

Abstract
Achieving an effective drug concentration in the brain is as important as targeting the right pathway when developing targeted agents for brain tumors. SAR405838 is a novel molecularly targeted agent that is in clinical trials for various solid tumors. Its application for tumors in the brain has not yet been examined, even though the target, the MDM2-p53 interaction, is attractive for tumors that could occur in the brain, including glioblastoma and brain metastases. In vitro and in vivo studies indicate that SAR405838 is a substrate of P-glycoprotein (P-gp). P-gp mediated active efflux at the blood-brain barrier plays a dominant role in limiting SAR405838 brain distribution. Even though the absence of P-gp significantly increases the drug exposure in the brain, the systemic exposure, including absorption and clearance processes, were unaffected by P-gp deletion. Model-based parameters of SAR405838 distribution across the blood-brain barrier indicate the CLout of the brain was approximately 40-fold greater than the CLin. The free fraction of SAR405838 in plasma and brain were found to be low, and subsequent Kpuu values were less than unity, even in P-gp/Bcrp knockout mice. These results indicate additional efflux transporters other than P-gp and Bcrp may be limiting distribution of SAR405838 to the brain. Concomitant administration of elacridar significantly increased brain exposure, also without affecting the systemic exposure. This study characterized the brain distributional kinetics of SAR405838, a novel MDM2 inhibitor, to evaluate its potential in the treatment of primary and metastatic brain tumors.
SIGNIFICANCE STATEMENT
This paper examined the brain distributional kinetics of a novel MDM2-p53 targeted agent, SAR405838, to see its possible application for brain tumors by using in vitro, in vivo, and in silico approaches. SAR405838 is found to be a substrate of P-glycoprotein (P-gp), which limits its distribution to the brain. Based on the findings in the paper, manipulation of the function of P-gp can significantly increase the brain exposure of SAR405838, which may give an insight on its potential benefit as a treatment for primary and metastatic brain cancer.
Introduction
The tumor suppressor p53 has been an attractive target in cancer therapeutics due to its crucial role in tumorigenesis (Hainaut and Hollstein, 2000; Vogelstein et al., 2000). The signaling pathway of p53 is found to be inactivated in various types of human cancers, often without a gene mutation in p53 itself (Wade et al., 2013). Therefore, it was a challenge to find ways to reactivate this protein in tumor cells for therapeutic purposes, until the role of the oncoprotein murine double minute 2 (MDM2) was discovered (Momand et al., 1992; Finlay, 1993). MDM2 has been identified as a major negative regulator of p53 by either direct binding or ubiquitination, leading to degradation (Momand et al., 2000; Wade et al., 2013). MDM2 is often amplified or overexpressed in various tumors, which leads to cancer development by downregulating p53 (Wade et al., 2013). Therefore, reactivation of p53 in tumors by the use of small molecule antagonists that target the interaction between MDM2 and p53 has been investigated as a novel molecularly targeted therapy for various cancers.
Currently there are several small molecule MDM2 antagonists under clinical investigation for various solid tumors. One of these, SAR405838 (Fig. 1), is a potent inhibitor that has high selectivity and affinity to MDM2 (Ki = 0.88 nM) (Wang et al., 2014). A previous study from our group has shown that SAR405838 was highly efficacious in a patient-derived xenograft model of glioblastoma (GBM) both in vitro and in heterotopic tumor implanted subcutaneously in the flank (Kim et al., 2018b). However, SAR405838 showed a lack of efficacy against an orthotopic tumor model, where the tumor was implanted intracerebrally. We conclude that this is likely due to limited delivery of SAR405838 to the tumor in the brain (Kim et al., 2018b). Interestingly, the in vivo orthotopic efficacy of SAR405838 was shown to be significantly increased in a tumor line where the BBB was disrupted by the overexpression of vascular endothelial growth factor A (Kim et al., 2018b). These data indicate that the brain distribution of this MDM2 inhibitor is a crucial factor in limiting treatment efficacy for infiltrative brain tumors, such as GBM (Sarkaria et al., 2018). Therefore, it is critical to understand the mechanisms that limit SAR405838 entry into the brain at an intact BBB. Clearly, adequate SAR405838 exposure in the brain depends on both systemic pharmacokinetic properties and distribution across the BBB, exemplified by SAR405838 efficacy in the patient-derived xenograft model of GBM dependence on the brain delivery of the compound (Kim et al., 2018b).
Fig. 1.

Chemical structure of SAR405838.
Brain distribution of many agents is often limited by the blood-brain barrier (BBB), which is characterized by tight and adherens junctions, that blocks the intercellular pathway for small molecules and by expression of efflux transporters that limit transcellular transport. Many of the molecularly targeted anti-cancer therapeutics are not able to penetrate the BBB and are subject to these active efflux transporters, including P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp). P-gp and Bcrp are highly expressed active efflux transporters at the BBB of the mouse (Agarwal et al., 2012) and human (Uchida et al., 2011). How these transporters influence the brain delivery of SAR405838 is critical in understanding the delivery and efficacy in the context of tumors in the brain, whether they be primary tumors, such as GBM (Cancer Genome Atlas Research Network, 2008), or metastatic brain tumors that may overexpress MDM2 and have areas with an intact BBB (Wade et al., 2013).
The objective of the current study was to examine the role of major efflux transporters, P-gp and Bcrp, on the brain distribution of SAR405838 using in vitro and in vivo methods. Moreover, a compartmental model was developed for quantitative and mechanistic understanding of the distributional kinetics of SAR405838 into and out of the brain in the presence and the absence of the major active efflux transporters at the BBB. These studies provide insights on the use of SAR405838 in brain tumor therapy.
