biliary cirrhosis. Gut, 51(2), 265–269. 10.1136/gut.51.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, T. T. , Makishima, M. , Repa, J. J. , Schoonjans, K. , Kerr, T. A. , Auwerx, J. , & Mangelsdorf, D. J. (2000). Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Molecular Cell, 6(3), 507–515. 10.1016/S1097-2765(00)00050-2 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Men, R. , Wen, M. , Dan, X. , Zhu, Y. , Wang, W. , Li, J. , … Yang, L. (2015). Nogo‐B: A potential indicator for hepatic cirrhosis and regulator in hepatic stellate cell activation. Hepatology Research, 45(1), 113–122. 10.1111/hepr.12324 [DOI] [PubMed] [Google Scholar]
- Morita, S. Y. , Kobayashi, A. , Takanezawa, Y. , Kioka, N. , Handa, T. , Arai, H. , … Ueda, K. (2007). Bile salt‐dependent efflux of cellular phospholipids mediated by ATP binding cassette protein B4. Hepatology, 46(1), 188–199. 10.1002/hep.21591 [DOI] [PubMed] [Google Scholar]
- Musso, G. , Cassader, M. , Paschetta, E. , & Gambino, R. (2017). Pioglitazone for advanced fibrosis in nonalcoholic steatohepatitis: New evidence, new challenges. Hepatology, 65(3), 1058–1061. [DOI] [PubMed] [Google Scholar]
- Nagy, L. , Tontonoz, P. , Alvarez, J. G. , Chen, H. , & Evans, R. M. (1998). Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell, 93(2), 229–240. [DOI] [PubMed] [Google Scholar]
- Neuschwander‐Tetri, B. A. , Loomba, R. , Sanyal, A. J. , Lavine, J. E. , Van Natta, M. L. , Abdelmalek, M. F. , et al. (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo‐controlled trial. Lancet, 385(9972), 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevens, F. , Andreone, P. , Mazzella, G. , Strasser, S. I. , Bowlus, C. , Invernizzi, P. , … POISE Study Group (2016). A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. The New England Journal of Medicine, 375(7), 631–643. 10.1056/NEJMoa1509840 [DOI] [PubMed] [Google Scholar]
- Park, J. K. , Shao, M. , Kim, M. Y. , Baik, S. K. , Cho, M. Y. , Utsumi, T. , … Iwakiri, Y. (2017). An endoplasmic reticulum protein, Nogo‐B, facilitates alcoholic liver disease through regulation of kupffer cell polarization. Hepatology, 65(5), 1720–1734. 10.1002/hep.29051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupon, R. E. , Poupon, R. , & Balkau, B. (1994). Ursodiol for the long‐term treatment of primary biliary cirrhosis. The UDCA‐PBC Study Group. The New England Journal of Medicine, 330(19), 1342–1347. [DOI] [PubMed] [Google Scholar]
- Ritzel, U. , Leonhardt, U. , Nather, M. , Schafer, G. , Armstrong, V. W. , & Ramadori, G. (2002). Simvastatin in primary biliary cirrhosis: Effects on serum lipids and distinct disease markers. Journal of Hepatology, 36(4), 454–458. [DOI] [PubMed] [Google Scholar]
- Seo, M. J. , & Oh, D. K. (2017). Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Progress in Lipid Research, 66, 50–68. [DOI] [PubMed] [Google Scholar]
- Shaw, L. M. , Stromme, J. H. , London, J. L. , & Theodorsen, L. (1983). International Federation of Clinical Chemistry. Scientific Committee, Analytical Section. Expert Panel on Enzymes. IFCC methods for measurement of enzymes. Part 4. IFCC methods for γ‐glutamyltransferase [(γ‐glutamyl)‐peptide: amino acid γ‐glutamyltransferase, EC 2.3.2.2]. IFCC Document, Stage 2, Draft 2, 1983‐01 with a view to an IFCC Recommendation. Clinica Chimica Acta, 135(3), 315F–338F. [PubMed] [Google Scholar]
- Stojakovic, T. , Putz‐Bankuti, C. , Fauler, G. , Scharnagl, H. , Wagner, M. , Stadlbauer, V. , … Trauner, M. (2007). Atorvastatin in patients with primary biliary cirrhosis and incomplete biochemical response to ursodeoxycholic acid. Hepatology, 46(3), 776–784. 10.1002/hep.21741 [DOI] [PubMed] [Google Scholar]
- Strautnieks, S. S. , Bull, L. N. , Knisely, A. S. , Kocoshis, S. A. , Dahl, N. , Arnell, H. , … Thompson, R. J. (1998). A gene encoding a liver‐specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nature Genetics, 20(3), 233–238. 10.1038/3034 [DOI] [PubMed] [Google Scholar]
