

Abstract
Background and Purpose
Inhibition of the T‐ and B‐cell interaction through the CD40/CD40 ligand (L) axis is a favourable approach for inflammatory disease treatment. Clinical studies of anti‐CD40L molecules in autoimmune diseases have met challenges because of thromboembolic events and adverse haemostasis. VIB4920 (formerly MEDI4920) is a novel CD40L antagonist and Tn3 fusion protein designed to prevent adverse haemostasis and immunopharmacology. We evaluated the pharmacokinetics, activity and toxicity of VIB4920 in monkeys.
Experimental Approach
Cynomolgus monkeys received i.v. or s.c. 5–300 mg·kg−1 VIB4920 or vehicle, once weekly for 1 month (Studies 1 and 2) or 28 weeks (Study 3). VIB4920 exposure and bioavailability were determined using pharmacokinetic analyses, and immune cell population changes via flow cytometry. Pharmacological activity was evaluated by measuring the animals' capacity to elicit an immune response to keyhole limpet haemocyanin (KLH) and tetanus toxoid (TT).
Key Results
VIB4920 demonstrated linear pharmacokinetics at multiple doses. Lymphocyte, monocyte, cytotoxic T‐cell and NK cell counts were not significantly different between treatment groups. B‐cell counts reduced dose‐dependently and the T‐cell dependent antibody response to KLH was suppressed by VIB4920 dose‐dependently. The recall response to TT was similar across treatment groups. No thromboembolic events or symptoms of immune system dysfunctionality were observed.
Conclusions and Implications
VIB4920 demonstrated an acceptable safety profile in monkeys. VIB4920 showed favourable pharmacokinetics, dose‐dependent inhibition of a neoantigen‐specific immune response and no adverse effects on immune function following long‐term use. Our data support the use of VIB4920 in clinical trials.
Abbreviations
- ADA
antidrug antibody
- AUEC
area under the effect curve
- Cmax
maximum observed concentration
- Ctrough
trough concentration
- DPBS
Dulbecco's PBS
- FITC
fluorescein isothiocyanate
- H&E
haematoxylin and eosin
- HSA
human serum albumin
- IACUC
Institutional Animal Care and Use Committees
- KLH
keyhole limpet haemocyanin
- L
ligand
- LLOQ
lower limit of quantification
- MSD®
Meso Scale Discover
- PBMC
peripheral blood mononuclear cells
- PD
pharmacodynamics
- PFA‐100
platelet function screening
- PK
pharmacokinetic
- TAT
thrombin–antithrombin
- TDAR
T‐cell‐dependent antibody response
- TT
tetanus toxoid
- USDA
U.S. Department of Agriculture
What is already known
Anti‐CD40L antibodies caused thromboembolic events in clinical trials.
Thromboembolic events may result from mAb Fc‐mediated cross‐linking interactions with platelets expressing CD40L.
What does this study add
VIB4920 does not contain an Fc, and no thromboembolic events were observed in any of the cyno repeat‐dose studies.
VIB4920 effectively blocks the CD40L/CD40 interaction similar to the anti‐CD40L mAbs.
What is the clinical significance
Because there were no thromboembolic events associated with VIB4920‐mediated CD40L blockade, VIB4920 is proceeding in clinical studies.
1. INTRODUCTION
Autoimmune disease pathology is driven, in part, by plasma cells that produce autoantibodies and inflammatory cytokines. Antigen‐presenting cells, including B cells, dendritic cells, macrophages, and non‐haematopoietic stem cells, express the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1874, a costimulatory protein and member of the TNF receptor family (Schonbeck & Libby, 2001), while the ligand for this receptor, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5077, is primarily expressed on activated T cells under inflammatory conditions (Kawabe, Matsushima, Hashimoto, Imaizumi, & Hasegawa, 2011). The interaction of CD40 and CD40L promotes B‐cell activation, proliferation, differentiation, and antibody production, as well as T‐cell activation and inflammatory cytokine production (Kawabe et al., 2011). Given its critical role in the immune response, the CD40/CD40L costimulatory pathway provides a valuable target for the treatment of patients with autoimmune disease.
In mouse models for human systemic sclerosis (Komura et al., 2008), multiple sclerosis (Howard, Dal Canto, & Miller, 2002; Howard, Neville, Haynes, Dal Canto, & Miller, 2003), chronic colitis (De Jong et al., 2000), and Sjogren's syndrome (Mahmoud et al., 2016), blockade of the CD40/CD40L pathway using an anti‐CD40L molecule reduced clinical disease symptoms. However, despite these promising results, clinical utility of anti‐CD40L molecules has proven challenging because of adverse haemostasis. Several studies were terminated early because of the incidence of clinically significant cardiovascular thromboembolic events (Boumpas et al., 2003; Huang et al., 2002). These thromboembolic events could be attributable to the platelet activation and aggregation caused by the interaction of the Fc region of the anti‐CD40L protein with the platelet receptor https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3017 and the antigen‐binding region of the anti‐CD40L protein with the platelet‐expressed CD40L (Mirabet, Barrabes, Quiroga, & Garcia‐Dorado, 2008; Robles‐Carrillo et al., 2010).
As the previously terminated studies featured anti‐CD40L molecules that contained intact Fc regions, it was suggested that utilising CD40L‐targeted molecules specifically engineered to lack the Fc region could prevent the occurrence of thromboembolic events. VIB4920 (formerly MEDI4920) is a CD40L antagonist that has been specifically designed without the Fc region and thus should avoid the risk of thromboembolic events without affecting the desired pharmacology. VIB4920 consists of two identical Tn3 modules, engineered forms of the Fn3 protein domain of human tenascin C, fused with polyglycine linkers to human serum albumin (HSA). Each Tn3 module is engineered to bind specifically to human CD40L (Oganesyan et al., 2013) and inhibits its interaction with the human CD40 receptor (Karnell et al., 2019). The t 1/2 of VIB4920 in humans is 7 days (Li, 2016, Albescu, 2016).
Preclinical pharmacology studies have demonstrated that VIB4920 binds specifically to human CD40L with high affinity. In both in vitro functional platelet assessment systems and in vivo, no platelet activation occurred following VIB4920 administration suggesting that VIB4920 does not induce platelet‐mediated thrombogenic activity in humans (Karnell et al., 2019). VIB4920 may act as an immune modulator and therefore has potential therapeutic activity for a range of autoimmune diseases through CD40/CD40L blockade, including rheumatoid arthritis, systemic lupus erythematosus, primary Sjögren's syndrome, and idiopathic thrombocytopenic purpura. Here, the toxicity, pharmacokinetics, and in vivo activity of VIB4920 in cynomolgus monkeys (Macaca fascicularis) has been evaluated to determine its safety for use in clinical trials.
2. METHODS
2.1. Test article and vehicle
The vehicle composition was 10‐mM sodium phosphate, 150‐mM sodium chloride, pH 7.0 (Study 1) or 10‐mM sodium phosphate, 250‐mM sucrose, 0.02% polysorbate 80, pH 7.4 (Studies 2 and 3). Vehicle solutions were stored between −50°C and − 90°C (Study 1) or between 2°C and 8°C (Studies 2 and 3), filtered through a 0.22‐μM filter unit and prepared for use on each individual day of dosing. VIB4920 (MedImmune/AstraZenec) was formulated in 10‐mM sodium phosphate, 150‐mM sodium chloride, pH 7.0 at 82.5 mg·ml−1 (Study 1) or 10‐mM sodium phosphate, 250‐mM sucrose, 0.02% polysorbate 80, pH 7.4 at a nominal concentration of 50 mg·ml−1 (Studies 2 and 3). Formulations were stored between −50°C and − 90°C and prepared for use on each day of dosing by serial dilution with the vehicle; the diluted formulations were stored between 2°C and 8°C when not in use.
2.2. Animal test system and husbandry
All study sites were accredited by the association for assessment and accreditation of laboratory animal care international, licensed by the United States Department of Agriculture (USDA), and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of each institution. all animal experiments were conducted in accordance with USDA Animal Welfare Act (9 CFR, parts 1, 2, and 3) and as described in the guide for the care and use of laboratory animals and were approved by the IACUC. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Purpose‐bred experimentally naive male and female cynomolgus monkeys were evaluated in these studies. The cynomolgus monkey was selected for preclinical safety studies as VIB4920 binds to and neutralises the pharmacological activity of human and cynomolgus monkey CD40L and does not cross‐react with murine CD40L.
In Study 1, animals were either pair‐housed or up to four animals were housed in four stainless steel cages with an outer extension that allowed them to move throughout. In Study 2, animals were either pair‐housed or housed in groups of three or four by sex. In Study 3, animals were pair‐housed socially and by sex. Animals were individually housed when required for individual data collection. For Studies 2 and 3, primary enclosures met criteria specified in the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes, U.S. Department of Agriculture Animal Welfare Act (9 CFR, Parts 1, 2, and 3) and the Guide for the Care and Use of Laboratory Animals (Academies, 2011).
Animals were kept under a 12‐hr light/12‐hr dark cycle, except when interrupted for designated procedures. Animals received a basal diet twice daily of Lab Diet® Certified Primate Diet #5048 (PMI Nutrition International), except during fasting periods. The diet was also supplemented with fruit or vegetables. Animals were socialised and provided with environmental enrichment throughout all studies. For procedures where animals were handled, sedation with ketamine was conducted when necessary.
All study animals were killed at their scheduled necropsy, or, if necessary, moribund animals were killed for humane reasons, thereby preventing the loss of tissues through autolysis. A routine necropsy was carried out on all killed animals. Animals were killed by administration of a euthanasia solution (under ketamine sedation if necessary) followed by exsanguination, according to the standard operating procedures of the testing facility.
2.3. Study design
The cynomolgus monkey was selected specifically for use in these studies as VIB4920 binds to and neutralises the pharmacological activity of human and cynomolgus monkey CD40L only. In Study 1, male cynomolgus monkeys were administered vehicle, i.v. bolus 5, 50, or 150 mg·kg−1 VIB4920, or s.c. 150 mg·kg−1 VIB4920, once weekly for 1 month, followed by a 20‐week recovery period (Table S1). The required number of animals on study was randomized into treatment groups using simple randomization. In Study 2, male and female cynomolgus monkeys received i.v. 50, 150, or 300 mg·kg−1 VIB4920 by slow infusion (30 min) or vehicle, once weekly for 1 month, followed by a 27‐week recovery period (Table S1). Animals were randomized by sex into treatment groups based on previous social grouping of three animals per group. In Study 3, male and female cynomolgus monkeys were administered i.v. and s.c. vehicle, or s.c. 125 or 250 mg·kg−1 VIB4920, or i.v. 150 or 300 mg·kg−1 VIB4920, once weekly for 28 weeks. Study 3 i.v. treatments were administered by 30‐min slow infusion. Animals then entered a 29‐week recovery period (Table S1). Previously established social pairs of animals were randomized and assigned to groups prior to transfer to study. Male and female pairs were randomized separately.