Materials and Methods
Chemicals and Reagents
(2'S,3′R,4'S,5′R)-6-chloro-4'-(3-chloro-2-fluoro-phenyl)-2'-(2,2-dimethyl-propyl)-2-oxo-1,2-dihydro-spiro (indole-3,3′-pyrrolidine)-5′-carboxylic acid (trans-4-hydroxy-cyclohexyl)-amide (SAR405838; Fig. 1) was obtained from Sanofi Pharmaceutical (Vitry-sur-Seine, France). N-(3-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide (PLX-4720) was purchased from Chemietek (Indianapolis, IN). N-[4-[2-(3,4-dihydro-6,7-dimethoxy-2(1H)isoquinolinyl)ethyl]phenyl]-9,10-dihydro-5-methoxy-9-oxo-4-acridinecarboxamide (elacridar) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). [3H]Prazosin was purchased from Perkin Elmer Life and Analytical Sciences (Waltham, MA). [3H]Vinblastine was purchased from Moravek Biochemicals (La Brea, CA). (3S,6S,12aS)1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino(19,29:1,6)pyrido(3,4-b)indole-3-propanoic acid 1,1-dimethylethyl ester (Ko143) was purchased from Tocris Bioscience (Ellisville, MO) and LY335979 (zosuquidar), (R)-4-((1aR,6R,10bS)-1,2-difluoro-1,1a,6,10b-tetrahydrodibenzo-(a,e)cyclopropa(c)cycloheptan-6-yl)-((5-quinoloyloxy)methyl)-1-piperazine ethanol, trihydrochloride (zosuquidar) was kindly provided Eli Lilly and Co. (Indianapolis, IN). All other chemicals and reagents used were high-performance liquid chromatography grade from Thermo Fisher Scientific). The rapid equilibrium dialysis (RED) base plate and membrane inserts (8 kDa molecular weight cut-off cellulose membrane) were purchased from Thermo Fisher Scientific.
Animals
An equal number of female and male Friend Leukemia Virus strain B (FVB) wild-type and transgenic mice lacking either or both efflux transporters, Mdr1 or/and Bcrp1 including Bcrp1(–/–), Mdr1a/b(–/–), and Mdr1a/b(–/–)Bcrp1(–/–) mice (Taconic Biosciences, Inc., Germantown, NY) at the age of 8–14 weeks were used for the experiments. Animal colonies were maintained and housed in Research Animal Resources (RAR) facility located at the Academic Health Center, University of Minnesota, following an established breeding protocol. Animal genotypes were routinely verified by conducting tail snip (TransnetYX, Cordova, TN). All protocols for the animal experiments were approved by University of Minnesota Institutional Animal Care and Use Committee and performed in accordance with the Guide for the Care and Use of Laboratory Animals established by the U.S. National Institutes of Health (Bethesda, MD).
In Vitro Cell Accumulation
Cell accumulation experiments were performed with Madin-Darby canine kidney II (MDCKII) cells that overexpress either human multidrug resistance protein 1 (P-glycoprotein, MDR1) or murine breast cancer resistance protein (Bcrp1) or vector-controlled cells. Bcrp1 transfected (MDCKII-BCRP1) and P-gp transfected (MDCKII-MDR1) cell lines were kindly provided by Dr. Alfred Schinkel and Dr. Piet Borst (The Netherlands Cancer Institute), respectively. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and antibiotics (penicillin, 100 U/ml; streptomycin, 100 mg/ml; amphotericin B, 250 ng/ml). Cells were seeded in 12-well polystyrene plates with a density of 4 × 105 cells/well 1 day prior to the experiment (over 80% confluent). Cells were washed with serum free cell assay buffer containing 10 mM HEPES and then preincubated with either buffer alone or with a selective inhibitor for P-gp (1 μM of LY335979) or Bcrp1 (0.2 μM of Ko-143) for 30 minutes. Cells were incubated with 2 μM of SAR405838 with or without the selective transporter inhibitor for 60 minutes at 37°C with 60 rpm of agitation in an orbital shaker. At the end of incubation, cells were washed with ice-cold PBS twice to quench transport prior to cell lysis with 1% Triton X-100. The activities of efflux transporters expressed in the cell were validated using positive controls, [3H]vinblastine for P-gp and [3H]prazosin for Bcrp. The lysates were stored in −80°C freezer until the analysis with liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), and intracellular concentration was normalized to the cellular protein content measured by the BCA assay.
Free Fraction in Mouse Plasma and Brain Homogenate
The free fractions of SAR405838 in mouse plasma and brain homogenate were determined by using a rapid equilibrium dialysis (RED) device according to the manufacturer’s protocol. Briefly, the brain homogenate was prepared by adding two volumes (w/v) of PBS (pH 7.4) followed by mechanical homogenization. SAR405838 was added to mouse plasma or brain homogenate to a final concentration of 5 μM containing 0.3% DMSO. The matrix with the drug was loaded into the sample chamber (300 μl) of the inserts first, and then 500 μl of blank PBS was added into the corresponding buffer chamber. The base plate was sealed with an adhesive lid and incubated at 37°C for 4 hours in an orbital shaker with a 300 rpm of agitation. At the end of incubation, samples were collected from both chambers and stored in −80°C freezer until LC-MS/MS analysis. Undiluted free fraction in the brain was calculated with the equation below reported previously (Kalvass and Maurer, 2002):
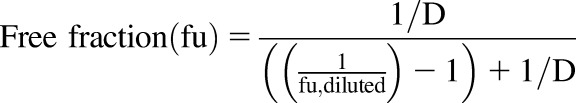 |
(1) |
The dilution factor (D) was 3 in the experiment described above.
The unbound (free) concentration partitioning to the brain was determined as shown below:
 |
(2) |
where Kpbrain is the ratio of brain-to-plasma areas under the total concentration time profile.
Systemic and Distributional Pharmacokinetics
Concentration-time Profile after a Single Oral or Intravenous Administration of SAR405838.
A single dose of SAR405838 was administered by oral gavage (25 mg/kg) or tail vein injection (5 mg/kg) in a solution to wild-type and genetic knockout FVB mice. The dosing solution was prepared with 98% of PEG200 (v/v) and 2% of TPGS (v/v) for the oral administration or 10% of PEG400 (v/v), 3% of Cremophor (v/v), and 87% of PBS (v/v) for the intravenous study. Blood and brain samples were collected at the predetermined time points ranging from 0.5 to 24 hours after oral administration or from 0.167 to 10 hours after intravenous administration (N = 4 at each time point). Mouse whole blood was collected via cardiac puncture using heparinized syringes after mice were euthanized in a carbon dioxide chamber. Plasma was separated by centrifuge at 3500 rpm at 4°C for 20 minutes. Plasma and brain samples were stored at −80°C until LC-MS/MS analysis.
