

Abstract
Objectives
Describe the histopathology of the temporal bones in MELAS (myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes) syndrome. The syndrome results from a known point mutation in mitochondrial DNA.
Methods
Histopathology analysis of a pair of temporal bones from the oldest surviving MELAS syndrome temporal bone donor. Histopathologic findings were correlated with known premortem clinical data.
Results
The inner ears showed severe but incomplete atrophy of the stria vascularis for the length of the cochleae. In contrast, the organ of Corti and inner hair cells appeared intact with some loss of outer hair cells. Other than moderate loss at the basal turn, spiral ganglion cells numbers were normal. The vestibular neuroepithelium was mostly normal with the exception of moderate degeneration of the macula sacculi and partial collapse of the saccular wall on the right. The cerebral cortex had infarct‐like lesions with adjacent gliosis.
Conclusion
This is an analysis of the oldest patient with MELAS syndrome to date, an addition to only two previously published patients. It supports the notion that hearing loss is a result of dysfunction of the stria vascularis and not loss of hair cells or neurons. Patterns of vestibular pathology are in agreement to in‐vivo measurements. These findings support auditory rehabilitation with cochlear implants and may be relevant to hearing loss due to other mitochondrial mutations.
Level of evidence
4
Keywords: genetics, MELAS syndrome, otology, otopathology, sensorineural hearing loss
1. INTRODUCTION
Hearing loss is common, and genetic abnormalities are a frequent cause. Mutations in mitochondrial DNA (mtDNA) are responsible for close to 1% of prelingual congenital deafness and approximately 5% of post lingual hearing loss in Caucasians.1 Mutations in mtDNA can cause both syndromic and isolated hearing loss.
MELAS is a syndrome typified by mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes.2 Although a number of mutations can cause the syndrome, the majority (80%) are caused by a point mutation of alanine and guanine at 3243.3 MELAS syndrome is the most common maternally inherited mitochondrial disease.
The goal of this article is to describe the histopathology of the temporal bones, in a patient with MELAS syndrome, and discuss the broader implications on mitochondrial hearing loss.
2. METHODS
2.1. Temporal bone acquisition
Temporal bone specimens were obtained in compliance with Human Subject Assurance No. FWA00006221 (exemption 4) from the Massachusetts Eye and Ear Infirmary.
2.2. Temporal bone preparation and light microscopy
Both temporal bones are available for study. The time elapsed between the death of the donor to tissue fixation was 21 hours. The specimens were fixed in 10% formalin, decalcified with ethylene diamine tetra‐acetic acid (EDTA), dehydrated in graded alcohols, and embedded in celloidin. Serial sections 20 μm thick were cut in the axial (horizontal) plane. Every tenth section was stained with hematoxylin and eosin (H&E) and mounted on glass slides. For analysis and quantification of cellular elements two‐dimensional reconstruction of the cochlea was done. Neurons were counted by identification of their nucleoli under 400× magnification. A correction factor of 0.9 was applied to the counted number of neurons to account for the possibility of double counting in two consecutive sections.4
3. RESULTS
3.1. Clinical history
A 42‐year‐old man first experienced hearing loss at the age of 25. The hearing loss was progressive and worse on the right. He used hearing aids, but an audiogram is not available. None of nine other siblings had a history of hearing loss or other mitochondria‐related diagnoses. At the age of 40, he developed systemic illness with headache, malaise, and fatigue for a week, followed by an acute onset of expressive aphasia. His language gradually improved. MRI demonstrated a left temporoparietal lesion and later subsided. The following month he had a tonic‐clonic seizure. These manifestations were preceded by subtle changes in personality and behavior. The combination of these symptoms with the patient's short stature and family history of type 1 diabetes raised the suspicion for MELAS syndrome. He tested positive for mtDNA mutation 3243A>G. He died at the age of 42 of respiratory failure.
3.2. Histopathological findings
Preservation of the temporal bones was good despite the 21 hours elapsed between death and fixation of tissue. Minor vesiculation of the neuroepithelium was seen in the ampullae (Figure 1). The most significant abnormality in the cochlea was severe atrophy of the stria vascularis (Figure 2) with basophilic inclusion cysts in the stria vascularis seen on both sides (Figure 3). The organ of Corti was mostly normal (Figure 2). There was mild loss of neurons of the spiral ganglion (Figure 4A) more pronounced at the basal turn (Figure 4B) and in the right temporal bone. The number of spiral ganglion neurons in the right temporal bone per cochlear segment was (in parenthesis—percentage of age matched controls): 1738 (49%), 6452 (63%), 3720 (57%), and 3312 (49%). For the left temporal bone the count was: 1638 (46%), 10 180 (99%), 4852 (75%), and 5926 (87%). Segment 1 is the basal quadrant of the cochlea and segment 4 the most apical. The vestibular neuroepithelium was normal with the exception of mild cellular loss in the saccule. The saccule was collapsed (Figure 5). In a cytocochleogram of the right and left temporal bones these findings are depicted along the cochlear duct (Figure 6A,B).
