

Artemisinin resistance has emerged in Southeast Asia, endangering the substantial progress in malaria elimination worldwide. It is associated with mutations in the PfK13 protein, but how PfK13 mediates artemisinin resistance is not completely understood. Here we used a new antibody against PfK13 to show that the PfK13 protein is expressed in all stages of the asexual intraerythrocytic cycle as well as in gametocytes and is partially localized in the endoplasmic reticulum. By introducing four PfK13 mutations into the 3D7 strain and reverting these mutations in field parasite isolates, we determined the impacts of these mutations identified in the parasite populations from northern Myanmar on the ring stage using the in vitro ring survival assay. The introduction of the N458Y mutation into the 3D7 background significantly increased the survival rates of the ring-stage parasites but at the cost of the reduced fitness of the parasites. Introduction of the F446I mutation, the most prevalent PfK13 mutation in northern Myanmar, did not result in a significant increase in ring-stage survival after exposure to dihydroartemisinin (DHA), but these parasites showed extended ring-stage development. Further, parasites with the F446I mutation showed only a marginal loss of fitness, partially explaining its high frequency in northern Myanmar. Conversely, reverting all these mutations, except for the C469Y mutation, back to their respective wild types reduced the ring-stage survival of these isolates in response to in vitro DHA treatment.
KEYWORDS: PfK13, Plasmodium falciparum, artemisinin resistance, China-Myanmar border, mutations, drug resistance
ABSTRACT
Mutations in the Plasmodium falciparum Kelch 13 (PfK13) protein are associated with artemisinin resistance. PfK13 is essential for asexual erythrocytic development, but its function is not known. We tagged the PfK13 protein with green fluorescent protein in P. falciparum to study its expression and localization in asexual and sexual stages. We used a new antibody against PfK13 to show that the PfK13 protein is expressed ubiquitously in both asexual erythrocytic stages and gametocytes and is localized in punctate structures, partially overlapping an endoplasmic reticulum marker. We introduced into the 3D7 strain four PfK13 mutations (F446I, N458Y, C469Y, and F495L) identified in parasites from the China-Myanmar border area and characterized the in vitro artemisinin response phenotypes of the mutants. We found that all the parasites with the introduced PfK13 mutations showed higher survival rates in the ring-stage survival assay (RSA) than the wild-type (WT) control, but only parasites with N458Y displayed a significantly higher RSA value (26.3%) than the WT control. After these PfK13 mutations were reverted back to the WT in field parasite isolates, all revertant parasites except those with the C469Y mutation showed significantly lower RSA values than their respective parental isolates. Although the 3D7 parasites with introduced F446I, the predominant PfK13 mutation in northern Myanmar, did not show significantly higher RSA values than the WT, they had prolonged ring-stage development and showed very little fitness cost in in vitro culture competition assays. In comparison, parasites with the N458Y mutations also had a prolonged ring stage and showed upregulated resistance pathways in response to artemisinin, but this mutation produced a significant fitness cost, potentially leading to their lower prevalence in the Greater Mekong subregion.
INTRODUCTION
The emergence and spread of artemisinin (ART)-resistant Plasmodium falciparum parasites in the Greater Mekong subregion (GMS) of Southeast Asia threaten the substantial progress made toward malaria elimination (1). Reports of ART resistance first surfaced from northwest Cambodia in about 2006 (2, 3), which was followed by a series of reports of ART-resistant parasites appearing in other parts of the GMS (4–10). Although resistance is not yet detected in Africa, cross-sectional surveys in Uganda detected P. falciparum parasites with elevated ex vivo ring-stage survival rates (11), prompting further follow-up studies in Africa. Since ART-based combination therapies (ACTs) are the frontline treatment for falciparum malaria, close surveillance of ART resistance is warranted. It is therefore a priority to track the evolution and spread of ART resistance so that strategies can be designed to prevent the further spread of the resistant parasites.
According to WHO guidelines, artemisinin resistance is defined as delayed parasite clearance following treatment with an artesunate monotherapy or with an artemisinin-based combination therapy (2, 12). ART-resistant parasites have a parasite clearance half-life of >5 h after ART treatment, whereas that for sensitive parasites is ∼2 h. As a result, patients infected with ART-resistant parasites often remain parasite positive 3 days after treatment (13). Variations in host immunity also influence parasite clearance after ART treatment and the interpretation of emerging ART resistance (14–18). The delayed-clearance phenotype cannot be captured by the traditional in vitro/ex vivo drug assays that measure parasite proliferation but is associated with the results of the ring-stage survival assay (RSA), which determines the percentage of early (0 to 3 h postinvasion) ring-stage parasites (RSA0–3 h) able to survive a single 6-h pulse of 700 nM dihydroartemisinin (DHA), the active metabolite of ART (19). Whereas genome-wide association studies, gene manipulation experiments, and studies of field isolates with resistant phenotypes have led to the identification of a number of genetic loci and proteins (e.g., ATG18, coronin, pfap2μ, falcipain 2a) predicted to play a role in ART resistance (10, 20–28), now the P. falciparum Kelch 13 (PfK13) protein appears to be a major player, as mutations in its propeller domain have been associated with ART resistance (29). Some of the PfK13 mutations, including Y493H, R539T, I543T, and C580Y, that were associated with clinical ART resistance have been genetically confirmed to confer elevated RSA0–3 h survival rates in vitro (30, 31). ART drugs are known to induce oxidative stress and cellular damage in P. falciparum, but how the mutated forms of the PfK13 protein allow the parasites to tolerate this oxidative assault is not yet clear. PfK13 has been shown to be essential for parasite survival (32, 33). Conditional knockout of PfK13 leads to rapid growth arrest at the ring stage, with parasites gradually turning into condensed forms, suggesting that PfK13 is required for the survival of the ring stage and/or for the ring stage-to-trophozoite transition in P. falciparum (32). Interestingly, PfK13 mutant parasites can reprogram their intraerythrocytic development cycle (IDC) to have a prolonged ring stage (34–36). Different mechanisms of ART resistance, which may act cooperatively, have been proposed, including the upregulation of the unfolded protein response pathway (37), decreased expression of genes involved in DNA replication (31), and an enhanced stress response, comprising the ubiquitin/proteasome pathway (34). Phosphorylation of the parasite α subunit of eukaryotic initiation factor 2 (eIF2α) and the interaction of phosphatidylinositol 3-kinase (PI3K) with PfK13 have also been proposed to play a role in ART resistance (38–40). Thus, it appears that PfK13 is central to multiple intracellular processes in P. falciparum.
More than 200 PfK13 mutations have been reported across the globe (7, 8, 41–43), but only some of these mutations (especially those prevalent in the GMS) have been associated with slow parasite clearance and reduced ART susceptibility in RSA. Even within geographical proximity of the GMS, PfK13 mutations are highly diverse and region specific, probably reflecting the different drug use histories and the divergent evolutionary trajectories of these parasites. PfK13 C580Y is the predominant mutation in parasites from the Thai-Cambodian border area, southern Laos, and Vietnam (7, 29, 44, 45), whereas the Y493H, R539T, and I543T mutations are more common in the Thai-Cambodian/Thai-Myanmar border areas (8, 9, 29, 46). The F446I mutation is most prevalent in northern Myanmar and the China-Myanmar border area, and some reports showed its association with ART resistance (43, 47, 48). In our earlier study, several mutations present sporadically in the China-Myanmar border area (e.g., N458Y, C469Y, and F495L) showed relatively high RSA values (43). The N458Y mutation was also identified in Thailand and Cambodia and showed a significant association with prolonged parasite clearance half-lives in clinical studies and elevated in vitro RSA values (42, 43, 49, 50). We identified one field isolate from the China-Myanmar border area with the C469Y mutation showing high in vitro RSA values (43, 51). It is noteworthy that this mutation was also reported recently from Uganda (11, 52–54). The F495L mutation was also lately reported from sub-Saharan Africa (47, 52, 53, 55, 56), but it was not associated with ART resistance. In the China-Myanmar border area, the F446I, N458Y, and C469Y mutations have all been associated with positive parasitemia on day 3 after treatment with an ACT (47). Here we aimed to determine whether these PfK13 mutations are associated with in vitro ART susceptibility by introducing them into laboratory strain 3D7 and reverting these mutations to the wild type (WT) in respective field isolates.
RESULTS
Expression and localization of PfK13.
To study the expression and localization of PfK13, we generated affinity-purified rabbit polyclonal antisera against the PfK13 peptide from amino acids (aa) 239 to 257 and a parasite line (GFP::PfK13) in the 3D7 strain with the N terminus of the endogenous PfK13 protein tagged with green fluorescent protein (GFP). To study PfK13 expression, Western blotting was performed with 100 μg of lysates from each asexual stage, including rings, trophozoites, and schizonts, of the 3D7 parasites using anti-PfK13 peptide antibodies. A specific band of ∼83 kDa was detected in the trophozoites and schizonts, corresponding to the predicted size of the endogenous PfK13 protein (Fig. 1A). In ring-stage parasites, however, in addition to the ∼83-kDa major protein band, the anti-PfK13 peptide antibodies also detected three smaller, less abundant protein bands, which may have been degraded or/and processed PfK13 or cross-reacting proteins expressed only in the ring-stage parasites. Compared with the expression of the aldolase protein as the protein loading control, PfK13 protein expression appeared to be relatively consistent throughout the IDC. For validation of the PfK13 expression results detected with the anti-PfK13 peptide antibodies, we tagged PfK13 with GFP at its N terminus using the selection-linked integration (SLI) approach (see Fig. S1A in the supplemental material), as PfK13 is refractory to GFP tagging at the C terminus (32). Using mixed asexual-stage parasites, we confirmed the successful integration of GFP by integration-specific PCR (Fig. S1B). In a Western blot, the anti-GFP antibody detected a major protein band of ∼165 kDa, which agreed with the predicted size of PfK13 fused with two FK506-binding protein (FKBP) domains and GFP, as well as some protein bands of lower molecular weights, suggestive of degraded protein products (Fig. S1C). Western blotting with protein lysates from synchronized rings, trophozoites, schizonts, and purified merozoites of the GFP::PfK13 parasite line using the anti-GFP antibodies consistently detected the presence of the 165-kDa band in all asexual stages (Fig. S2A). Although lower-molecular-weight protein products were detected in all stages, bands that were exclusive to the ring stage were not detected in the GFP::PfK13 parasites.
FIG 1.
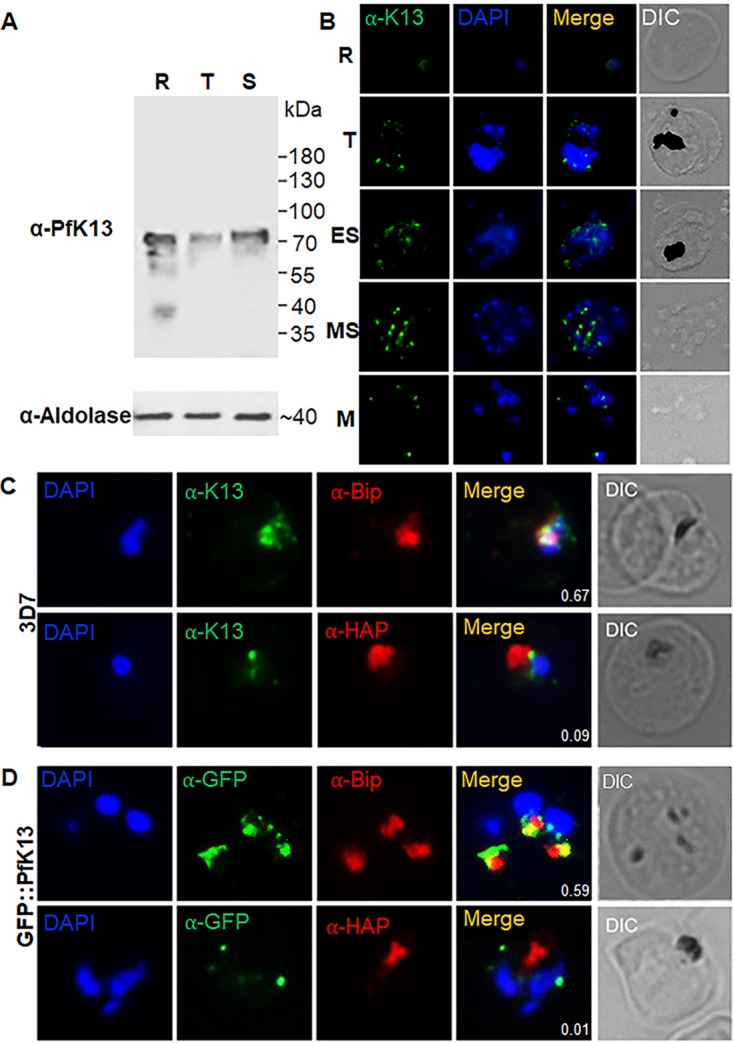
Expression and localization of PfK13 during asexual erythrocytic growth. (A) Western blots of PfK13 expression at the ring (R), trophozoite (T), and schizont (S) stages of 3D7 parasites with anti-PfK13 antibodies. Aldolase was used as the control for equal protein loading from different stages. (B) Localization of PfK13 in the ring (R), trophozoite (T), early schizont (ES), mature schizont (MS), and merozoite (M) stages by IFA with anti-PfK13 antibodies. (C) Colocalization analysis of PfK13 with BiP (an ER marker) and HAP (a food vacuole marker) in 3D7 using anti-PfK13 antibodies. (D) Colocalization analysis of PfK13 with BiP and HAP in GFP::PfK13 parasites using anti-GFP antibodies. DIC, differential interference contrast.
