

Abstract
In recent years, several novel aspects of GPCR pharmacology have been described, which are thought to play a role in determining the in vivo efficacy of a compound. Fluorescent ligands have been used to study many of these, which have also required the development of new experimental approaches. Fluorescent ligands offer the potential to use the same fluorescent probe to perform a broad range of experiments, from single‐molecule microscopy to in vivo BRET. This review provides an overview of the in vitro use of fluorescent ligands in further understanding emerging pharmacological paradigms within the GPCR field, including ligand‐binding kinetics, allosterism and intracellular signalling, along with the use of fluorescent ligands to study physiologically relevant therapeutic agents.
Abbreviations
- BRET
Bioluminescence resonance energy transfer
- FCS
fluorescence correlation spectroscopy
- HILO
highly inclined and laminated optical sheet
- RET
resonance energy transfer
- TR‐FRET
time‐resolved FRET
1. INTRODUCTION
GPCRs are membrane proteins characterised by seven transmembrane spanning domains, an extracellular N‐terminus, and an intracellular C‐terminus and are responsible for the transduction of an extracellular stimulus into an intracellular response. Accounting for approximately 4% of the protein‐encoding human genome (Bjarnadottir et al., 2006), they represent the largest family of proteins targeted by drugs in clinical use, with approximately 30% of all drugs targeting a GPCR. However, these targeted GPCRs represent only 30% of the non‐olfactory GPCR population, and approximately 16% of all GPCRs in the human genome (Sriram & Insel, 2018), providing ample further opportunity for drug development.
Over the past 20 years, a number of novel facets of GPCR pharmacology have been described, which considerably extend the ternary complex model of GPCR activation (De Lean, Stadel, & Lefkowitz, 1980) and which play a significant role in ultimately determining the in vivo efficacy of a given compound. These include biased agonism (Kenakin & Christopoulos, 2013), binding kinetics (Swinney, Haubrich, Van Liefde, & Vauquelin, 2015), allosterism (Wang, Martin, Brenneman, Luttrell, & Maudsley, 2009), receptor dimerisation (Guidolin, Agnati, Marcoli, Borroto‐Escuela, & Fuxe, 2015), and signalling from intracellular compartments (Thomsen, Jensen, Hicks, & Bunnett, 2018; Figure 1). The study of each of these phenomena has required the development of new experimental approaches, which, in many cases, have utilised fluorescently tagged ligands.
Figure 1.

Schematic of emerging paradigms in GPCR pharmacology. (a) Biased agonism, whereby a ligand binding to the receptor stabilises a receptor conformation that selectively couples one effector protein over another, in this case β‐arrestin over heterotrimeric G proteins. (b) Ligand binding to the same receptor can have different binding rate constants, and there is a growing understanding that many clinically used drugs have non‐equilibrium binding kinetics. (c) Receptor dimerisation (either homodimers or heterodimers) can alter the localisation, ligand affinity, and signalling of the respective protomers. (d) Finally, there is increasing evidence of GPCRs signalling from intracellular compartments. This has been shown at the early endosome following receptor internalisation. (e) Interestingly, there is also evidence of active GPCRs signalling from the Golgi apparatus prior to trafficking to cell membrane
Fluorescent ligands for GPCRs are composed of an agonist or antagonist that is chemically linked to a fluorescent molecule (Vernall, Hill, & Kellam, 2014). The attachment of a fluorescent molecule can drastically alter the pharmacology of a compound, such as a change in selectivity (Vernall et al., 2013) or a significant loss in affinity compared with the parent compound (Baker et al., 2010; Sexton et al., 2011). A variety of fluorophores can be attached to the parent molecule, depending on the final application of the ligand, and these can vary significantly in size (Kuder & Kieć‐Kononowicz, 2014; Vernall et al., 2014). In many cases, fluorophores that emit in near‐IR wavelengths are favoured, such as Cy5 and BODIPY630/650. This avoids interference with autofluorescence from endogenous proteins and spectral crossover with commonly used fluorescent proteins such as GFP. With the development of better molecular models, based on the high‐resolution structural data for a range of GPCRs, there is a greater understanding of where a fluorescent molecule can be attached to extend out of the binding pocket and minimise its effect on ligand affinity (Stoddart et al., 2018). Although many fluorescent ligands are still custom‐synthesised in academic and industrial settings, a number are now available commercially. This increase in availability, coupled with improvements in affinity and selectivity, has widened their suitability for a range of techniques from single‐molecule microscopy to high‐throughput binding assays, leading to a significant rise in their use. Although there have been several reviews on fluorescent ligands for GPCRs (Sridharan, Zuber, Connelly, Mathew, & Dumont, 2014; Stoddart, Kilpatrick, Briddon, & Hill, 2015), the rapid progress of research in both ligand design and GPCR pharmacology warranted an updated discussion, and therefore, this review will focus on publications in this area from the last eight years. We discuss the diverse ways that fluorescent ligands have been used experimentally to probe emerging paradigms in GPCR pharmacology with an emphasis on ligand‐binding kinetics, allosteric modulation, and ligand bias.
2. EQUILIBRIUM BINDING AND COMPOUND LIBRARY SCREENING
One of the initial steps in the identification of a new drug‐like molecule for a GPCR is to determine its affinity for the receptor of interest. Affinities of unlabelled compounds are often determined through the use of equilibrium competition ligand‐binding experiments, whereby the effect of increasing concentrations of a test compound on the binding of a labelled compound is measured (Hulme & Trevethick, 2010). The principles of competition binding assays are the same whether a radiolabelled or fluorescently labelled ligand is used, and the ultimate aim is to estimate the amount of ligand bound. For radioligands, the amount of ligand bound is typically measured using scintillation counting, which often requires separation of bound and free ligand, whereas there are many ways to measure the amount of fluorescent ligand bound (Figure 2). Methods using fluorescent ligands generally fall into two categories: direct measurement of fluorescence or indirect measurement using resonance energy transfer (RET; Stoddart, White, Nguyen, Hill, & Pfleger, 2016). Both of these techniques have been applied to measure the binding of known ligands for a variety of receptors and in general have found good agreement of affinity constants with radioligand‐binding assays (Bosma et al., 2019; Leyris et al., 2011; Nederpelt et al., 2016; Soave, Stoddart, Brown, Woolard, & Hill, 2016). This has allowed fluorescence‐based assays to be accepted as a robust way to measure affinity.
Figure 2.

Measuring ligand binding using fluorescence‐ or radioactive‐based approaches. Using untagged receptors, radioligand‐binding assays rely on the use of scintillation proximity technologies, or the separation of bound ligand from free ligand, followed by the detection of bound ligand using scintillation counters. Fluorescent ligands offer more versatility in the approaches that can be taken. Binding to unlabelled GPCRs can be monitored through microscopy, using plate readers to detect fluorescence, or through flow cytometry. Alternatively, the binding of fluorescent ligands to SNAP‐tagged or NanoLuc‐tagged GPCRs can be detected with RET approaches (FRET and BRET, respectively). These techniques often rely on plate readers to detect ligand binding, opening up the possibility of increasing the experimental throughput and miniaturisation
Drug discovery often relies on the ability to screen large compound libraries at a protein or receptor target. Although efforts in the literature have not been focused on miniaturisation, fluorescence‐based binding assays can be easily used in both 96‐well (Stoddart et al., 2012) and 384‐well formats (Zwier et al., 2010). It has been noted that RET‐based methods such as time‐resolved FRET (TR‐FRET) and BRET may be more suited to high‐throughput screening due to their higher signal‐to‐noise ratios and lower non‐specific signal compared with direct fluorescence measurement‐based methods (Cottet et al., 2011). These high signal‐to‐noise ratios mean fluorescent ligands can be used at much higher concentrations than radioligands, and consequently, ligands with relatively low affinity (K D ~500 nM) have been successfully used in a BRET‐based binding assay for the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=324 (Conroy et al. 2018). Fluorescent ligands have been used in a number of small‐scale screens (~1,000 compounds) to successfully identify novel compounds that bind to the https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=23 (Young et al., 2005), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=36 (Iturrioz et al., 2010), and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=21 (Arruda et al., 2017). These studies demonstrate the potential for fluorescent ligands to be used in screening large compound libraries at a GPCR, without the need for radioligands.
Fragment‐based drug discovery is becoming a widely used tool in drug discovery and is based on screening a large number of low molecular weight (MW) compounds (typically less than 300 Da), with the aim of chemically expanding hit molecules into a lead compound with a high complexity with a relatively lower MW (Hopkins, Groom, & Alex, 2004). Fluorescence‐based assays have been used to screen a small library of fragment‐like molecules at the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=18 and A3 receptors and were successful in identifying a number of fragment molecules with moderate affinity for both receptors (Stoddart et al., 2012). In addition, this fluorescence‐based study used intact, living cells for screening compounds in contrast to traditional fragment‐based drug discovery methods that often used purified receptors. The use of purified receptors can expose sites in the receptor that are normally unavailable for ligand binding when the protein is within a membrane environment and lead to higher levels of false positive results. The use of a live‐cell system can overcome some of these limitations and determine compound binding to a receptor in its native environment. The ability to screen low MW compounds using fluorescence assays has been further expanded to identify compounds that are selective for the A3 receptor over the A1 receptor (Ranganathan, Stoddart, Hill, & Carlsson, 2015). Using in silico models of the A3 receptor and A1 receptor, based on the then available crystal structure of the closely related https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=19, >0.5 million fragments were docked into these models. From this in silico modelling, a very small number of identified binders (21) were screened in the live‐cell fluorescence ligand‐binding assay, and eight were found to have medium affinity at the A3 receptor (K i < 10 μM) with one compound displaying 29‐fold selectivity over the A1 receptor. Further elaboration of the hit compound using the in silico models resulted in a molecule with high affinity for the A3 receptor and over 100‐fold selectivity over the A1 receptor (Ranganathan et al., 2015). A similar approach has recently used virtual screening that was used to identify compounds that bind to the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=71 (Adlere et al., 2019). In this study, the virtual hits were confirmed using a combination of radioligand and BRET ligand‐binding assays, and there was a good correlation between the binding affinities obtained from both assays. Low MW compounds are thought to be a more attractive starting point in the development of a candidate molecule for clinical trials, and through the use of fluorescent ligand, it has been shown that they can be identified to bind to GPCRs in live cells, and when coupled with in silico modelling, this may be a powerful tool to develop new therapeutically active molecules.