The s.c. and i.v. routes of administration were selected to reflect the intended delivery routes for human exposure. Dose levels were selected following human dose‐projection modelling and evaluation of data from previous pharmacokinetic (PK)/pharmacodynamics (PD) studies in cynomolgus monkeys.
For all studies, each animal was randomly assigned a number, which was recalled via a microchip or permanent chest tattoo. Data were recorded in the Provantis data collection system.
2.4. Pharmacokinetic analysis
The plasma concentration of VIB4920 was determined using a quantitative electrochemiluminescent assay. Serial blood samples were collected pre‐ and post‐dose at appropriate time points (Table S2) and processed to obtain platelet poor plasma. A Meso Scale Discover (MSD®) high binding plate coated with anti‐FLAG®‐tag antibody (Agilent, Santa Clara, CA, USA [Study 1]; Sigma Aldrich, St. Louis, MO, USA [Studies 2 and 3]) was blocked with 5% casein buffer. FLAG®‐labelled‐MEGACD40L (Enzo Life Sciences, Farmingdale, NY, USA) was then added to the plate to capture VIB4920 from the sample. Calibrators were prepared in 100% plasma and then diluted to 1:100 (Study 1) or 1% plasma in assay buffer (Studies 2 and 3). Calibrators prepared in undiluted matrix, quality controls, and unknown samples were diluted to a minimum of 1:100 prior to plate addition; calibrators prepared in 1% plasma were added directly to the plate. Samples were incubated on the plate for 2 hr. To complete the binding complex, Sulfo‐TAG™ (ruthenium)‐labelled anti‐HSA antibody (Abcam, Cambridge, MA, USA; labelling carried out at MedImmune, Gaithersburg, MD, USA) was then added to the plate for 1 hr and MSD® 2× Read Buffer T (a Tris‐based buffer containing tripropylamine as a co‐reactant for light generation in electrochemiluminescence immunoassays) was used to quantitatively measure the binding complex.
The plate was read on an MSD® Sector™ 6000 reader (MSD, Gaithersburg, MD, USA), and results were analysed using SoftMax Pro® software version 5.2 or higher (SoftMax Pro Data Acquisition and Analysis Software, RRID:SCR_014240; link). Data were fitted using a weighted (1/y2) 5‐ (Study 1) or 4‐ (Studies 2 and 3) parameter logistic function, and concentrations of VIB4920 were measured by interpolation from the standard curve. The quantitative range for the assay was 0.08 to 10.00 μg·ml−1 (Study 1) or 0.16 to 10.00 μg·ml−1 (Studies 2 and 3). PK parameters were derived using noncompartmental methods with Phoenix® WinNonlin® Version 6.3 (Certara Corp, St Louis, MO, USA). All descriptive and inferential statistical computations were performed using Phoenix® WinNonlin® Version 6.3 (Phoenix, RRID:SCR_003163; link).
Anti‐drug antibodies (ADAs) were evaluated in all three repeat‐dose studies. Detailed methods for this analysis are presented in Supporting Information.
2.5. Immunophenotyping
The antibody based procedures used in this srudy comply with the recommendations made by the British Journal of Pharmacology. For Studies 1 and 2, peripheral blood immunophenotyping samples were collected from all animals pretest, on Day 15, prior to the terminal (Day 31 [Study 1]; Day 32 [Study 2]) and recovery (Day 170 [Study 1]; Day 224 [Study 2 i.v. 300 mg·kg−1 VIB4920 group]) necropsies, and prior to the Day 136 tetanus toxoid (TT) injection (Study 1). In Study 3, peripheral blood immunophenotyping samples were collected pre‐dose at intervals on Days 15–191 and during recovery on Days 218, 246, 274, 302, 330, 358, and 386.
Lymphocyte populations were identified and gated using forward side scatter/side–side scatter dot plots. The forward scatter/side–side scatter lymphocyte gate was applied to additional dot plots and histograms to identify and quantify the immune cell populations analysed (Table 1). Aliquots of 50‐μl (Studies 1 and 2) or 100‐μl (Study 3) blood samples were mixed with one or more fluorochrome‐labelled antibodies (Table S3) and incubated in the dark for 30 min. VersaLyse™ solution (Beckman Coulter, USA; Studies 1 and 2) or TQ‐Prep using Immunoprep reagents (Coulter, Miami, FL, USA) were utilised to lyse red blood cells and to stabilise any remaining intact cells. To allow the real‐time quantification of immune cell populations, 50‐μl (Studies 1 and 2) or 100‐μl (Study 3) aliquots of flow‐count fluorospheres were added to each sample tube.
Table 1.
Immunophenotyping: Flow cytometry antigens and cell populations
| Antigen marker(s) | Cell population identifieda | |
|---|---|---|
| Studies 1 and 2 | Study 3 | |
| CD45+ | CD2+/CD20+ | All lymphocytesb |
| CD45+/CD20+ | CD20+ | B lymphocytes |
| CD45+/CD3+ | CD3+ | T lymphocytes |
| CD45+/CD3+/CD4+ | CD3+/CD4+ | T‐helper lymphocytes |
| CD45+/CD3+/CD8+ | CD3+/CD8+ | T‐cytotoxic lymphocytes |
| CD45+/CD159a+ | CD3−/CD16+ | NK cells |
| CD45+/CD14+ | CD3−/CD14+ | Monocytes |
Absolute count and percent of baseline/gated were calculated and reported.
In Study 3, a lymphocyte purity estimate (CD2+/CD20+) was calculated but not reported.
Samples were analysed using the Beckman–Coulter FC500 Flow Cytometer and MXP (Studies 1 and 2) or CXP 2.2 (Study 3) Software. Absolute cell counts and percentage of gated cells were calculated. The mean (SD) and/or total cell counts were calculated by treatment group and compared with the vehicle group.
2.6. Peripheral blood mononuclear cell (PBMC) isolation
Cynomolgus monkey blood was collected into Cyto‐Chex Blood Collection Tubes and shipped at 4°C. Samples were allowed to equilibrate to room temperature for 1 hr. PBMCs were isolated using ACCUSPIN™ (Sigma) tubes. Briefly, ACCUSPIN™ tubes were preloaded with 3 ml of lymphocyte separation medium (diluted with 10% Dulbecco's PBS [DPBS]) below the frit and 3 ml of DPBS above. Following the equilibration period, 4 ml of Cyto‐Chex preserved whole blood was gently added on top of the frit. Tubes were spun at room temperature for 20 min at 800×g. Following centrifugation, PBMCs were poured off the frit and transferred to a 15‐ml conical tube and filled with cold RPMI 1640+ media (Gibco®). Cells were then centrifuged for 10 min at room temperature at 250×g with the centrifuge brake set to 1 or “slow.” Following aspiration of the supernatant, the remaining mononuclear cells were washed once with and resuspended in 3 ml of cold RPMI 1640+ media.
2.7. Flow cytometric evaluation
A total of 250‐μl cell suspension was aliquoted into 96‐well U‐bottom plates, spun down, and Fc receptors were blocked for 15 min in 20‐μl Human Tru‐stain FcX (BioLegend) diluted 1:4 in FACS buffer containing 10% wt/vol sodium azide and 0.2% wt/vol BSA in DPBS. Cells were then stained with 30‐μl surface antibody cocktail containing anti‐IgD‐fluorescein isothiocyanate (FITC; Dako Cytometry), anti‐CD40 PerCP 5.5, anti‐CD20‐Alexa Fluor 700, anti‐CD27‐phycoerhthrin, anti‐CD196 BV421, and anti‐CD4 BV785. The cells were stained for 30 min at 4°C, washed, fixed in 4% paraformaldehyde for 17 min, and resuspended in 100‐μl FACS buffer for storage overnight at 4°C. The following day, cells were permeabilised with 1× FoxP3 Perm Buffer (eBiosciences) for 15 min, washed, and resuspended in 50‐μl of intracellular Perm Buffer containing anti‐Ki‐67‐Alexa Fluor 647. The cells were stained for 60 min at 4°C, washed and resuspended in 100 μL FACS Buffer. All samples were run on an LSR II flow cytometer.
2.8. T‐cell‐dependent antibody response to keyhole limpet haemocyanin (KLH) and TT
Animals were immunised with 1‐ml s.c. KLH (10 mg·ml−1 in saline; Pierce, ThermoFisher Scientific, Waltham, MA, USA [Studies 1 and 2]; Pierce, ThermoFisher Scientific, Rockford, IL, USA [Study 3]) on prestudy Day −28 and 1‐hr post‐VIB4920 administration on Day 8 and with an intramuscular dose of TT (0.5 ml; Super‐Tet; Intervet, HeartLand Vet Supply, Hastings, NE, USA [Studies 1 and 2]; Intervet, Millsboro, DE, USA [Study 3]) on prestudy Day −28 and during VIB4920 washout on Day 136 (Studies 1 and 2) or 351 (Study 3). Serial blood samples were collected from the femoral vein at various time points for PD analyses (Table S2). Serum samples were analysed by elisa to determine anti‐KLH and anti‐TT IgG and IgM titres.
2.9. In‐life evaluations
A daily qualitative assessment of food intake and appetite was conducted for all animals. Urine volume, specific gravity, and pH were measured in all animals for urinalysis evaluations; physical, biochemical, and microscopic urinary components were also analysed. Body weight was measured and recorded at transfer and at regular intervals. In Studies 1 and 2, the BP of each animal was measured, and an ECG examination was conducted pre‐test, 24 hr after receipt of the first and last or third dose of VIB4920 and during the last week of the recovery period. In Study 3, BP measurements and ECG examinations were recorded pre‐test, on Days 92 and 176, and once prior to recovery necropsy. Ophthalmoscopic examinations were conducted on all animals at Week 28 (Study 3), pre‐test and prior to the terminal or recovery necropsies (all studies). Blood samples were collected pre‐test, on Days 1 and 22 (Studies 1 and 2) and on Days 1, 29, 57, 85, 113, 141, 169, and 190 (Study 3) via the femoral vein for https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2362–anti‐thrombin (TAT) complex analysis.