Pharmacological Inhibition of Efflux Transporters
Elacridar (a dual inhibitor of P-glycoprotein and Bcrp) and LY335979 (zosuquidar, a selective P-gp inhibitor) were prepared in a microemulsion formulation as described previously (Sane et al., 2013). Both inhibitors formulated in the microemulsion were diluted with two volumes of sterile water to a final concentration of 1 mg/ml. Vehicle control was formulated in the same manner, including all components of the microemulsion, but without any inhibitor. Wild-type FVB mice received either vehicle control or 5 mg/kg of inhibitor, either elacridar or LY335979 (zosuquidar) by tail vein injection. A dose of 25 mg/kg of SAR405838 was administered orally 1 hour after the administration of either vehicle control or inhibitors. Blood and brain samples were collected as described in pharmacokinetic experiment, 2 hours following the administration of SAR405838, and stored at −80°C until LC-MS/MS analysis.
LC-MS/MS Bioanalysis
An LC-MS/MS method was developed by using reverse-phase liquid chromatography (Waters AQUITY ultra performance liquid chromatography system; Waters, Milford, MA) interfaced with a Waters Micromass Quattro Ultima triple quadruple mass spectrometer (Waters) with an electrospray interface in negative ion mode. Chromatographic separation was performed by injecting 5 μl of reconstituted sample onto an ZORBAX Eclipse XDB-C18 column (Rapid Resolution HT 4.6 × 50 mm 1.8 μm; Agilent, Santa Clara, CA). Mobile phase was composed of aqueous phase (A) of 55% distilled and filtered water with 0.1% formic acid and organic phase (B) of 45% acetonitrile with 0.1% formic acid using an isocratic method. The total assay run time was 8 minutes, while the retention time for SAR405838 and internal standard (PLX-4720) were 1.95 and 5.45 minutes, respectively. SAR405838 and internal standard were detected with the mass transition of 560 > 305.9 and 411.9 > 304.86, respectively. These methods were sensitive and linear over the range of 1–5000 ng/ml with coefficient of variation of less than 15% (weighting factor of 1/Y2). All the specimen concentrations measured were within the range of calibration curve.
Pharmacokinetic Data Analysis
Non-Compartmental Analysis.
Concentration-time profiles in plasma and brain after a single oral or intravenous dose of SAR405838 were analyzed by using Phoenix WinNonlin version 6.4 (Certara USA Inc., Princeton, NJ). Pharmacokinetic parameters and metrics were calculated by non-compartmental analysis (NCA). Areas under the curve (AUCs) from time 0 to infinity for plasma and brain were calculated by log-linear trapezoidal integration, and the extrapolation for AUC from last time point to infinite time was calculated by dividing the last concentration measured by the terminal elimination rate constant, determined from the last three to four points in the concentration-time profiles. Other pharmacokinetic parameters, including systemic clearance (CL), volume of distribution, and t1/2 (half-life), were calculated by using NCA. The brain concentrations were not corrected by residual blood volume of microvasculature in the brain, because the correction resulted in negative concentration in the brain of wild-type and Bcrp1−/− mice (Fridén et al., 2010).
The brain-to-plasma partition coefficient (Kpbrain) was calculated as below:
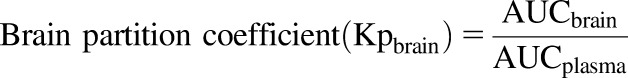 |
(3) |
where AUCbrain is an area under the curve from time zero to infinity of brain concentration-time profile ([AUC0→∞, brain]) and AUCplasma is an area under the curve plasma concentration-time profile ([AUC0→∞, plasma]).
The brain partition coefficient of free drug was calculated as described above in methods for free fraction. The distribution advantage (DA) to the brain resulting from lack of P-gp- and/or Bcrp-mediated efflux at the BBB was determined by the ratio of Kpknockout to Kpwild-type to understand the magnitude of the role efflux transporters play in the brain distribution of SAR405838. The oral bioavailability of SAR405838 was calculated by the following equation:
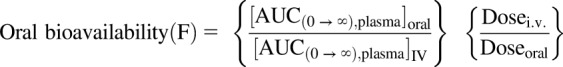 |
(4) |
Where the 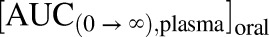 is the area under the curve from time zero to infinity of plasma concentration-time profile following a single oral dose and
is the area under the curve from time zero to infinity of plasma concentration-time profile following a single oral dose and 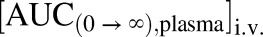 is the area under the curve from time zero to infinity of plasma concentration-time profile following a single intravenous dose.
is the area under the curve from time zero to infinity of plasma concentration-time profile following a single intravenous dose.
Compartmental Analysis with a BBB Model.
A compartmental model that includes a brain compartment (BBB model) was used to quantitatively assess the rate and extent of SAR405838 distribution into and out of the mouse brain (Liu et al., 2005; Laramy et al., 2018). The model was fit to the data in two steps. First, a one-compartment model was fit to wild-type (WT) and triple knockout (TKO) mean pooled plasma concentration-time data from a single intravenous bolus (Fig. 2A), given that there was no difference between WT and TKO plasma concentration profiles and individual model fits yielded the same disposition parameters for each genotype. These systemic disposition parameters, i.e., clearance (CL), the volume of distribution (Vc), and elimination rate constant from the central compartment (Ke) for wild-type and triple-knockout FVB mice were determined using the one-compartment model fit to the data obtained following a single intravenous administration. Then, absorption rate constants for each genotype were estimated from the model fitted to the observed plasma data following a single oral administration. In the second step, a compartmental model that includes a brain compartment was fit to the observed brain concentration-time profile data either from an intravenous bolus or an oral administration (Fig. 2B). A forcing function, comprised of the systemic disposition parameters obtained in step 1, including the absorption rate constant when appropriate, was implemented to describe the plasma concentration in the central compartment and used as an input function into the compartmental BBB model. Simulation and model fitting for systemic disposition and brain distribution were performed by using SAAM II (version 2.3; The Epsilon Group, Charlottesville, VA).