Figure 1.
Post mortem changes: vesiculation in the neuroepithelium of the ampulla of the lateral semi‐circular canal. Left ear. (hematoxylin and eosin staining ×200 magnification)
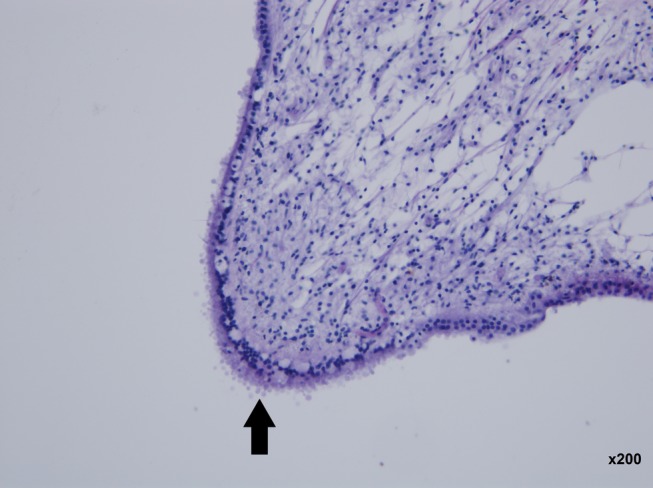
Figure 2.
Severe atrophy of the stria vascularis (black arrow). Preservation of the organ of Corti (white arrow). Left ear, ascending basal turn. (hematoxylin and eosin staining ×200 magnification)
Figure 3.
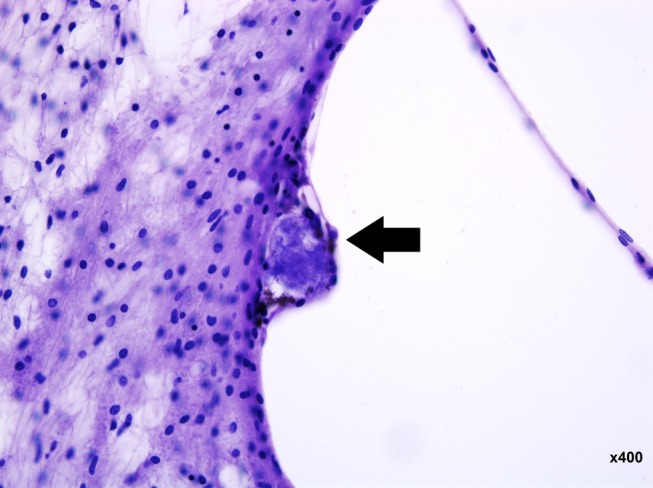
Basophilic inclusion cyst in the stria vascularis of the ascending basal turn of the left ear. (hematoxylin and eosin staining ×400 magnification)