(A) Schematic showing the replacement of endogenous PfK13 with N-terminal GFP/PTP-tagged full-length recoded PfK13 (WT or mutated) using the selection-linked integration (SLI) method. Genomic integration of the plasmid results in the expression of a second resistance marker (yeast dihydroorotate dehydrogenase [yDHODH]) that is flanked by two skip peptides (2A), generating separate proteins. Arrows labeled 1 to 5 indicate the primers used for integration-specific PCR. Arrows 6 and 7 indicate the hsp86 and the PfK13 endogenous promoters, respectively. hDHFR, human dihydrofolate reductase; FKBP, FK506-binding protein; GFP, green fluorescent protein; PTP, protein C-tobacco etch virus-protein A. (B) PCR performed using genomic DNA from different parasite lines and the primers indicated in panel A to show integration of the plasmid into the correct endogenous locus with primers 1 and 2 (top) and the intact endogenous locus with primers 1 and 4 (bottom). (C) Western blots detecting the full-length PfK13 protein (165 kDa) with an N-terminal 2×FKBP-GFP-2×FKBP tag (left) and 2×FKBP-PTP-2×FKBP tag (right) using anti-GFP and anti-protein C (1:1,000; catalog number A00637; GenScript) antibodies, respectively. (D) Schematic showing the replacement of endogenous PfK13 with N-terminal GFP/PTP-tagged full-length recoded WT PfK13 using selection-linked integration. Genomic integration of the plasmid results in the expression of a second resistance marker (yeast dihydroorotate dehydrogenase [yDHODH]) that is flanked by two skip peptides (2A). Arrows labeled 1 to 5 indicate the primers used for performing integration-specific PCR. Arrows 6 and 7 indicate the hsp86 and the PfK13 endogenous promoters, respectively. hDHFR, human dihydrofolate reductase; FKBP, FK506-binding protein; GFP, green fluorescent protein; PTP, protein C-tobacco etch virus-protein A. (E) PCR performed using genomic DNA from different parasite lines and the primers indicated in panel A to show integration into the correct endogenous locus with primers 1 and 5 (top) and the intact endogenous locus with primers 1 and 4 (bottom). (F) IFAs detecting the expression of GFP::PfK13 WT in transgenic field isolates using anti-GFP antibodies. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). Download FIG S1, PPT file, 1.0 MB (1MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression and localization of GFP::PfK13 during asexual erythrocytic stages. (A) Western blots of PfK13 expression in GFP::PfK13 probed with anti-GFP antibodies. Aldolase was used as the control for equal parasite protein loading. Lysates were prepared from synchronized ring (R), trophozoite (T), schizont (S), and merozoite (M) stages of GFP::PfK13 parasites and mixed asexual stages of the 3D7 parasite. (B) IFA of asexual-stage parasites of the GFP::PfK13 line probed with anti-GFP antibodies. (C). IFA of stage II to V gametocytes of the GFP::PfK13 line probed with anti-GFP antibodies. PfK13 was detected in all asexual stages and gametocytes and was localized in punctate structures in the cytoplasm. The number of puncta increases from the ring to the schizont stage. Download FIG S2, PPT file, 1.2 MB (1.2MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To investigate the subcellular localization of PfK13, an indirect immunofluorescence assay (IFA) and live-cell imaging were performed with both asexual-stage parasites and gametocytes. Probing of the 3D7 parasites with the anti-PfK13 antibodies showed punctate staining in the cytoplasm of the parasites, and the number of puncta increased from the ring stage to the schizont stage (Fig. 1B). Live-cell imaging of the GFP::PfK13 line showed a similar distribution pattern of the GFP fluorescent puncta (Fig. S2B). These puncta did not overlap with the parasite nuclei, suggesting that they were localized in the cytoplasm. Similarly, live-cell imaging of gametocytes in the GFP::PfK13 parasite line identified multiple punctate green fluorescent foci throughout gametocyte development (Fig. S2C). To further define the PfK13 localization, we performed colocalization analysis of PfK13 with an endoplasmic reticulum (ER) marker (heat shock protein 70 [BiP]) and a food vacuole marker (histoaspartic protease [HAP]). The results obtained using anti-PfK13 antibodies in 3D7 parasites showed a partial colocalization of PfK13 with the ER marker (Pearson’s correlation coefficient, 0.575) and an insignificant overlap with the food vacuole marker (Pearson’s correlation coefficient, <0.3) (Fig. 1C). IFA performed in the GFP::PfK13 line further confirmed a partial colocalization with the ER marker (Pearson’s correlation coefficient, 0.524) and much less overlap with the food vacuole marker (Pearson’s correlation coefficient, <0.3) (Fig. 1D).
Proteins potentially associated with PfK13.
To identify proteins that are associated with PfK13, we performed immunoprecipitation (IP) with protein lysates of asexual blood-stage parasites from the GFP::PfK13 line using GFP-Trap beads followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Using a false discovery rate (FDR) of 1%, 34 proteins that were common in all three biological replicates were identified (Table 1). Among them, 10 proteins are involved in the unfolded protein response pathway. Other proteins are predicted to play roles in transcription, translation, and other biosynthetic pathways. Three proteins, BiP, protein disulfide isomerase (ERp72), and the putative endoplasmin (GRP94), are potentially involved in the Plasmodium reactive oxidative stress complex. Interestingly, among these proteins possibly associated with PfK13, 7 and 4 proteins were found in the up- and downregulated gene categories, respectively, in the transcriptome analysis of ART-resistant field isolates (Table 1) (37).
TABLE 1.
Proteins identified from affinity purification of GFP::PfK13 parasites followed by mass spectrometrya
| Protein name | Identifier | Predicted function |
|---|---|---|
| Kelch protein K13 | PF3D7_1343700 | Unfolded protein response/protein binding |
| Heat shock protein 70 (BiP) | PF3D7_0917900 | Unfolded protein response |
| DnaJ protein | PF3D7_0629200 | Unfolded protein response/protein binding |
| Protein disulfide isomerase (ERp72) |
PF3D7_0827900 | Protein folding |
| 40S ribosomal protein S16 | PF3D7_0813900 | Translation/ structural constituent of ribosome |
| 40S ribosomal protein S19 | PF3D7_0422400 | Organelle assembly/translation/structural constituent of ribosome |
| 40S ribosomal protein S11 | PF3D7_0516200 | Organelle assembly/translation/structural constituent of ribosome |
| Plasmepsin II | PF3D7_1408000 | Aspartic protease |
| 14-3-3 protein | PF3D7_0818200 | Protein binding |
| DNA/RNA-binding protein Alba 3 |
PF3D7_1006200 | Transcription factor/protein binding/mRNA binding |
|
S-Adenosylmethionine synthase |
PF3D7_0922200 | Cellular biosynthetic process/methionine adenosyltransferase activity |
| Parasitophorous vacuolar protein 1 |
PF3D7_1129100 | Food vacuole protein/protein binding |
| Heat shock protein 70 | PF3D7_0818900 | Unfolded protein response/ATPase activity |
| Heat shock protein 70 | PF3D7_1134000 | Unfolded protein response |
| Endoplasmin homolog (GRP94) |
PF3D7_1222300 | Unfolded protein response |
| Merozoite surface protein 1 | PF3D7_0930300 | Protein binding/invasion/protein-containing complex binding |
| High-molecular-weight rhoptry protein 2 |
PF3D7_0929400 | Protein binding/invasion/protein-containing complex binding |
| T-complex protein 1 subunit gamma |
PF3D7_1229500 | Protein folding/chaperonin/unfolded protein binding |
| 26S protease regulatory subunit 8 |
PF3D7_1248900 | Transcription preinitiation complex assembly |
| 26S protease regulatory subunit 6B |
PF3D7_0413600 | Transcription preinitiation complex assembly |
| Elongation factor 1-alpha | PF3D7_1357000 | Translation elongation factor/RNA binding |
| Elongation factor 2 | PF3D7_1451100 | Translation elongation factor/RNA binding |
| Eukaryotic initiation factor 4A | PF3D7_1468700 | Translation initiation/mRNA binding |
| 40S ribosomal protein S3a | PF3D7_0322900 | Translation/structural constituent of ribosome |
| 40S ribosomal protein S15A | PF3D7_0316800 | Translation/structural constituent of ribosome |
| DNA/RNA-binding protein Alba 1 |
PF3D7_0814200 | Transcription factor/translation regulator activity, nucleic acid binding/mRNA binding |
| 60S acidic ribosomal protein P0 |
PF3D7_1130200 | Organelle assembly |
| 60S ribosomal protein L12 | PF3D7_0517000 | Organelle assembly/structural molecule activity |
| Heat shock protein 60 | PF3D7_1015600 | Unfolded protein response/ATPase activity |
| Glyceraldehyde-3-phosphate dehydrogenase |
PF3D7_1462800 | Pyruvate metabolic process/oxidoreductase activity |
| GTP-binding nuclear protein | PF3D7_1117700 | GTPase activity |
| Protein DJ-1 | PF3D7_0627500 | Unfolded protein response/protein deglycase activity |
| Acyl coenzyme A synthetase | PF3D7_0525100 | Ligase |
| l-Lactate dehydrogenase | PF3D7_1324900 | Tricarboxylic acid cycle/lactate dehydrogenase activity |
| 60S ribosomal protein L10 | PF3D7_1414300 | Formation of actively translating ribosomes/structural constituent of ribosome |
Light and dark shading indicate genes up- and downregulated, respectively, in ART-resistant isolates, as reported by Mok et al. (37). All proteins and peptides have an FDR of <1%.
Introduction of PfK13 mutations and drug sensitivity.
The KARMA consortium identified 108 nonsynonymous mutations in the PfK13 propeller domain in P. falciparum populations worldwide (7). In the GMS, two areas (one including Cambodia, Vietnam, and Laos and the other including western Thailand, Myanmar, and China) have nonoverlapping, region-specific distributions of PfK13 mutations (7, 42, 43). We focused on four PfK13 mutations (F446I, N458Y, C469Y, and F495L) that were prevalent or specific to isolates from northern Myanmar and the China-Myanmar border area (43), and some of these have been associated with delayed parasite clearance (29, 42, 47), day 3 parasitemia, and increased RSA values (43). To test whether these PfK13 mutations are linked to altered ART sensitivity, we introduced these four mutations as well as the C580Y mutation as a positive control into the 3D7 strain using the SLI method together with N-terminal GFP tagging. We also used WT PfK13 as the transfection control. Integration of the different PfK13 versions was confirmed by PCR, Sanger sequencing, and Western blotting (Fig. S1B and S3A).
Western blot (A), flow cytometry (B), and IFA (C) data showing no significant difference in the level of expression between GFP::WT PfK13 and GFP::mutant PfK13 (F446I, N458Y, C469Y, F495L, and C580Y) in asexual stages. To obtain the mean fluorescence intensities with flow cytometry, 50,000 cells were counted at the trophozoite stage for each parasite line in 2 independent experiments. (D) IFA of GFP::PfK13 in stage III gametocytes of mutant transgenic parasites showing the similar expression and distribution patterns of mutant PfK13. For Western blots and IFA, GFP::PfK13 was detected by anti-GFP antibodies. Download FIG S3, PPT file, 1.8 MB (1.9MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The WT and mutant PfK13 proteins showed expression levels and localization patterns similar to those of the WT recombinant when analyzed by Western blotting, flow cytometry, and live-cell imaging (Fig. S3A to C). The punctate GFP localization patterns of the WT and PfK13 mutant lines also appeared to be similar in gametocyte stages (Fig. S3D). Since PfK13 was reported to concentrate in phosphatidylinositol 3-phosphate (PI3P) vesicles in P. falciparum (40), we wanted to determine whether the PfK13 mutations examined here may affect the distribution of PfK13 and PI3P. Colocalization analysis of GFP::PfK13 with PI3P revealed at least one green fluorescent dot close to the site of PI3P staining, but no overlap of PfK13 staining with PI3P in the WT or the mutant parasite lines was observed at late asexual stages, with Pearson’s correlation coefficient being <0.24 (Fig. S4). The mouse anti-PI3P antibodies used here were unable to detect PI3P at the early ring stage.
Colocalization analysis of WT and mutated PfK13 with PI3P. Anti-GFP rabbit antibodies and anti-PI3P mouse antibodies were used to probe GFP::PfK13 (green) and PI3P (red) in transgenic 3D7 parasites at the trophozoite (A) and schizont (B) stages. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). Also shown are the bright-field (BF) and merged images. Download FIG S4, PPT file, 2.1 MB (2.1MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine whether the introduced PfK13 mutations conferred ART resistance in the transfected 3D7 parasite lines, RSA was performed for each parasite line with three biological replicates (Table S2A). Given that the study design introduces two drug selection cassettes into the 3D7 parasites, which could lead to altered drug responses, a transfection control with the WT PfK13 was included for comparison. RSA results showed that the transfection-control parasites with WT PfK13 had ring survival (1.9%) slightly but not significantly higher than that for the parent 3D7 line (0.4%), whereas introduction of PfK13 with the C580Y mutation in the 3D7 line as a positive control conferred an RSA level (30.6%) significantly higher than that for the transfection control (P < 0.001, nonparametric Wilcoxon matched-pairs test; Fig. 2A). Parasites with F495L, F446I, and C469Y all had RSA values greater than 1% (6.2%, 4.8%, and 3.5%, respectively), albeit they were not statistically significantly higher than those for 3D7 or the WT PfK13 transfection control. Only the N458Y parasites showed a significantly higher ring survival rate (26.3%) than both 3D7 and the transfection control (P < 0.0001, nonparametric Wilcoxon matched-pairs test; Fig. 2A).