2.10. Clinical pathology
Blood was collected for haematology, clinical chemistry, and coagulation analyses. Specialised clinical pathology evaluations (D‐dimer, TAT, PFA‐100) were conducted to assess the risk of incidence of haemostatic or thromboembolic activity. Animals were fasted prior to clinical chemistry blood collections. Detailed methods for these evaluations are provided in Supporting Information.
2.11. Necropsy
All animals were subjected to a complete gross examination at necropsy that included evaluation of the carcass and musculoskeletal system, all external surfaces and orifices, cranial cavity and external surfaces of the brain, and thoracic, abdominal, and pelvic cavities with their associated organs and tissues.
2.12. Histopathology
All protocol‐designated tissues were fixed in 10% neutral buffered formalin, except for the eye (including the optic nerve) and testes, which were fixed using modified Davidson's fixative. Tissues were processed to paraffin blocks, sectioned at 5 μM, and stained with haematoxylin and eosin (H&E) for microscopic evaluation.
Microscopic examination of fixed H&E‐stained paraffin tissue sections was performed on at least 50 protocol‐designated tissues (Table S4). The lung, kidney, spleen, and liver were also stained with phosphotungstic acid haematoxylin to specifically identify thrombi in these highly vascular organs. Grades from 1 (minimal) to 5 (severe) were used to determine the severity of histopathological findings.
2.13. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. In all studies, B‐cell counts, anti‐KLH IgG titre, and CCR+/−Ki67 + CD20+ lymphocyte frequency, ANOVA followed by Dunnett's test was used to test for difference between control and treatment groups. If test assumptions were not satisfied, Kruskal–Wallis followed by Dunn's test was used to test for differences between groups. In Study 2, B‐cell counts for VIB4920‐dosed animals for each time point were compared to vehicle using the Mann–Whitney test to assess differences in median rank. In all studies, the anti‐TT IgG titre for VIB4920‐dosed animals was compared to vehicle, following VIB4920 washout, using a two‐way ANOVA repeated measures model (with Greenhouse–Geisser correction) followed by Dunnett's multiple comparisons test. Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Prism, RRID:SCR_002798; link). A P value < .05 was considered statistically significant.
2.14. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and, where relevant, are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019; Alexander et al., 2019; Alexander, Christopoulos et al., 2019).
3. RESULTS
3.1. Baseline characteristics
Of the 126 animals randomised to treatment across all three studies, 30 (males) were evaluated in Study 1, 36 in Study 2 (18 male and 18 female), and 60 in Study 3 (30 male and 30 female). Male animals weighing 3.1 to 5.0, 4.3 to 6.5, and 4.5 to 10.6 kg received the study drug and were evaluated in Studies 1, 2 and 3, respectively. Female animals, who were only evaluated in Studies 2 and 3, had comparable weight ranges of 2.8 to 5.9 and 2.9 to 5.7 kg, respectively. At baseline, animals were aged between 3.2 and 8.8 years across all studies.
3.2. Pharmacokinetic analysis
VIB4920 plasma concentrations measured in Study 1 declined in a biphasic manner, initially decreasing rapidly followed by a slower elimination phase (Figure 1a). In general, VIB4920 displayed linear PK. Concentrations declined below the lower limit of quantification (LLOQ; 0.08 μg·ml−1) at 120 days post‐dose in all four treatment groups. Following the first dose of i.v. (bolus) VIB4920 and at steady state (after 5 weeks), Cmax, AUCτ and Ctrough all increased dose‐proportionally across the dose range of 5 to 150 mg·kg−1 (Table 2A). The t 1/2 of VIB4920 was approximately 6 days regardless of dose (Table 2A), and bioavailability was high, reported as 83% in the s.c. 150 mg·kg−1 weekly treatment group.
Figure 1.
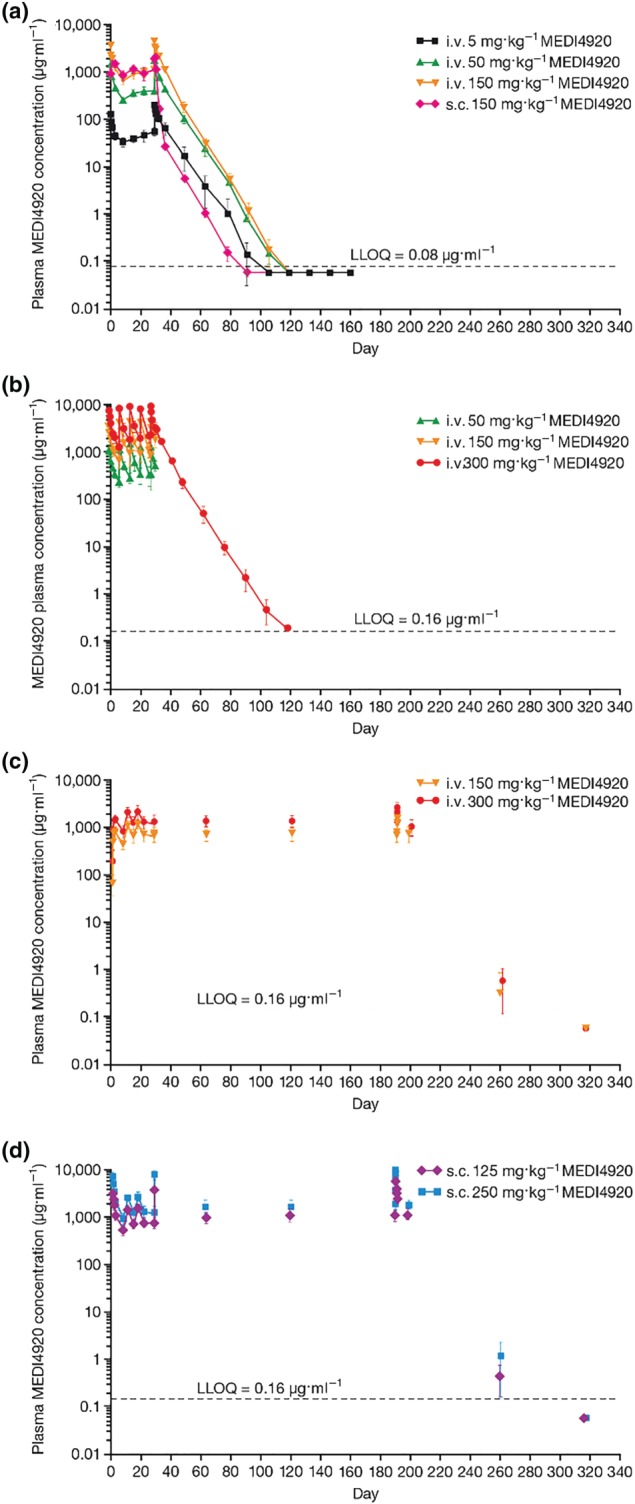
Mean (SD) plasma concentration‐time profiles of VIB4920 for Study 1 (a) where animals were given five weekly doses of VIB4920 followed by a 20‐week recovery period (for all groups, dosing phase N = 6, recovery phase N = 3), Study 2 (b) where animals were given five weekly doses of VIB4920 followed by a 27‐week recovery period (for vehicle and 300 mg·kg−1 groups, dosing phase N = 12, recovery phase N = 6; for 50 and 150 mg·kg−1 groups, dosing phase N = 6, recovery phase N = 3), and Study 3 where animals were given 28 weekly i.v. (c) or s.c. (d) doses of VIB4920 followed by a 29‐week recovery period (for all groups, dosing phase N = 12, recovery phase N = 6). i.v., intravenous; s.c., subcutaneous
Table 2A.
PK parameters of VIB4920 after administration of the first dose of VIB4920 and at steady state by study: Study 1
| Parameter | VIB4920 dose | |||
|---|---|---|---|---|
| 5 mg·kg−1 i.v. (N = 6) | 50 mg·kg−1 i.v. (N = 6) | 150 mg·kg−1 i.v. (N = 6) | 150 mg·kg−1 s.c. (N = 6) | |
| Tmax (days) | 0.02 (0.00) | 0.02 (0.00) | 0.02 (0.00) | 1.83 (0.75) |
| Cmax (μg·ml − 1) | 130 (15) | 1,560 (327) | 3,400 (200) | 1,720 (164) |
| Ctrough (μg·ml − 1) | 34 (7) | 262 (16) | 649 (81) | 858 (184) |
| AUCτ (μg·day·ml − 1) | 366 (44) | 3,930 (327) | 9,210 (906) | 8,720 (916) |
| Tmax ss (days) | 0.18 (0.40) | 0.02 (0.00) | 0.02 (0.00) | 1.33 (1.29) |
| Cmax ss (μg·ml − 1) | 193 (44) | 1,870 (179) | 4,240 (853) | 2,050 (323) |
| Ctrough ss (μg·ml−1) | 56 (11) | 412 (62) | 1,070 (339) | 1,140 (176) |
| AUCτ ss (μg·day·ml−1) | 745 (103) | 6,150 (26) | 14,700 (463) | 12,200 (1,560) |
| t 1/2 (days) | 6.19 (0.99) | 6.02 (0.20) | 5.55 (0.30) | 5.54 (0.21) |
Note. All values are presented as mean (SD).
Abbreviations: Cmax, maximum observed concentration; Ctrough, trough concentration; i.v., intravenous; PK, pharmacokinetic; s.c., subcutaneous; ss, steady state; Tmax, time of maximum observed concentration.
Mean VIB4920 plasma concentrations over time in Study 2 are presented in Figure 1b. All animals treated with VIB4920 at 50 or 150 mg·kg−1 weekly were terminated at the end of the dosing phase on Day 32. VIB4920 displayed a linear terminal phase during the first dosing interval, with low intersubject variability in concentrations observed up to Day 32 for the 50 and 150 mg·kg−1 groups and up to Day 115 across the remaining treatment groups. A linear terminal phase (up to Day 115) was demonstrated in animals treated with 300 mg·kg−1 VIB4920 weekly. In the vehicle group, all VIB4920 concentrations were below the LLOQ (0.16 μg·ml−1). Cmax increased dose‐proportionally following the first dose of i.v. (slow infusion) VIB4920 and after delivery of the fifth dose on Day 29; AUCτ and Ctrough also increased dose‐proportionally after the initial dose was given (Table 2B). Accumulation of VIB4920 was minimal, and the t 1/2 and volume of distribution at steady state were comparable to equivalent doses evaluated in Study 1 (data not shown).