3. LIGAND‐BINDING KINETICS
There is a growing understanding that measuring the affinity at equilibrium may not be the best predictor of the in vivo efficacy of a compound, as a number of clinically approved drugs show non‐equilibrium modes of binding (Hothersall, Brown, Dale, & Rawlins, 2016; Kola & Landis, 2004; Schuetz et al., 2017; Waring et al., 2015). Equilibrium affinity is a function of the association and dissociation rate constants (K D = k off/k on) of a molecule binding to a receptor, and compounds with the same affinity can have different association and dissociation rates. Therefore, understanding a molecule's ligand‐binding kinetics and incorporating this into the early stages of drug discovery may improve predictions of in vivo efficacy (Copeland, 2016; Schuetz et al., 2017). In some cases, such as for the use of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 agonists in treatment of chronic obstructive pulmonary disease, compounds with slow k off have been shown to have better clinical outcomes (Beeh et al., 2015). Whereas in other clinical settings, such as in the targeting of the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 in the treatment of schizophrenia, anti‐psychotics with fast k off are thought to avoid long‐term side effects (Kapur & Seeman, 2001). As with competition binding assays, ligand‐binding kinetics has historically been quantified using radioligands. Multiple time points are essential to estimate kinetic rates, and in a radioligand‐binding assay, a separate assay point is required for each time point. This can make them time consuming to perform and not well suited to high‐throughput screening. Both direct measurement of fluorescence and RET‐based fluorescent ligand‐binding assays can be performed in real time using microplate readers or confocal microscopes with measurements as frequently as every second, if required, and have been widely adopted in the past 5 years to measure binding kinetics at GPCRs (Table 1 and references within table).
Table 1.
Summary of GPCRs at which binding kinetics have been measured using fluorescent ligands
| Receptor | Fluorescent ligand | Assay format | Kinetics of unlabelled ligands determined? | Reference |
|---|---|---|---|---|
| Adenosine A1 receptor | CA200645 | NanoBRET | No | Stoddart, Johnstone, et al., 2015 |
| Adenosine A2B receptor | XAC‐X‐BY630 | NanoBRET | No | White et al., 2019 |
| Adenosine A3 receptor | XAC‐ser‐tyr‐X‐BY630, AV039 | NanoBRET | Yes | Bouzo‐Lorenzo et al., 2019 |
| β2‐Adrenoceptor | Propranolol‐ala‐ala‐BY630 | NanoBRET | No | Alcobia et al., 2018 |
| Calcitonin receptor | sCT8‐32:AF568 | Fluorescence polarisation | Yes | Furness et al., 2016 |
| Dopamine D2 receptor | PPHT‐Red | TR‐FRET | Yes | Klein Herenbrink et al., 2016; Sykes et al., 2017 |
| Formyl peptide receptors | Fluorescent fMLP derivative | Flow cytometry | Yes | Sklar et al., 1985 |
| Free fatty acid receptor 1 | Fluorescent derivative of TUG905 | NanoBRET | No | Christiansen, Hudson, Hansen, Milligan, & Ulven, 2016 |
| Free fatty acid receptor 2 | Fluorescent derivative of TUG‐1609 | NanoBRET | Yes | Hansen et al., 2017 |
| Gonadotropin‐releasing hormone receptor | Fluorescent buserelin | TR‐FRET | Yes | Nederpelt et al., 2016 |
| Histamine H1 receptor | mepyramine‐ala‐ala‐BY630 (AV082), Gmep | NanoBRET, TR‐FRET | Yes | Schiele et al., 2015; Stoddart et al., 2018; Bosma et al., 2019 |
| Histamine H3 receptor | Bodilisant | Fluorescence polarisation | Yes | Reiner & Stark, 2019 |
| Histamine H4 receptor | Clobenpropit‐BY630 | NanoBRET | Yes | Mocking, Verweij, Vischer, & Leurs, 2018 |
| Melanocortin 4 receptor | UTBC101, UTBC102 | Fluorescence anisotropy | Yes | Link et al., 2017 |
| Relaxin family peptide receptor 1 | TAMRA‐relaxin | NanoBRET | No | Hoare et al., 2019 |
| Oxytocin receptor | Fluorescein and DY647 derivatives of PF‐3274167 | TR‐FRET | No | Karpenko et al., 2015 |
Fluorescent ligand‐based techniques have made extensive use of the framework developed by Motulsky and Mahan (1984) to measure the binding kinetics of unlabelled ligands. In fact, one of the first applications of this method used a fluorescent peptide ligand to determine the binding kinetics of ligands to the formyl peptide receptor endogenously expressed on human neutrophils using flow cytometry (Sklar, Sayre, McNeil, & Finney, 1985). The Motulsky and Mahan method requires the kinetic (k on and k off) parameters of a labelled ligand (radiolabelled or fluorescent‐labelled) to be predetermined, and then by measuring the effect of an unlabelled ligand on these rates, the binding kinetics of the unlabelled ligand can be determined (Figure 3; see Bosma et al., 2019, for the Motulsky–Mahan equation used to simulate these data). Due to the ease of collecting larger datasets with fluorescence techniques, there have been several recent papers comparing the binding kinetics of unlabelled ligands measured using radioligand‐ and fluorescence‐based methods (Bosma et al., 2019; Bouzo‐Lorenzo et al., 2019; Nederpelt et al., 2016). Using the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=256, Nederpelt et al. (2016) compared the binding kinetics of a number of agonists measured in radioligand and TR‐FRET assays. In this study, measured dissociation rates (k off) at the gonadotropin‐releasing hormone receptor were compared well across both assays, whereas the correlation in association rates (k on) was poor. For the adenosine A3 receptor, a comparison of radioligand and NanoBRET (BRET with the small bright luciferease Nanoluc as the donor) assays using labelled ligands with markedly different binding kinetics suggested that it is difficult to determine the kinetics of fast binding unlabelled ligands when using a probe with a very slow off rate (Bouzo‐Lorenzo et al., 2019). Using the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=262 as a model system, Bosma et al. (2019) compared the binding kinetics of a number of unlabelled antagonists using two radioligands and both NanoBRET and TR‐FRET fluorescence‐based assays. This large dataset allowed multiple comparisons to be made and found that for the H1 receptor, the association rates (k on) compared well across all three assay set‐ups, and there was good correlation between the dissociation rates measured in the radioligand and TR‐FRET assays but not with the NanoBRET assays. By examining the accuracy of the binding rate constants (using the magnitude of error in the fit of the data to the non‐linear equations), it was found that the error for both k on and k off of unlabelled ligands increased if they had considerably faster or slower binding rate constants than the labelled ligand (Bosma et al., 2019).
Figure 3.
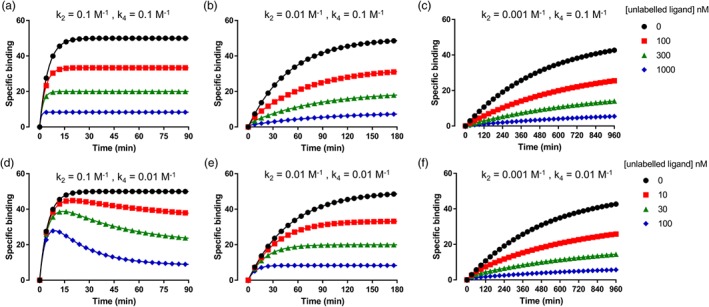
Simulated competition association binding curves with increasing k off values of the labelled ligand simulations were generated in GraphPad Prism using the Motulsky and Mahan equation. k on (both k 1 and k 3) was set to 1 × 106 M−1·min−1 for all simulated data (see Bosma et al., 2019, for full equation on the Motulsky–Mahan model). For (a, d), k off (M−1) of the labelled ligand (k 2) was set to 0.1, (b, e) to 0.01, and (c, f) to 0.001, simulating a 10‐fold increase in k off each time and a “fast,” “slow,” and “very slow” labelled ligand. The concentration of labelled ligand (L) was set to the K D of each ligand (100 nM for a and d, 10 nM for b and e, and 1 nM for c and f). For (a–c), k off of the unlabelled ligand (k 4) was set to 0.1, and for (d‐f), it was set to 0.01, simulating an unlabelled ligand with a “fast” and “slow” k off. The concentration of the unlabelled ligand was set to 1, 3, and 10 times its K D (100, 300, and 1,000 nM for a–c and 10, 30, and 100 nM for d‐f). In (d), the classic “overshoot” of an unlabelled ligand with a slower k off than the labelled ligand can be observed, whereas when a labelled ligand with a slower k off value is used, it becomes more difficult to distinguish between “slow” and “fast” unlabelled ligand
These data from both the H1 receptor and the A3 receptor indicate that the kinetics of the probe ligand can mask the binding kinetics of the unlabelled ligand if they are significantly different from those of the probe. This may help to explain the data with the gonadotropin‐releasing hormone receptor, as there was a 50‐fold difference in the association rate of the radioligand compared with the fluorescent ligand. As demonstrated by the simulated data in Figure 3 using the Motulsky and Mahan equations, binding rates of unlabelled ligands with significantly faster kinetics than the labelled ligand can be difficult to distinguish even with 10‐fold difference in their k off values. As the Motulsky and Mahan method is the most commonly used method to measure binding kinetics of unlabelled ligands, the above examples emphasise the importance of selecting a labelled probe with the appropriate kinetics for accurate determination of test compounds. It is also becoming clear that a probe with fast binding kinetics may be more generally applicable for measuring the kinetics of an unlabelled ligand (Bosma et al., 2019; Sykes, Jain, & Charlton, 2019). As radioligands are only subtly different from the parent pharmacophore, differences between radioligand probe binding kinetics are often less pronounced. Fluorescent probes differ much more widely in their chemical structure as a result of using chemically diverse fluorophores and linkers. Consequently, marked differences have been observed in the binding kinetics of fluorescent ligands with the same pharmacophore but differing fluorescent labels and linker compositions (Bouzo‐Lorenzo et al., 2019). With increasing knowledge of the structural and chemical factors influencing ligand‐binding kinetics, rationale design of fluorescent ligands with the required binding kinetics for specific compound sets using the Motulsky and Mahan method may become possible.