Figure 4.
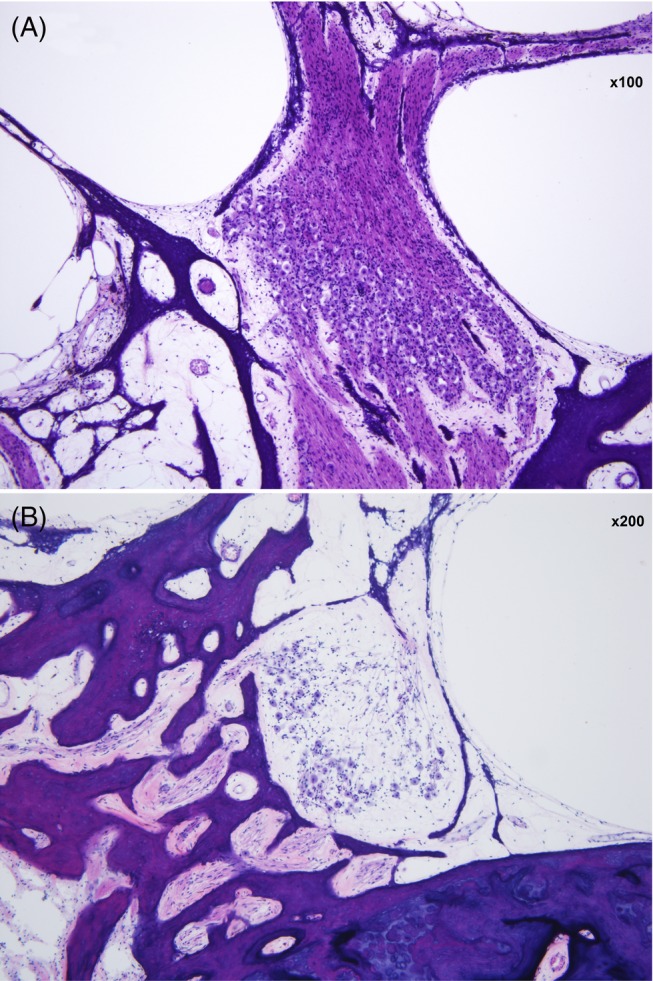
Spiral ganglion neurons of the left ear. A, Mid‐modiolar section with a good complement of neurons (×100 magnification). B, Moderate loss at the basal turn (×200 magnification). (hematoxylin and eosin staining)
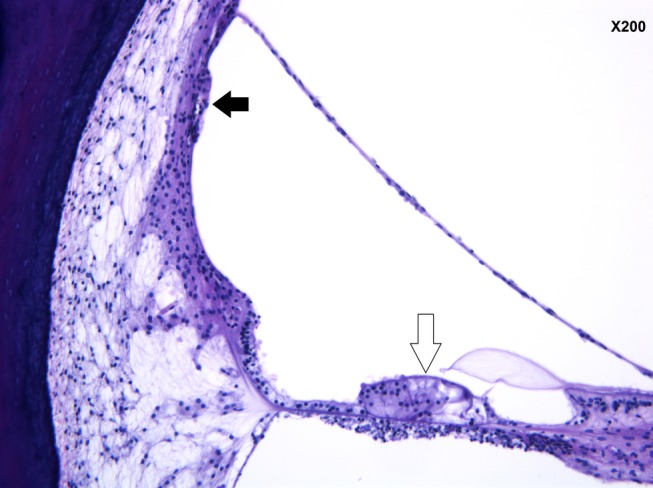
Figure 5.
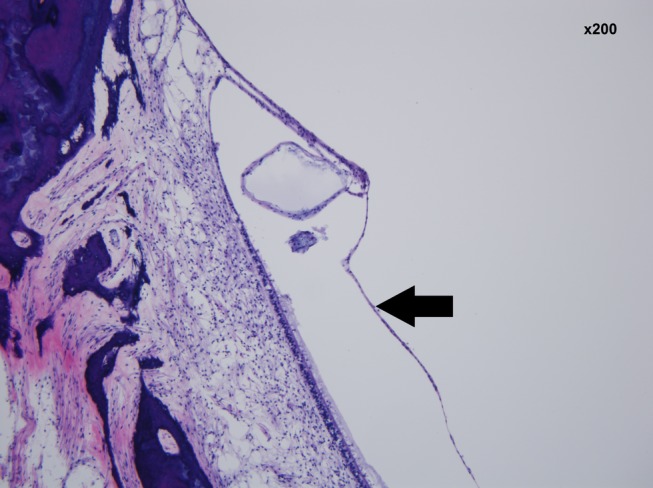
Collapse of the membrane of the saccule—left ear (black arrow). (hematoxylin and eosin staining ×200 magnification)
Figure 6.