FIG 2.
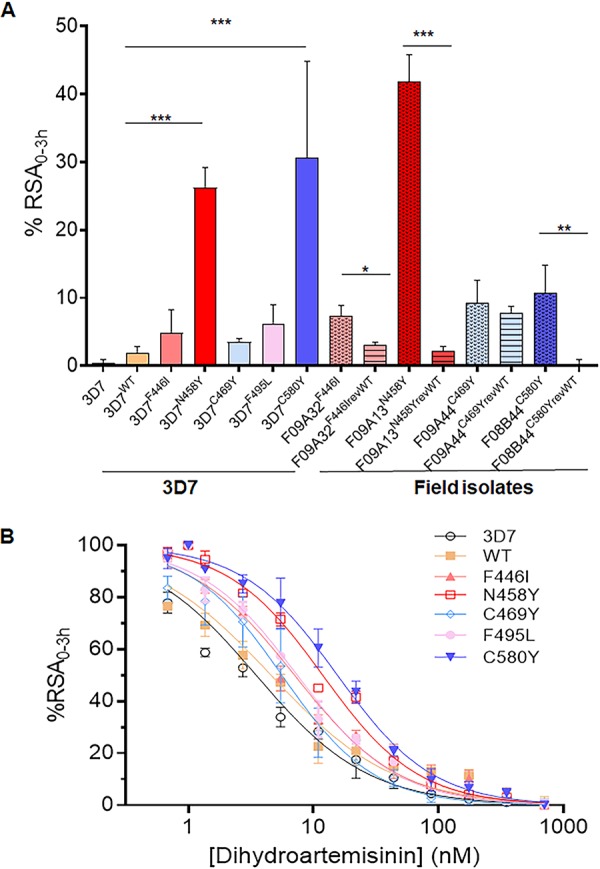
Ring-stage survival assay (RSA) of WT and mutant PfK13 parasites. (A) Survival for 0 to 3 h of early-ring-stage parasites of 3D7, the transfection control with the WT PfK13 gene (3D7WT), and 3D7 with one of five mutations (3D7F446I, 3D7N458Y, 3D7C469Y, 3D7F495L, 3D7C580Y), along with the transgenic field isolates before and after removal of PfK13 mutations (F09A32F446I, F09A32F446IrevWT, F09A13N458Y, F09A13N458YrevWT, F09A44C469Y, F09A44C469YrevWT, F08B44C580Y, F08B44C580YrevWT), was determined using the standard RSA0–3 h. The results for each parasite strain or clone were measured in three independent experiments, and the RSA values (in percent) are shown as the mean and standard deviation. Comparison of the results was done using a nonparametric Wilcoxon t test, and the P values are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) RSA0–3 h50% of the transgenic 3D7 parasites with WT and mutant PfK13. Parasites were exposed to a 4-h pulse of DHA ranging from 0.6 to 700 nM, and the survival of the parasites under this treatment condition was plotted against the drug concentration. The sigmoid parasite drug response curves were used to estimate the concentration of DHA causing 50% death of the ring-stage parasites (see Table S3 in the supplemental material). Each data point is the mean percent survival from at least three independent experiments performed in duplicate. As a vehicle control, parasites were treated with DMSO.
To confirm these findings in isogenic 3D7 parasite lines, we reverted these PfK13 mutations to the WT in the respective field isolates from the China-Myanmar border area (F09A32F446I, F09A13N458Y, F09A44C469Y, F08B44C580Y) using the same SLI method (Fig. S1D). These revertant parasites were verified by integration-specific PCR as well as live-cell imaging of GFP fluorescence in asexual stages (Fig. S1E and F). Whereas isolate F09A44C469Y reverted back to the WT (F09A44C469YrevWT) showed only a slight, insignificant decrease in the RSA value compared to that for F09A44C469Y, all other field strains showed significant decreases when the PfK13 alleles were reverted back to the WT (Fig. 2A). Specifically, RSA values for F09A32F446I, F09A13N458Y, and F08B44C580Y decreased from 7.3%, 42%, and 10% to 3.1%, 2.2%, and 0.3%, respectively (P < 0.05) (Fig. 2A).
Since RSA0–3 h measures the ring-stage survival only after exposure to 700 nM DHA, we further evaluated the in vitro sensitivity of the isogenic 3D7 parasites to a gradient of DHA concentrations ranging from 0.6 to 700 nM for 4 h in the RSA0–3 h50% (57–59), which determines the amount of DHA required to kill 50% of the parasites. Again, all parasite lines with introduced PfK13 mutations showed increased RSA0–3 h50% values compared to those for 3D7 (3.5 nM) and the WT PfK13 transfection control (4.6 nM) (Fig. 2B), but only parasites with the C580Y and N458Y mutations had RSA0–3 h50% values (15.6 and 11.9 nM, respectively; n = 3) significantly higher than those for 3D7 or the WT PfK13 transfection control (Table S3).
To determine if altered DHA sensitivity also affected in vitro susceptibility to other antimalarial drugs, we determined the 50% inhibitory concentration (IC50) values of some commonly used antimalarial drugs for the 3D7 parasite lines carrying the introduced PfK13 mutations using the traditional SYBR green I-based drug sensitivity assay (Table 2). Compared to both 3D7 and the PfK13 transfection-control parasites, some parasites with PfK13 mutations appeared to be slightly more sensitive to chloroquine (CQ) (F495L) and mefloquine (MFQ) (F495L and N458Y), but none of these changes were statistically significant. Thus, introduction of these PfK13 mutations alone did not alter the sensitivities of these transgenic 3D7 parasites to other antimalarial drugs.
TABLE 2.
IC50 values of 10 different antimalarial drugs for parasites with mutated and WT PfK13
| Parasite | IC50 (nM)a
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AMQ | AS | AM | DHA | CQ | LMF | MFQ | PND | PPQ | QN | |
| 3D7 | 11 ± 5 | 1.7 ± 0.2 | 1.80 ± 0.01 | 0.7 ± 0.4 | 18.7 ± 0.8 | 3.2 ± 0.4 | 16.10 ± 0.04 | 1.7 ± 0.4 | 18 ± 2 | 25 ± 6 |
| WT | 13 ± 4 | 1.5 ± 0.2 | 1.8 ± 0.1 | 0.6 ± 0.4 | 19 ± 2 | 5 ± 2 | 15 ± 1 | 2.0 ± 0.1 | 19.2 ± 0.3 | 23.8 ± 0.6 |
| F446I | 12 ± 4 | 1.4 ± 0.1 | 1.60 ± 0.03 | 0.8 ± 0.4 | 17.5 ± 0.1 | 5 ± 1 | 14 ± 2 | 1.8 ± 0.3 | 18.90 ± 0.04 | 22 ± 8 |
| C469Y | 11 ± 4 | 1.5 ± 0.1 | 1.6 ± 0.3 | 0.6 ± 0.1 | 16.8 ± 0.4 | 10 ± 9 | 18 ± 8 | 1.90 ± 0.01 | 19.8 ± 0.5 | 23 ± 4 |
| F495L | 12 ± 4 | 1.4 ± 0.1 | 1.6 ± 0.1 | 0.8 ± 0.3 | 15.40 ± 0.03 | 4.0 ± 0.2 | 11.96 ± 0.6 | 1.88 ± 0.07 | 17.4 ± 0.8 | 19 ± 5 |
| C580Y | 9 ± 8 | 1.2 ± 0.5 | 1.8 ± 0.2 | 0.8 ± 0.4 | 17.1 ± 0.3 | 4.1 ± 0.3 | 16 ± 3 | 2.0 ± 0.2 | 18.1 ± 0.1 | 17 ± 19 |
| N458Y | 12 ± 6 | 1.3 ± 0.1 | 1.7 ± 0.1 | 0.9 ± 0.5 | 19 ± 1 | 5.4 ± 0.5 | 13.7 ± 0.6 | 1.9 ± 0.1 | 20 ± 3 | 30 ± 12 |
The IC50 values (mean ± SD) of 10 different antimalarial drugs were measured in 72-h proliferation assays, with the final parasitemia being determined from the fluorescence intensities measured on a FLUOstar Optima microplate fluorometer using SYBR green staining. Results from three independent experiments performed in duplicate are presented for 3D7 and parasites with WT and mutated (F446I, C469Y, F495L, C580Y, and N458Y) PfK13. AMQ, amodiaquine dihydrochloride dihydrate; AS, artesunate; AM, artemether; DHA, dihydroartemisinin; CQ, chloroquine diphosphate; LMF, lumefantrine; MFQ, mefloquine hydrochloride; PND, pyronaridine; PPQ, piperaquine; QN, quinine.
In vitro fitness of transgenic parasites.
3D7 parasites with PfK13 mutations did not show a noticeable difference in gross morphology from the WT. To analyze their in vitro growth in more detail, we first followed the growth of tightly synchronized parasites at a 3-h interval in a single IDC. Our results showed that parasites with the PfK13 F446I, N458Y, and C580Y mutations had ring-stage development extended for 3 to 4 h. However, subsequent trophozoite development in these parasites was shortened, and they completed schizogony at approximately the same time as the WT PfK13 parasites (Fig. 3). When cultures of all parasite clones were initiated at a 0.1% ring-stage parasitemia, those with the mutant PfK13 alleles grew more slowly than the WT PfK13 parasites (Fig. 4A). The difference became noticeable on day 4 of culture, and on day 8, parasites carrying the N458Y and F495L mutations had significantly lower parasitemias than the WT PfK13 parasites (P < 0.05, nonparametric Wilcoxon matched-pairs test; Fig. 4A). To determine if the difference in asexual growth of the transgenic parasites also extended to gametocyte development, we measured gametocytemia in the WT and the transgenic 3D7 parasite lines on day 3 and day 10 after the induction of gametocytogenesis. On both days, the gametocytemias were not significantly different among the WT 3D7 and transgenic parasite lines, albeit the WT, C469Y, and N458Y parasites had higher gametocytemias than the other parasite lines tested (Fig. S5).
FIG 3.

Comparison of intraerythrocytic development patterns between parasites with the introduced WT and mutant PfK13 genes. Purified schizonts from tightly synchronized parasite lines were used to obtain early-ring-stage parasites. The cultures were then maintained in drug-free medium after sorbitol treatment. Blood smears were made every 3 h, and the percentage of each stage was counted (y axis). Ring, trophozoite, and schizont stages are distinguished by different colors, while WT and 3D7 transfected parasites or mutant transfected parasites are shown as continuous and dashed lines, respectively.
FIG 4.
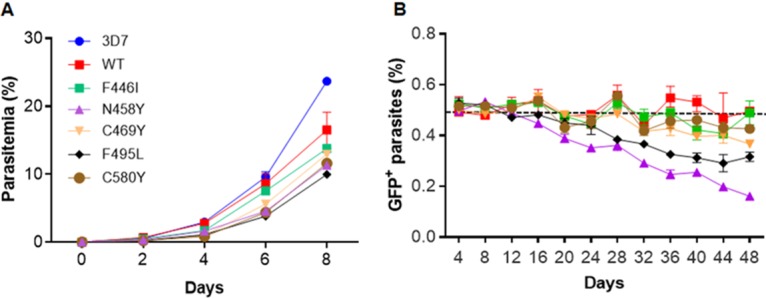
In vitro growth of parasites with the WT or mutant PfK13 gene. (A) In vitro growth curves of 3D7 and parasites with introduced PfK13 genes (WT and five mutations). The data represent parasitemia counts from three independent experiments performed in duplicate. All parasites were started at 0.1% ring-stage parasitemia, and parasitemia was monitored every other day by using Giemsa-stained smears. (B) Relative growth of the parasite lines carrying the WT and mutant PfK13 genes under in vitro competition conditions. All the GFP-tagged WT and mutant PfK13 (F446I, C469Y, F495L, C580Y, and N458Y) parasites were mixed with PTP::WT PfK13 parasites at a 1:1 ratio and cocultured for a period of 48 days in drug-free medium. The cultures were sampled every 3 to 4 days by flow cytometry to determine the proportions of GFP+ parasites and total parasites (Deep Red MitoTracker). The y axis indicates the average values and standard deviations for the percentage of GFP+ cells from two independent assays performed in triplicate. Values of about 50% (dashed line) indicate that the mutated parasites were able to compete well with the PTP::WT PfK13 parasites, whereas numbers <50% indicate a fitness cost.