Investigating binding kinetics using fluorescence ligands can also reveal interesting aspects of GPCR pharmacology. The https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=351 (RXFP1) contains leucine‐rich repeats in its N‐terminus with which the endogenous ligand https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1989 interacts (Bullesbach & Schwabe, 2005). Additional interaction points have been identified in an extracellular linker region (Sethi et al., 2016). To understand the interplay of these regions on binding of the ligand, Hoare et al. (2019) used a fluorescently labelled relaxin in a kinetic NanoBRET assay. Using kinetic assays, the authors uncovered the multi‐step binding of relaxin to the RXFP1 receptor. By first assuming a single‐step binding mechanism, they noticed a difference in the affinity of relaxin obtained under kinetic association experiments compared with the equilibrium saturation assays. Upon further investigation, relaxin was shown to exhibit a two‐phase dissociation profile at the RXFP1 receptor, indicative of a multi‐step binding process. In addition, the authors demonstrated that the receptor had at least two relaxin‐bound states and that, when the linker region was removed, a single‐step binding mechanism was achieved confirming that this region is important for ligand binding (Hoare et al., 2019). The ability to gain detailed time‐resolved NanoBRET data on fluorescent ligand binding to the receptor was crucial for the interpretation of these data, and this would have been much more challenging to obtain with a traditional radioligand‐binding assay. An important recent study of dopamine D2 receptor related binding kinetics to drug effects at this receptor. The D2 receptor is the main target of antipsychotic drugs used in the treatment of schizophrenia, but extrapyramidal motor side effects caused by these compounds often limit their use (Miyamoto, Miyake, Jarskog, Fleischhacker, & Lieberman, 2012). Sykes et al. (2017) measured the binding kinetics of a wide range of antipsychotic drugs using a fluorescent D2‐antagonist in a high‐throughput TR‐FRET‐based method and correlated these measured to side effects. They demonstrated that side effects correlated with association rates rather than dissociation rates (Sykes et al., 2017). This knowledge may help to direct future development of novel antipsychotics with optimal binding kinetics with the aim of reducing side effects. The increase in throughput associated with fluorescence‐based kinetic assays allows much larger data sets to be generated and enables insights into both basic and therapeutic pharmacology of GPCRs.
4. INFLUENCE OF BINDING KINETICS ON BIASED AGONISM
Many GPCRs have been suggested to exhibit biased agonism (Chen et al., 2012; Chen et al., 2013; Rankovic, Brust, & Bohn, 2016), namely, the ability of ligands acting at the same GPCR to stabilise different active conformations of the receptor. Typically, this has been characterised by the ability of a ligand to preferentially signal via G protein‐ or β‐arrestin‐mediated pathways, often measured with endpoint assays after a given stimulation time. The ability to use biased ligands to activate a given pathway has been potentially linked to improved therapeutic outcomes (Bohn et al., 1999; Rominger, Cowan, Gowen‐MacDonald, & Violin, 2014).
A number of studies have described biased ligands at the D2 receptor, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=34, as having improved antipsychotic efficacy, but there have been contradictory studies as to the exact nature of this signalling bias (Allen et al., 2011; Masri et al., 2008; Szabo, Klein Herenbrink, Christopoulos, Lane, & Capuano, 2014; Urban, Vargas, von Zastrow, & Mailman, 2007). Many of these studies compare multiple endpoint signalling readouts from assays performed using different incubation times. To reconcile these discrepancies, Klein Herenbrink et al. (2016) considered the influence of ligand‐binding kinetics on perceived biased agonism. Using a fluorescently labelled D2 receptor agonist in a TR‐FRET‐based experimental system, it was possible to monitor the binding kinetics of a panel of dopamine ligands. The ligands with slower dissociation kinetics as determined with this approach (including aripiprazole) changed their apparent G protein versus β‐arrestin bias profile over time (Klein Herenbrink et al., 2016). The data presented by Klein Herenbrink and colleagues suggested that the perceived biased agonism of compounds such as aripiprazole at the D2 receptor was in fact due to differences in ligand‐binding kinetics and differences in timings of the endpoint assays.
This is not a universal phenomenon, however, and not all observations of ligand bias can be explained by differences in binding kinetics. For the human https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=48&familyId=11&familyType=GPCR, it has been shown that the human and salmon variants of the endogenous peptide https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=685 display differential potency and efficacy at the cAMP pathway (Andreassen et al., 2014). In contrast to the example of the D2 receptor, kinetic assays using a fluorescent version of salmon calcitonin and chimeric peptides confirmed that the differential effects of human and salmon calcitonin were not due to differences in their binding kinetics (Furness et al., 2016). Signalling differences between these two variants of calcitonin arose instead from agonist‐dependent differences in the conformational change induced in the receptor–G protein complex. These two examples highlight the importance of first determining ligand‐binding kinetics before ascribing biased agonism. The relative ease of measuring binding kinetics using fluorescent ligands should allow binding kinetics to be evaluated routinely for ligands suggested to display biased agonism in endpoint signalling assays.
5. GPCR ALLOSTERISM
Ligands that bind to a GPCR on a topographically distinct site from the orthosteric ligand are termed allosteric modulators. They can elicit a conformational change in the receptor that modulates the binding of orthosteric ligands (Kenakin, 2012; May, Leach, Sexton, & Christopoulos, 2007). This so‐called cooperativity between the two ligand‐binding sites is a central principle of allosterism (May et al., 2007), and the effect of the allosteric modulator on the affinity or efficacy of an orthosteric ligand is reciprocal. Furthermore, the reciprocity of the interaction gives rise to the phenomenon of probe dependence whereby the extent and nature of allosteric modulation can differ depending on the orthosteric ligand used (Kenakin, 2012).
As with biased agonism, research into GPCR allosterism has been facilitated by specialised receptor‐specific fluorescent ligand probes. For the adenosine A1 receptor, negative allosteric modulators have a potential therapeutic application in conditions where high levels of adenosine are released, such as after an ischaemic incident. To understand how these molecules interact with the A1 receptor bound to compounds with different intrinsic efficacies, Cooper et al. (2019) used NanoBRET saturation binding assays to directly measure the effect of the positive allosteric modulators VCP171 (Aurelio et al., 2009) and PD 81,723 (Bruns & Fergus, 1990) on fluorescent agonist and antagonist binding to human and rat A1 receptors. The fluorescent agonists used in this study were derived from the endogenous A1 receptor ligand, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2844 (ABA‐X‐BY630) or from the full agonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=377 (ABEA‐X‐BY630, and AAG‐ABEA‐X‐BY630), and the fluorescent antagonist was based on the non‐selective adenosine receptor antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=404 (CA200645). While the allosteric modulators had little effect on the binding of the fluorescent antagonist, they enhanced the affinity of both fluorescent and non‐fluorescent agonists. Interestingly, at the rat A1 receptor, VCP171 increased the maximal binding capacity (B max) with all three fluorescent agonists but only significantly increased the affinity of AAG‐ABEA‐X‐BY630 (Cooper et al., 2019). This increase in agonist‐specific affinity suggested that the allosteric modulators were allowing a larger proportion of the receptor population to adopt a high‐affinity, R*, confirmation. The availability of fluorescent ligands with differing intrinsic efficacy (i.e., agonists and antagonists) was the cornerstone of the study and with the development of more fluorescent ligands will allow similar studies to be performed on different GPCR subtypes.
The five https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=23 (M1–M5) have a principal role in the central and parasympathetic nervous system and as such have been the target of a number of drug development efforts over the past decades, including for the treatment of schizophrenia, Alzheimer's disease, and chronic obstructive pulmonary disease (Felder et al., 2018; Melancon, Tarr, Panarese, Wood, & Lindsley, 2013). These efforts have been hampered, as many of the developed ligands lack subtype selectivity due to the high conservation of the orthosteric binding site in these receptors (Kruse et al., 2013), and emphasis has been placed on the discovery of novel allosteric modulators that target less conserved allosteric sites (Felder et al., 2018). In one approach, the Ilien group developed a series of fluorescent bitopic ligands against the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13&familyId=2&familyType=GPCR. These ligands were based on the orthosteric antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=328 (Daval et al., 2013) or the known M1 allosteric modulator https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=289 (Daval et al., 2012). Pharmacological characterisation of this ligand series with FRET suggested a bitopic mode of ligand binding, with the fluorescent ligands able to occupy both the orthosteric and allosteric ligand‐binding pockets on the same M1 receptor. Altering the linker length between the pharmacophore and the fluorophore conferred differential abilities to occupy the allosteric binding site (Daval et al., 2013), demonstrating the application of allosteric fluorescent molecules for GPCR drug discovery.
However, it is not only low MW ligands that are allosteric modulators—proteins that interact with the receptor and change the pharmacology of a ligand are also considered to be allosteric modulators (Kenakin, 2012). This intermolecular allosterism can occur as a result of the receptor interacting with other membrane bound proteins, such as receptor activity‐modifying proteins (Gingell et al., 2016; Hay & Pioszak, 2016), or can be applied to intracellular signalling proteins (such as G proteins or β‐arrestin) as they also bind the receptor and result in changes in affinity or efficacy of particular agonists (Hill, May, Kellam, & Woolard, 2014; Kenakin, 2012). Of note for this review, the interactions between two or more receptors forming dimers or higher order oligomers can also be allosteric (Gherbi, May, Baker, Briddon, & Hill, 2015; Hill et al., 2014; Hiller, Kuhhorn, & Gmeiner, 2013; May, Bridge, Stoddart, Briddon, & Hill, 2011). Fluorescent ligands have been successfully used to detect the presence of GPCR heterodimers on the cell surface (Hounsou et al., 2014; Loison et al., 2012). This relies on the ability to use a fluorescent ligand to specifically label one protomer within a dimer whilst the other protomer is labelled with a fluorescent tag. However, a more nuanced approach is required for homodimers, as the fluorescent ligand in question has the potential to bind each protomer equally if there is no cooperativity. This obstacle has been tackled through the application of fluorescent ligands to continuously monitor the dissociation kinetics of the fluorescent ligand in real time using confocal microscopy coupled to continued perfusion of buffer in the presence or absence of other ligands (Gherbi et al., 2015; May et al., 2011). By using low concentrations of the fluorescent probe, it was possible to only occupy one of the protomers within a dimer. This particular methodology is highly sensitive to detecting cooperativity between two orthosteric binding sites that may exist within a GPCR dimer. May et al. (2011) used a fluorescently labelled adenosine receptor agonist (ABA‐X‐BY630) to monitor cooperativity across adenosine A1 and A3 receptor homodimers. Under these conditions, the dissociation rate constant of the fluorescent agonist was enhanced when an A3 receptor homodimer was exposed to a second ligand. This was strong evidence for negative cooperativity across the dimer interface. Mutating a key residue in the orthosteric ligand‐binding pocket of one of the dimer protomers abolished this cooperativity (May et al., 2011).