- Sutendra, G. , Dromparis, P. , Wright, P. , Bonnet, S. , Haromy, A. , Hao, Z. , … Michelakis, E. D. (2011). The role of Nogo and the mitochondria‐endoplasmic reticulum unit in pulmonary hypertension. Science Translational Medicine, 3(88), 88ra55 10.1126/scitranslmed.3002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro, K. , Satoh, A. , Utsumi, T. , Chung, C. , & Iwakiri, Y. (2013). Absence of Nogo‐B (reticulon 4B) facilitates hepatic stellate cell apoptosis and diminishes hepatic fibrosis in mice. The American Journal of Pathology, 182(3), 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam, G. M. , & Gips, C. H. (1997). Primary biliary cirrhosis in The Netherlands. An analysis of associated diseases, cardiovascular risk, and malignancies on the basis of mortality figures. Scandinavian Journal of Gastroenterology, 32(1), 77–83. [DOI] [PubMed] [Google Scholar]
- Yerushalmi, B. , Dahl, R. , Devereaux, M. W. , Gumpricht, E. , & Sokol, R. J. (2001). Bile acid‐induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology, 33(3), 616–626. [DOI] [PubMed] [Google Scholar]
- Yu, L. , Hammer, R. E. , Li‐Hawkins, J. , Von Bergmann, K. , Lutjohann, D. , Cohen, J. C. , & Hobbs, H. H. (2002). Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proceedings of the National Academy of Sciences of the United States of America, 99(25), 16237–16242. 10.1073/pnas.252582399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Li‐Hawkins, J. , Hammer, R. E. , Berge, K. E. , Horton, J. D. , Cohen, J. C. , & Hobbs, H. H. (2002). Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. The Journal of Clinical Investigation, 110(5), 671–680. 10.1172/JCI0216001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Utsumi, T. , Huang, H. C. , Gao, L. , Sangwung, P. , Chung, C. , … Iwakiri, Y. (2011). Reticulon 4B (Nogo‐B) is a novel regulator of hepatic fibrosis. Hepatology, 53(4), 1306–1315. 10.1002/hep.24200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Su, H. , Li, Y. , Fan, Y. , Wang, Q. , Jiang, J. , … Qiu, F. (2018). Different effects of ursodeoxycholic acid on intrahepatic cholestasis in acute and recovery stages induced by α‐naphthylisothiocyanate in mice. Toxicology and Applied Pharmacology, 342, 69–78. 10.1016/j.taap.2018.01.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of Nogo family members in HepG2 cells, human and mouse serum A: total cellular proteins were extracted from Ctrl‐cas9 and Nogo‐B‐cas9 HepG2 cells. Expression of Nogo family members in HepG2 cells and human serum was determined by Western blot; B: serum samples were colleted from littermate control and Nogo‐B KO mice, followed by determination of Nogo family member protein expression by Western blot. Both Nogo‐B2 and Nogo‐B can be determined in HepG2 cells, while only Nogo‐B was detectable in both human and mouse serum.
Figure S2. Rosiglitazone induces expression of molecules for bile homeostasis in mouse liver.
Total proteins were extracted from a piece of mouse liver after treatment as indicated in Figure 1A. Expression of ABCB4, ABCG5, BSEP, CYP7A1 and FXR protein was determined by Western blot with quantification of band density.*p<0.05 vs. Vehicle, n=6.
Figure S3. ANIT induces Nogo‐B expression in PPARγfl/fl and hepatocyte PPARγ KO mouse liver.
Total proteins were extracted from a piece of liver of PPARγfl/fl and hepatocyte PPARγ KO mice after ANIT treatment as indicated in Figure 3A. Expression of Nogo‐B in the liver was determined by Western blot with quantification of band density.*,^p<0.05 vs. Vehicle in corresponding type of mice, n=5 for Vehicle groups, n=7 for ANIT groups.
Figure S4. Nogo‐B deficiency induces PPARγ protein expression in HepG2 cells and mouse liver.
Total proteins were extracted from Ctrl‐cas9 andNogo‐B‐cas9 HepG2 cells, ora piece of liver of littermate control and Nogo‐B KO mice, followed by determination of PPARγ protein expression by Western blot with quantification of band density.*p<0.05 vs. Ctrl‐cas9 cells or littermate control mice, n=6.
Figure S5. The model depicts anti‐intrahepatic cholestasis properties of rosiglitazone and Nogo‐B deficiency.
In wildtype mice, ANIT induces intrahepatic cholestasis by inhibiting expression of molecules for bile homeostasis, which is associated with reduction of 15d‐PGJ2and increase of Nogo in circulation. Treatment of cholestatic mice with rosiglitazone activates PPARγ to correct ANIT‐inhibited hepatic expression of molecules for bile homeostasis and circulating 15d‐PGJ2and Nogo, thereby reducing intrahepatic cholestasis. In Nogo KO mice, lack of Nogo‐B expression in the liver enhances 15d‐PGJ2production to activate PPARγ, therefore, the basal expression of molecules for bile homeostasis is activated which results in that Nogo KO mice are resistant to intrahepatic cholestasis naturally.