Table 2B.
PK parameters of VIB4920 after administration of the first dose of VIB4920 and at steady state by study: Study 2
| Parameter | VIB4920 dose | ||
|---|---|---|---|
| 50 mg·kg−1 i.v. (N = 6) | 150 mg·kg−1 i.v. (N = 6) | 300 mg·kg−1 i.v. (N = 12) | |
| Tmax (days) | 0.07 (0.13) | 0.02 (0.00) | 0.05 (0.09) |
| Cmax, Day 1 (μg·ml−1) | 1,094.4 (223.4) | 3,321.2 (1,045.5) | 6,896.4 (1,542.7) |
| Ctrough, Day 8 (μg·ml−1) | 223.7 (56.2) | 576.7 (176.7) | 1,188.2 (341.7) |
| AUCDay 1–Day 8 (μg·day·ml−1) | 2,982.8 (471.3) | 7,982.3 (1,454.8) | 17,142 (3,793.7) |
| Tmax ss (days) | 0.02 (0.00) | 0.02 (0.00) | 0.05 (0.09) |
| Cmax, Day 29 (μg·ml−1) | 1,125.5 (159.6) | 4,109.6 (918.1) | 8,245.3 (1,541.2) |
| Ctrough, Day 36 (μg·ml−1) | NC | NC | 1,485.5 (225.1) |
| AUCDay 29–Day 36 (μg·day·ml−1) | NC | NC | 22,713.1 (2,461.4) |
| t 1/2 (days) | NC | NC | 6.08 (0.39) |
Note. All values are presented as mean (SD).
Abbreviations: Cmax, maximum observed concentration; Ctrough, trough concentration; i.v., intravenous; NC, not calculated; PK, pharmacokinetic; ss, steady state; Tmax, time of maximum observed concentration.
The mean VIB4920 plasma concentration over time during 28 weeks of i.v. (30‐min infusion) and s.c. repeat‐dosing during Study 3 is shown in Figure 1c,d. Peak concentrations were observed at the end of the i.v. infusion and between 30 min and 3 days following s.c. VIB4920 administration. Following the first dose of i.v. or s.c. VIB4920 (Day 1) and at Day 190, Cmax and AUC0 − t increased dose proportionally across the dose ranges analysed. As with the previously reported studies, t 1/2 was approximately 6 days regardless of dose, and accumulation of VIB4920 was minimal (Table 2C). The bioavailability of s.c. VIB4920 in Study 3 (73% and 78% for VIB4920 125 and 250 mg·kg−1, respectively) was slightly lower than the value reported in Study 1 and exposure to VIB4920 was similar between male and female animals.
Table 2C.
PK parameters of VIB4920 after administration of the first dose of VIB4920 and at steady state by study: Study 3
| Parameter | VIB4920 dose | |||
|---|---|---|---|---|
| 125 mg·kg−1 s.c. (N = 12) | 250 mg·kg−1 s.c. (N = 12) | 150 mg·kg−1 i.v. (N = 12) | 300 mg·kg−1 i.v. (N = 12) | |
| Tmax (days)a | 3.0 (1.0 to 3.0) | 3.0 (1.0 to 3.0) | 0.02 (0.02 to 0.02) | 0.02 (0.02 to 0.02) |
| Cmax, Day 1 (μg·ml−1) | 870 (209) | 1,590 (241) | 3,330 (520) | 7,250 (1,680) |
| AUC0–t (μg·day·ml−1) | 4,600 (864) | 8,600 (1,370) | 8,830 (776) | 17,200 (3,610) |
| Tmax Day 190 (days)a | 1.00 (0.50 to 1.00) | 1.00 (0.50 to 3.00) | 0.02 (0.02 to 0.02) | 0.02 (0.02 to 0.35) |
| Cmax, Day 190 (μg·ml−1) | 1,590 (297) | 2,870 (481) | 4,640 (696) | 7,920 (1,590) |
| AUC0–t, Day 190 (μg·day·ml−1) | 10,300 (15,800) | 23,500 (25,500) | 21,800 (20,200) | 35,900 (35,600) |
| CL (ml·day−1·kg−1) | NC | NC | 11.1 (1.2) | 12.7 (3.2) |
| t 1/2 (days) | 6.02 (ID) | 5.20 (1.37) | 5.66 (0.30) | 5.96 (0.48) |
Note. All values are presented as mean (SD) unless stated otherwise.
Median (range).
Abbreviations: CL, systemic clearance; Cmax, maximum observed concentration; i.v., intravenous; NC, not calculated; PK, pharmacokinetic; s.c., subcutaneous; Tmax, time of maximum observed concentration.
ADAs were present in several animals in all three studies; however, this had no effect on exposure to VIB4920. Detailed results of the ADA analyses for all repeat‐dose studies with VIB4920 are provided in Supporting Information.
3.3. Immunophenotyping
B‐cell counts for individual animals, normalised to baseline (pre‐dose), for each study, are shown in Figure 2. For all studies, B‐cell counts for untreated animals, as measured by flow cytometry, show great variability during the dosing and recovery phases. Counts spread across a wide range. This variability is particularly exaggerated toward the lower end of the range for Study 2. For these reasons, we have graphed all the individual data points for each study and have drawn a line through the median for each group for each time point.
Figure 2.
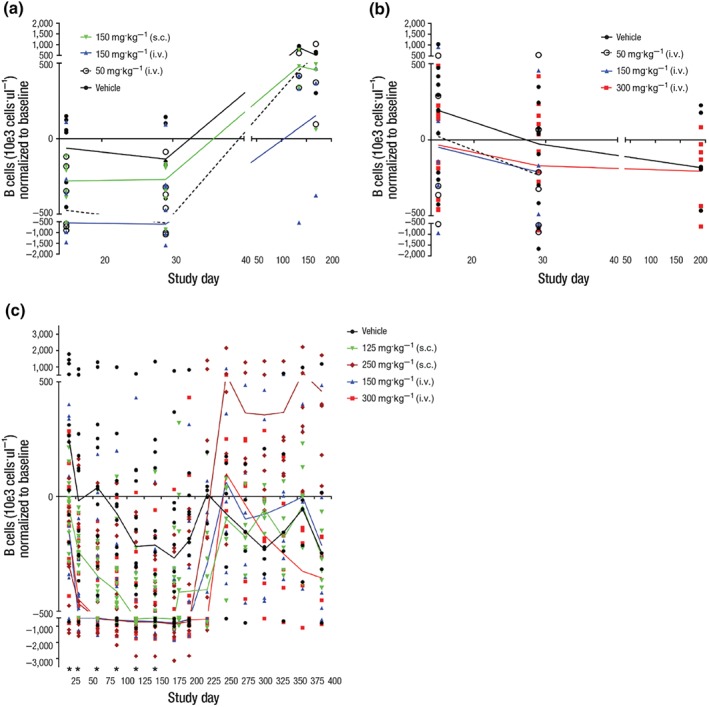
The change over time in B‐cell count for individual animals treated with vehicle or VIB4920: (a) Study 1, five weekly doses of vehicle, VIB4920 at 5, 50, 150 mg·kg−1 i.v., 150 mg·kg−1 s.c. (for all groups, dosing phase N = 6, recovery phase N = 3); (b) Study 2, five weekly doses of vehicle, VIB4920 at 50, 150, 300 mg·kg−1 i.v. (for vehicle and 300 mg·kg−1 groups, dosing phase N = 12, recovery phase N = 6; for 50 and 150 mg·kg−1 groups, dosing phase N = 6, recovery phase N = 3); (c) Study 3, 28 weekly doses of vehicle, VIB 4920 at 150, 300 mg·kg−1 i.v., 125, 250 mg·kg−1 s.c. (for all groups, dosing phase N = 12, recovery phase N = 6). The line represents the median for each group. *P ≤ .05, significantly different from vehicle group; one‐way ANOVA with Dunnetts multiple comparisons test with a single pooled variance or Kruskal–Wallis test with Dunns multiple comparisons test or Mann–Whitney test. i.v., intravenous; s.c., subcutaneous
In all studies, there is a trend toward dose‐related lower B‐cell counts in the VIB4920‐treatment groups, which lasts through the dosing phase. However, statistical analysis shows that there are no significant differences between B‐cell counts for untreated and VIB4920‐treated animals in either of the 1‐month studies. Difference was significant for all dose groups in the 6‐month study on Day 15. Significant difference for 250 mpk s.c., 150 mpk i.v., and 300 mpk i.v. was shown for Study Days 29, 57, 85, 113, and 141 only. Upon drug withdrawal, the B‐cell counts return to levels shown for the untreated animals in all studies.
3.4. Anti‐KLH T‐cell‐dependent antibody response
The KLH‐specific IgG titre, following primary administration on pre‐study Day −28 and secondary administration 1 hr following VIB4920 administration on Day 8, is shown in Figure 3 for each dose group. The secondary KLH response was suppressed by VIB4920 for all dose groups. Specifically, in Study 1, VIB4920 suppressed the secondary KLH‐specific IgG expression in a dose‐dependent fashion (Figure 3b,c) compared with the vehicle group (Figure 3a). Animals treated with vehicle had a sixfold higher IgG AUC after the second administration of KLH relative to the initial administration. As expected, the KLH‐specific IgM titre after primary challenge was threefold to 13‐fold higher than after the secondary challenge (data not shown). IgG AUC was suppressed by 18%, 77%, 86%, and 81% in animals treated with i.v. 5 mg·kg−1 (data not shown), i.v. 50 mg·kg−1 (Figure 3b), i.v. 150 mg·kg−1 (Figure 3c), and s.c. 150 mg·kg−1 VIB4920 weekly (Figure 3e), respectively, compared with the vehicle group. KLH‐specific IgM titre expression in VIB4920‐treated animals followed a similar trend in suppression.
Figure 3.