Comparison of gametocytemias in WT and mutated PfK13 parasite lines. Daily gametocytemia of all transgenic lines and 3D7 was determined by counting Giemsa-stained gametocytes under a microscope from day 3 through day 10 after induction. Data are shown as the mean ± standard deviation from three replicates at days 3 and 7. None of the values for mutant parasites were significantly different from those for either 3D7 or WT parasites (P > 0.1, one-way analysis of variance). Download FIG S5, PPT file, 0.1 MB (122KB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We next determined the potential fitness cost of the PfK13 mutations in the transgenic 3D7 parasites using an in vitro growth competition assay. As a control, we used another parasite line (PTP::PfK13) with N-terminal tagging of PfK13 with a protein C-tobacco etch virus-protein A (PTP) tag (Fig. S1C). The WT PfK13 transfection control did not show any fitness cost, as both PTP::WT PfK13 and GFP::WT PfK13 parasites, when mixed at a 1:1 ratio, maintained an approximately 1:1 ratio during in vitro growth for 48 days (Fig. 4B). Values below the dashed line in Fig. 4B, indicating 50% GFP-positive (GFP+) parasites, indicate that these parasites grew more slowly and were less fit than the WT PfK13 parasites. All parasites with PfK13 mutant alleles displayed variable degrees of fitness costs compared to the WT transgenic control and 3D7. Whereas parasite lines with the F446I and C580Y mutations showed only marginal reductions in fitness, those with the N458Y and F495L mutations were significantly less fit (P < 0.001, nonparametric Wilcoxon matched-pairs test; Fig. 4B). In particular, parasites with the N458Y mutation decreased by 30% during the 48-day growth competition assay.
PfK13 mutations and cellular stress responses.
ART treatment is known to result in less accumulation of ubiquitinated proteins in ART-resistant parasites, consistent with engagement of the ubiquitin-proteasome system in ART resistance (34). To determine whether introduction of PfK13 mutations altered the parasites’ stress responses, we measured the levels of protein ubiquitination in WT PfK13 and transgenic parasites at the trophozoite stage in response to a 90-min exposure to DHA. Protein ubiquitination levels in trophozoites of 3D7 and WT PfK13 transfection-control parasites increased after DHA exposure, confirming the involvement of the ubiquitin-proteasome system in response to the ART family of drugs (Fig. 5A and B). Compared with the results for the WT PfK13 control and 3D7, parasites with the N458Y, C580Y, and F495L mutations showed lower levels of accumulation of ubiquitinated proteins after exposure to DHA. However, this level of reduction in protein ubiquitination was not evident in parasites with the F446I and C469Y mutations (Table S2B). To further delve into the mechanism of the stress response in these parasites, we tested the transcript levels of some of the genes which were significantly upregulated in ART-resistant field isolates (37). Compared to the level of expression of PHISTa by the transgenic WT control, increased expression of PHISTa was shown in all variants except the F446I mutant (Fig. 5C). The N458Y parasites exhibited elevated levels of 4 out of the 6 genes tested, while the C580Y line presented higher levels of transcripts for the peptidyl-prolyl cis-trans isomerase and PHISTa genes.
FIG 5.

Oxidative stress response in parasites with the WT and mutant PfK13 genes. (A) Ubiquitination of P. falciparum proteins following ART treatment. Representative Western blots of parasites show protein ubiquitination after ART treatment. Uninfected RBCs or trophozoite-stage parasites of the 3D7, WT, F446I, C469Y, F495L, C580Y, and N458Y strains were treated with 6 μM ART at 3% hematocrit for 90 min at 37°C. Cell extracts were separated by SDS-PAGE and probed with anti-ubiquitin IgG or anti-aldolase antibodies. (B) Quantitation of ubiquitination in parasites after ART treatment. The graph shows the results of a densitometry analysis of the anti-ubiquitin signal normalized to the anti-aldolase signal from three independent experiments (***, P <0.005, nonparametric Wilcoxon t test; the raw data are presented in Table S2B in the supplemental material). (C) Quantification of relative expression of genes upregulated in ART-resistant field isolates at the early ring stage. RT-quantitative PCR analysis was performed at the early ring stage of WT and isogenic transfected 3D7 parasites carrying the F446I, C469Y, F495L, N458Y, and C580Y mutations for the transcripts of six genes which are upregulated in the ART-resistant parasites (PF3D7_0827900, ERp72; PF3D7_0917900, BiP; PF3D7_1010700, dolichyl-phosphate-mannose protein mannosyl transferase; PF3D7_1372000, plasmodium exported protein [PHISTa]; PF3D7_1108600, endoplasmic reticulum-resident calcium binding protein; and PF3D7_1115600, peptidyl-prolyl cis-trans isomerase). Expression levels were normalized to the level for the housekeeping gene seryl-tRNA synthetase, and relative expression (fold change) was calculated using the expression levels in the WT parasites. Values of more than 1 (dashed line) indicate upregulation.
DISCUSSION
In this study, we aimed to genetically determine whether four PfK13 Kelch domain mutations (F446I, N458Y, C469Y, and F495L) present in P. falciparum parasite populations from the China-Myanmar border area confer in vitro resistance to ART drugs. We introduced these four mutations into the 3D7 genetic background using the SLI approach, with the recoded PfK13 gene being driven by its endogenous promoter, which allowed us to compare the WT with the isogenic mutants. Though the introduction of all PfK13 mutations into the 3D7 genetic background increased in vitro RSA values, only the N458Y mutation conferred ART resistance with a significantly increased in vitro RSA value similar to that for the positive transfection control with the C580Y mutation. These findings were further reinforced by the RSA0-3 h50% assay, which showed a trend similar to that for RSA0-3 h: all parasites with introduced PfK13 mutations had increased RSA0-3 h50% values, but the increase was significant only in parasites with the N458Y and C580Y mutations. Further validation of the contribution of these PfK13 mutations to ART resistance came from the reciprocal removal of these mutations from field isolates, which showed that reverting the mutated PfK13 to the WT increased the sensitivity to DHA in these field isolates in the case of all mutations tested except C469Y. Although some of the discrepancies between the findings for field isolates from the China-Myanmar border region where ART resistance has evolved and those for the 3D7 parasite genetic background cannot be reconciled with the current evidence, it is very likely that the genetic backgrounds are a determinant factor. It is noteworthy that the evolution of ART resistance in the GMS is accompanied by several additional mutations in the genome (26) and that the same PfK13 mutations in different genetic backgrounds confer different levels of ART resistance (31).
To elucidate the molecular mechanism underlying PfK13-mediated resistance to ART drugs, we first attempted to identify potential cellular processes in which PfK13 might be involved. PfK13 is consistently expressed during the IDC and localized in distinct cytoplasmic foci (32). The results of a colocalization study were also consistent with a recent report showing the predominant localization of PfK13 to the ER (40). Yet, the discrepancy in the findings of PfK13-PI3P colocalization studies could possibly be due to the use in the previous study of tagged reporter proteins that bind selectively to PI3P to visualize their sites of concentration in live (and fixed) cells, as opposed to the use of commercial anti-PI3P antibodies and conventional IFA in this study with detergents (or methanol) to permeabilize the cells, which may result in the loss of lipids (40). To further investigate the cellular function of PfK13, the PfK13-associated proteome was identified by affinity purification and mass spectrometry (MS) analysis, which revealed a number of proteins potentially involved in the unfolded protein response pathway, protein folding, protein binding, and translation, consistent with the proposed mechanism of PfK13’s involvement in upregulated unfolded protein and oxidative stress responses (37, 40). We also found elevated expression for some of these genes (probably regulating resistance) in our transgenic parasites. Significantly, we detected protein disulfide isomerase (ERp72) from the PfK13 pull-down, and the gene for this enzyme was highly upregulated in the N458Y parasites, hinting at a role in lowering ART sensitivity. ERp72 functions in cell redox homeostasis, protein folding, and the ER stress response (60), but direct evidence of its function in P. falciparum is lacking. Another gene that was upregulated in four of our mutant lines was PHISTa. PHIST-family proteins are known to have variable expression in resistant parasites (61) and are central to host cell remodeling, along with PfEMP1 (62). We also tested for eIF2α phosphorylation in our isogeneic parasite lines, as enhanced phosphorylated eIF2α was shown to correlate with high rates of recrudescence following ART (38). We could not detect any differences in the total and phosphorylated forms of eIF2α in these parasites using commercial phosphospecific antibodies for eIF2α (data not shown).
Recent studies demonstrated that a reduced PfK13 abundance in resistant parasites impaired hemoglobin uptake or catabolism (63, 64), but we were unable to detect any significant changes in PfK13 expression or localization after introduction of the PfK13 mutations in the 3D7 strain, as determined by Western blotting, live-cell imaging, and flow cytometry, a finding which is consistent with a few other earlier findings (37, 40). However, by an unknown mechanism, parasites with the PfK13 N458Y and F495L mutations were able to more strongly tolerate the oxidative stress induced by ART drugs, as indicated by lower levels of protein ubiquitination. This result aligns well with earlier findings showing that clinical ART-resistant parasites have an upregulated unfolded protein response pathway (37) and that ART-resistant parasites harboring certain PfK13 mutations have an enhanced stress response involving the ubiquitin/proteasome pathways (34). It is interesting to note that ART-resistant parasites obtained from in vitro selection also exhibited genome-wide upregulation of the antioxidant pathways (65).
Ring-stage dormancy and decelerated ring-stage development have been invoked to explain the unique ART resistance phenomenon (36, 66). Since ring-stage parasites are metabolically less active and more able to endure an oxidative onslaught than later stages, it is conceivable that extended ring stages would enable the parasites to better survive ART treatment, which has a short half-life. Population transcriptomics revealed that ART-resistant parasites had decelerated ring-stage development (37). By using highly synchronous ring-stage cultures, we observed a prolonged ring phase in parasites with the F446I, N458Y, and C580Y mutations compared to that in 3D7 and the WT transfectant, even in the absence of drug pressure. Of note, the extended ring stage was not as drastic as that reported in the resistant field isolates, where the ring-stage parasites could persist for up to 30 h, but this phenotype could be more pronounced in the presence of drug, as shown earlier (35, 36).
PfK13 mutations have been shown to incur a fitness cost on the parasites, manifested as lower growth rates as well as reduced fitness in an in vitro growth competition assay, as summarized in Table S4 in the supplemental material (57, 67, 68). Different PfK13 alleles show considerable variations in their impact on parasite fitness, which appear to be governed by the genetic backgrounds of the strains. Straimer et al. found the C580Y mutation to be fitness neutral in recent Cambodian field isolates compared to the effect of the R539T or I543T mutation, whereas the C580Y mutation exerted much higher growth disadvantages in a strain that was culture adapted in 1976, long before ART deployment (57). Two later studies determined that the C580Y mutation carries a greater competitive fitness burden than other PfK13 alleles tested in field isolates obtained from the Thailand-Myanmar border area in 2008 and 2011, respectively (67, 68). Interestingly, the C580Y isolate obtained in 2011 is a clinically resistant parasite strain with a long parasite clearance half-life of 7.84 h (67). In the 3D7 genetic background used in this study, although all mutations introduced brought about various fitness costs, parasites with the C580Y and F446I mutations showed only a marginal reduction in fitness. While this correlated well with the high prevalence of these two mutations in different regions of the GMS (69, 70), the current study also highlights the lack of genetic barriers for the evolution and spread of such mutations in African parasites. It is worrisome that the independent emergence of the C580Y mutation has been detected in South America (71) and Papua New Guinea (72). Furthermore, parasites with all mutations tested in this study appeared to form gametocytes normally, suggesting the potential transmission of these mutant parasites through mosquitoes.
F446I is the most prevalent PfK13 mutation present in northern Myanmar and the China-Myanmar border region (42, 52). This mutation has been associated with in vivo delayed parasite clearance (47, 73) and day 3 positive parasitemia (43). Our transgenic 3D7F446I parasites showed RSA values exceeding 1%, a cutoff value used to define ART resistance among field isolates, but these values were not significantly different from the value for the transgenic WT control (19). In a recent study, when the F446I mutation was introduced into the 3D7 and FCC1/HN strains, no statistically significant increase in the RSA value was detected at 700 nM DHA (none of the RSA values were more than 1%), although exposure to lower DHA concentrations did present survival rates significantly higher than those for the WT control (48). In this study, we found an increase in ART sensitivity in one of the field isolates from the China-Myanmar border region after F446I was reverted to the WT, suggesting that the impact of F446I on ART sensitivity varies greatly depending on the genetic backgrounds of the parasites. Moreover, the F446I parasites showed a prolonged ring stage and almost no reduction in fitness, which may contribute to the high prevalence of this mutation in western GMS.
Introduction of the N458Y mutation into 3D7 conferred high RSA values, and reciprocal removal of this mutation in one of the field isolates from the China-Myanmar border region increased the parasite’s sensitivity to DHA. However, the high-level resistance conferred by the N458Y mutation may come with a compromised fitness cost, leading to its low prevalence in areas where malaria is endemic, as observed for the R539T and I543T mutations (57). The N458Y mutation was extremely rare in the population in the China-Myanmar border region, with only 1 isolate with this mutation being identified among 191 clinical isolates (52), and this parasite had the highest RSA value of ∼60% among the field isolates examined in our earlier study (43). The N458Y mutation was also present at a low frequency in the Thai-Myanmar border region, and it was significantly associated with a prolonged parasite clearance half-life of >5 h, day 3 parasite positivity, and an RSA value of >1% in field isolates (49, 50, 74). Our results provide solid genetic evidence supporting these field and laboratory observations associating the N458Y mutation with ART resistance as well as potential reasons for the low prevalence of this mutation in the GMS.