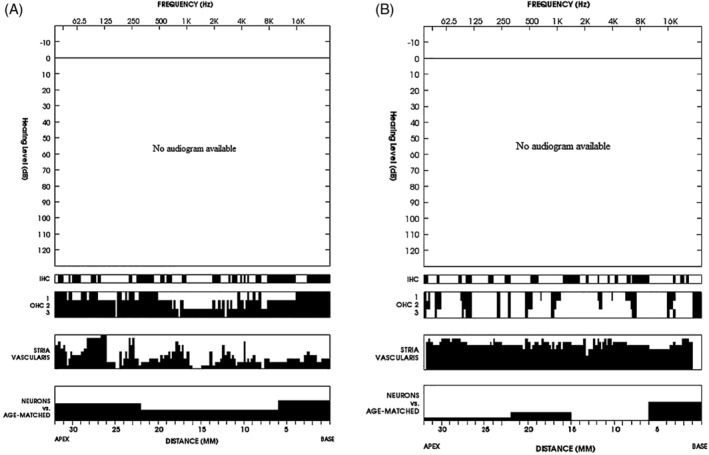
Cytocochleogram of the right (A) and left ear (B). Graphic display of the findings: atrophy of stria vascularis was the most prominent pathology. Black symbols a missing cochlear component
4. DISCUSSION
The mitochondria is the intracellular organelle responsible for generating energy required for normal function of cells and organs by oxidative phosphorylation. A second role of the mitochondria is control of reactive oxygen species (ROS). ROS are reactive derivatives of O2 metabolism, including superoxide anion, hydrogen peroxide, and nitric oxide. In addition, mitochondria play an important role in cell apoptosis.5
The mtDNA is 16 568 bp long, double stranded, circular, and transcribed from both the guanine‐rich heavy (H) strand and the cytosine rich light (L) strand, encoding 22 tRNAs, 2 rRNAs, and 13 mRNAs. Mitochondrial genes are located close to each other (37 genes on 16.5kb). The mtDNA has no introns, and only a small noncoding region.6 As all protein‐encoding mitochondrial genes encode proteins involved in oxidative phosphorylation, all diseases caused by mutations in the mitochondrial genome are characterized by defective oxidative phosphorylation. In most cases, this leads to multisystem disorders, but some mitochondrial diseases can cause dysfunction in one organ, often the cochlea.1 In addition some mitochondrial proteins are encoded by nuclear DNA. The nuclear genes encoding mitochondrial proteins are involved in all mitochondrial functions mentioned, and the expression of mt DNA itself. Therefore, the spectrum of mitochondrial genetic diseases caused by inherited mutations in nuclear genes is broad.
Given the significant energy consumption of the cochlea, mitochondrial disease can cause syndromic or isolated hearing loss. The prevalence of mtDNA‐related deafness is less than 1% of patients with congenital deafness but is close to 5% in Caucasians with postlingual deafness. MELAS is a syndrome typified by mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes.2 It is the most common maternally inherited mitochondrial disease. Although a number of mutations can cause the syndrome, the majority (80%) are caused by a point mutation of alanine and guanine at 3243.3 This particular mutation can be identified in other mitochondrial diseases with hearing loss such as MIDD (Maternally Inherited Diabetes mellitus with Deafness) and Kearns‐Sayre syndrome.7
Hearing loss is present in 30%‐75% of patients with MELAS syndrome.8 It typically presents before the age of 40 and can vary from mild loss to severe loss meeting the criteria for cochlear implantation. About 20% of patients with MELAS syndrome have severe to profound sensorineural hearing loss. Hearing loss is symmetric, commonly starts at the high tones progressing to involve the mid‐ and low tones. However, it can be the same across frequencies. Hearing loss can be the first and initially the only manifestation of MELAS syndrome. The diagnosis of a mitochondrial cause for hearing loss can have several clinical implications such as avoidance of exposure to noise and aminoglycosides. In the case presented hearing loss was the sole symptom of MELAS syndrome for more than a decade. In a family with no history of either hearing loss or mitochondrial disease, a patient with such long duration of deafness is unlikely to be identified with mitochondrial deafness. The patient died at the age of 42 years. Previous descriptions of temporal bone histopathology are of patients who died at the age of 23 and 30 years, making the reported case the oldest one with MELAS syndrome.9, 10
The main histopathology finding in the studied case is severe atrophy of the stria vascularis in both cochleae with preservation of the organ of Corti and most spiral ganglion neurons. Spiral ganglion population exceeded 50% of the normal age‐matched complement even in the worse hearing right ear. By itself, this level of neuronal population usually is not associated with pure‐tone thresholds shifts.
These observations were seen in two previously reported temporal bones.9, 11 Why do the various cochlear cells demonstrate such a wide variability in the impact of the mutation on their survival? Processed temporal bones were used to correlate the mutation rate in various cochlear cells with the extent of damage seen. LASER capture can allow harvesting of a single cell from the cochlea. The rate of mutated mtDNA as a fraction of the total mtDNA in cochlear cells was measured. No correlation between the rate of mutated cell and cellular damage was detected.9, 11 Conceivably, different cochlear tissues have different energy needs, and the stria vascularis could be more sensitive to mitochondrial injury as compared to other cochlear cells. Alternatively, damage to the stria vascularis may not be a primary event but rather a secondary one. The production of cytochrome oxidase complex 4 is modified, resulting in an increase in ROS that induce cochlear damage. ROS acts differently on different structures of the cochlea, possibly explaining the variability in intracochlear damage.