A similar approach was later taken at the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=28 to determine if the observed secondary low‐affinity conformation was a result of receptor oligomerisation. Non‐conventional β‐adrenoceptor ligands, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=532 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=91, antagonised catecholamine‐mediated responses with high affinity and were also able to stimulate a partial agonist response at much higher ligand concentrations. Gherbi et al. (2015) used BODIPY‐TMR‐CGP (a fluorescent analogue of CGP 12177; Heithier et al., 1994) to investigate whether this ability to exhibit high‐affinity antagonism with low‐affinity partial agonism was a result of cooperativity across β1‐adrenoceptor dimers. As with the adenosine A3 receptor, the dissociation rate constant of the fluorescent ligand was increased when in the presence of second “orthosteric” ligand. This negative cooperativity was abolished by mutating residues in the transmembrane domain 4 of the β1‐adrenoceptor to that of the β2‐adrenoceptor (Gherbi et al., 2015). These data suggested that the secondary low‐affinity pharmacology of the β1‐adrenoceptor was a result of negative cooperativity across a homodimer. These two studies were possible only with the ability to continuously monitor the dissociation kinetics of the tracer ligand in real time, an experimental facet that would have been technically challenging with radioligands.
6. INTRACELLULAR SIGNALLING OF GPCRs
Another exciting recent development in the GPCR field has been the discovery that receptors can signal from intracellular compartments, including endosomes (Calebiro, Godbole, Lyga, & Lohse, 2015; Irannejad et al., 2013) or the trans‐Golgi network (Godbole, Lyga, Lohse, & Calebiro, 2017). Fluorescent ligands have been used to monitor GPCR internalisation and have found a niche in labelling untagged or endogenously expressed GPCRs (Brand, Klutz, Jacobson, Fredholm, & Schulte, 2008; Lam et al., 2018; Margathe et al., 2016; Stoddart, Vernall, Briddon, Kellam, & Hill, 2015). It is important to note that the trafficking and organisation of GPCRs can change depending on the expression level (Ward, Pediani, Godin, & Milligan, 2015) and can be cell‐type specific (Eichel & von Zastrow, 2018; Werthmann, Volpe, Lohse, & Calebiro, 2012). Calebiro et al. (2015) used a fluorescently labelled https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4395 (TSH) to monitor the internalisation of the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=255 in primary murine thyroid cells. The TSH receptor is a Gs‐coupled GPCR, and thus, activation of these receptors results in an increase in cAMP production. They combined the fluorescent ligand with highly inclined and laminated optical sheet (HILO) microscopy to achieve the high degree of spatio‐temporal resolution required to visualise internalisation of TSH receptors at endogenous levels of expression. Stimulation of the TSH receptor results in a sustained increase in intracellular cAMP levels, and by blocking internalisation through the use of dynamin inhibitors, this sustained response was lost (Calebiro et al., 2015). Further work demonstrated that this sustained response mediated by internalised receptors was occurring in the trans‐Golgi network, and this localised perinuclear cAMP signalling was required for efficient CREB‐mediated gene transcription (Godbole et al., 2017). Taken together, these data revealed the ability for the TSH receptor to signal from the endosomes and Golgi apparatus in primary cells, providing real insight into the mechanisms of TSH receptor action in an endogenous system. This study was possible due to the ability to use fluorescent TSH ligand to label TSH receptors with a high degree of specificity.
7. FLUORESCENT LIGANDS FOR PHYSIOLOGICALLY RELEVANT SYSTEMS
The advent of CRISPR/Cas9‐mediated genome engineering (Cong et al., 2013) has been a major breakthrough in cell biology. CRISPR/Cas9 has enabled the genetic modification of endogenously expressed proteins, including appending bioluminescent or fluorescent tags to the C‐terminus of endogenous GPCRs (Figure 4a; White, Vanyai, See, Johnstone, & Pfleger, 2017). White, Johnstone, See, and Pfleger (2019) used CRISPR/Cas9 to generate an N‐terminally NanoLucTM‐tagged https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=20 and performed NanoBRET ligand binding in HEK293 cells. The endogenous expression level of the A2B receptor in HEK293 cells has been shown to be sufficient for a robust functional responses upon ligand stimulation (Goulding, May, & Hill, 2018). White et al. (2019) found that the luminescence generated in the genome‐edited cells was similar to those transiently transfected with a very low amount of NLuc‐A2B cDNA. However, despite this low luminescence, robust fluorescent ligand binding could be measured to the genome‐edited cells with NanoBRET. The resulting fluorescent ligand affinity was similar to that measured with HEK 293 cells transiently transfected with NLuc‐A2B cDNA (White et al., 2019). Furthermore, competition and kinetic NanoBRET experiments yielded similar ligand affinities to those determined in cells exogenously expressing NLuc‐A2B. This study represents the first time NanoBRET ligand binding has been performed at receptors expressed under endogenous promotion. This opens up the possibilities of using fluorescent ligands to interrogate ligand binding and binding kinetics to receptors expressed at endogenous levels rather than in an overexpression system.
Figure 4.
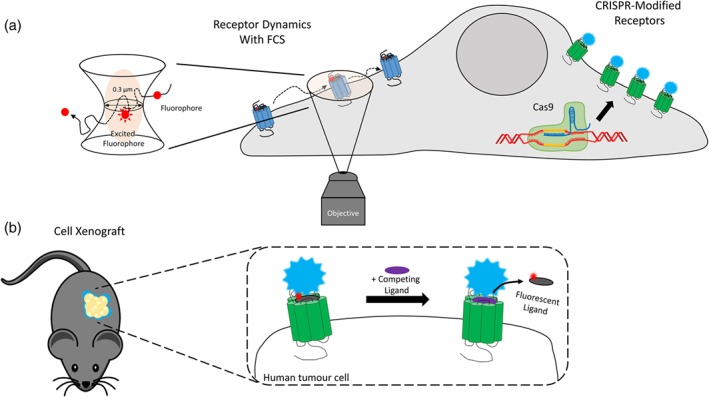
Uses of fluorescent ligands to study GPCR pharmacology in vitro at endogenously levels of expression and in vivo. in vitro uses shown in (a) include using fluorescence correlation spectroscopy to measure receptor dynamics of an endogenously expressed GPCR labelled with a fluorescent ligand. This continues with using CRISPR/Cas9 to genetically modify the GPCR with NanoLuc to perform NanoBRET binding studies. In (b) human, tumour cells stably transfected with NanoLuc‐tagged GPCRs were implanted into immune‐compromised mice as a xenograft. NanoBRET binding studies were performed to monitor fluorescent ligand binding to these GPCRs in vivo
Fluorescent ligands have been used to elucidate new roles for endogenously express GPCRs in cellular responses. In neutrophils, the adenosine A3 receptor has been shown to be a key regulator of function in response to chemoattractant stimulation and, in fixed cells, the receptor was localised in a highly polarised manner to the leading edge of the cell (Chen et al., 2006). Using a fluorescent antagonist for the A3 receptor allowed the localisation of the receptor to be monitored in live cells and further investigate the role of the receptor (Corriden et al., 2013). It was shown that the A3 receptor was localised predominantly in plaques at the base of cytonemes—filopodia‐like projections that neutrophils use to tether bacteria ahead of phagocytosis. These experiments showed the A3 receptor to have an integral role in the formation of these projections.
However, studying GPCRs at endogenous levels is technically challenging, often due to the low expression of these proteins found in many recombinant cell lines. Better understanding of GPCR dynamics requires the use of highly sensitive techniques to overcome these technical difficulties. The sensitive imaging technique of fluorescence correlation spectroscopy (FCS) represents a useful alternative experimental approach for monitoring the dynamics of GPCRs at low expression levels (Briddon & Hill, 2007; Briddon et al., 2018). Paired with selective fluorescent ligands, FCS can quantify the pharmacology of GPCRs at the subcellular level (Figure 4a). Rose, Briddon, and Hill (2012) used a fluorescent derivative of the histamine H1 receptor antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1227 to characterise endogenous H1 receptors in HeLa cells with FCS. This ligand showed high affinity for the H1 receptor, but it also displayed high levels of non‐specific binding and intracellular uptake of the ligand, which made the ligand unsuitable for the conventional confocal microscopy‐based ligand‐binding assays discussed previously (Arruda et al., 2017; Stoddart et al., 2012). However, the high sensitivity and diffraction‐limited measuring volume of FCS allowed measurements to be taken in small areas of the cell membrane in the absence of significant cytosolic contamination and detect specific binding of ligand to the endogenous H1 receptor (Rose et al., 2012). FCS measurements of the fluorescent ligand‐bound H1 receptor suggested a difference in the macromolecular organisation of the receptors in HeLa cells compared with that measured in CHO cells when the H1 receptor was overexpressed. In an orthogonal approach to study endogenous expression of GPCRs, Soethoudt et al. (2018) utilised a modified antagonist for the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=57 that contained both a photoaffinity label and a ligation handle. The photoaffinity label covalently cross‐linked the antagonist to the receptor upon UV irradiation, and the ligation handle allowed the conjugation of the antagonist, which is attached to the receptor, to a fluorophore. This permanent labelling approach allowed the expression of CB2 receptors in peripheral blood mononuclear cells to be profiled using FACS analysis, and they demonstrated that the CB2 receptor was highly expressed in CD19+ B cells. These studies demonstrate that through the use of advanced imaging and labelling technologies, fluorescent ligands can be used to study GPCRs in endogenously expressing systems.