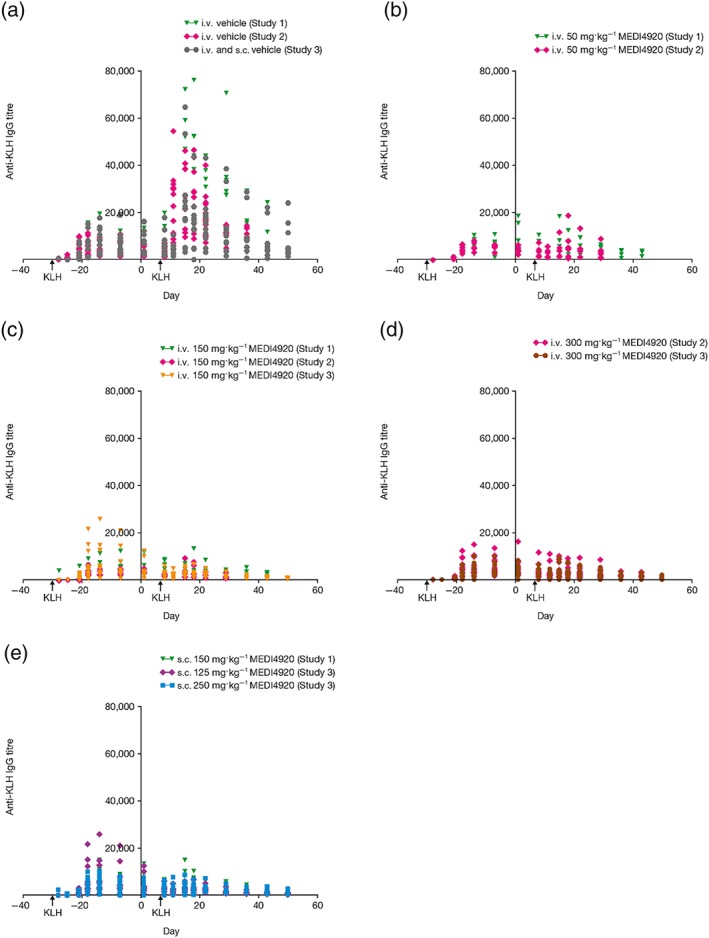
The anti‐KLH IgG titre over time (pre‐ and post‐treatment) for animals treated with vehicle N = 30 (a), i.v. 50 mg·kg−1 VIB4920 N = 12 (b), i.v. 150 mg·kg−1 VIB4920 N = 24 (c), i.v. 300 mg·kg−1 VIB4920 N = 24 (d), and s.c. VIB4920 (e) across all three studies; N = 6 for 150 mg·kg−1 s.c., N = 12 for 125 mg·kg−1 s.c., N = 12 for 250 mg·kg−1 s.c. KLH immunisation was given on pre‐study Day −28 and 1 hr post‐VIB4920 administration on Day 8. IgG titres for VIB4920‐dosed groups were significantly different (P ≤ .05) for all groups, compared to vehicle, on Days 15, 18, 22, 29, 36, 43, and 50; one‐way ANOVA with Dunnetts multiple comparisons test with a single pooled variance or Kruskal–Wallis test with Dunns multiple comparisons test. IgG, immunoglobulin G; i.v., intravenous; KLH, keyhole limpet haemocyanin; s.c., subcutaneous
In Study 2, IgG AUC after secondary KLH re‐immunisation on Day 8 was suppressed by 79.2%, 87.0%, and 86.1% in the 50 mg·kg−1, 150 mg·kg−1, and 300 mg·kg−1 VIB4920 weekly groups, respectively (Figure 3b–d, compared with vehicle (Figure 3a). IgM titre following secondary KLH immunisation was less pronounced compared with the response following the initial administration. The peak titres of KLH‐specific IgM after the secondary immunisation were suppressed by 68.0%, 54.6%, and 57.1% at 50 mg·kg−1, 150 mg·kg−1, and 300 mg·kg−1 VIB4920 weekly, respectively, compared with the vehicle group.
In Study 2, expression of Ki‐67 (a marker for cell proliferation) after the second KLH immunisation was increased in both https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=63+ and CCR6− https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2628 + B cells in vehicle‐treated animals but not in the VIB4920‐treated animals as shown in Figure 4. CCR6 is essential for memory B cells to mount an effective secondary response upon antigen re‐challenge (Elgueta et al., 2015). Additionally, the post‐switch memory circulating B cells from VIB4920‐treated animals did not show an increase at the time point (Day 15) following the secondary KLH administration (data not shown). Thus, VIB4920 treatment abolished the ability of peripheral B cells to proliferate in response to secondary KLH immunisation.
Figure 4.
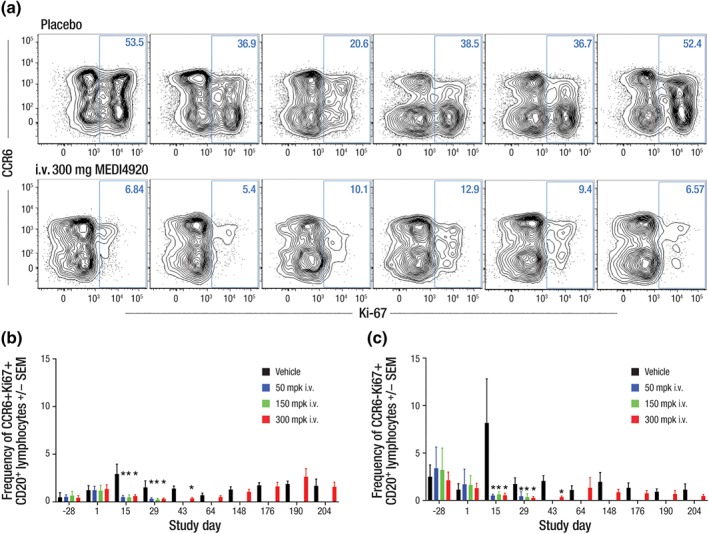
Ki‐67 expression on CCR6+/−CD20+ B cells following KLH administration in animals treated with vehicle or i.v. 300 mg·kg−1 VIB4920 in Study 2 (N = 12). VIB4920 treatment inhibits the B‐cell proliferative response following secondary KLH administration. (a) Flow cytometry plots are pre‐gated on CD20+ lymphocytes and show the percent CCR6+/−Ki‐67+ for six representative animals; the change over time in circulating (b) CCR6+ (c) CCR6− Ki‑67+ B‐cell count (as frequency of gated CD20+) in animals treated with vehicle, or i.v. 50, 150, or 300 mg·kg−1 VIB4920. Proliferation is significantly affected on Days 15 and 29 of the dosing phase and on Day 43 of the recovery phase. *P ≤ .05, significantly different from vehicle group; one‐way ANOVA with Dunnetts multiple comparisons test with a single pooled variance and the Mann–Whitney test to assess differences in mean rank. CD, cluster of differentiation; i.v., intravenous; KLH, keyhole limpet haemocyanin
In Study 3, all animals produced a primary humoral anti‐KLH IgM (data not shown) and IgG response (Figure 3) following immunisation with KLH on Day −28 (prior to VIB4920 administration). There was a dose‐dependent decrease in anti‐KLH IgG antibody expression in all VIB4920‐treated animals following the secondary KLH immunisation (Figure 3c,d,e) compared with the vehicle group (Figure 3a). The secondary anti‐KLH IgM response was lower than the primary anti‐KLH IgM response for all VIB4920‐treated animals (data not shown).
3.5. Anti‐TT T‐cell dependent antibody response (TDAR)
TT‐specific IgG responses following primary and secondary TT immunisation were similar across all treatment groups. The secondary TT‐specific antibody response was measured during recovery following VIB4920 washout. The TT‐specific IgG titre is shown in Figure 5 for each dose group. In each of the dose groups, the overall immune response was not affected by chronic exposure to VIB4920.
Figure 5.
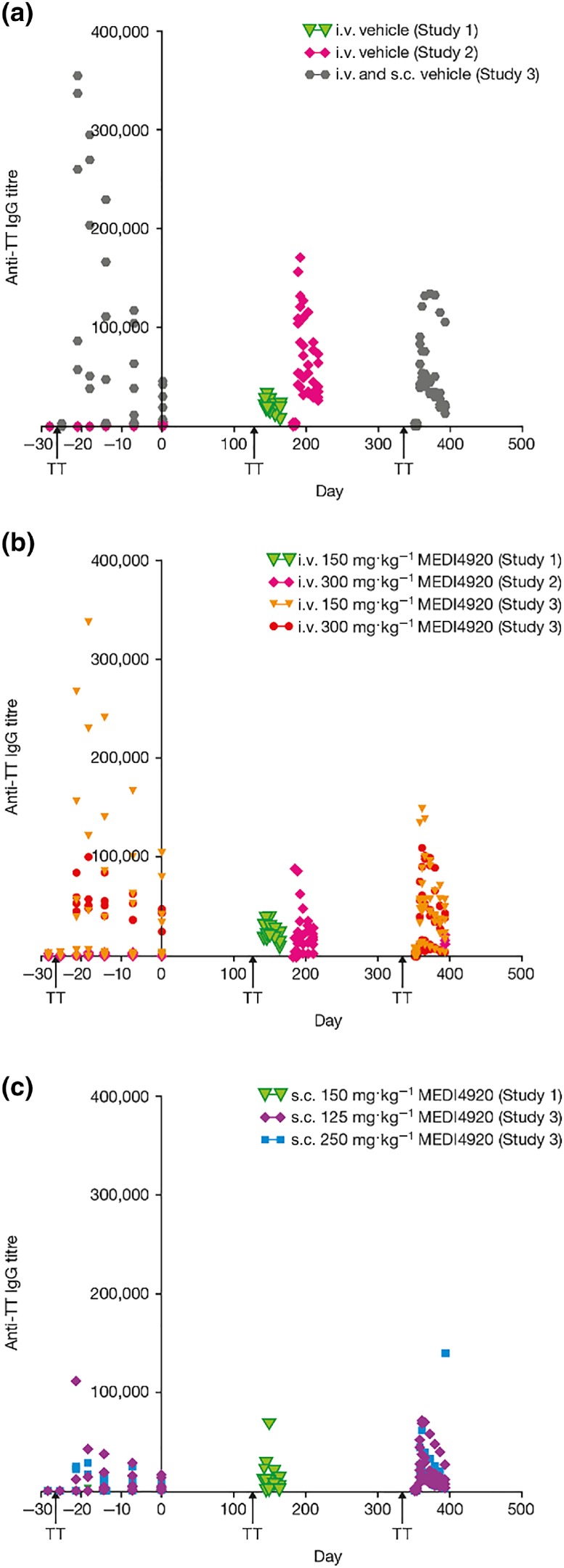
The anti‐TT IgG titre over time (pre‐ and post‐treatment) for animals treated with vehicle (a) N = 30 pre‐treatment, N = 18 post‐treatment, i.v. VIB4920 (b) for vehicle and 300 mg·kg−1 N = 18 pre‐treatment and N = 9 post‐treatment, for 150 mg·kg−1 dose group N = 18 pre‐treatment and N = 6 post‐treatment, and s.c. VIB4920 (c) N = 18 pre‐treatment, N = 0 post‐treatment in all three studies. TT immunisation was given on pre‐study Day −28 and following VIB4920 washout on Day 136 (Studies 1 and 2) or 351 (Study 3). Treatment with VIB4920 did not have a long‐term effect on the immune response as the secondary TT response was restored after VIB4920 washout. Two‐way ANOVA repeated measures model (with Greehouse–Geisser correction) followed by Dunnetts multiple comparisons test. IgG, immunoglobulin G; i.v., intravenous; s.c., subcutaneous; TT, tetanus toxoid
Specifically, in Study 1, TT‐specific IgG AUEC after the secondary immunisation on Day 136 was approximately 20‐ to 56‐fold higher relative to the primary immunisation (Day −28) for all groups. A small increase (1.5‐ to 4.5‐fold) in IgM AUEC was seen after the primary TT immunisation, relative to the secondary immunisation for all groups (data not shown). Results were comparable for all groups in Study 2 (IgM data not shown). In Study 3, all animals produced a primary humoral anti‐TT IgM and IgG response following TT immunisation on Day −28. There were no changes in titres of anti‐TT IgM (data not shown) or IgG antibodies (Figure 5b,c), compared to vehicle, following secondary TT immunisation at Day 351.