Fig. 2.

A compartmental blood-brain barrier (BBB) model to describe a concentration-time profile in the central (plasma) and brain compartment after a single intravenous bolus or oral dose. (A) A one-compartment model to describe the total concentration-time profile in plasma and to get systemic parameters for a forcing function. (B) A compartmental BBB model to describe the total concentration-time profile in brain. Cb, concentration in brain; Cp, concentration in plasma; Ka, absorption rate constant; Ke, the elimination rate constant from the central compartment; Kin, tissue transfer rate constant into the brain; Kout, tissue transfer rate constant out of the brain; Vb, the apparent volume of distribution in brain; Vc, the volume of distribution in central compartment.
The changes in total brain concentration with respect to time were described by using the following differential equation:
| (5) |
where Vbrain is the apparent volume of distribution in the brain, Kin and Kout are the first-order rate constants that describe the rates into and out of the brain, and Vc is the volume of distribution of total drug in the central compartment. In this model, Cplasma is the predicted total drug concentration in plasma under the model from step 1, and Cbrain is the observed total drug concentrations in brain. Given that the total concentration of drug was measured in the brain as the reference concentration to relate to the total amount in brain, the Vbrain for SAR405838 was estimated to be the same as the anatomic volume of mouse brain that was obtained from the overall average of our in vivo experiments, i.e., 0.42 ± 0.034 ml/g brain.
The clearances into and out of the brain were calculated with the model estimated Kin and Kout of the brain by using following equations:
| (6) |
| (7) |
The exposure of brain tissue to SAR405838 was also quantified by the mean transit time (MTT) using the following equation (Kong and Jusko, 1988):
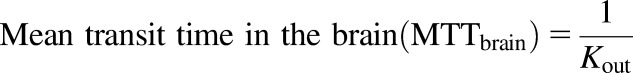 |
(8) |
Statistical Analysis
All data are presented as means ± S.D. or means ± S.E. of the estimate (S.E.). Comparison between two groups was tested by using an unpaired two-sample t test with GraphPad Prism version 6.04 (GraphPad, La Jolla, CA) software. A significance level of P < 0.05 was used for the test.
Results
In Vitro Cell Accumulation Assay.
The role of the two efflux transporters that are highly expressed on the luminal membrane in the endothelial cell of brain microvasculature, P-glycoprotein (P-gp, ABCB1) and breast cancer resistance protein (Bcrp, ABCG2), on the brain distribution of SAR405838 was initially examined using an in vitro cell accumulation assay in MDCKII wild-type, MDCKII-MDR1-overexpressing, and MDCKII-BCRP1-overexpressing cell lines. [3H]-Vinblastine and [3H]-prazosin were used as positive controls to check the functionality of P-gp and Bcrp, respectively, in the transfected cell lines. The intracellular accumulation of these positive control substrates was significantly lower in transporter-overexpressing cells compared with their normalized vector-controlled wild-type cells (Fig. 3) [wild-type (MDR1-vector control): 100% ± 32.18%, Mdr1: 36.7% ± 12.26%, P < 0.05; wild-type (BCRP-vector control): 100% ± 11.0%, Bcrp1: 29.83% ± 9.91%, P < 0.01]. When LY335979, a selective inhibitor of P-gp, and Ko-143, a selective inhibitor of Bcrp, were coincubated with their respective substrates, the intracellular accumulation was similar to the vector control due to inhibition of the respective efflux transporter (Fig. 3) [wild-type (MDR1) + LY335979: 130% ± 12.2%, MDR1 + LY335979: 158.3% ± 21.81%, (N.S.); wild-type (BCRP1) + Ko-143: 103.1% ± 13.7%, BCRP1 + Ko-143: 90.8% ± 19.9%, (N.S.)]. The accumulation of SAR405838 in MDCKII-MDR1 cells was only 35.2% of the corresponding vector-controlled cells, and this difference was abolished in the presence of the P-gp selective inhibitor, LY335979 (Fig. 3A) [139.8% ± 22.4%, P < 0.0001]. However, no significant difference was observed in the accumulation of SAR405838 between Bcrp vector control and Bcrp overexpressing cells (Fig. 3B) [Bcrp1: 121.5% ± 26.6%, (N.S.)]. These in vitro results indicate that SAR405838 is a substrate of P-gp, but not of Bcrp, suggesting that P-gp may play a significant role in limiting the brain distribution of SAR405838. The use of a selective and potent P-gp inhibitor, such as LY335979, was able to significantly diminish the function of P-gp, and increase the intracellular accumulation of SAR405838 in these in vitro experiments.
Fig. 3.

Cell accumulation of SAR405838. (A) The intracellular accumulation of vinblastine (positive control) and SAR405838 in MDCKII vector control and MDR1-transfected cells in the presence and absence of P-gp inhibitor, LY335979 (1 μM). (B) The intracellular accumulation of prazosin (positive control) and SAR405838 in MDCKII vector control and Bcrp-transfected cells in the presence and absence of Bcrp inhibitor, Ko-143 (0.2 μM). Data presented as mean ± S.D. where N = 3 for all groups. *P < 0.05; **P < 0.01; ****P < 0.001.
SAR405838 Disposition following Intravenous Dose.