The C469Y mutation was reported at a very low frequency in the China-Myanmar border area and was recently reported from Uganda (1, 11, 43, 52, 54). Only one isolate carrying this mutation from the China-Myanmar border area was found to have day 3 positivity and had an ∼20% in vitro ring-stage survival rate (43), but its introduction into 3D7 did not confer a significant increase in ring-stage survival rates. Conversely, the reciprocal removal of C469Y from this field isolate did not significantly alter the parasite’s sensitivity to DHA. Data from the RSA0-3 h50%, the growth phenotype, and the proteasome stress response to drug treatment further suggest that C469Y is not associated with ART resistance in different genetic backgrounds.
The F495L mutation has been reported at low frequencies from parasite populations in the China-Myanmar border area, Mayotte, and the Democratic Republic of Congo (43, 47, 55, 56), suggesting that it is not associated with ART resistance. Neither clinical nor laboratory studies established any association of F495L with the parasite clearance half-life or in vitro phenotype. Although this mutation is near the Y493H mutation, which was described to be associated with delayed parasite clearance (29), it did not produce significantly increased ring survival when introduced into the 3D7 strain. At the same time, the fitness of parasites of the 3D7 background with the F495L mutation was also substantially compromised.
In conclusion, we analyzed the impact of four PfK13 mutations found in northern Myanmar and the China-Myanmar border area on ART resistance using transgenics in 3D7 and mutation reversion in field isolates (Table S5). Our results demonstrated that the introduction of N458Y and C580Y into the 3D7 background significantly elevated the RSA values of the transgenic parasites, whereas other mutations only insignificantly increased the RSA values of the transfectants. In mutation revertants of field isolates, revertants with all mutations except C469Y showed significant decreases in RSA values, highlighting the important role of the genetic backgrounds of the parasites in mediating ART resistance. Compared to C580Y parasites, N458Y parasites showed similar phenotypes in terms of RSA and RSA0-3 h50%, higher parasite clearance half-lives, a longer ring stage, an elevated proteasomal stress response, and the upregulation of ART resistance-related genes (42, 43, 49, 50), but the mutation had a considerable fitness cost, partially explaining the low prevalence of this mutation in the GMS. While F495L and F446I are potentially involved in ART resistance (Table S5), they alone may not be enough to induce a strong ART resistance phenotype. Generation of these mutations in the African 3D7 background suggests that these mutations may potentially evolve in Africa, emphasizing the importance of heightened surveillance in areas of P. falciparum hyperendemicity.
MATERIALS AND METHODS
Parasite culture.
Asexual blood-stage parasites were maintained in O+ human red blood cells (RBCs) and a humidified 5% CO2 incubator at 37°C as previously described (75). Briefly, parasites were grown in RPMI 1640 with 25 mM NaHCO3, 11 mM glucose, 25 mM HEPES (pH 7.4), 0.367 mM hypoxanthine, and 5 μg/liter gentamicin supplemented with 0.5% AlbuMAX II lipid-rich bovine serum albumin (Thermo Fisher Scientific, MA). Synchronization was performed at the ring stage by 5% d-sorbitol treatment (76).
Plasmid construction and transfection.
pSLI-N-sandwich-loxP (K13) was a gift from Tobias Spielmann. The pSLI-N-sandwich-loxP (PfK13) plasmid contains a recoded PfK13 gene with its N terminus tagged with the green fluorescent protein (GFP) sandwiched between two FK506-binding protein (FKBP) sequences (2×FKBP::GFP-2×FKBP::K13) (32). To introduce the point mutations (F446I, N458Y, C469Y, F495L, and C580Y) into the recoded PfK13 gene in this plasmid, site-directed mutagenesis was performed using a Q5 site-directed mutagenesis kit (New England Biolabs, MA) with the primers listed in Table S1A in the supplemental material. To tag the PfK13 gene with a protein C-tobacco etch virus-protein A (PTP) tag (77), the GFP gene in the pSLI-N-sandwich-loxP (K13) plasmid was replaced with the PTP-coding sequence with the primers listed in Table S1A using an In-Fusion HD cloning kit (TaKaRa Bio USA, Inc., CA). Transfection of the 3D7 parasite and in vitro selection were performed as described previously (32, 78, 79). Successful integration was obtained using WR99210 (which selects for the human dihydrofolate reductase [hDHFR]) and DSM1 (which is a dihydroorotate dehydrogenase inhibitor) drug selection. GFP-positive cells were sorted on a Beckman Coulter MoFlo Astrios (Brea, CA) system with 488-nm laser excitation. Correct editing of the PfK13 gene in the genetically manipulated parasites was verified by PCR, sequencing, and Western blotting.
(A) Primers used for site-directed mutagenesis to introduce the point mutations and to replace GFP with PTP. Lowercase letters indicate the mutations introduced. (B) Primers used for RT-PCR. FP, forward primer; RP, reverse primer; BiP, PF3D7_0917900; DPMPM (dolichyl-phosphate-mannose protein mannosyl transferase), PF3D7_1010700; ERCaBP (endoplasmic reticulum-resident calcium binding protein), PF3D7_1108600; PDI (protein disulfide isomerase; ERp72), PF3D7_0827900; PHISTa (plasmodium exported protein), PF3D7_1372000; PPCTI (peptidyl-prolyl cis-trans isomerase), PF3D7_1115600. Download Table S1, DOCX file, 0.02 MB (16.6KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Raw data for ring-stage survival assay. Percent survival was calculated as (DHA/NE) × 100, where DHA is the number of viable parasites in DHA-exposed wells, and NE is the number of viable parasites in the nonexposed/DMSO-treated well. (B) Raw values from densitometry analysis of Western blots probed with antiubiquitin antibodies for the protein ubiquitination experiment. Data from 3 independent experiments are provided. Download Table S2, DOCX file, 0.01 MB (15.3KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
IC50 values obtained in an RSA0–3 h50% assay showing the percentage of early-ring-stage parasites (0 to 3 h postinvasion) surviving a 4-h pulse of DHA ranging from 0.6 to 700 nM. *, the IC50 values for parasite lines carrying the PfK13 N458Y and C580Y mutations were significantly different from those for 3D7 and WT PfK13 transfection-control parasites (P < 0.05, nonparametric Wilcoxon t test). Download Table S3, DOCX file, 0.01 MB (15.8KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effect of different K13 mutations on parasite fitness. Download Table S4, XLSX file, 0.01 MB (12.6KB, xlsx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Summary of the P. falciparum phenotypes observed with different PfK13 mutations. Download Table S5, DOCX file, 0.1 MB (65.1KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Western blotting.
Western blotting was performed to detect native or GFP-tagged PfK13 protein expression in the WT 3D7 and GFP::PfK13 transgenic lines. Equal amounts of proteins from different stages were separated on a 10% SDS-PAGE gel, transferred to a nitrocellulose membrane, and probed with monoclonal anti-GFP antibodies (Roche, IN), followed by detection with anti-rabbit immunoglobulin horseradish peroxidase (HRP)-conjugated antibody (Sigma-Aldrich, MO) at 1:5,000. The detected proteins were visualized using an enhanced chemiluminescence (ECL) kit (Invitrogen, MA). Rabbit anti-Plasmodium aldolase antibodies (catalog number ab207494; Abcam, Cambridge, UK) were used as a loading control at a 1:3,000 dilution. We also generated antibodies against three different peptides of the K13 protein (which were custom generated using keyhole limpet hemocyanin-conjugated peptides), but only the antibodies generated against amino acids (aa) 239 to 257 showed high titers and detected a single band in Western blots. The PfK13 peptide antibodies were used at a 1:1,000 dilution.
Indirect immunofluorescence assay (IFA).
For PfK13 localization, 3D7 and the GFP-tagged transgenic parasites at different asexual and sexual stages were fixed using methanol and probed with either rabbit anti-PfK13 antibodies (1:300) or monoclonal anti-GFP antibodies (Roche, IN), followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG antibodies. Parasite nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and the slides were mounted with an antifade reagent (catalog number H-1500; VectaLabs, Australia). For colocalization experiments, 3D7 or GFP::PfK13 parasites were fixed and probed with anti-BiP mouse antisera as the endoplasmic reticulum (ER) marker, anti-HAP mouse sera as the food vacuole marker (catalog number MRA-811A; BEI Resources, VA), or anti-PI3P mouse antibodies (Echelon Biosciences Inc., UT) at a 1:200 dilution (80). Images were acquired on an Olympus FluoView FV1000 epifluorescence microscope. At least 20 images were captured for each colocalization experiment, and Pearson’s correlation coefficients were calculated.
Protein IP.
Protein immunoprecipitation (IP) from the GFP::K13 parasite line was performed using a GFP-Trap-A kit (Chromotek, Germany) according to the manufacturer’s instructions. Briefly, GFP::PfK13 or 3D7 (control) parasites were harvested after saponin lysis. The pellets were resuspended in a cold lysis buffer (30 mM Tris, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride [PMSF], Roche protease inhibitors) and incubated on ice for 5 min. Then, the parasites were manually lysed by 60 to 100 strokes in a Dounce homogenizer with a tight pestle and centrifuged at 16,000 × g for 20 min at 4°C. The supernatant was incubated with GFP-Trap beads for 4 h or overnight at 4°C, and the beads were washed 3 times with the lysis buffer (without NP-40). The bound proteins were eluted using the elution buffer from the Pierce co-IP kit (Thermo Fisher Scientific). IP eluates were prepared for mass spectrometry (MS)-based proteomics using filter-aided sample preparation. Briefly, proteins were alkylated with iodoacetamide, buffer exchanged with urea followed by ammonium bicarbonate, and finally, digested with trypsin/Lys-C overnight at 37°C. The peptides were eluted and subsequently desalted using C18 SPE cartridges (Waters, MA) with a vacuum manifold. Desalted peptides were dried in a vacuum concentrator. The peptides were resuspended in 0.1% formic acid for liquid chromatography-tandem MS (LC-MS/MS) analysis.
Peptides were separated using a 75-μm by 50-cm C18 reversed-phase high-performance liquid chromatography column on an Ultimate 3000 ultra-high-performance liquid chromatograph (Thermo Fisher Scientific) with a 120-min gradient (2 to 32% acetonitrile with 0.1% formic acid) and analyzed on a hybrid quadrupole-Orbitrap instrument (Q Exactive Plus; Thermo Fisher Scientific). Full MS survey scans were acquired at a resolution of 70,000. The top 10 most abundant ions were selected for MS/MS analysis.
Raw data files were processed in MaxQuant software (www.maxquant.org) and searched against the UniprotKB P. falciparum protein sequence database. Search parameters included constant modification of cysteine by carbamidomethylation and the variable modification, as well as methionine oxidation. Proteins were identified using the filtering criteria of a 1% protein and peptide false discovery rate (FDR) and at least two unique peptides. Gene Ontology (GO) term enrichment of select gene groups or clusters was carried out using the analysis tools at http://www.PlasmoDB.org.
Real-time RT-PCR.
For reverse transcriptase (RT) PCR analysis, cDNA was synthesized from 1 μg of total RNA using SuperScript III RT (Invitrogen) and an oligo(dT)12–17 primer, and the reaction mixture was diluted to 100 μl. Real-time RT-PCR was performed using a SYBR green PCR kit (Roche, IN) with 1 μl of the cDNA and the primers listed in Table S1B. The relative expression levels of the selected genes at the ring stage were determined using the 2−ΔΔCT method, with the seryl-tRNA synthetase (STS) gene (PF07_0073) being used as the internal reference. All expression values were further normalized with the respective values in the transgenic WT parasites. Data analysis was performed and the threshold cycle (CT) value was determined as described previously (81).
Ring-stage survival assays (RSA0–3 h and RSA0–3 h50%).
RSA was performed as previously described (19, 27, 43, 57). Briefly, schizonts were purified from tightly synchronized cultures over a gradient of 75% Percoll (Sigma-Aldrich), washed once in RPMI 1640 incomplete medium, and allowed to rupture and invade fresh RBCs for 3 h. The cultures were synchronized again using sorbitol to select for early rings and to eliminate the remaining schizonts. For RSA0–3 h, ring-stage parasites (0 to 3 h postinvasion) at 1% parasitemia and 1% hematocrit were exposed to 700 nM DHA for 6 h, followed by a single wash. After culture for 66 h, ∼10,000 RBCs were blind counted on thin blood smears to count viable parasites. The RSA0–3 h50% value was also evaluated for these parasites and was defined as the DHA concentration required to kill 50% of the parasites when ring-stage parasites (0 to 3 h postinvasion) are exposed to increasing DHA concentrations (0.6 to 700 nM). After 4 h of incubation, the cells were washed four times, followed by two transfers of cells to new 96-well plates as described previously (57, 58). Parasite survival was then assessed by counting 100,000 total cells by flow cytometry on a Beckman Coulter MoFlo Astrios flow cytometer using the GFP signal and MitoTracker Deep Red dye staining (Invitrogen). For all assays, parallel dimethyl sulfoxide (DMSO)-treated controls (0.1%) were used, and survival rates were expressed as the ratios of viable parasites in DHA-exposed and DMSO-exposed samples. RSA0–3 h50% values were calculated by nonlinear curve fitting of log-transformed data using the GraphPad Prism (v5) program (La Jolla, CA).
In vitro drug sensitivity assays.