Although fluorescent ligands have been used to probe GPCR pharmacology in in vitro systems with great success, their ability to be used in vivo has been limited. Recently, however, several groups have used fluorescent ligands to monitor GPCR target engagement in vivo using emerging imaging technologies and new advances in fluorescent moieties. In one approach, Ma et al. (2016) developed an α1‐https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4 antagonist conjugated to a caged Cy5 moiety that would fluoresce brightly in the near‐IR range (700–3,000 nm), which increases tissue penetrance of fluorescence in vivo. The quinazoline‐conjugated Cy5 displayed a 10‐fold increase in fluorescence upon binding α1‐adrenoceptors in vitro. The fluorescent ligand (compound 3a in this study) bound α1‐adrenoceptors located in ex vivo slices of murine prostate, demonstrating the ability to measure endogenous levels of receptor expression with compound 3a. Additionally, compound 3a was able to bind tumour xenografts that were implanted into mice containing cell lines that expressed the α1‐adrenoceptor and found that compound was 3a was localised in organs with high α1‐adrenoceptor expression when administered intravenously. Building upon the development of NanoBRET to monitor GPCR–ligand binding in living cells using the bright luciferase NanoLuc, Alcobia et al. (2018) sought to implement NanoBRET in vivo. They used a fluorescent analogue of the β‐adrenoceptor antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=564 (Prop‐BY630) that had previously been characterised at β‐adrenoceptors with NanoBRET in vitro (Soave et al., 2016; Stoddart et al., 2015). Alcobia et al. (2018) used the metastatic triple‐negative breast cancer MDA‐MB‐231 cell line stably transfected with the NLuc‐β2‐adrenoceptor as their model cell since propranolol has been shown to prevent breast cancer metastasis in murine models (Sloan et al., 2010). In a murine tumour xenograft model, the brightness of NanoLuc enabled the detection of luminescence from within a tumour cell mass upon the intravenous administration of the NanoLuc substrate, furimazine, and when Prop‐BY630 was administered intratumourally, an increase in the BRET signal was observed (Figure 4b). The specificity of this signal was confirmed by the ability of the β2‐adrenoceptor selective antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=543 to reduce the BRET ratio (Alcobia et al., 2018). This study demonstrated the ability to monitor target engagement in vivo using a fluorescent ligand and NanoBRET, and this approach could be implemented for other targets, including those involved with cancer progression, such as the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1813&familyId=324&familyType=CATALYTICRECEPTOR (Kilpatrick et al., 2017) or intracellular kinases (Robers et al., 2015).
8. FUTURE DIRECTIONS
Advances in the field of GPCR pharmacology and drug discovery have been facilitated by the use of fluorescent ligands. With the increasing emphasis on improving efficacy rates in clinical trials, it is more important than ever to fully validate the pharmacology of lead candidates and to quantify this under non‐equilibrium (kinetic) conditions. The kinetic approaches described above show the RET technologies can quantify ligand‐binding kinetics in whole cells and membranes and how ligand‐binding kinetics can potentially relate to side effects in the clinic. The combination of CRISPR/Cas9 and BRET techniques also offers an avenue to study ligand–receptor interactions at endogenous levels. Furthermore, improving subtype selectivity of fluorescent ligands will enable the labelling and monitoring of specific receptors in primary cells. These represent more physiologically relevant models to study these emerging paradigms of GPCR pharmacology. With improved physiochemical properties of fluorescent ligands, such as improved hydrophilicity and fluorescence emission, these techniques can be applied in vivo as a powerful tool for monitoring target engagement and residence time.
8.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Christopoulos et al., 2019; Alexander, Fabbro et al., 2019).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENT
The authors' work is supported by the Medical Research Council (Grant MR/N020081/1).
Soave M, Briddon SJ, Hill SJ, Stoddart LA. Fluorescent ligands: Bringing light to emerging GPCR paradigms. Br J Pharmacol. 2020;177:978–991. 10.1111/bph.14953
REFERENCES
- Adlere, I. , Sun, S. , Zarca, A. , Roumen, L. , Gozelle, M. , Viciano, C. P. , … Leurs, R. (2019). Structure‐based exploration and pharmacological evaluation of N‐substituted piperidin‐4‐yl‐methanamine CXCR4 chemokine receptor antagonists. European Journal of Medicinal Chemistry, 162, 631–649. 10.1016/j.ejmech.2018.10.060 [DOI] [PubMed] [Google Scholar]
- Alcobia, D. C. , Ziegler, A. I. , Kondrashov, A. , Comeo, E. , Mistry, S. , Kellam, B. , … Sloan, E. K. (2018). Visualizing ligand binding to a GPCR in vivo using NanoBRET. Science, 6, 280–288. 10.1016/j.isci.2018.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , … CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, J. A. , Yost, J. M. , Setola, V. , Chen, X. , Sassano, M. F. , Chen, M. , … Jin, J. (2011). Discovery of β‐arrestin‐biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proceedings of the National Academy of Sciences of the United States of America, 108(45), 18488–18493. 10.1073/pnas.1104807108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen, K. V. , Hjuler, S. T. , Furness, S. G. , Sexton, P. M. , Christopoulos, A. , Nosjean, O. , … Henriksen, K. (2014). Prolonged calcitonin receptor signaling by salmon, but not human calcitonin, reveals ligand bias. PLoS ONE, 9(3), e92042 10.1371/journal.pone.0092042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda, M. A. , Stoddart, L. A. , Gherbi, K. , Briddon, S. J. , Kellam, B. , & Hill, S. J. (2017). A non‐imaging high throughput approach to chemical library screening at the unmodified adenosine‐A3 receptor in living cells. Frontiers in Pharmacology, 8, 908 10.3389/fphar.2017.00908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio, L. , Valant, C. , Flynn, B. L. , Sexton, P. M. , Christopoulos, A. , & Scammells, P. J. (2009). Allosteric modulators of the adenosine A1 receptor: Synthesis and pharmacological evaluation of 4‐substituted 2‐amino‐3‐benzoylthiophenes. Journal of Medicinal Chemistry, 52(14), 4543–4547. 10.1021/jm9002582 [DOI] [PubMed] [Google Scholar]
- Baker, J. G. , Middleton, R. , Adams, L. , May, L. T. , Briddon, S. J. , Kellam, B. , & Hill, S. J. (2010). Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands: Themed section: Imaging in pharmacology research paper. British Journal of Pharmacology, 159, 772–786. 10.1111/j.1476-5381.2009.00488.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeh, K. M. , Westerman, J. , Kirsten, A. M. , Hebert, J. , Gronke, L. , Hamilton, A. , … Derom, E. (2015). The 24‐h lung‐function profile of once‐daily tiotropium and olodaterol fixed‐dose combination in chronic obstructive pulmonary disease. Pulmonary Pharmacology & Therapeutics, 32, 53–59. 10.1016/j.pupt.2015.04.002 [DOI] [PubMed] [Google Scholar]
- Bjarnadottir, T. K. , Gloriam, D. E. , Hellstrand, S. H. , Kristiansson, H. , Fredriksson, R. , & Schioth, H. B. (2006). Comprehensive repertoire and phylogenetic analysis of the G protein‐coupled receptors in human and mouse. Genomics, 88(3), 263–273. 10.1016/j.ygeno.2006.04.001 [DOI] [PubMed] [Google Scholar]
- Bohn, L. M. , Lefkowitz, R. J. , Gainetdinov, R. R. , Peppel, K. , Caron, M. G. , & Lin, F. T. (1999). Enhanced morphine analgesia in mice lacking β‐arrestin 2. Science, 286(5449), 2495–2498. [DOI] [PubMed] [Google Scholar]
- Bosma, R. , Stoddart, L. A. , Georgi, V. , Bouzo‐Lorenzo, M. , Bushby, N. , Inkoom, L. , … Leurs, R. (2019). Probe dependency in the determination of ligand binding kinetics at a prototypical G protein‐coupled receptor. Scientific Reports, 9(1), 7906 10.1038/s41598-019-44025-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzo‐Lorenzo, M. , Stoddart, L. A. , Xia, L. , IJzerman, A. P. , Heitman, L. H. , Briddon, S. J. , & Hill, S. J. (2019). A live cell NanoBRET binding assay allows the study of ligand‐binding kinetics to the adenosine A3 receptor. Purinergic Signal, 15(2), 139–153. 10.1007/s11302-019-09650-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, F. , Klutz, A. M. , Jacobson, K. A. , Fredholm, B. B. , & Schulte, G. (2008). Adenosine A2A receptor dynamics studied with the novel fluorescent agonist Alexa488‐APEC. European Journal of Pharmacology, 590(1‐3), 36–42. 10.1016/j.ejphar.2008.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon, S. J. , & Hill, S.J. (2007). Pharmacology under the microscope: the use of fluorescence correlation spectroscopy to determine the properties of ligand‐receptor complexes. Trends in Pharmacological Sciences, 28(12), 637–645. 10.1016/j.tips.2007.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon, S. J. , Kilpatrick, L. E. , & Hill, S. J. (2018). Studying GPCR Pharmacology in Membrane Microdomains: Fluorescence Correlation Spectroscopy Comes of Age. Trends in Pharmacological Sciences, 39(2), 158–174. 10.1016/j.tips.2017.11.004 [DOI] [PubMed] [Google Scholar]