The brain and plasma concentration-time profiles were examined at multiple time points up to 10 hours after a single intravenous administration of SAR405838 in wild-type (WT) and triple knockout FVB mice (Mdr1a/1b−/−Bcrp1−/−) that lack both Mdr1a/b and Bcrp (Fig. 4, A and B). Concentrations of SAR405838 in plasma and brain were measured in the specimens that were collected at the predetermined time points after administration of 5 mg/kg SAR405838 by tail vein injection. The plasma concentrations over time [AUC(0→tlast)] of SAR405838 in Mdr1a/1b−/−Bcrp1−/− mice were not significantly different than in wild-type FVB mice (Table 1, plasma AUC WT = 15,851 ± 542, plasma AUC Mdr1a/1b−/−Bcrp1−/− = 15,033 ± 761, N.S.). Importantly, this was not the case in the distribution of SAR405838 to the brain, where concentrations of SAR405838 in the brain were significantly higher in Mdr1a/1b−/−Bcrp1−/− mice compared with wild-type mice at all time points (P < 0.05). Both plasma and brain concentrations exhibit a mono-exponential decline with respect to time for both wild-type and Mdr1a/1b−/−Bcrp1−/− mice. Brain-to-plasma ratios of SAR405838 were significantly higher in Mdr1a/1b−/−Bcrp1−/− than wild type and increased over time in the Mdr1a/1b−/−Bcrp1−/− genotype (P < 0.05) but did not increase after the second measurement (30 minutes) in the wild-type (N.S.) (Fig. 4C). Correspondingly, a plateau in the brain-to-plasma ratio was observed early post dose (30 minutes) in wild type, but it was not reached in Mdr1a/1b−/−Bcrp1−/− even after 10 hours after the dose. Plasma pharmacokinetic parameters and metrics were calculated in both wild type and Mdr1a/1b−/−Bcrp1−/− (Table 1). The wild type and Mdr1a/1b−/−Bcrp1−/− mice had a terminal elimination half-life of 2.25 and 2.76 hours, volume of distribution (Vd) of 973 and 1227 ml/kg and systemic clearance of 300 and 308 (ml/h)/kg, respectively, indicating that there are no differences in the systemic elimination of SAR405838 between these two transporter genotypes. Also, the brain partition coefficient, calculated using AUC(0→∞), plasma, and AUC(0→∞), brain, was over 45-fold higher in Mdr1a/1b−/−Bcrp1−/− than wild type [0.0275 in wild type and 1.29 in Mdr1a/1b−/−Bcrp1−/−] (Table 1), indicating that P-gp (Mdr1) is critical in limiting the BBB permeability and brain distribution of SAR405838.
Fig. 4.

Pharmacokinetic profile of SAR405838 following a single intravenous administration. (A) Concentration-over-time in plasma, (B) concentration-over-time in brain, and (C) brain-to-plasma ratio over time in wild-type and Mdr1a/b–/–Bcrp1–/– mice. Data presented as mean ± S.D. where N = 3–5 for each time point.
TABLE 1.
Pharmacokinetic/metric parameters and brain partition coefficients determined by non-compartmental analysis following a single intravenous dose of SAR405838 (5 mg/kg) in wild-type and Mdr1a/b–/–Bcrp1–/– FVB mice
In TKO, equilibrium between plasma and brain was not reached until the last time point (10 h). Values are means ± SE.
| Plasma | Brain | |||
|---|---|---|---|---|
| Wild Type | Mdr1a/b−/− Bcrp1−/− | Wild Type | Mdr1a/b−/− Bcrp1−/− | |
| t1/2 (hour) | 2.25 | 2.76 | 3.10 | 7.71a |
| AUC0→tlast [(h*ng)/ml] | 15,851 ± 542.0 | 15,033 ± 761.3 | 414 ± 11.9 | 11,925 ± 609 |
| AUC0→∞ [(h*ng)/ml] | 16,715 | 16,280 | 460 | 20,973 |
| Vd (ml/kg) | 973 | 1227 | — | — |
| CL [(ml/h)/kg] | 300 | 308 | — | — |
| Kpbrain | — | — | 0.0275 | 1.29 |
| D) | — | — | 1 | 46.8 |
AUC0-tlast, area under the curve from zero to the time of last measured concentration; AUC0–∞, area under the curve from zero to time infinity; CL, clearance; DA, distribution advantage, the ratio of Kpknockout to Kpwild-type; Kpbrain (AUC ratio), the ratio of AUC(0–∞,brain) to AUC(0–∞,plasma) using total drug concentrations; t1/2, half-life; Vd, volume of distribution.
Half-life was determined by the slope of last four time points in concentration-time profile.
SAR405838 Absorption and Disposition following Single Oral Dose.
The brain and plasma concentration-time profiles were determined after a single oral dose of SAR405838 (25 mg/kg) in four different genotypes of mice, including wild type, Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– (Fig. 5). The plasma concentration-time profiles of SAR405838 were very similar among the different genotypes (Fig. 5A), even though there was some variability in AUC(0→∞), plasma. The brain concentrations of SAR405838 in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– were higher compared with wild-type and Bcrp1–/– at all time points (Fig. 5B, P < 0.05). The brain-to-plasma ratios, shown in Fig. 5C, were consistently greater in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– than wild type and Bcrp1–/– (significantly different at all time points in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– compared with wild type and Bcrp1–/–, P < 0.05). In all four genotypes, the brain to plasma ratio increased over time and reached an early plateau in wild-type and Bcrp1−/−mice, but the distributional equilibrium plateau was observed at much later times in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– mice. The Kp values calculated from the brain and plasma AUCs after oral administration were much greater in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– mice (2.35 and 1.53, respectively) than in wild type and Bcrp1−/− (0.0218 and 0.0285, respectively), suggesting the dominant influence of P-gp on the brain exposure of SAR405838. The corresponding brain distribution advantage achieved by eliminating the efflux mechanism was calculated in Bcrp1–/–, Mdr1a/b–/–, and Mdr1a/b–/–Bcrp1–/– mice compared with wild-type mice and were 1.31, 108, and 70.1, respectively, after a single oral dose (Table 2).
Fig. 5.

Pharmacokinetic profile of SAR405838 following a single oral administration. (A) Concentration-over-time in plasma, (B) concentration-over-time in brain, and (C) brain-to-plasma ratio over time in wild-type, Mdr1a/b–/–, Bcrp1–/–, and Mdr1a/b–/–Bcrp1–/– FVB mice. Data presented as mean ± S.D. where N = 3–5 for each time point.
TABLE 2.