The in vitro susceptibilities of the WT and mutant parasites to 10 antimalarial drugs, amodiaquine dihydrochloride dihydrate (AQ), artemether (AM), chloroquine (CQ), piperaquine (PPQ), mefloquine (MFQ), quinine (QN), lumefantrine (LMF), pyronaridine (PND), artesunate (AS), and DHA, were tested using a SYBR green I-based assay as described previously (43, 82, 83). PPQ was purchased from Chongqing Kangle Pharmaceutical Co. (Chongqing, China), CQ, MFQ, QN, and ART drugs were purchased from Sigma-Aldrich, while LMF and PND were from Kunming Pharmaceutical Co. (Kunming, Yunnan, China). The stock solutions were prepared as described before (43). To determine the 50% inhibitory concentration (IC50), synchronized ring-stage parasites were cultured with serial dilutions of each drug at 1% hematocrit and 0.5% parasitemia in a 96-well plate. Wells with no drug and only RBCs were used as a positive control and the background, respectively. After 72 h of incubation, the plates were wrapped and frozen at −20°C for at least 16 h. The cultures were then thawed and lysed using 100 μl lysis buffer (20 mM Tris-HCl [pH 7.5], 5 mM EDTA, 0.08% Triton X-100, 0.008% saponin in phosphate-buffered saline [PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4], 0.2 μl SYBR green I) (84). The plates were incubated at 37°C for 1 h in the dark after thorough mixing, and fluorescence intensities were measured using a FLUOstar Optima microplate reader (BMG Labtech Inc, NC) at excitation and emission wavelengths of 485 and 520 nm, respectively. Percent growth was calculated against the positive control after subtracting the RBC background signal. IC50s were calculated using the GraphPad Prism (v5) program by constructing a dose-response curve. Each experiment was performed three times independently, each with two technical replicates. 3D7 was included in all experiments as an internal reference strain.
Phenotype analysis.
To measure parasite propagation, schizont-stage parasites from tightly synchronized cultures were purified using 75% Percoll and allowed to rupture and invade erythrocytes for 2 h. Unruptured schizonts were eliminated using sorbitol treatment. Synchronized ring-stage cultures were cultured in 24-well plates at 0.1% rings and 2% hematocrit. The medium was replenished every 24 h, and parasitemia was examined daily for 8 days using Giemsa-stained smears and flow cytometry. After two cycles, the cultures were maintained at 0.2% hematocrit to sustain high parasitemias. Cell cycle progression was monitored using similarly synchronized cultures with a starting parasitemia of 1%. Giemsa-stained smears were read every 3 h for 60 h (85). Three independent biological repeats were performed. For comparing the levels of gametocytemia in mutant and WT parasites, a modified method of gametocyte induction was followed to obtain highly synchronous gametocyte cultures (86). Gametocytemia was determined by counting the gametocytes in at least 5,000 RBCs on Giemsa-stained thin smears for each parasite line.
Protein ubiquitination in parasites.
Ubiquitinated proteins were analyzed as described previously (34). We used the less potent parent drug ART for treatment, as it was shown to produce a more pronounced response in ubiquitination (34). Briefly, 5% trophozoite parasites from WT and PfK13 mutant strains were treated with 6 μM ART for 90 min at 37°C, followed by washing with PBS containing 20 mM N-ethylmaleimide, 2 mM PMSF, an antiprotease mixture, 0.5 mM EDTA, and the cOmplete Mini EDTA-free protease inhibitor mixture (Roche). The RBC membrane was lysed by saponin treatment. Parasite pellets were lysed and separated on 4 to 12% gradient SDS-PAGE gels. Western blotting was performed using polyclonal rabbit antiubiquitin antibodies (1:500 dilution; catalog number 631634; EMD Millipore Corp., MA), followed by goat anti-rabbit immunoglobulin HRP-conjugated antibodies (Sigma-Aldrich) at 1:5,000. The blots were visualized using an enhanced chemiluminescence (ECL) kit (Invitrogen). Rabbit anti-Plasmodium aldolase antibodies were used as loading controls at 1:3,000. Densitometry analysis was performed for each lane of the gel using ImageJ software, and all data were normalized according to the aldolase signal.
In vitro growth competition assay.
To determine the potential fitness cost associated with the PfK13 mutations, we performed a mixed-culture competition assay using 7 different parasite lines: PTP::WT, GFP::WT, GFP::F446I, GFP::N458Y, GFP::C469Y, GFP::F495L, and GFP::C580Y. All GFP-tagged parasites (WT or mutant) were mixed with the PTP::WT parasites in a 1:1 ratio at a 3% ring-stage parasitemia. One-fourth of the parasites were used for flow cytometry every 4 days. Cultures were reduced to half and replenished with fresh blood at 50% hematocrit. The ratio of GFP+ parasites to the total parasitemia measured by 100 nM MitoTracker Deep Red staining was determined using flow cytometry for an average of 48 days. Experiments were conducted independently three times in duplicate, and the percentage of GFP+ parasites was plotted over time. Each time, a total of 50,000 events were read per well.
Statistical analysis.
Statistical analysis was performed using the GraphPad Prism (v5) program. The geometric mean of the IC50 and the 95% confidence interval (CI) were calculated by fitting the drug response data to a sigmoid curve. A nonparametric Wilcoxon matched-pairs test or one-way analysis of variance was used to compare the mean values between treatment groups. Differences were considered significant at a P value of <0.05.
ACKNOWLEDGMENTS
The anti-P. falciparum histoaspartic protease (HAP) monoclonal antibody was obtained from BEI Resources (catalog number MRA-811A) and was contributed by Daniel E. Goldberg. The anti-BiP antibody was a kind gift from Scott Lindner from Pennsylvania State University.
This study was supported by grants U19AI 089672 and R01AI128940 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Y.C. is supported by grant 81761128017 from National Natural Science Foundation of China.
Footnotes
Citation Siddiqui FA, Boonhok R, Cabrera M, Mbenda HGN, Wang M, Min H, Liang X, Qin J, Zhu X, Miao J, Cao Y, Cui L. 2020. Role of Plasmodium falciparum Kelch 13 protein mutations in P. falciparum populations from northeastern Myanmar in mediating artemisinin resistance. mBio 11:e01134-19. https://doi.org/10.1128/mBio.01134-19.
REFERENCES
- 1.Imwong M, Suwannasin K, Kunasol C, Sutawong K, Mayxay M, Rekol H, Smithuis FM, Hlaing TM, Tun KM, van der Pluijm RW, Tripura R, Miotto O, Menard D, Dhorda M, Day NPJ, White NJ, Dondorp AM. 2017. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis 17:491–497. doi: 10.1016/S1473-3099(17)30048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium. 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 4.Bustos MD, Wongsrichanalai C, Delacollette C, Burkholder B. 2013. Monitoring antimalarial drug efficacy in the Greater Mekong subregion: an overview of in vivo results from 2008 to 2010. Southeast Asian J Trop Med Public Health 44(Suppl 1):201–230. [PubMed] [Google Scholar]
- 5.Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, Tan JC, Al Saai S, Phyo AP, Moo CL, Lwin KM, McGready R, Ashley E, Imwong M, Stepniewska K, Yi P, Dondorp AM, Mayxay M, Newton PN, White NJ, Nosten F, Ferdig MT, Anderson TJ. 2012. A major genome region underlying artemisinin resistance in malaria. Science 336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hien TT, Thuy-Nhien NT, Phu NH, Boni MF, Thanh NV, Nha-Ca NT, Thai Le H, Thai CQ, Toi PV, Thuan PD, Long Le T, Dong Le T, Merson L, Dolecek C, Stepniewska K, Ringwald P, White NJ, Farrar J, Wolbers M. 2012. In vivo susceptibility of Plasmodium falciparum to artesunate in Binh Phuoc Province, Vietnam. Malar J 11:355. doi: 10.1186/1475-2875-11-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ménard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, Rahim-Awab G, Barnadas C, Berry A, Boum Y, Bustos MD, Cao J, Chen J-H, Collet L, Cui L, Thakur G-D, Dieye A, Djallé D, Dorkenoo MA, Eboumbou-Moukoko CE, Espino F-E-CJ, Fandeur T, Ferreira-da-Cruz M-F, Fola AA, Fuehrer H-P, Hassan AM, Herrera S, Hongvanthong B, Houzé S, Ibrahim ML, Jahirul-Karim M, Jiang L, Kano S, Ali-Khan W, Khanthavong M, Kremsner PG, Lacerda M, Leang R, Leelawong M, Li M, Lin K, Mazarati J-B, Ménard S, Morlais I, Muhindo-Mavoko H, Musset L, Na-Bangchang K, Nambozi M, Niaré K, Noedl H, et al. 2016. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med 374:2453–2464. doi: 10.1056/NEJMoa1513137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nyunt MH, Hlaing T, Oo HW, Tin-Oo LL, Phway HP, Wang B, Zaw NN, Han SS, Tun T, San KK, Kyaw MP, Han ET. 2015. Molecular assessment of artemisinin resistance markers, polymorphisms in the K13 propeller, and a multidrug-resistance gene in the eastern and western border areas of Myanmar. Clin Infect Dis 60:1208–1215. doi: 10.1093/cid/ciu1160. [DOI] [PubMed] [Google Scholar]
- 9.Takala-Harrison S, Jacob CG, Arze C, Cummings MP, Silva JC, Dondorp AM, Fukuda MM, Hien TT, Mayxay M, Noedl H, Nosten F, Kyaw MP, Nhien NT, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Ariey F, Mercereau-Puijalon O, Menard D, Newton PN, Khanthavong M, Hongvanthong B, Starzengruber P, Fuehrer HP, Swoboda P, Khan WA, Phyo AP, Nyunt MM, Nyunt MH, Brown TS, Adams M, Pepin CS, Bailey J, Tan JC, Ferdig MT, Clark TG, Miotto O, MacInnis B, Kwiatkowski DP, White NJ, Ringwald P, Plowe CV. 2015. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 211:670–679. doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Cabrera M, Yang J, Yuan L, Gupta B, Liang X, Kemirembe K, Shrestha S, Brashear A, Li X, Porcella SF, Miao J, Yang Z, Su XZ, Cui L. 2016. Genome-wide association analysis identifies genetic loci associated with resistance to multiple antimalarials in Plasmodium falciparum from China-Myanmar border. Sci Rep 6:33891. doi: 10.1038/srep33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda M, Kaneko M, Tachibana SI, Balikagala B, Sakurai-Yatsushiro M, Yatsushiro S, Takahashi N, Yamauchi M, Sekihara M, Hashimoto M, Katuro OT, Olia A, Obwoya PS, Auma MA, Anywar DA, Odongo-Aginya EI, Okello-Onen J, Hirai M, Ohashi J, Palacpac NMQ, Kataoka M, Tsuboi T, Kimura E, Horii T, Mita T. 2018. Artemisinin-resistant Plasmodium falciparum with high survival rates, Uganda, 2014–2016. Emerg Infect Dis 24:718–726. doi: 10.3201/eid2404.170141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WHO. 2018. Status report on artemisinin resistance and ACT efficacy. https://www.who.int/malaria/publications/atoz/artemisinin-resistance-august2018/en/. Accessed 17 July 2019.