- Bruns, R. F. , & Fergus, J. H. (1990). Allosteric enhancement of adenosine A1 receptor binding and function by 2‐amino‐3‐benzoylthiophenes. Molecular Pharmacology, 38(6), 939–949. [PubMed] [Google Scholar]
- Bullesbach, E. E. , & Schwabe, C. (2005). The trap‐like relaxin‐binding site of the leucine‐rich G‐protein‐coupled receptor 7. The Journal of Biological Chemistry, 280(14), 14051–14056. 10.1074/jbc.M500030200 [DOI] [PubMed] [Google Scholar]
- Calebiro, D. , Godbole, A. , Lyga, S. , & Lohse, M. J. (2015). Trafficking and function of GPCRs in the endosomal compartment. Methods in Molecular Biology, 1234, 197–211. 10.1007/978-1-4939-1755-6_16 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Sassano, M. F. , Zheng, L. , Setola, V. , Chen, M. , Bai, X. , … Jin, J. (2012). Structure–functional selectivity relationship studies of β‐arrestin‐biased dopamine D2 receptor agonists. Journal of Medicinal Chemistry, 55(16), 7141–7153. 10.1021/jm300603y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. T. , Pitis, P. , Liu, G. , Yuan, C. , Gotchev, D. , Cowan, C. L. , … Yamashita, D. S. (2013). Structure–activity relationships and discovery of a G protein biased μ opioid receptor ligand, [(3‐methoxythiophen‐2‐yl)methyl]({2‐[(9R)‐9‐(pyridin‐2‐yl)‐6‐oxaspiro‐[4.5]decan‐9‐yl]ethyl})amine (TRV130), for the treatment of acute severe pain. Journal of Medicinal Chemistry, 56(20), 8019–8031. 10.1021/jm4010829 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Corriden, R. , Inoue, Y. , Yip, L. , Hashiguchi, N. , Zinkernagel, A. , … Junger, W. G. (2006). ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science, 314(5806), 1792–1795. 10.1126/science.1132559 [DOI] [PubMed] [Google Scholar]
- Christiansen, E. , Hudson, B. D. , Hansen, A. H. , Milligan, G. , & Ulven, T. (2016). Development and characterization of a potent free fatty acid receptor 1 (FFA1) fluorescent tracer. Journal of Medicinal Chemistry, 59(10), 4849–4858. 10.1021/acs.jmedchem.6b00202 [DOI] [PubMed] [Google Scholar]
- Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , … Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339(6121), 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy, S. , Kindon, N. D. , Glenn, J. , Stoddart, L. A. , Lewis, R. J. , Hill, S. J. , … Stocks, M. J. (2018). Synthesis and evaluation of the first fluorescent antagonists of the human P2Y2 receptor based on AR‐C118925. Journal of Medicinal Chemistry, 61, 3089–3113. 10.1021/acs.jmedchem.8b00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, S. L. , Soave, M. , Jorg, M. , Scammells, P. J. , Woolard, J. , & Hill, S. J. (2019). Probe dependence of allosteric enhancers on the binding affinity of adenosine A1‐receptor agonists at rat and human A1‐receptors measured using NanoBRET. British Journal of Pharmacology, 176(7), 864–878. 10.1111/bph.14575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland, R. A. (2016). The drug‐target residence time model: A 10‐year retrospective. Nature Reviews. Drug Discovery, 15(2), 87–95. 10.1038/nrd.2015.18 [DOI] [PubMed] [Google Scholar]
- Corriden, R. , Self, T. , Akong‐Moore, K. , Nizet, V. , Kellam, B. , Briddon, S. J. , & Hill, S. J. (2013). Adenosine‐A3 receptors in neutrophil microdomains promote the formation of bacteria‐tethering cytonemes. EMBO Reports, 14(8), 726–732. 10.1038/embor.2013.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottet, M. , Faklaris, O. , Zwier, J. M. , Trinquet, E. , Pin, J.‐P. , & Durroux, T. (2011). Original fluorescent ligand‐based assays open new perspectives in G‐protein coupled receptor drug screening. Pharmaceuticals, 4(1), 202–214. [Google Scholar]
- Daval, S. B. , Kellenberger, E. , Bonnet, D. , Utard, V. , Galzi, J. L. , & Ilien, B. (2013). Exploration of the orthosteric/allosteric interface in human M1 muscarinic receptors by bitopic fluorescent ligands. Molecular Pharmacology, 84(1), 71–85. 10.1124/mol.113.085670 [DOI] [PubMed] [Google Scholar]
- Daval, S. B. , Valant, C. , Bonnet, D. , Kellenberger, E. , Hibert, M. , Galzi, J. L. , & Ilien, B. (2012). Fluorescent derivatives of AC‐42 to probe bitopic orthosteric/allosteric binding mechanisms on muscarinic M1 receptors. Journal of Medicinal Chemistry, 55(5), 2125–2143. 10.1021/jm201348t [DOI] [PubMed] [Google Scholar]
- De Lean, A. , Stadel, J. M. , & Lefkowitz, R. J. (1980). A ternary complex model explains the agonist‐specific binding properties of the adenylate cyclase‐coupled beta‐adrenergic receptor. The Journal of Biological Chemistry, 255(15), 7108–7117. [PubMed] [Google Scholar]
- Eichel, K. , & von Zastrow, M. (2018). Subcellular organization of GPCR signaling. Trends in Pharmacological Sciences, 39(2), 200–208. 10.1016/j.tips.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder, C. C. , Goldsmith, P. J. , Jackson, K. , Sanger, H. E. , Evans, D. A. , Mogg, A. J. , & Broad, L. M. (2018). Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology, 136(Pt C), 449–458. 10.1016/j.neuropharm.2018.01.028 [DOI] [PubMed] [Google Scholar]
- Furness, S. G. B. , Liang, Y. L. , Nowell, C. J. , Halls, M. L. , Wookey, P. J. , Dal Maso, E. , … Sexton, P. M. (2016). Ligand‐dependent modulation of G protein conformation alters drug efficacy. Cell, 167(3), 739–749. 10.1016/j.cell.2016.09.021 [DOI] [PubMed] [Google Scholar]
- Gherbi, K. , May, L. T. , Baker, J. G. , Briddon, S. J. , & Hill, S. J. (2015). Negative cooperativity across β1‐adrenoceptor homodimers provides insights into the nature of the secondary low‐affinity CGP 12177 β1‐adrenoceptor binding conformation. The FASEB Journal, 29, 2859–2871. 10.1096/fj.14-265199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell, J. , Simms, J. , Barwell, J. , Poyner, D. R. , Watkins, H. A. , Pioszak, A. A. , … Hay, D. L. (2016). An allosteric role for receptor activity‐modifying proteins in defining GPCR pharmacology. Cell Discovery, 2, 16012 10.1038/celldisc.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbole, A. , Lyga, S. , Lohse, M. J. , & Calebiro, D. (2017). Internalized TSH receptors en route to the TGN induce local Gs‐protein signaling and gene transcription. Nature Communications, 8(1), 443 10.1038/s41467-017-00357-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding, J. , May, L. T. , & Hill, S. J. (2018). Characterisation of endogenous A2A and A2B receptor‐mediated cyclic AMP responses in HEK 293 cells using the GloSensor biosensor: Evidence for an allosteric mechanism of action for the A2B‐selective antagonist PSB 603. Biochemical Pharmacology, 147, 55–66. 10.1016/j.bcp.2017.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidolin, D. , Agnati, L. F. , Marcoli, M. , Borroto‐Escuela, D. O. , & Fuxe, K. (2015). G‐protein‐coupled receptor type A heteromers as an emerging therapeutic target. Expert Opinion on Therapeutic Targets, 19(2), 265–283. 10.1517/14728222.2014.981155 [DOI] [PubMed] [Google Scholar]
- Hansen, A. H. , Sergeev, E. , Pandey, S. K. , Hudson, B. D. , Christiansen, E. , Milligan, G. , & Ulven, T. (2017). Development and characterization of a fluorescent tracer for the free fatty acid receptor 2 (FFA2/GPR43). Journal of Medicinal Chemistry, 60, 5638–5645. 10.1021/acs.jmedchem.7b00338 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, D. L. , & Pioszak, A. A. (2016). Receptor activity‐modifying proteins (RAMPs): New insights and roles. Annual Review of Pharmacology and Toxicology, 56, 469–487. 10.1146/annurev-pharmtox-010715-103120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithier, H. , Hallmann, D. , Boege, F. , Reilander, H. , Dees, C. , Jaeggi, K. A. , … Helmreich, E. J. (1994). Synthesis and properties of fluorescent beta‐adrenoceptor ligands. Biochemistry, 33(31), 9126–9134. 10.1021/bi00197a015 [DOI] [PubMed] [Google Scholar]
- Hill, S. J. , May, L. T. , Kellam, B. , & Woolard, J. (2014). Allosteric interactions at adenosine A1 and A3 receptors: New insights into the role of small molecules and receptor dimerization. British Journal of Pharmacology, 171(5), 1102–1113. 10.1111/bph.12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, C. , Kuhhorn, J. , & Gmeiner, P. (2013). Class A G‐protein‐coupled receptor (GPCR) dimers and bivalent ligands. Journal of Medicinal Chemistry, 56(17), 6542–6559. 10.1021/jm4004335 [DOI] [PubMed] [Google Scholar]
- Hoare, B. L. , Bruell, S. , Sethi, A. , Gooley, P. R. , Lew, M. J. , Hossain, M. A. , … Bathgate, R. A. D. (2019). Multi‐component mechanism of H2 relaxin binding to RXFP1 through NanoBRET kinetic analysis. Science, 11, 93–113. 10.1016/j.isci.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, A. L. , Groom, C. R. , & Alex, A. (2004). Ligand efficiency: A useful metric for lead selection. Drug Discovery Today, 9(10), 430–431. [DOI] [PubMed] [Google Scholar]
- Hothersall, J. D. , Brown, A. J. , Dale, I. , & Rawlins, P. (2016). Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discovery Today, 21, 90–96. 10.1016/j.drudis.2015.07.015 [DOI] [PubMed] [Google Scholar]
- Hounsou, C. , Margathe, J. F. , Oueslati, N. , Belhocine, A. , Dupuis, E. , Thomas, C. , … Durroux, T. (2014). Time‐resolved FRET binding assay to investigate hetero‐oligomer binding properties: Proof of concept with dopamine D1/D3 heterodimer. ACS Chemical Biology, 10, 466–474. 10.1021/cb5007568 [DOI] [PubMed] [Google Scholar]