Pharmacokinetic/metric parameters determined by non-compartmental analysis following a single oral dose of SAR405838 (25 mg/kg) in wild-type, Mdr1a/b–/–, Bcrp1–/–, and Mdr1a/b–/–Bcrp1–/– FVB mice
Values are means ± SE.
| Plasma | Brain | |||||||
|---|---|---|---|---|---|---|---|---|
| Wild Type | Mdr1a/b−/− | Bcrp1−/− | Mdr1a/b−/− Bcrp1−/− | Wild Type | Mdr1a/b−/− | Bcrp1−/− | Mdr1a/b−/− Bcrp1−/− | |
| t1/2 (hour) | 3.26 | 4.18 | 3.02 | 5.08 | 4.03 | 10.1 | 3.36 | 12.1 |
| Tmax (hour) | 8 | 8 | 4 | 2 | 8 | 16 | 4 | 8 |
| Cmax (ng/ml) | 4651 | 3582 | 7209 | 6164 | 161 | 4176 | 159 | 4554 |
| AUC0→tlast [(h*ng)/ml] | 60,425 ± 3584 | 40,490 ± 2559 | 68,107 ± 9193 | 62,490 ± 7253 | 1995 ± 132 | 74,213 ± 5908 | 1925 ± 249 | 75,281 ± 9392 |
| AUC0→∞ [(h*ng)/ml] | 61,195 | 41,382 | 68,681 | 65,867 | 1335 | 97,283 | 1956 | 100,663 |
| F oral | 0.732 | NA | NA | 0.809 | — | — | — | — |
| Vd/F (ml/kg) | 1922 | 3642 | 1585 | 2778 | — | — | — | — |
| CL/F (ml/kg) | 409 | 604 | 364 | 379 | — | — | — | — |
| Kpbrain | — | — | — | — | 0.0218 | 2.35 | 0.0285 | 1.53 |
| DA | — | — | — | — | 1 | 108 | 1.31 | 70.1 |
AUC0-tlast, area under the curve from zero to the time of last measured concentration; AUC0–∞, area under the curve from zero to time infinity; CL/F, apparent clearance; Cmax, observed maximum concentration; DA, distribution advantage, the ratio of Kpknockout to Kpwild-type; F, absolute bioavailability, ratio of the dose corrected AUC(0–∞) following oral administration to dose corrected AUC(0–∞) following intravenous administration; Kpbrain (AUC ratio), the ratio of AUC(0–∞,brain) to AUC(0–∞,plasma) using total drug concentrations; Tmax, time to reach the maximum concentration; Vd/F, apparent volume of distribution.
The systemic oral bioavailability was calculated in both wild type and Mdr1a/b–/–Bcrp1–/– and were 73.2% and 81%, respectively (Table 2). These similar values in bioavailability in these genotypes indicate that P-gp and Bcrp do not have a profound influence on the bioavailability of SAR405838, even though efflux transport significantly changes the brain exposure.
Plasma and Brain Unbound Fraction.
The unbound fraction (fu) of SAR405838 in plasma and brain homogenate was determined by using rapid equilibrium dialysis after a 4-hour incubation that was shown to be adequate time to reach equilibrium in pilot experiments. The fu of SAR405838 in the plasma was extremely low (0.059% ± 0.034%, N = 9); however, it was approximately three times higher (P < 0.01) than that in the brain (0.015% ± 0.0035%, N = 9) (Table 3). The free (unbound) brain-to-plasma ratio (Kpuu) values were calculated based on these fu values and are summarized in Table 3. The Kpuu, brain values are 0.006 and 0.007 in wild type and Bcrp1–/– after a single oral administration, respectively, indicating that efflux mechanisms play a highly significant role in the brain penetration of SAR405838 in these genotypes. The Kpuu, brain values in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– (0.598 and 0.389, respectively) were higher than those in wild type and Bcrp1–/– due to lack of the dominant efflux transporter, P-gp, at the blood-brain barrier. Even though both P-gp and Bcrp were genetically deleted, the values of Kpuu, brain still did not reach unity, suggesting the possibility that other efflux transporters may be involved in the brain penetration of SAR405838 or other elimination processes may exist in the brain, such as enzymatic degradation or metabolism as well as possible mechanisms of clearance involving bulk flow (Hammarlund-Udenaes et al., 2008).
TABLE 3.
Free fraction (fu) values, partition coefficient of brain (Kpbrain and Kpuu, brain), and distribution advantage
Data presented as mean ± S.D.
| Intravenous | Oral | |||||
|---|---|---|---|---|---|---|
| Wild Type | Mdr1a/1b−/− Bcrp1−/− | Wild Type | Mdr1a/1b−/− | Bcrp1−/− | Mdr1a/1b−/− Bcrp1−/− | |
| Kpbrain | 0.0275 | 1.29 | 0.0218 | 2.35 | 0.0285 | 1.53 |
| fu, plasma | 0.00059 ± 0.00034 | |||||
| fu, brain | 0.00015 ± 0.000035 | |||||
| Kpuu, brain | 0.007 | 0.328 | 0.006 | 0.598 | 0.007 | 0.389 |
| DAtotal | 1 | 46.8 | 1 | 108 | 1.31 | 70.1 |
DA, distribution advantage, the ratio of Kpknockout to Kpwild-type; fu,brain, free fraction of SAR405838 in brain homogenate determined by rapid equilibrium dialysis (N = 9); fu,plasma, free fraction of SAR405038 in plasma determined by rapid equilibrium dialysis (N = 9); Kp (AUC ratio), the ratio of AUC(0–∞,brain) to AUC(0–∞,plasma) using total drug concentrations; Kpuu (AUC ratio), the ratio of AUC(0–∞,brain) to AUC(0–∞,plasma) using free drug concentrations.
Pharmacological Inhibition of Efflux on the Brain Distribution of SAR405838.