Treatment of mitochondrial disease is challenging. Deficiency in coenzyme Q10, is the only condition that is ameliorated with dietary supplements. For MELAS, arginine and citrulline can provide some symptomatic relief. The patient reported herein was treated with these supplements.12 Both arginine and citrulline act as nitrous oxide precursors. Their administration can result in increased nitrous oxide availability and may lessen the stroke‐like episodes typical of MELAS syndrome. While great advancements are made with treatment of nuclear genetic defects, gene therapy for suppressing or replacing mutated mtDNA is not feasible at this stage. Hearing loss in MELAS can be helped with hearing aids. If hearing worsens to nonaidable level, cochlear implantation can be considered. Despite the ubiquitous insult to the central nervous system in MELAS syndrome, cochlear implantation can be effective.13, 14, 15, 16
Based on the observed findings of good preservation of auditory neurons effective stimulation with a cochlear implant could be expected. This finding may have implications for patients with hearing loss associated with other mitochondrial disease.
In summary, MELAS syndrome is caused by a point mutation in mtDNA. The most significant cochlear pathology is atrophy of the stria vascularis with good preservation of the organ of Corti and spiral ganglion neurons. These observations have implications for the rehabilitation of hearing loss in this and other mitochondrial diseases.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Handzel O, Ungar OJ, Lee DJ, Nadol JB Jr. Temporal bone histopathology in MELAS syndrome. Laryngoscope Investigative Otolaryngology. 2020;5:152–156. 10.1002/lio2.344
Funding information Margaret and Leo Meyer and Hans M. Hirsch Foundation
REFERENCES
- 1. Kokotas H, Peterson MB, Willems PJ. Mitochondrial deafness. Clin Genet. 2007;71:379‐391. [DOI] [PubMed] [Google Scholar]
- 2. Corrado A. Genetic neuromuscular disorders: a case based approach; Switzerland: Springer, 2014:233‐237. [Google Scholar]
- 3. Di Stadio A, Pegoraro V, Giaretta L, Dipietro L, Marozzo R, Angelini C. Hearing impairment in MELAS: new prospective in clinical use of miRNA: a systemic review. Orphanet J Rare Dis. 2018;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merchant SN, Joseph B, Nadol JB Jr. Schuknecht's pathology of the ear. Vol 3e USA: PMPH; 2010:29. [Google Scholar]
- 5. Smith RA, Hartley RC, Cochemé HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012. Jun;33(6):341‐352. [DOI] [PubMed] [Google Scholar]
- 6. Van der Wijst MG, van Tilburg AY, Ruiters MH, Rots MG. Experimental mitochondria‐targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci Rep. 2017;7:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sinnathuray AR, Raut V, Toner JG, Magee A. Cochlear implantation in a profoundly deaf patient with MELAS syndrome. J Neurol Neurosurg Psych. 2002;73:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vandana VP, Bindu PS, Sonam K, et al. Audiological manifestation in MELAS syndrome. Clin Neurol Neurosurg. 2016;148:17‐21. [DOI] [PubMed] [Google Scholar]
- 9. Takahashi K, Merchant SN, Miyazawa T, et al. Temporal bone histopathological and quantitative analysis of mitochondrial DNA in MELAS. Laryngoscope. 2003;113:1362‐1368. [DOI] [PubMed] [Google Scholar]
- 10. Nadol JB Jr, Merchant SN. Histopathology and molecular genetics of hearing loss in humans. Int J Ped Otolaryngol. 2001;61:1‐15. [DOI] [PubMed] [Google Scholar]
- 11. Koda H, Kimura Y, Ishige I, Eishi Y, Iino Y, Kitamura K. Quantitative cellular level analysis of mitochondrial DNA 3243A>G mutation in individual tissues from archival temporal bones of a MELAS patient. Acta Otolaryngologica. 2010;130:344‐350. [DOI] [PubMed] [Google Scholar]
- 12. El‐Hattab AW, Almannai M, Scaglia F. Arginine and citrulline for the treatment of MELAS syndrome. J Inborn Errors Metab Screen. 2017. Jan;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karkos PD, Anari S, Johnson IJ. Cochlear implantation in patients with MELAS syndrome. Eur Arch Otorhinolaryngol. 2005. Apr;262(4):322‐324. [DOI] [PubMed] [Google Scholar]
- 14. Scarpelli M, Zappini F, Filosto M, Russignan A, Tonin P, Tomelleri G. Mitochondrial sensorineural hearing loss: a retrospective study and a description of cochlear implantation in a MELAS patient. Genet Res Int. 2012;2012:287432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hill D, Wintersgill S, Stott L, Cadge B, Graham J. Cochlear implantation in a profoundly deaf patient with MELAS syndrome. J Neurol Neurosurg Psychiatry. 2001;71(2):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosenthal EL, Kileny PR, Boerst A, Telian SA. Successful cochlear implantation in a patient with MELAS syndrome. Am J Otol. 1999. Mar;20(2):187‐190. [PubMed] [Google Scholar]