- Hulme, E. C. , & Trevethick, M. A. (2010). Ligand binding assays at equilibrium: Validation and interpretation. British Journal of Pharmacology, 161(6), 1219–1237. 10.1111/j.1476-5381.2009.00604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad, R. , Tomshine, J. C. , Tomshine, J. R. , Chevalier, M. , Mahoney, J. P. , Steyaert, J. , … von Zastrow, M. (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature, 495(7442534‐+), 534–538. 10.1038/nature12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturrioz, X. , Alvear‐Perez, R. , de Mota, N. , Franchet, C. , Guillier, F. , Leroux, V. , … Llorens‐Cortes, C. (2010). Identification and pharmacological properties of E339‐3D6, the first nonpeptidic apelin receptor agonist. The FASEB Journal, 24(5), 1506–1517. 10.1096/fj.09-140715 [DOI] [PubMed] [Google Scholar]
- Kapur, S. , & Seeman, P. (2001). Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. The American Journal of Psychiatry, 158(3), 360–369. 10.1176/appi.ajp.158.3.360 [DOI] [PubMed] [Google Scholar]
- Karpenko, I. A. , Margathe, J. F. , Rodriguez, T. , Pflimlin, E. , Dupuis, E. , Hibert, M. , … Bonnet, D. (2015). Selective nonpeptidic fluorescent ligands for oxytocin receptor: Design, synthesis, and application to time‐resolved FRET binding assay. Journal of Medicinal Chemistry, 58, 2547–2552. 10.1021/jm501395b [DOI] [PubMed] [Google Scholar]
- Kenakin, T. , & Christopoulos, A. (2013). Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nature Reviews. Drug Discovery, 12(3), 205–216. 10.1038/nrd3954 [DOI] [PubMed] [Google Scholar]
- Kenakin, T. P. (2012). Biased signaling and allosteric machines: New vistas and challenges for drug discovery. British Journal of Pharmacology, 165, 1659–1669. 10.1111/j.1476-5381.2011.01749.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick, L. E. , Friedman‐Ohana, R. , Alcobia, D. C. , Riching, K. , Peach, C. J. , Wheal, A. J. , … Hill, S. J (2017). Real‐time analysis of the binding of fluorescent VEGF 165 a to VEGFR2 in living cells: Effect of receptor tyrosine kinase inhibitors and fate of internalized agonist‐receptor complexes. Biochemical Pharmacology, 136, 62–75. 10.1016/j.bcp.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink, C. , Sykes, D. A. , Donthamsetti, P. , Canals, M. , Coudrat, T. , Shonberg, J. , … Lane, J. R. (2016). The role of kinetic context in apparent biased agonism at GPCRs. Nature Communications, 7, 10842 10.1038/ncomms10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola, I. , & Landis, J. (2004). Can the pharmaceutical industry reduce attrition rates? Nature Reviews. Drug Discovery, 3(8), 711–715. 10.1038/nrd1470 [DOI] [PubMed] [Google Scholar]
- Kruse, A. C. , Ring, A. M. , Manglik, A. , Hu, J. , Hu, K. , Eitel, K. , … Kobilka, B. K. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature, 504(7478), 101–106. 10.1038/nature12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuder, K. J. , & Kieć‐Kononowicz, K. (2014). Fluorescent GPCR ligands as new tools in pharmacology—update, years 2008–early 2014. Current Medicinal Chemistry, 21(34), 3962–3975. 10.2174/0929867321666140826120058 [DOI] [PubMed] [Google Scholar]
- Lam, R. , Gondin, A. B. , Canals, M. , Kellam, B. , Briddon, S. J. , Graham, B. , & Scammells, P. J. (2018). Fluorescently labeled morphine derivatives for bioimaging studies. Journal of Medicinal Chemistry, 61(3), 1316–1329. 10.1021/acs.jmedchem.7b01811 [DOI] [PubMed] [Google Scholar]
- Leyris, J. P. , Roux, T. , Trinquet, E. , Verdié, P. , Fehrentz, J. A. , Oueslati, N. , … Marie, J. (2011). Homogeneous time‐resolved fluorescence‐based assay to screen for ligands targeting the growth hormone secretagogue receptor type 1a. Analytical Biochemistry, 408(2), 253–262. 10.1016/j.ab.2010.09.030 [DOI] [PubMed] [Google Scholar]
- Link, R. , Veiksina, S. , Rinken, A. , & Kopanchuk, S. (2017). Characterisation of ligand binding to melanocortin 4 receptors using fluorescent peptides with improved kinetic properties. European Journal of Pharmacology, 799, 58–66. 10.1016/j.ejphar.2017.01.040 [DOI] [PubMed] [Google Scholar]
- Loison, S. , Cottet, M. , Orcel, H. , Adihou, H. , Rahmeh, R. , Lamarque, L. , … Bonnet, D. (2012). Selective fluorescent nonpeptidic antagonists for vasopressin V2 GPCR: Application to ligand screening and oligomerization assays. Journal of Medicinal Chemistry, 55(20), 8588–8602. 10.1021/jm3006146 [DOI] [PubMed] [Google Scholar]
- Ma, Z. , Lin, Y. , Cheng, Y. , Wu, W. , Cai, R. , Chen, S. , … Li, M. (2016). Discovery of the first environment‐sensitive near‐infrared (NIR) fluorogenic ligand for α1‐adrenergic receptors imaging in vivo. Journal of Medicinal Chemistry, 59(5), 2151–2162. 10.1021/acs.jmedchem.5b01843 [DOI] [PubMed] [Google Scholar]
- Margathe, J. F. , Iturrioz, X. , Regenass, P. , Karpenko, I. A. , Humbert, N. , de Rocquigny, H. , … Bonnet, D. (2016). Convenient access to fluorescent probes by chemoselective acylation of hydrazinopeptides: Application to the synthesis of the first far‐red ligand for apelin receptor imaging. Chemistry, 22(4), 1399–1405. 10.1002/chem.201503630 [DOI] [PubMed] [Google Scholar]
- Masri, B. , Salahpour, A. , Didriksen, M. , Ghisi, V. , Beaulieu, J. M. , Gainetdinov, R. R. , & Caron, M. G. (2008). Antagonism of dopamine D2 receptor/β‐arrestin 2 interaction is a common property of clinically effective antipsychotics. Proceedings of the National Academy of Sciences of the United States of America, 105(36), 13656–13661. 10.1073/pnas.0803522105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, L. T. , Bridge, L. J. , Stoddart, L. A. , Briddon, S. J. , & Hill, S. J. (2011). Allosteric interactions across native adenosine‐A3 receptor homodimers: Quantification using single‐cell ligand‐binding kinetics. FASEB Journal, 25(10), 3465–3476. 10.1096/fj.11-186296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, L. T. , Leach, K. , Sexton, P. M. , & Christopoulos, A. (2007). Allosteric modulation of G protein‐coupled receptors. Annual Review of Pharmacology and Toxicology, 47, 1–51. 10.1146/annurev.pharmtox.47.120505.105159 [DOI] [PubMed] [Google Scholar]
- Melancon, B. J. , Tarr, J. C. , Panarese, J. D. , Wood, M. R. , & Lindsley, C. W. (2013). Allosteric modulation of the M1 muscarinic acetylcholine receptor: Improving cognition and a potential treatment for schizophrenia and Alzheimer's disease. Drug Discovery Today, 18(23‐24), 1185–1199. 10.1016/j.drudis.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, S. , Miyake, N. , Jarskog, L. F. , Fleischhacker, W. W. , & Lieberman, J. A. (2012). Pharmacological treatment of schizophrenia: A critical review of the pharmacology and clinical effects of current and future therapeutic agents. Molecular Psychiatry, 17(12), 1206–1227. 10.1038/mp.2012.47 [DOI] [PubMed] [Google Scholar]
- Mocking, T. A. M. , Verweij, E. W. E. , Vischer, H. F. , & Leurs, R. (2018). Homogeneous, real‐time NanoBRET binding assays for the histamine H3 and H4 receptors on living cells. Molecular Pharmacology, 94(6), 1371–1381. 10.1124/mol.118.113373 [DOI] [PubMed] [Google Scholar]
- Motulsky, H. J. , & Mahan, L. C. (1984). The kinetics of competitive radioligand binding predicted by the law of mass action. Molecular Pharmacology, 25(1), 1–9. [PubMed] [Google Scholar]
- Nederpelt, I. , Georgi, V. , Schiele, F. , Nowak‐Reppel, K. , Fernandez‐Montalvan, A. E. , IJzerman, A. P. , & Heitman, L. H. (2016). Characterization of 12 GnRH peptide agonists—A kinetic perspective. British Journal of Pharmacology, 173(1), 128–141. 10.1111/bph.13342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan, A. , Stoddart, L. A. , Hill, S. J. , & Carlsson, J. (2015). Fragment‐based discovery of subtype‐selective adenosine receptor ligands from homology models. Journal of Medicinal Chemistry, 58(24), 9578–9590. 10.1021/acs.jmedchem.5b01120 [DOI] [PubMed] [Google Scholar]
- Rankovic, Z. , Brust, T. F. , & Bohn, L. M. (2016). Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorganic & Medicinal Chemistry Letters, 26(2), 241–250. 10.1016/j.bmcl.2015.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner, D. , & Stark, H. (2019). Ligand binding kinetics at histamine H3 receptors by fluorescence‐polarization with real‐time monitoring. European Journal of Pharmacology, 848, 112–120. 10.1016/j.ejphar.2019.01.041 [DOI] [PubMed] [Google Scholar]
- Robers, M. B. , Dart, M. L. , Woodroofe, C. C. , Zimprich, C. A. , Kirkland, T. A. , Machleidt, T. , … Wood, K. V. (2015). Target engagement and drug residence time can be observed in living cells with BRET. Nature Communications, 6(1). 10.1038/ncomms10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rominger, D. H. , Cowan, C. L. , Gowen‐MacDonald, W. , & Violin, J. D. (2014). Biased ligands: Pathway validation for novel GPCR therapeutics. Current Opinion in Pharmacology, 16, 108–115. 10.1016/j.coph.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Rose, R. H. , Briddon, S. J. , & Hill, S. J. (2012). A novel fluorescent histamine H1 receptor antagonist demonstrates the advantage of using fluorescence correlation spectroscopy to study the binding of lipophilic ligands. British Journal of Pharmacology, 165(6), 1789–1800. 10.1111/j.1476-5381.2011.01640.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiele, F. , Ayaz, A. , & Fernandez‐Montalvan, A. (2015). A universal homogeneneous assay for high‐throughput determination of binding kinetics. Analytical Biochemistry, 468, 42–49. 10.1016/j.ab.2014.09.007 [DOI] [PubMed] [Google Scholar]