We examined the effect of elacridar, a dual inhibitor of both P-gp and Bcrp, and LY335979, a selective inhibitor of P-gp, on the brain distribution of SAR405838 in mice following co-dosing of inhibitors and SAR405838. Concentrations of SAR405838 in plasma with vehicle control were not different from the inhibitor group at both 2 and 6 hours after the administration of SAR405838 (Fig. 6A). However, brain concentrations of SAR405838 were 8.7 times higher at 2 hours and 3.8 times higher at 6 hours with elacridar (P < 0.01 for both) compared with the corresponding vehicle control at each time point (Fig. 6A). Interestingly, brain concentrations of SAR405838 with LY335979 were not different from vehicle control group at 2 and 6 hours after the dosing. The brain-to-plasma ratio (Kpbrain) at 2-hour post SAR405838 dosing, was also significantly higher (P < 0.005) than vehicle control with elacridar (dual inhibitor), but there was no difference with LY335979 (selective P-gp inhibitor) (Fig. 6B).
Fig. 6.

The effect of a pharmacological inhibitor of efflux transport, elacridar, on the plasma and brain concentration of SAR405838. (A) Concentrations in plasma and brain at 2- and 6-hour postdose with coadministration of either vehicle control or inhibitor, either LY335979 or elacridar. (B) Brain-to-plasma ratio at 2- and 6-hour postdose. Data presented as mean ± S.D. where N = 3–5 for each group. **P < 0.01; ***P < 0.005.
Brain Distributional Kinetics of SAR405838 Using BBB Modeling.
A one-compartment model was fit to mean pooled total plasma concentrations to describe the plasma concentration-time profile following a single intravenous administration and to yield systemic disposition parameters to use as a forcing function in the BBB model. The model predicted plasma concentration-time profiles, and the observed plasma concentrations from the experiments for intravenous administration are shown in Fig. 7. The systemic volume of distribution was estimated to be 1166 ml/kg, and the elimination rate constant (Ke) from the central compartment was estimated to be 0.269 hour−1 in Mdr1a/b–/–Bcrp1–/– animals. Initial models for both wild-type and triple-knockout animals were separately fit to the data obtained from each genotype and there were no differences in these systemic parameters between wild type and triple knockouts. Therefore, systemic parameters obtained from the mean pooled data were used for all genotypes and are summarized in Table 4. All parameter estimates were precisely estimated and had a coefficient of variation (CV) of less than 10%. With these parameter estimates, the systemic clearance of SAR405838 [314 (ml/h)/kg], half-life (t1/2) (2.57 hours), and plasma concentration at time zero (Cp0) (4288 ng/ml) were calculated (Table 4). These parameter estimates were then used in the plasma concentration-time forcing function to then estimate distribution parameters across the BBB.
Fig. 7.

Observed (squares and circles) and model predicted (solid line and dotted line) plasma (red) and brain (blank) concentrations of SAR405838 following a single intravenous bolus administration (5 mg/kg) in wild-type (A) and Mdr1a/b–/–Bcrp1–/– (B) FVB mice. The observed data are presented as the mean S.D. where N = 3 to 4 for each time point.
TABLE 4.
Pharmacokinetic parameters estimated from one-compartment model that describes the total concentration-time profile from each genotype following a single intravenous bolus (5 mg/kg) administration
| Mean | CV (%) | 95% CI | |
|---|---|---|---|
| Estimated parameters | |||
| Vc (ml/kg) | 1166 | 6.07 | (984.1, 1348.1) |
| Ke (h−1) | 0.269 | 4.40 | (0.239, 0.300) |
| Calculated parameters | |||
| CLsystem [(ml/h)/kg] | 314 | 4.24 | (279.8, 348.2) |
| t1/2 (h) | 2.57 | 4.4 | (2.28, 2.86) |
| Cp0 | 4288 | 6.07 | (3619, 4957) |
CLsystem, clearance from the systemic circulation; Cp0, initial concentration of SAR405838 in the central compartment at time 0; Ke, elimination rate constant from the central compartment; t1/2, half-life; Vc, volume of distribution of a drug in the central compartment.
One-compartment models for oral administration were individually fit to the total plasma concentration-time data of each genotype following an oral administration of SAR405838 to compare the absorption rate constants in wild-type and transgenic mice. For the models of oral administration, the volume of distribution for the central compartment and the systemic clearance from the central compartment were fixed as described in Table 5. The model predicted plasma concentration-time profiles for each genotype and the observed plasma concentration-time profiles are presented in Fig. 8. The absorption rate constants for each genotype were estimated to be 0.265 hour−1 in wild-type, 0.290 hour−1 in Bcrp1–/–, 0.240 hour−1 in Mdr1a/b–/–, and 0.258 hour−1 in Mdr1a/b–/–Bcrp1–/– mice (Table 5). All absorption rate constant parameter estimates had a coefficient of variation (CV) of less than 20%. Overall, the model predicted plasma concentration-time profiles for each genotype visually matched well with the observed plasma concentration-time data after both intravenous bolus and oral administration (Figs. 7 and 8).
TABLE 5.
Pharmacokinetic parameters estimated from one-compartment model that describes the total concentration-time profile from each genotype following a single oral (25 mg/kg) administration
| Estimated Parameters | Mean | CV (%) | 95% C) |
|---|---|---|---|
| Vc (ml/kg) | 1166 | ** fixed ** | |
| Ke (h−1) | 0.269 | ** fixed ** | |
| Ka, WT (h−1) | 0.265 | 6.1 | (0.225, 0.304) |
| Ka, BKO (h−1) | 0.290 | 15.7 | (0.179, 0.401) |
| Ka, PKO (h−1) | 0.240 | 10.1 | (0.177, 0.302) |
| Ka, TKO (h−1) | 0.258 | 14.3 | (0.221, 0.460) |
BKO, Bcrp1−/−; Ka, absorption rate constant after oral dosing in different genotypes; Ke, elimination rate constant from the central compartment (obtained value from mean pooled analysis of all genotypes); PKO, Mdr1a/b−/−; TKO, Mdr1a/b−/−Bcrp1−/−; Vc, volume of distribution of a drug in the central compartment (obtained value from mean pooled analysis of all genotypes); WT, wild type.
Fig. 8.