3.6. Additional in‐life evaluations
In total, all 126 animals across the three studies did not have any VIB4920‐related changes in food consumption, urinalysis parameters, body weight, BP, electrocardiology, ophthalmology, or TAT complex concentrations.
On Day 92, a female in the Study 3 i.v. 150 mg·kg−1 VIB4920 weekly dose group was killed because of long‐term viability concerns and poor clinical condition despite supportive care. This female lost 0.3 kg of body weight between Days 56 and 70 and presented with slightly sunken eyes and skin turgor. The animal received lactated Ringers solution; however, after further physical examination, observations of dehydration and sunken eyes were reported.
On Day 149, a female in the Study 3 i.v. 300 mg·kg−1 VIB4920 weekly dose group presented with splenomegaly during a routine physical examination. This animal was diagnosed with an opportunistic fungal infection attributed to the immunosuppressive effects of VIB4920. The fungus was identified as Talaromyces (Penicillium) marneffei; the clinical pathology and histopathological findings in this animal associated with this infection have previously been reported (Iverson et al., 2018).
3.7. Clinical pathology
In Study 1, animals receiving i.v. and s.c. 150 mg kg−1 VIB4920 weekly experienced small decreases in mean triglyceride concentrations relative to the vehicle group (−34% and − 42%, respectively). These changes were mostly resolved and, although considered related to VIB4920 treatment, were not deemed biologically relevant based on their small magnitude and transient nature. No VIB4920‐related changes in coagulation, specialised clinical pathology, or haematology parameters were reported in Study 1.
In Study 2, animals receiving greater doses of VIB4920 (≥150 mg·kg−1 weekly) demonstrated increased platelet counts at Day 15 and at termination (up to 47%), compared with the vehicle group. These increases were considered treatment related but had resolved by the recovery interval. Similar findings were observed for control female animals. All other mean and individual haematology values were considered to be within an acceptable range for biological variation. There were no test article‐related effects on coagulation times, D‐dimer, TAT complex generation, or PFA‐100 closure times (data provided in Supporting Information).
In Study 3, VIB4920‐related changes in haematology parameters were limited to dose‐independent, minimal to mild increased platelet counts in males and females at all doses at most time points during the dosing phase with recovery. Changes in coagulation parameters included increased fibrinogen in animals receiving i.v. 300 mg·kg−1 VIB4920 and increased D‐dimer in some animals receiving i.v. 150 or 300 mg·kg−1 VIB4920; parameters normalised during the recovery period.
3.8. Mortality
No mortalities were reported in any of the studies. Two animals from Study 3 were killed early. One female receiving i.v. 150 mg·kg−1 VIB4920 weekly was killed on Day 92 because of long‐term viability concerns and poor clinical condition despite supportive care. The exact cause of this animal's condition could not be determined but was considered related to VIB4920. A female in the vehicle group of Study 3 was killed on Day 243 following an accidental injury to the right foot.
3.9. Necropsy
Across all studies, macroscopic observations were infrequent, sporadic, and scattered in several groups, with no evidence of a dose response. In Study 1, nine (30%) animals had red areas noted at the i.v. injection site, which correlated with a microscopic observation of haemorrhage in some cases. There were no VIB4920‐related changes in organ weights. In both Studies 2 and 3, no VIB4920‐related macroscopic observations were reported.
3.10. Histopathology
Injection‐site reactions were observed in all three studies. In Study 1, polymorphonuclear cell infiltrates of severity Grade 1 (minimal) to 3 (moderate) and/or haemorrhage were present at injection sites in nine i.v. VIB4920‐treated animals and in one vehicle‐treated animal. These changes were generally perivascular and were considered to be related to the dosing procedure and were not present in any of the recovery animals. In Study 2, both treated and control animals sometimes had injection‐site haemorrhage with infiltration mixed leukocytes or mononuclear cells; the incidence and severity were greater in VIB4920‐treated animals with no relationship to dose. In Study 3, two females in the s.c. 250 mg·kg−1 VIB4920 weekly treatment group had residual lesions in injection sites due to VIB4920 administration (data not shown).
VIB4920‐related findings were observed in various tissues for all three studies. In Study 1, three animals receiving higher VIB4920 doses (150 mg·kg−1 i.v. [n = 2] and 150 mg·kg−1 s.c. [n = 1] weekly) had minimal Kupffer cell hypertrophy, which is commonly seen during high‐dose protein administration (Maclaren, Levin, Lowman, & Trikha, 2017; Regenass‐Lechner et al., 2016).
At the end of the dosing phase (Day 32), in Study 2, a greater number of VIB4920‐treated animals (n = 14 [77.8%]) had slightly more severe red pulp hyperplasia in the spleen compared with vehicle‐treated animals (n = 1 [16.7%]), a finding commonly observed following the administration of foreign proteins. Consistent with the pharmacology of VIB4920, there was a dose‐dependent decrease in splenic germinal centre size in all but one of the VIB4920‐treated animals (n = 17 [94.4%]). The effect of VIB4920 on splenic germinal centres in animals evaluated in Study 2 is shown in Figure 6. Germinal centres were present in the spleen of animals treated with vehicle and absent in 300 mg·kg−1 VIB4920‐treated animals. These treatment‐related changes were not present in the recovery animals indicating restoration of the germinal centres.
Figure 6.
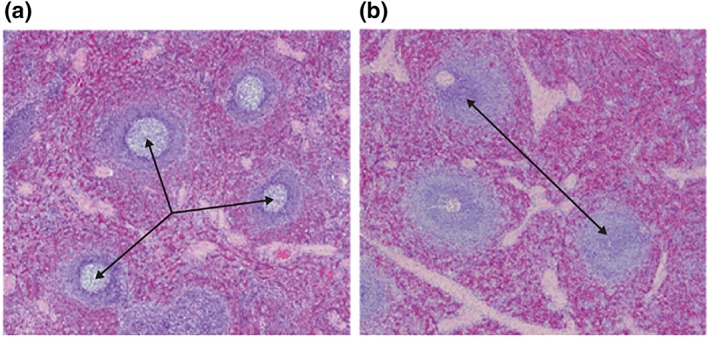
Haematoxylin and eosin stained splenic tissue from animals at Day 32 following treatment with vehicle (a) or i.v. 300 mg·kg−1 VIB4920 (b) in Study 2. The arrows show the germinal centres present in splenic cells of control‐treated animals (a). Germinal centres are absent in VIB4920‐treated animals (b). i.v., intravenous
In Study 3, at the terminal kill on Day 192, all VIB4920‐treated animals (n = 30) had dose‐dependent moderate to complete decreases in germinal centres in the spleen, lymph nodes, and https://en.wikipedia.org/wiki/Gut-associated_lymphoid_tissue, consistent with the pharmacological profile of VIB4920. There were no VIB4920‐related changes in tissues of any of the animals at the end of the recovery period. Microscopic findings in the animal killed early included mononuclear cell infiltrates in multiple organs, moderate increases in cortical lymphocytes in the mandibular and mesenteric lymph nodes, increases in globulins and fibrinogen, and decreases in albumin, suggestive of a chronic viral infection. However, no pathogens were identified with the special testing performed (special stains and PCR of frozen tissue samples). A fungal infection (Talaromyces (Penicillium) marneffei) was identified in one of the infected animals (Iverson et al., 2018).
4. DISCUSSION
Here, we report results from three in vivo studies, evaluating the safety of CD40L blockade by VIB4920 and the pharmacological effect of VIB4920 (5 to 300 mg·kg−1 weekly) on immune cell subsets and the TDAR in cynomolgus monkeys. VIB4920 given once weekly via i.v. or s.c. administration displayed linear PK at multiple doses. t 1/2 for VIB4920, in the in vivo studies, was approximately 6 days regardless of dose. The accumulation of VIB4920 was minimal, and its bioavailability in cynomolgus monkeys was >70% following s.c. administration (125 to 250 mg·kg−1 weekly), suggesting that human exposure to the drug following administration at potentially efficacious doses would not pose a risk of cumulative toxicity.
VIB4920 has recently completed clinical studies for the evaluation of PK and efficacy (Karnell et al., 2019). In other clinical trials, single dose administration of CDP7657, an anti‐CD40L PEG‐Fab (dapirolizumab pegol), showed a t ½ of 6.25–16.1 days across the dose groups of 0.5–5 mg·kg−1 for healthy individuals and a t ½ of 8.59–14.6 days across the dose groups of 5–60 mg·kg−1 for patients with SLE (Tocoian et al., 2015). The estimated t 1/2 of dapirolizumab pegol was in the range of 7.8–14 days following every 2 weeks for 10 weeks (total of five doses; Chamberlain et al., 2017).