- 13.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ataide R, Ashley EA, Powell R, Chan J-A, Malloy MJ, O'Flaherty K, Takashima E, Langer C, Tsuboi T, Dondorp AM, Day NP, Dhorda M, Fairhurst RM, Lim P, Amaratunga C, Pukrittayakamee S, Hien TT, Htut Y, Mayxay M, Faiz MA, Beeson JG, Nosten F, Simpson JA, White NJ, Fowkes FJI. 2017. Host immunity to Plasmodium falciparum and the assessment of emerging artemisinin resistance in a multinational cohort. Proc Natl Acad Sci U S A 114:3515–3520. doi: 10.1073/pnas.1615875114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hastings IM, Kay K, Hodel EM. 2015. How robust are malaria parasite clearance rates as indicators of drug effectiveness and resistance? Antimicrob Agents Chemother 59:6428–6436. doi: 10.1128/AAC.00481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopera-Mesa TM, Doumbia S, Chiang S, Zeituni AE, Konate DS, Doumbouya M, Keita AS, Stepniewska K, Traore K, Diakite SA, Ndiaye D, Sa JM, Anderson JM, Fay MP, Long CA, Diakite M, Fairhurst RM. 2013. Plasmodium falciparum clearance rates in response to artesunate in Malian children with malaria: effect of acquired immunity. J Infect Dis 207:1655–1663. doi: 10.1093/infdis/jit082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odhiambo G, Bergmann-Leitner E, Maraka M, Wanjala CNL, Duncan E, Waitumbi J, Andagalu B, Jura W, Dutta S, Angov E, Ogutu BR, Kamau E, Ochiel D. 2019. Correlation between malaria-specific antibody profiles and responses to artemisinin combination therapy for treatment of uncomplicated malaria in western Kenya. J Infect Dis 219:1969–1979. doi: 10.1093/infdis/jiz027. [DOI] [PubMed] [Google Scholar]
- 18.Wojnarski M, Mouri O, Chambrion C, Roussel C, Chartrel N, Smith B, Smith P, Thellier M, Buffet P, Ndour PA. 2019. Plasmodium falciparum clearance is pitting-dependent with artemisinin-based drugs but pitting-independent with atovaquone-proguanil or mefloquine. J Infect Dis 220:535–539. doi: 10.1093/infdis/jiz115. [DOI] [PubMed] [Google Scholar]
- 19.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WR, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassett MR, Sternberg AR, Roepe PD. 2017. Inhibition of human class I vs class III phosphatidylinositol 3′-kinases. Biochemistry 56:4326–4334. doi: 10.1021/acs.biochem.7b00413. [DOI] [PubMed] [Google Scholar]
- 21.Rocamora F, Zhu L, Liong KY, Dondorp A, Miotto O, Mok S, Bozdech Z. 2018. Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites. PLoS Pathog 14:e1006930. doi: 10.1371/journal.ppat.1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breglio KF, Amato R, Eastman R, Lim P, Sa JM, Guha R, Ganesan S, Dorward DW, Klumpp-Thomas C, McKnight C, Fairhurst RM, Roberts D, Thomas C, Simon AK. 2018. A single nucleotide polymorphism in the Plasmodium falciparum atg18 gene associates with artemisinin resistance and confers enhanced parasite survival under nutrient deprivation. Malar J 17:391. doi: 10.1186/s12936-018-2532-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demas AR, Sharma AI, Wong W, Early AM, Redmond S, Bopp S, Neafsey DE, Volkman SK, Hartl DL, Wirth DF. 2018. Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc Natl Acad Sci U S A 115:12799–12804. doi: 10.1073/pnas.1812317115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henriques G, van Schalkwyk DA, Burrow R, Warhurst DC, Thompson E, Baker DA, Fidock DA, Hallett R, Flueck C, Sutherland CJ. 2015. The Mu subunit of Plasmodium falciparum clathrin-associated adaptor protein 2 modulates in vitro parasite response to artemisinin and quinine. Antimicrob Agents Chemother 59:2540–2547. doi: 10.1128/AAC.04067-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klonis N, Crespo-Ortiz MP, Bottova I, Abu-Bakar N, Kenny S, Rosenthal PJ, Tilley L. 2011. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A 108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siddiqui FA, Cabrera M, Wang M, Brashear A, Kemirembe K, Wang Z, Miao J, Chookajorn T, Yang Z, Cao Y, Dong G, Rosenthal PJ, Cui L. 2018. Plasmodium falciparum falcipain-2a polymorphisms in Southeast Asia and their association with artemisinin resistance. J Infect Dis 218:434–442. doi: 10.1093/infdis/jiy188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee A, Bopp S, Magistrado P, Wong W, Daniels R, Demas A, Schaffner S, Amaratunga C, Lim P, Dhorda M, Miotto O, Woodrow C, Ashley EA, Dondorp AM, White NJ, Wirth D, Fairhurst R, Volkman SK. 2017. Artemisinin resistance without pfkelch13 mutations in Plasmodium falciparum isolates from Cambodia. Malar J 16:195. doi: 10.1186/s12936-017-1845-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Ménard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Ménard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. 2014. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 31.Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Ménard D, Fidock DA. 2015. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birnbaum J, Flemming S, Reichard N, Soares AB, Mesen-Ramirez P, Jonscher E, Bergmann B, Spielmann T. 2017. A genetic system to study Plasmodium falciparum protein function. Nat Methods 14:450–456. doi: 10.1038/nmeth.4223. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, Brown J, Li S, Swanson J, Rayner JC, Jiang RHY, Adams JH. 2018. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360:eaap7847. doi: 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, Chotivanich K, Kenny S, Gnadig N, Straimer J, Bozdech Z, Fidock DA, Simpson JA, Dondorp AM, Foote S, Klonis N, Tilley L. 2015. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 13:e1002132. doi: 10.1371/journal.pbio.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale JC, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hott A, Casandra D, Sparks KN, Morton LC, Castanares GG, Rutter A, Kyle DE. 2015. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob Agents Chemother 59:3156–3167. doi: 10.1128/AAC.00197-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, Chotivanich K, Imwong M, Pukrittayakamee S, Dhorda M, Nguon C, Lim P, Amaratunga C, Suon S, Hien TT, Htut Y, Faiz MA, Onyamboko MA, Mayxay M, Newton PN, Tripura R, Woodrow CJ, Miotto O, Kwiatkowski DP, Nosten F, Day NP, Preiser PR, White NJ, Dondorp AM, Fairhurst RM, Bozdech Z. 2015. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347:431–435. doi: 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Gallego-Delgado J, Fernandez-Arias C, Waters NC, Rodriguez A, Tsuji M, Wek RC, Nussenzweig V, Sullivan WJ Jr.. 2017. Inhibiting the Plasmodium eIF2alpha kinase PK4 prevents artemisinin-induced latency. Cell Host Microbe 22:766–776.e4. doi: 10.1016/j.chom.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mbengue A, Bhattacharjee S, Pandharkar T, Liu H, Estiu G, Stahelin RV, Rizk SS, Njimoh DL, Ryan Y, Chotivanich K, Nguon C, Ghorbal M, Lopez-Rubio JJ, Pfrender M, Emrich S, Mohandas N, Dondorp AM, Wiest O, Haldar K. 2015. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520:683–687. doi: 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhattacharjee S, Coppens I, Mbengue A, Suresh N, Ghorbal M, Slouka Z, Safeukui I, Tang HY, Speicher DW, Stahelin RV, Mohandas N, Haldar K. 2018. Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood 131:1234–1247. doi: 10.1182/blood-2017-11-814665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, Fukuda MM, Nosten F, Noedl H, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Socheat D, Ariey F, Phyo AP, Starzengruber P, Fuehrer HP, Swoboda P, Stepniewska K, Flegg J, Arze C, Cerqueira GC, Silva JC, Ricklefs SM, Porcella SF, Stephens RM, Adams M, Kenefic LJ, Campino S, Auburn S, MacInnis B, Kwiatkowski DP, Su XZ, White NJ, Ringwald P, Plowe CV. 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A 110:240–245. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tun KM, Imwong M, Lwin KM, Win AA, Hlaing TM, Hlaing T, Lin K, Kyaw MP, Plewes K, Faiz MA, Dhorda M, Cheah PY, Pukrittayakamee S, Ashley EA, Anderson TJ, Nair S, McDew-White M, Flegg JA, Grist EP, Guerin P, Maude RJ, Smithuis F, Dondorp AM, Day NP, Nosten F, White NJ, Woodrow CJ. 2015. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect Dis 15:415–421. doi: 10.1016/S1473-3099(15)70032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z, Wang Y, Cabrera M, Zhang Y, Gupta B, Wu Y, Kemirembe K, Hu Y, Liang X, Brashear A, Shrestha S, Li X, Miao J, Sun X, Yang Z, Cui L. 2015. Artemisinin resistance at the China-Myanmar border and association with mutations in the K13 propeller gene. Antimicrob Agents Chemother 59:6952–6959. doi: 10.1128/AAC.01255-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson TJ, Nair S, McDew-White M, Cheeseman IH, Nkhoma S, Bilgic F, McGready R, Ashley E, Pyae Phyo A, White NJ, Nosten F. 2017. Population parameters underlying an ongoing soft sweep in Southeast Asian malaria parasites. Mol Biol Evol 34:131–144. doi: 10.1093/molbev/msw228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MalariaGEN Plasmodium falciparum Community Project. 2016. Genomic epidemiology of artemisinin resistant malaria. Elife 5:e08714. doi: 10.7554/eLife.08714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thriemer K, Hong NV, Rosanas-Urgell A, Phuc BQ, Ha DM, Pockele E, Guetens P, Van NV, Duong TT, Amambua-Ngwa A, D'Alessandro U, Erhart A. 2014. Delayed parasite clearance after treatment with dihydroartemisinin-piperaquine in Plasmodium falciparum malaria patients in central Vietnam. Antimicrob Agents Chemother 58:7049–7055. doi: 10.1128/AAC.02746-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang F, Takala-Harrison S, Jacob CG, Liu H, Sun X, Yang H, Nyunt MM, Adams M, Zhou S, Xia Z, Ringwald P, Bustos MD, Tang L, Plowe CV. 2015. A single mutation in K13 predominates in southern China and is associated with delayed clearance of Plasmodium falciparum following artemisinin treatment. J Infect Dis 212:1629–1635. doi: 10.1093/infdis/jiv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Huang Y, Zhao Y, Ye R, Zhang D, Pan W. 2018. Introduction of F446I mutation in the K13 propeller gene leads to increased ring survival rates in Plasmodium falciparum isolates. Malar J 17:248. doi: 10.1186/s12936-018-2396-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boulle M, Witkowski B, Duru V, Sriprawat K, Nair SK, McDew-White M, Anderson TJ, Phyo AP, Menard D, Nosten F. 2016. Artemisinin-resistant Plasmodium falciparum K13 mutant alleles, Thailand-Myanmar border. Emerg Infect Dis 22:1503–1505. doi: 10.3201/eid2208.160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talundzic E, Okoth SA, Congpuong K, Plucinski MM, Morton L, Goldman IF, Kachur PS, Wongsrichanalai C, Satimai W, Barnwell JW, Udhayakumar V. 2015. Selection and spread of artemisinin-resistant alleles in Thailand prior to the global artemisinin resistance containment campaign. PLoS Pathog 11:e1004789. doi: 10.1371/journal.ppat.1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nyunt MH, Soe MT, Myint HW, Oo HW, Aye MM, Han SS, Zaw NN, Cho C, Aung PZ, Kyaw KT, Aye TT, San NA, Ortega L, Thimasarn K, Bustos MDG, Galit S, Hoque MR, Ringwald P, Han ET, Kyaw MP. 2017. Clinical and molecular surveillance of artemisinin resistant falciparum malaria in Myanmar (2009–2013). Malar J 16:333. doi: 10.1186/s12936-017-1983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Shrestha S, Li X, Miao J, Yuan L, Cabrera M, Grube C, Yang Z, Cui L. 2015. Prevalence of K13-propeller polymorphisms in Plasmodium falciparum from China-Myanmar border in 2007–2012. Malar J 14:168. doi: 10.1186/s12936-015-0672-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fairhurst RM, Dondorp AM. 2016. Artemisinin-resistant Plasmodium falciparum malaria. Microbiol Spectr 4:EI10-0013-2016. doi: 10.1128/microbiolspec.EI10-0013-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye R, Hu D, Zhang Y, Huang Y, Sun X, Wang J, Chen X, Zhou H, Zhang D, Mungthin M, Pan W. 2016. Distinctive origin of artemisinin-resistant Plasmodium falciparum on the China-Myanmar border. Sci Rep 6:20100. doi: 10.1038/srep20100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torrentino-Madamet M, Collet L, Lepère JF, Benoit N, Amalvict R, Ménard D, Pradines B. 2015. K13-propeller polymorphisms in Plasmodium falciparum isolates from patients in Mayotte in 2013 and 2014. Antimicrob Agents Chemother 59:7878–7881. doi: 10.1128/AAC.01251-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mvumbi DM, Bobanga TL, Kayembe JN, Mvumbi GL, Situakibanza HN, Benoit-Vical F, Melin P, De Mol P, Hayette MP. 2017. Molecular surveillance of Plasmodium falciparum resistance to artemisinin-based combination therapies in the Democratic Republic of Congo. PLoS One 12:e0179142. doi: 10.1371/journal.pone.0179142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Straimer J, Gnadig NF, Stokes BH, Ehrenberger M, Crane AA, Fidock DA. 2017. Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. mBio 8:e00172-17. doi: 10.1128/mBio.00172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang T, Xie SC, Cao P, Giannangelo C, McCaw J, Creek DJ, Charman SA, Klonis N, Tilley L. 2016. Comparison of the exposure time dependence of the activities of synthetic ozonide antimalarials and dihydroartemisinin against K13 wild-type and mutant Plasmodium falciparum strains. Antimicrob Agents Chemother 60:4501–4510. doi: 10.1128/AAC.00574-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siriwardana A, Iyengar K, Roepe PD. 2016. Endoperoxide drug cross-resistance patterns for Plasmodium falciparum exhibiting an artemisinin delayed-clearance phenotype. Antimicrob Agents Chemother 60:6952–6956. doi: 10.1128/AAC.00857-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellgaard L, Molinari M, Helenius A. 1999. Setting the standards: quality control in the secretory pathway. Science 286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 61.Zhu L, Tripathi J, Rocamora FM, Miotto O, van der Pluijm R, Voss TS, Mok S, Kwiatkowski DP, Nosten F, Day NPJ, White NJ, Dondorp AM, Bozdech Z, Tracking Resistance to Artemisinin Collaboration I. 2018. The origins of malaria artemisinin resistance defined by a genetic and transcriptomic background. Nat Commun 9:5158. doi: 10.1038/s41467-018-07588-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warncke JD, Vakonakis I, Beck HP. 2016. Plasmodium helical interspersed subtelomeric (PHIST) proteins, at the center of host cell remodeling. Microbiol Mol Biol Rev 80:905–927. doi: 10.1128/MMBR.00014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Birnbaum J, Scharf S, Schmidt S, Jonscher E, Hoeijmakers WAM, Flemming S, Toenhake CG, Schmitt M, Sabitzki R, Bergmann B, Fröhlke U, Mesén-Ramírez P, Blancke Soares A, Herrmann H, Bártfai R, Spielmann T. 2020. A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 367:51–59. doi: 10.1126/science.aax4735. [DOI] [PubMed] [Google Scholar]