- Schuetz, D. A. , de Witte, W. E. A. , Wong, Y. C. , Knasmueller, B. , Richter, L. , Kokh, D. B. , … Ecker, G. F. (2017). Kinetics for drug discovery: An industry‐driven effort to target drug residence time. Drug Discovery Today, 22, 896–911. 10.1016/j.drudis.2017.02.002 [DOI] [PubMed] [Google Scholar]
- Sethi, A. , Bruell, S. , Patil, N. , Hossain, M. A. , Scott, D. J. , Petrie, E. J. , … Gooley, P. R. (2016). The complex binding mode of the peptide hormone H2 relaxin to its receptor RXFP1. Nature Communications, 7, 11344 10.1038/ncomms11344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton, M. , Woodruff, G. , Horne, E. A. , Lin, Y. H. , Muccioli, G. G. , Bai, M. , … Stella, N. (2011). NIR‐mbc94, a fluorescent ligand that binds to endogenous CB2 receptors and is amenable to high‐throughput screening. Chemistry & Biology, 18(5), 563–568. 10.1016/j.chembiol.2011.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar, L. A. , Sayre, J. , McNeil, V. M. , & Finney, D. A. (1985). Competitive binding kinetics in ligand–receptor–competitor systems. Rate parameters for unlabeled ligands for the formyl peptide receptor. Molecular Pharmacology, 28(4), 323–330. [PubMed] [Google Scholar]
- Sloan, E. K. , Priceman, S. J. , Cox, B. F. , Yu, S. , Pimentel, M. A. , Tangkanangnukul, V. , … Cole, S. W. (2010). The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Research, 70(18), 7042–7052. 10.1158/0008-5472.Can-10-0522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soave, M. , Stoddart, L. A. , Brown, A. , Woolard, J. , & Hill, S. J. (2016). Use of a new proximity assay (NanoBRET) to investigate the ligand‐binding characteristics of three fluorescent ligands to the human β1‐adrenoceptor expressed in HEK‐293 cells. Pharmacology Research & Perspectives, 4(5), e00250 10.1002/prp2.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt, M. , Stolze, S. C. , Westphal, M. V. , van Stralen, L. , Martella, A. , van Rooden, E. J. , … van der Stelt, M. (2018). Selective photoaffinity probe that enables assessment of cannabinoid CB2 receptor expression and ligand engagement in human cells. Journal of the American Chemical Society, 140, 6067–6075. 10.1021/jacs.7b11281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan, R. , Zuber, J. , Connelly, S. M. , Mathew, E. , & Dumont, M. E. (2014). Fluorescent approaches for understanding interactions of ligands with G protein coupled receptors. Biochimica et Biophysica Acta, 1838, 15–33. 10.1016/j.bbamem.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram, K. , & Insel, P. A. (2018). G protein‐coupled receptors as targets for approved drugs: How many targets and how many drugs? Molecular Pharmacology, 93(4), 251–258. 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart, L. A. , Johnstone, E. K. M. , Wheal, A. J. , Goulding, J. , Robers, M. B. , Machleidt, T. , … Pfleger, K. D. G. (2015). Application of BRET to monitor ligand binding to GPCRs. Nature Methods, 12(7), 661–663. 10.1038/nmeth.3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart, L. A. , Kilpatrick, L. E. , Briddon, S. J. , & Hill, S. J. (2015). Probing the pharmacology of G protein‐coupled receptors with fluorescent ligands. Neuropharmacology, 98, 48–57. 10.1016/j.neuropharm.2015.04.033 [DOI] [PubMed] [Google Scholar]
- Stoddart, L. A. , Vernall, A. J. , Bouzo‐Lorenzo, M. , Bosma, R. , Kooistra, A. J. , de Graaf, C. , … Hill, S. J. (2018). Development of novel fluorescent histamine H1‐receptor antagonists to study ligand‐binding kinetics in living cells. Scientific Reports, 8(1), 1572 10.1038/s41598-018-19714-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart, L. A. , Vernall, A. J. , Briddon, S. J. , Kellam, B. , & Hill, S. J. (2015). Direct visualisation of internalization of the adenosine A3 receptor and localization with arrestin3 using a fluorescent agonist. Neuropharmacology, 98, 68–77. 10.1016/j.neuropharm.2015.04.013 [DOI] [PubMed] [Google Scholar]
- Stoddart, L. A. , Vernall, A. J. , Denman, J. L. , Briddon, S. J. , Kellam, B. , & Hill, S. J. (2012). Fragment screening at adenosine‐A3 receptors in living cells using a fluorescence‐based binding assay. Chemistry & Biology, 19(9), 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart, L. A. , White, C. W. , Nguyen, K. , Hill, S. J. , & Pfleger, K. D. (2016). Fluorescence‐ and bioluminescence‐based approaches to study GPCR ligand binding. British Journal of Pharmacology, 173(20), 3028–3037. 10.1111/bph.13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney, D. C. , Haubrich, B. A. , Van Liefde, I. , & Vauquelin, G. (2015). The role of binding kinetics in GPCR drug discovery. Current Topics in Medicinal Chemistry, 15(24), 2504–2522. [DOI] [PubMed] [Google Scholar]
- Sykes, D. A. , Jain, P. , & Charlton, S. J. (2019). Investigating the influence of tracer kinetics on competition–kinetic association binding assays: Identifying the optimal conditions for assessing the kinetics of low‐affinity compounds. Molecular Pharmacology, 96(3), 378–392. 10.1124/mol.119.116764 [DOI] [PubMed] [Google Scholar]
- Sykes, D. A. , Moore, H. , Stott, L. , Holliday, N. , Javitch, J. A. , Lane, J. R. , & Charlton, S. J. (2017). Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nature Communications, 8(1), 763 10.1038/s41467-017-00716-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, M. , Klein Herenbrink, C. , Christopoulos, A. , Lane, J. R. , & Capuano, B. (2014). Structure–activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D2 receptor. Journal of Medicinal Chemistry, 57(11), 4924–4939. 10.1021/jm500457x [DOI] [PubMed] [Google Scholar]
- Thomsen, A. R. B. , Jensen, D. D. , Hicks, G. A. , & Bunnett, N. W. (2018). Therapeutic targeting of endosomal G‐protein‐coupled receptors. Trends in Pharmacological Sciences, 39(10), 879–891. 10.1016/j.tips.2018.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban, J. D. , Vargas, G. A. , von Zastrow, M. , & Mailman, R. B. (2007). Aripiprazole has functionally selective actions at dopamine D2 receptor‐mediated signaling pathways. Neuropsychopharmacology, 32(1), 67–77. 10.1038/sj.npp.1301071 [DOI] [PubMed] [Google Scholar]
- Vernall, A. J. , Hill, S. J. , & Kellam, B. (2014). The evolving small‐molecule fluorescent‐conjugate toolbox for Class A GPCRs. British Journal of Pharmacology, 171(5), 1073–1084. 10.1111/bph.12265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernall, A. J. , Stoddart, L. A. , Briddon, S. J. , Ng, H. W. , Laughton, C. A. , Doughty, S. W. , … Kellam, B. (2013). Conversion of a non‐selective adenosine receptor antagonist into A3‐selective high affinity fluorescent probes using peptide‐based linkers. Organic & Biomolecular Chemistry, 11, 5673–5682. 10.1039/c3ob41221k [DOI] [PubMed] [Google Scholar]
- Wang, L. , Martin, B. , Brenneman, R. , Luttrell, L. M. , & Maudsley, S. (2009). Allosteric modulators of G protein‐coupled receptors: Future therapeutics for complex physiological disorders. The Journal of Pharmacology and Experimental Therapeutics, 331(2), 340–348. 10.1124/jpet.109.156380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, R. J. , Pediani, J. D. , Godin, A. G. , & Milligan, G. (2015). Regulation of oligomeric organization of the serotonin 5‐hydroxytryptamine 2C (5‐HT2C) receptor observed by spatial intensity distribution analysis. The Journal of Biological Chemistry, 290(20), 12844–12857. 10.1074/jbc.M115.644724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring, M. J. , Arrowsmith, J. , Leach, A. R. , Leeson, P. D. , Mandrell, S. , Owen, R. M. , … Weir, A. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nature Reviews. Drug Discovery, 14(7), 475–486. 10.1038/nrd4609 [DOI] [PubMed] [Google Scholar]
- Werthmann, R. C. , Volpe, S. , Lohse, M. J. , & Calebiro, D. (2012). Persistent cAMP signaling by internalized TSH receptors occurs in thyroid but not in HEK293 cells. FASEB Journal, 26(5), 2043–2048. 10.1096/fj.11-195248 [DOI] [PubMed] [Google Scholar]
- White, C. W. , Johnstone, E. K. M. , See, H. B. , & Pfleger, K. D. G. (2019). NanoBRET ligand binding at a GPCR under endogenous promotion facilitated by CRISPR/Cas9 genome editing. Cellular Signalling, 54, 27–34. 10.1016/j.cellsig.2018.11.018 [DOI] [PubMed] [Google Scholar]
- White, C. W. , Vanyai, H. K. , See, H. B. , Johnstone, E. K. M. , & Pfleger, K. D. G. (2017). Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome‐edited protein in real‐time. Scientific Reports, 7(1), 3187 10.1038/s41598-017-03486-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, S. M. , Bologa, C. , Prossnitz, E. R. , Oprea, T. I. , Sklar, L. A. , & Edwards, B. S. (2005). High‐throughput screening with HyperCyt® flow cytometry to detect small molecule formylpeptide receptor ligands. Journal of Biomolecular Screening, 10(4), 374–382. 10.1177/1087057105274532 [DOI] [PubMed] [Google Scholar]
- Zwier, J. M. , Roux, T. , Cottet, M. , Durroux, T. , Douzon, S. , Bdioui, S. , … Trinquet, E. (2010). A fluorescent ligand‐binding alternative using Tag‐lite® technology. Journal of Biomolecular Screening, 15(10), 1248–1259. 10.1177/1087057110384611 [DOI] [PubMed] [Google Scholar]