The T‐cell/B‐cell interaction is essential for the immune response. The interaction between CD40L on T cells and CD40 on B cells exerts profound effects on B cells, which rapidly proliferate and differentiate into plasmablasts that secrete germline‐encoded IgG and IgM (Danese, Sans, & Fiocchi, 2004; De Silva & Klein, 2015; Elgueta et al., 2009). Assessment of the TDAR to KLH demonstrates the effect of VIB4920 on the B‐cell antibody response to KLH in the preclinical studies.
In line with these findings, treatment with VIB4920, an inhibitor of CD40L, demonstrated blockade of the T‐cell interaction with B cells in all three studies. VIB4920 also demonstrated pharmacological effects on the B‐cell population. In our studies, we observed a recoverable decrease in peripheral B‐cell counts following prolonged treatment with VIB4920. Blockade of the CD40/CD40L pathway may have immune modulating effects on immune cell types, which may result in changes in chemokine expression patterns. This could lead to changes in homing/migration patterns of B cells between periphery and tissue, reflected by a drop in peripheral B‐cell counts. This finding from our cynomolgus studies is being investigated in the clinical setting.
VIB4920 effectively inhibited the KLH‐specific IgG and IgM antibody responses to secondary immunisation in all three studies. This finding was subsequently reproduced in the Phase 1a clinical study in healthy volunteers (Karnell et al., 2019). A recent Keystone Symposia talk (Ettinger, 2019) featured our cynomolgus study showing that VIB4920 inhibited the primary IgM response to KLH.
Additionally, B‐cell proliferation, notably memory B cells, in response to secondary immunisation with KLH was reduced in animals treated with VIB4920, when compared with the effects of vehicle. We showed that proliferation of both the CCR6+ and CCR6− B lymphocytes was affected. CCR6 expression by B cells has been tied to several important roles in mouse and man. Most interestingly, it has been linked to proper B‐cell positioning in lymphoid tissues where it is thought to be a marker for memory B cell precursors and necessary for support of memory B‐cell recall responses (Elgueta et al., 2015; Suan et al., 2017). These findings are likely to reflect the interaction of VIB4920 with T cells, resulting in their defective interaction with B cells.
In all three studies, following washout of VIB4920, both TT‐specific IgG and IgM titres followed a similar trend between treatment groups, with no statistically significant difference observed, compared with vehicle for most time points analysed. This suggests that VIB4920 does not have a long‐term generalised immunosuppressive effect.
The CD40/CD40L interaction also plays a role in the formation and maintenance of germinal centres. These are sites within https://en.wikipedia.org/wiki/Lymph_node and the spleen where mature https://en.wikipedia.org/wiki/B_cell proliferate and differentiate, undergo somatic hypermutation to achieve higher affinity antibodies and class switching from https://en.wikipedia.org/wiki/IgM to https://en.wikipedia.org/wiki/IgG during a normal immune response (Natkunam, 2007; Thorbecke, Amin, & Tsiagbe, 1994). During the sustained blockade of CD40L by VIB4920, splenic germinal centres appeared decreased in size in all but one VIB4920‐treated animal in both Studies 2 and 3. This was the result of a change in the architecture of these germinal centres and indicates that in the presence of VIB4920‐related T (CD40L)‐ and B (CD40)‐cell interactions, B‐cell homeostasis within the germinal centres may be perturbed. The germinal centres were completely restored in the recovery animals, indicating that the effects of CD40L blockade are reversible.
The decreased B‐cell population concentration, loss of ability of the B cells to proliferate and produce antigen‐specific antibodies, and the effect of chronic CD40L blockade by VIB4920 on the germinal centres may have all contributed to a state of immune suppression in cynomolgus monkeys. We observed two potentially opportunistic infections in Study 3 where animals were treated with VIB4920 weekly for 28 weeks. In one case, we were able to identify the pathogen as Talaromyces (Penicillium) marneffei. In the other case, the clinical course and histopathology was consistent with a chronic viral infection, but no causative pathogen was identified with the tests that were used in the study.
Cardiovascular thromboembolic events were associated with the administration of an anti‐CD40L antibody (BG9588/hu5c8), and their incidence led to the termination of the Phase 2 clinical study (Boumpas et al., 2003; Huang et al., 2002). Thromboembolism formation is likely to be caused by the cross‐linking interaction of the IgG receptor (FcγRIIa) with the Fc region of the antibody, and CD40L on the platelet cell surface (Mirabet et al., 2008; Robles‐Carrillo et al., 2010).
CD40L itself is up‐regulated and released as sCD40L from the activated platelet surface (Aloui et al., 2014) and promotes platelet‐driven production of inflammatory molecules such as reactive oxygen and nitrogen species (RONS; Chakrabarti, Varghese, Vitseva, Tanriverdi, & Freedman, 2005) and the induction of fibrinogen binding as well as the formation of platelet microparticles. CD40L activates platelets via independent receptors, including CD40 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2579&familyId=760&familyType=CATALYTICRECEPTOR. (Henn et al., 1998; Inwald, 2003; André et al., 2002; Prasad et al., 2003).
VIB4920 was specifically designed as an Fc‐deficient, CD40L‐binding protein, to avoid the risk of thromboembolic events. Karnell et al. (2019) demonstrated that VIB4920 does not induce platelet aggregation by evaluating its effect, compared to that of an anti‐CD40L mAb, on washed human platelets in vitro. The anti‐CD40L mAb, when complexed with sCD40L, showed rapid induction of platelet aggregation. Anti‐CD40L mAb‐mediated platelet aggregation was blocked upon incubation with an anti‐FcγRIIa antibody. VIB4920 complexed with sCD40L showed no platelet aggregation in this assay.
Preclinical studies of other anti‐CD40L therapeutic agents lacking a functional Fc fragment also demonstrated that these agents did not induce thromboembolic events or pulmonary thrombovasculopathy in rhesus and cynomolgus monkeys (Cordoba et al., 2015; Shock et al., 2015). The inhibition of either sCD40L or αIIbβ3 attenuated the platelet‐mediated generation of RONS (Chakrabarti et al., 2005).
However, it is theoretically possible that ADAs to VIB4920 could complex with VIB4920 and mediate platelet aggregation through the Fc region. Although, the pharmacological effect of VIB4920 should preclude ADA formation at high doses, ADAs were observed for several animals in all of the repeat‐dose studies but did not affect exposure to VIB4920. In our studies, none of the animals presented with a thromboembolic event. There were no adverse VIB4920‐related changes in any of the specialised clinical pathology parameters designed to detect evidence of thromboembolic events, nor was thrombosis noted in the clinical studies with VIB4920 (Karnell et al., 2019), further supporting the hypothesis that molecules lacking an intact Fc region (such as VIB4920) do not directly or indirectly (via interaction with ADA) induce such events.
In conclusion, our results support the hypothesis that Fc‐deficient anti‐CD40L proteins minimise the risk of thromboembolic events associated with Fc‐enabled anti‐CD40L molecules. The pharmacological effect of VIB4920 was demonstrated in vivo through the dose‐dependent inhibition of antigen‐specific immune responses and the effects on B cells. VIB4920 showed a favourable safety and PK profile in our studies supporting the clinical investigation of VIB4920 in ongoing trials of autoimmune disease (NCT02151110; NCT02780388; Albulescu, 2016; Karnell et al., 2019).
AUTHOR CONTRIBUTIONS
S.M.N. contributed to the development of the protocol, study design, and data acquisition and interpretation. K.C. contributed to the development of the protocol, study design, and data acquisition, analysis and interpretation. M.G. contributed to data acquisition, analysis, and interpretation. W.I. contributed to the development of the protocol (including review of the pathology requirements), study design, and anatomical pathology data acquisition, analysis, and interpretation. S.D. contributed to the development of the protocol (including the notion to acquire flow data following cellular subsets for potential biomarker identification), study design, and data analysis and interpretation. H.C. contributed to data analysis and interpretation (preparation of KLH graphs and statistical analysis and interpretation of these data). S.S. contributed to the development of the protocol, study design, and data acquisition, analysis, and interpretation. S.K. contributed to the development of the protocol, study design, and data analysis and interpretation. T.O'D. contributed to the data interpretation and statistical analysis of the data. R.D. contributed to the development of the protocol, study design, and data interpretation. P.C.R. contributed to the development of the protocol, study design, and data interpretation. All authors critically revised the manuscript's content, approved the final version of the manuscript and agreed to be accountable for all aspects of work presented.
CONFLICT OF INTEREST
S.M.N., T.O'D., and P.C.R. are employees of MedImmune/AstraZeneca and hold stock in AstraZeneca (AZ). W.I., S.S., and S.K. are employees of MedImmune/AstraZeneca. K.C. holds stock in AZ and was an employee of MedImmune/AstraZeneca at the time that the original study and this analysis were conducted; her current affiliation is Bristol‐Meyers Squibb. M.G. holds stock in AZ and was an employee of MedImmune/AstraZeneca at the time that the original study and this analysis were conducted; her current affiliation is Viela Bio, and she holds stock in Viela Bio. R.D. holds stock in AZ and was an employee of MedImmune/AstraZeneca at the time that the original study and this analysis were conducted; his current affiliation is Bionavigen. S.D. was an employee of MedImmune/AstraZeneca at the time that the original study and this analysis were conducted; her current affiliation is Gilead Sciences. S.D. has a patent in WO 2013055745 A3 Cd40L‐specific Tn3‐derived scaffolds and the methodology for their use. H.C. was an employee of MedImmune/AstraZeneca at the time that the original study and this analysis were conducted; her current affiliation is Reaction Biology Corporation.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Data S1. Supporting Information
ACKNOWLEDGEMENTS
The authors wish to thank Joyce Heward, MS, DABT, and James A. Ford, Jr., PhD, for their critical roles as study directors in Studies 1 and 2. The studies were funded by MedImmune/AstraZeneca. The authors would like to acknowledge Rebecca Plant, MSc, of QXV Comms, Macclesfield, an Ashfield Company, part of UDG Healthcare plc, for medical writing support that was funded by MedImmune/AstraZeneca, Cambridge, UK, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Nicholson SM, Casey KA, Gunsior M, et al. The enhanced immunopharmacology of VIB4920, a novel Tn3 fusion protein and CD40L antagonist, and assessment of its safety profile in cynomolgus monkeys. Br J Pharmacol. 2020;177:1061–1076. 10.1111/bph.14897
Present Address: Kerry A. Casey, Bristol‐Meyers Squibb, Princeton, NJ, USA.