- 64.Yang T, Yeoh LM, Tutor MV, Dixon MW, McMillan PJ, Xie SC, Bridgford JL, Gillett DL, Duffy MF, Ralph SA, McConville MJ, Tilley L, Cobbold SA. 2019. Decreased K13 abundance reduces hemoglobin catabolism and proteotoxic stress, underpinning artemisinin resistance. Cell Rep 29:2917–2928.e5. doi: 10.1016/j.celrep.2019.10.095. [DOI] [PubMed] [Google Scholar]
- 65.Cui L, Wang Z, Miao J, Miao M, Chandra R, Jiang H, Su XZ, Cui L. 2012. Mechanisms of in vitro resistance to dihydroartemisinin in Plasmodium falciparum. Mol Microbiol 86:111–128. doi: 10.1111/j.1365-2958.2012.08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teuscher F, Gatton ML, Chen N, Peters J, Kyle DE, Cheng Q. 2010. Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J Infect Dis 202:1362–1368. doi: 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tirrell AR, Vendrely KM, Checkley LA, Davis SZ, McDew-White M, Cheeseman IH, Vaughan AM, Nosten FH, Anderson TJC, Ferdig MT. 2019. Pairwise growth competitions identify relative fitness relationships among artemisinin resistant Plasmodium falciparum field isolates. Malar J 18:295. doi: 10.1186/s12936-019-2934-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nair S, Li X, Arya GA, McDew-White M, Ferrari M, Nosten F, Anderson T. 2018. Fitness costs and the rapid spread of kelch13-C580Y substitutions conferring artemisinin resistance. Antimicrob Agents Chemother 62:e00605-18. doi: 10.1128/AAC.00605-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamilton WL, Amato R, van der Pluijm RW, Jacob CG, Quang HH, Thuy-Nhien NT, Hien TT, Hongvanthong B, Chindavongsa K, Mayxay M, Huy R, Leang R, Huch C, Dysoley L, Amaratunga C, Suon S, Fairhurst RM, Tripura R, Peto TJ, Sovann Y, Jittamala P, Hanboonkunupakarn B, Pukrittayakamee S, Chau NH, Imwong M, Dhorda M, Vongpromek R, Chan XHS, Maude RJ, Pearson RD, Nguyen T, Rockett K, Drury E, Goncalves S, White NJ, Day NP, Kwiatkowski DP, Dondorp AM, Miotto O. 2019. Evolution and expansion of multidrug-resistant malaria in southeast Asia: a genomic epidemiology study. Lancet Infect Dis 19:943–951. doi: 10.1016/S1473-3099(19)30392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van der Pluijm RW, Imwong M, Chau NH, Hoa NT, Thuy-Nhien NT, Thanh NV, Jittamala P, Hanboonkunupakarn B, Chutasmit K, Saelow C, Runjarern R, Kaewmok W, Tripura R, Peto TJ, Yok S, Suon S, Sreng S, Mao S, Oun S, Yen S, Amaratunga C, Lek D, Huy R, Dhorda M, Chotivanich K, Ashley EA, Mukaka M, Waithira N, Cheah PY, Maude RJ, Amato R, Pearson RD, Goncalves S, Jacob CG, Hamilton WL, Fairhurst RM, Tarning J, Winterberg M, Kwiatkowski DP, Pukrittayakamee S, Hien TT, Day NP, Miotto O, White NJ, Dondorp AM. 2019. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis 19:952–961. doi: 10.1016/S1473-3099(19)30391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chenet SM, Akinyi Okoth S, Huber CS, Chandrabose J, Lucchi NW, Talundzic E, Krishnalall K, Ceron N, Musset L, Macedo de Oliveira A, Venkatesan M, Rahman R, Barnwell JW, Udhayakumar V. 2016. Independent emergence of the Plasmodium falciparum Kelch propeller domain mutant allele C580Y in Guyana. J Infect Dis 213:1472–1475. doi: 10.1093/infdis/jiv752. [DOI] [PubMed] [Google Scholar]
- 72.Prosser C, Meyer W, Ellis J, Lee R. 2018. Resistance screening and trend analysis of imported falciparum malaria in NSW, Australia (2010 to 2016). PLoS One 13:e0197369. doi: 10.1371/journal.pone.0197369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tun KM, Jeeyapant A, Imwong M, Thein M, Aung SS, Hlaing TM, Yuentrakul P, Promnarate C, Dhorda M, Woodrow CJ, Dondorp AM, Ashley EA, Smithuis FM, White NJ, Day NP. 2016. Parasite clearance rates in Upper Myanmar indicate a distinctive artemisinin resistance phenotype: a therapeutic efficacy study. Malar J 15:185. doi: 10.1186/s12936-016-1240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phyo AP, Ashley EA, Anderson TJC, Bozdech Z, Carrara VI, Sriprawat K, Nair S, White MM, Dziekan J, Ling C, Proux S, Konghahong K, Jeeyapant A, Woodrow CJ, Imwong M, McGready R, Lwin KM, Day NPJ, White NJ, Nosten F. 2016. Declining efficacy of artemisinin combination therapy against P. falciparum malaria on the Thai-Myanmar border (2003–2013): the role of parasite genetic factors. Clin Infect Dis 63:784–791. doi: 10.1093/cid/ciw388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 76.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 77.Schimanski B, Nguyen TN, Gunzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell 4:1942–1950. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liang X, Hart KJ, Dong G, Siddiqui FA, Sebastian A, Li X, Albert I, Miao J, Lindner SE, Cui L. 2018. Puf3 participates in ribosomal biogenesis in malaria parasites. J Cell Sci 131:jcs212597. doi: 10.1242/jcs.212597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rug M, Maier AG. 2013. Transfection of Plasmodium falciparum. Methods Mol Biol 923:75–98. doi: 10.1007/978-1-62703-026-7_6. [DOI] [PubMed] [Google Scholar]
- 80.Siddiqui FA, Dhawan S, Singh S, Singh B, Gupta P, Pandey A, Mohmmed A, Gaur D, Chitnis CE. 2013. A thrombospondin structural repeat containing rhoptry protein from Plasmodium falciparum mediates erythrocyte invasion. Cell Microbiol 15:1341–1356. doi: 10.1111/cmi.12118. [DOI] [PubMed] [Google Scholar]
- 81.Miao J, Fan Q, Cui L, Li J, Li J, Cui L. 2006. The malaria parasite Plasmodium falciparum histones: organization, expression, and acetylation. Gene 369:53–65. doi: 10.1016/j.gene.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 82.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48:1803–1806. doi: 10.1128/aac.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. 2004. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob Agents Chemother 48:1807–1810. doi: 10.1128/aac.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bacon DJ, Latour C, Lucas C, Colina O, Ringwald P, Picot S. 2007. Comparison of a SYBR green I-based assay with a histidine-rich protein II enzyme-linked immunosorbent assay for in vitro antimalarial drug efficacy testing and application to clinical isolates. Antimicrob Agents Chemother 51:1172–1178. doi: 10.1128/AAC.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reilly HB, Wang H, Steuter JA, Marx AM, Ferdig MT. 2007. Quantitative dissection of clone-specific growth rates in cultured malaria parasites. Int J Parasitol 37:1599–1607. doi: 10.1016/j.ijpara.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, Swales CA, Sutherland CJ, Baker DA. 2007. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol 154:119–123. doi: 10.1016/j.molbiopara.2007.04.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic showing the replacement of endogenous PfK13 with N-terminal GFP/PTP-tagged full-length recoded PfK13 (WT or mutated) using the selection-linked integration (SLI) method. Genomic integration of the plasmid results in the expression of a second resistance marker (yeast dihydroorotate dehydrogenase [yDHODH]) that is flanked by two skip peptides (2A), generating separate proteins. Arrows labeled 1 to 5 indicate the primers used for integration-specific PCR. Arrows 6 and 7 indicate the hsp86 and the PfK13 endogenous promoters, respectively. hDHFR, human dihydrofolate reductase; FKBP, FK506-binding protein; GFP, green fluorescent protein; PTP, protein C-tobacco etch virus-protein A. (B) PCR performed using genomic DNA from different parasite lines and the primers indicated in panel A to show integration of the plasmid into the correct endogenous locus with primers 1 and 2 (top) and the intact endogenous locus with primers 1 and 4 (bottom). (C) Western blots detecting the full-length PfK13 protein (165 kDa) with an N-terminal 2×FKBP-GFP-2×FKBP tag (left) and 2×FKBP-PTP-2×FKBP tag (right) using anti-GFP and anti-protein C (1:1,000; catalog number A00637; GenScript) antibodies, respectively. (D) Schematic showing the replacement of endogenous PfK13 with N-terminal GFP/PTP-tagged full-length recoded WT PfK13 using selection-linked integration. Genomic integration of the plasmid results in the expression of a second resistance marker (yeast dihydroorotate dehydrogenase [yDHODH]) that is flanked by two skip peptides (2A). Arrows labeled 1 to 5 indicate the primers used for performing integration-specific PCR. Arrows 6 and 7 indicate the hsp86 and the PfK13 endogenous promoters, respectively. hDHFR, human dihydrofolate reductase; FKBP, FK506-binding protein; GFP, green fluorescent protein; PTP, protein C-tobacco etch virus-protein A. (E) PCR performed using genomic DNA from different parasite lines and the primers indicated in panel A to show integration into the correct endogenous locus with primers 1 and 5 (top) and the intact endogenous locus with primers 1 and 4 (bottom). (F) IFAs detecting the expression of GFP::PfK13 WT in transgenic field isolates using anti-GFP antibodies. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). Download FIG S1, PPT file, 1.0 MB (1MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression and localization of GFP::PfK13 during asexual erythrocytic stages. (A) Western blots of PfK13 expression in GFP::PfK13 probed with anti-GFP antibodies. Aldolase was used as the control for equal parasite protein loading. Lysates were prepared from synchronized ring (R), trophozoite (T), schizont (S), and merozoite (M) stages of GFP::PfK13 parasites and mixed asexual stages of the 3D7 parasite. (B) IFA of asexual-stage parasites of the GFP::PfK13 line probed with anti-GFP antibodies. (C). IFA of stage II to V gametocytes of the GFP::PfK13 line probed with anti-GFP antibodies. PfK13 was detected in all asexual stages and gametocytes and was localized in punctate structures in the cytoplasm. The number of puncta increases from the ring to the schizont stage. Download FIG S2, PPT file, 1.2 MB (1.2MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Western blot (A), flow cytometry (B), and IFA (C) data showing no significant difference in the level of expression between GFP::WT PfK13 and GFP::mutant PfK13 (F446I, N458Y, C469Y, F495L, and C580Y) in asexual stages. To obtain the mean fluorescence intensities with flow cytometry, 50,000 cells were counted at the trophozoite stage for each parasite line in 2 independent experiments. (D) IFA of GFP::PfK13 in stage III gametocytes of mutant transgenic parasites showing the similar expression and distribution patterns of mutant PfK13. For Western blots and IFA, GFP::PfK13 was detected by anti-GFP antibodies. Download FIG S3, PPT file, 1.8 MB (1.9MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Colocalization analysis of WT and mutated PfK13 with PI3P. Anti-GFP rabbit antibodies and anti-PI3P mouse antibodies were used to probe GFP::PfK13 (green) and PI3P (red) in transgenic 3D7 parasites at the trophozoite (A) and schizont (B) stages. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). Also shown are the bright-field (BF) and merged images. Download FIG S4, PPT file, 2.1 MB (2.1MB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of gametocytemias in WT and mutated PfK13 parasite lines. Daily gametocytemia of all transgenic lines and 3D7 was determined by counting Giemsa-stained gametocytes under a microscope from day 3 through day 10 after induction. Data are shown as the mean ± standard deviation from three replicates at days 3 and 7. None of the values for mutant parasites were significantly different from those for either 3D7 or WT parasites (P > 0.1, one-way analysis of variance). Download FIG S5, PPT file, 0.1 MB (122KB, ppt) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Primers used for site-directed mutagenesis to introduce the point mutations and to replace GFP with PTP. Lowercase letters indicate the mutations introduced. (B) Primers used for RT-PCR. FP, forward primer; RP, reverse primer; BiP, PF3D7_0917900; DPMPM (dolichyl-phosphate-mannose protein mannosyl transferase), PF3D7_1010700; ERCaBP (endoplasmic reticulum-resident calcium binding protein), PF3D7_1108600; PDI (protein disulfide isomerase; ERp72), PF3D7_0827900; PHISTa (plasmodium exported protein), PF3D7_1372000; PPCTI (peptidyl-prolyl cis-trans isomerase), PF3D7_1115600. Download Table S1, DOCX file, 0.02 MB (16.6KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Raw data for ring-stage survival assay. Percent survival was calculated as (DHA/NE) × 100, where DHA is the number of viable parasites in DHA-exposed wells, and NE is the number of viable parasites in the nonexposed/DMSO-treated well. (B) Raw values from densitometry analysis of Western blots probed with antiubiquitin antibodies for the protein ubiquitination experiment. Data from 3 independent experiments are provided. Download Table S2, DOCX file, 0.01 MB (15.3KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
IC50 values obtained in an RSA0–3 h50% assay showing the percentage of early-ring-stage parasites (0 to 3 h postinvasion) surviving a 4-h pulse of DHA ranging from 0.6 to 700 nM. *, the IC50 values for parasite lines carrying the PfK13 N458Y and C580Y mutations were significantly different from those for 3D7 and WT PfK13 transfection-control parasites (P < 0.05, nonparametric Wilcoxon t test). Download Table S3, DOCX file, 0.01 MB (15.8KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effect of different K13 mutations on parasite fitness. Download Table S4, XLSX file, 0.01 MB (12.6KB, xlsx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Summary of the P. falciparum phenotypes observed with different PfK13 mutations. Download Table S5, DOCX file, 0.1 MB (65.1KB, docx) .
Copyright © 2020 Siddiqui et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.