Michele Gunsior, Viela Bio, Gaithersburg, MD, USA.
Stacey Drabic, Gilead Sciences, Foster City, CA, USA.
Hailey Cook, Reaction Biology Corporation, Malvern, PA, USA.
Rakesh Dixit, Bionavigen, Gaithersburg, MD, USA.
Contributor Information
Simone M. Nicholson, Email: simone.nicholson@astrazeneca.com.
Michele Gunsior, Email: gunsiorm@vielabio.com.
REFERENCES
- ACADEMIES, N. R. C. O. T. N (2011). Guide for the care and use of laboratory animals. Washington D.C.: The National Academies Press. [Google Scholar]
- Albulescu, M. (2016). Safety, tolerability, and dose‐dependent inhibition of T‐cell‐dependent antibody response with MEDI4920, a novel, engineered CD40L antagonist: Results of a single‐ascending dose study in healthy volunteers. American College of Rheumatology. Abstract 1610 [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , & CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Faccenda, E. , … Collaborators, C. G. T. P. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloui, C. , Prigent, A. , Sut, C. , Tariket, S. , Hamzeh‐Cognasse, H. , Pozzetto, B. , … Garraud, O. (2014). The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. International Journal of Molecular Sciences, 15, 22342–22364. 10.3390/ijms151222342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- André, P. , Prasad, K. S. S. , Denis, C. V. , He, M. , Papalia, J. M. , Hynes, R. O. , … Wagner, D. D. (2002). CD40L stabilizes arterial thrombi by a β3 integrin‐dependent mechanism. Nature Medicine, 8, 247–252. [DOI] [PubMed] [Google Scholar]
- Boumpas, D. T. , Furie, R. , Manzi, S. , Illei, G. G. , Wallace, D. J. , Balow, J. E. , … GROUP, B. G. L. N. T (2003). A short course of BG9588 (anti‐CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis and Rheumatism, 48, 719–727. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, S. , Varghese, S. , Vitseva, O. , Tanriverdi, K. , & Freedman, J. E. (2005). CD40 ligand influences platelet release of reactive oxygen intermediates. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 2428–2434. [DOI] [PubMed] [Google Scholar]
- Chamberlain, C. , Colman, P. J. , Ranger, A. M. , Burkly, L. C. , Johnston, G. I. , Otoul, C. , … Hiepe, F. (2017). Repeated administration of dapirolizumab pegol in a randomised phase 1 study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Annals of Rheumatic Disease, 76, 1837–1844. [DOI] [PubMed] [Google Scholar]
- Cordoba, F. , Wieczorek, G. , Audet, M. , Roth, L. , Schneider, M. A. , Kunkler, A. , … Rush, J. S. (2015). A novel, blocking, Fc‐silent anti‐CD40 monoclonal antibody prolongs nonhuman primate renal allograft survival in the absence of B cell depletion. American Journal of Transplantation, 15, 2825–2836. [DOI] [PubMed] [Google Scholar]
- Danese, S. , Sans, M. , & Fiocchi, C. (2004). The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut, 53, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong, Y. P. , Comiskey, M. , Kalled, S. L. , Mizoguchi, E. , Flavell, R. A. , Bhan, A. K. , & Terhorst, C. (2000). Chronic murine colitis is dependent on the CD154/CD40 pathway and can be attenuated by anti‐CD154 administration. Gastroenterology, 119, 715–723. [DOI] [PubMed] [Google Scholar]
- De Silva, N. S. , & Klein, U. (2015). Dynamics of B cells in germinal centres. Nature Reviews. Immunology, 15, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgueta, R. , Benson, M. J. , De Vries, V. C. , Wasiuk, A. , Guo, Y. , & Noelle, R. J. (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunological Reviews, 229, 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgueta, R. , Marks, E. , Nowak, E. , Menezes, S. , Benson, M. , Raman, V. S. , … Noelle, R. J. (2015). CCR6‐dependent positioning of memory B cells is essential for their ability to mount a recall response to antigen. Journal of Immunology, 194, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger, R. (2019). VIB4920, a novel CD40L antagonist, decreased disease activity and improved biomarkers of immune activation in a phase 1b, proof‐of‐concept study in theumatoid arthritis. Keystone Symposia, Colorado USA: B Cell‐T Cell Interactions. [Google Scholar]
- Harding, S. , Sharman, J. , Faccenda, E. , Southan, C. , Pawson, A. , Ireland, S. , et al. (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn, V. , Slupsky, J. R. , Grafe, M. , Anagnostopoulos, I. , Förster, R. , Müller‐Berghaus, G. , & Kroczek, R. A. (1998). CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature, 391, 591–594. [DOI] [PubMed] [Google Scholar]
- Howard, L. M. , Dal Canto, M. C. , & Miller, S. D. (2002). Transient anti‐CD154‐mediated immunotherapy of ongoing relapsing experimental autoimmune encephalomyelitis induces long‐term inhibition of disease relapses. Journal of Neuroimmunology, 129, 58–65. [DOI] [PubMed] [Google Scholar]
- Howard, L. M. , Neville, K. L. , Haynes, L. M. , Dal Canto, M. C. , & Miller, S. D. (2003). CD154 blockade results in transient reduction in Theiler's murine encephalomyelitis virus‐induced demyelinating disease. Journal of Virology, 77, 2247–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W. , Sinha, J. , Newman, J. , Reddy, B. , Budhai, L. , Furie, R. , … Davidson, A. (2002). The effect of anti‐CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis and Rheumatism, 46, 1554–1562. [DOI] [PubMed] [Google Scholar]
- Inwald, D. P. (2003). CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circulation Research, 92, 1041–1048. [DOI] [PubMed] [Google Scholar]
- Iverson, W. , Karanth, S. , Wilcox, A. , Pham, C. , Lockhart, S. , & Nicholson, S. (2018). Talaromycosis (penicilliosis) in a cynmolgus macaque. Veterinary Pathology, 55, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell, J. , Albulescu, M. , Drabic, S. , Wang, L. , Moate, R. , Baca, M. , … Drappa, J. (2019). A CD40L‐targeting protein, reduces autoantibodies and improves disease activity in autoimmune patents. Science Translational Medicine, 11, 489–500. [DOI] [PubMed] [Google Scholar]
- Kawabe, T. , Matsushima, M. , Hashimoto, N. , Imaizumi, K. , & Hasegawa, Y. (2011). CD40/CD40 ligand interactions in immune responses and pulmonary immunity. Nagoya Journal of Medical Science, 73, 69–78. [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komura, K. , Fujimoto, M. , Yanaba, K. , Matsushita, T. , Matsushita, Y. , Horikawa, M. , … Sato, S. (2008). Blockade of CD40/CD40 ligand interactions attenuates skin fibrosis and autoimmunity in the tight‐skin mouse. Annals of the Rheumatic Diseases, 67, 867–872. [DOI] [PubMed] [Google Scholar]
- Li, J. (2016). Pharmacokinetics, pharmacodynamics, and immunogenicity of MEDI4920, a novel, engineered CD40 ligand antagonist, in healthy volunteers. American College of Rheumatology. Abstract 1620 [Google Scholar]
- Maclaren, A. , Levin, N. , Lowman, H. , & Trikha, M. (2017). Trph‐222, a novel anti‐CD22 antibody drug conjugate (ADC), has significant anti‐tumor activity in NHL xenografts and is well tolerated in non‐human primates. Blood, 130(Issue Supplement 1), 4105. [Google Scholar]
- Mahmoud, T. I. , Wang, J. , Karnell, J. L. , Wang, Q. , Wang, S. , Naiman, B. , … Ettinger, R. (2016). Autoimmune manifestations in aged mice arise from early‐life immune dysregulation. Science Translational Medicine, 8, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabet, M. , Barrabes, J. A. , Quiroga, A. , & Garcia‐Dorado, D. (2008). Platelet pro‐aggregatory effects of CD40L monoclonal antibody. Molecular Immunology, 45, 937–944. [DOI] [PubMed] [Google Scholar]
- Natkunam, Y. 2007. The biology of the germinal center. Hematology Am Soc Hematol Educ Program , 210–5. [DOI] [PubMed]
- Oganesyan, V. , Ferguson, A. , Grinberg, L. , Wang, L. , Phipps, S. , Chacko, B. , … Baca, M. (2013). Fibronectin type III domains engineered to bind CD40L: Cloning, expression, purification, crystallization and preliminary X‐ray diffraction analysis of two complexes. Acta Crystallographica. Section F, Structural Biology and Crystallization Communications, 69, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenass‐Lechner, F. , Staack, R. F. , Mary, J. L. , Richter, W. F. , Winter, M. , Jordan, G. , … Atzpodien, E. A. (2016). Immunogenicity, inflammation, and lipid accumulation in cynomolgus monkeys infused with a lipidated tetranectin‐ApoA‐I fusion protein. Toxicological Sciences, 150, 378–389. [DOI] [PubMed] [Google Scholar]
- Robles‐Carrillo, L. , Meyer, T. , Hatfield, M. , Desai, H. , Davila, M. , Langer, F. , … Amirkhosravi, A. (2010). Anti‐CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. Journal of Immunology, 185, 1577–1583. [DOI] [PubMed] [Google Scholar]
- Schonbeck, U. , & Libby, P. (2001). The CD40/CD154 receptor/ligand dyad. Cellular and Molecular Life Sciences, 58, 4–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock, A. , Burkly, L. , Wakefield, I. , Peters, C. , Garber, E. , Ferrant, J. , … Weir, N. (2015). CDP7657, an anti‐CD40L antibody lacking an Fc domain, inhibits CD40L‐dependent immune responses without thrombotic complications: An in vivo study. Arthritis Research & Therapy, 17, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan, D. , Krautler, N. J. , Maag, J. L. V. , Butt, D. , Bourne, K. , Hermes, J. R. , … Brink, R. (2017). CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light‐zone location and predominant low antigen affinity. Immunity, 47, 1142–1153. [DOI] [PubMed] [Google Scholar]
- Thorbecke, G. J. , Amin, A. R. , & Tsiagbe, V. K. (1994). Biology of germinal centers in lymphoid tissue. The FASEB Journal, 8, 832–840. [DOI] [PubMed] [Google Scholar]
- Tocoian, A. , Buchan, P. , Kirby, H. , Soranson, J. , Zmzcona, M. , Walley, R. , … Oliver, R. (2015). First‐in‐human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti‐CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus, 24, 1045–1056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information