Observed (squares and circles) and model predicted (solid line and dotted line) plasma (red) and brain (blank) concentrations of SAR405838 following a single oral administration (25 mg/kg) in wild-type (A), Bcrp1–/– (B), Mdr1a/b–/– (C), and Mdr1a/b–/–Bcrp1–/– (D) FVB mice. The observed data are presented as the mean S.D. where N = 3 to 4 for each time point.
As indicated above, to improve the estimation of the brain distribution parameters, estimated systemic disposition parameters from the one-compartment model were used in a forcing function to create a plasma concentration-time profile in the central compartment for the implementation of a BBB model. The model was fit to the concentration-time data by using eq. 5. The initial results using the BBB model confirmed that the tissue transfer rate constants into the brain (Kin) of each genotype are not different from one another. Therefore, given the initial results regarding Kin values, and to simplify the model by reducing the number of estimated parameters to improve precision, Kin values were fixed for all four genotypes using the value estimated in Mdr1a/b–/–Bcrp1–/– animals. The results from the BBB model were presented in Table 6. The tissue transfer rate constants into the brain (Kin) were estimated by the BBB model to be 1.12 × 10−4 hour−1 following intravenous bolus administration and 1.18 × 10−4 hour−1 following oral administration, and these values were not significantly different (P > 0.05). The estimated tissue transfer rate constants out of the brain (Kout) were much greater, ranging from 0.282 (PKO) to 0.300 (TKO) hour−1 in the P-gp-deficient genotypes, and the transfer rate constants out of brain from wild-type and Bcrp knockout mice were 16.8 and 11.3 hour−1, respectively, about 40–60 times higher than the P-gp deficient mice (Table 6). The resulting clearances into the brain (CLin) were estimated by eq. 6 to be 0.131 (ml/h)/kg in and intravenous study and 0.138 (ml/h)/kg in an oral dosing study. The resulting clearances out of the brain (CLout), estimated using eq. 7, in p-gp deficient mice (PKO and TKO) were similar to the clearance into the brain, however, that in wild-type and Bcrp knockout mice were much higher than the clearance into the brain, as expected, mainly due to the efflux by P-gp (see Table 6).
TABLE 6.
The changes in tissue transfer rate and clearance values with the total concentration model
| Route of Administration | Intravenous | Oral | |||||
|---|---|---|---|---|---|---|---|
| Genotype | Wild Type | Mdr1a/b−/−Bcrp1−/− | Wild Type | Bcrp1−/− | Mdr1a/b−/− | Mdr1a/b−/−Bcrp1−/− | |
| Kin (h−1) | Mean | 1.12E−04 | 1.12E−04 | 1.18E−04 | 1.18E−04 | 1.18E−04 | 1.18E−04 |
| CV (%) | — | 16.5 | — | — | — | 18.5 | |
| Kout (h−1) | Mean | 12.3 | 0.432 | 16.8 | 11.3 | 0.282 | 0.300 |
| CV (%) | 7.14 | 19.0 | 14.1 | 10.9 | 8.25 | 18.5 | |
| CLin (ml/h per kilogram) | Mean | 0.131 | 0.131 | 0.138 | 0.138 | 0.138 | 0.138 |
| CV (%) | — | 16.5 | — | — | — | 17.5 | |
| CLout (ml/h per kilogram) | Mean | 5.1 | 0.181 | 7.1 | 4.73 | 0.118 | 0.126 |
| CV (%) | 7.14 | 19.0 | 14.1 | 10.9 | 8.25 | 18.5 | |
| MTT (h) | 0.082 | 2.315 | 0.060 | 0.089 | 3.55 | 3.33 | |
| CLin/Clout | 0.026 | 0.72 | 0.019 | 0.029 | 1.17 | 1.10 | |
CLin, total drug clearance into the brain; CLout, total drug clearance out of the brain; Kin, tissue transfer rate constant into the brain; Kout, tissue transfer rate constant out of the brain; MTT, mean transit time in the brain: calculated by 1/Kout.
The mean transit time in the brain (MTT) was calculated by eq. 8 to quantify the exposure time of brain to SAR405838. As expected, the brain exposure to SAR405838, as quantified by MTT was significantly longer in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– genotypes following both an intravenous bolus (2.32 hours in Mdr1a/b–/–Bcrp1–/–) and an oral administration (3.55 and 3.33 hours, respectively) than in wild type and Bcrp1–/– (0.082 hours after an intravenous bolus; 0.060 and 0.089 hours after an oral dose) as summarized in Tables 6 and 7. In conclusion, the total drug exposure time in the brain is significantly increased in the absence of P-gp (Mdr1), the efflux system that plays a leading role at the BBB in preventing SAR405838 access to the brain.
TABLE 7.
The Kp and Kpuu values predicted from the compartmental BBB model describing the brain and plasma concentration-time profile following either intravenous or oral administration of SAR405838
| Route of Administration | Intravenous | Oral | Mdr1a/b−/− Bcrp1−/− | |||
|---|---|---|---|---|---|---|
| Wild Type | Mdr1a/b−/− Bcrp1−/− | Wild Type | Bcrp1−/− | Mdr1a/b−/− | ||
| AUC0→∞, plasma | 15,915 | 15,915 | 63,748 | 63,955 | 63,503 | 64,165 |
| AUC0→∞, brain | 404 | 11,462 | 1243 | 1859 | 71,308 | 68,317 |
| Kppreda | 0.025 | 0.72 | 0.019 | 0.029 | 1.12 | 1.06 |
| DApredb | — | 28 | — | 1.5 | 58 | 55 |
| Clin | 0.131 | 0.131 | 0.138 | 0.138 | 0.138 | 0.138 |
| Clout | 5.1 | 0.181 | 7.1 | 4.7 | 0.118 | 0.126 |
| CLin/CLoutc | 0.026 | 0.72 | 0.019 | 0.029 | 1.17 | 1.10 |
| DApredd | — | 28 | — | 1.5 | 60 | 56 |
Kppred, the ratio of model predicted AUC0→∞, brain to model predicted AUC0→∞, plasma.
DApred (distribution advantage), the ratio of Kpknockout to Kpwild-type.
The ratio of model predicted clearance into the brain to model predicted clearance out of the brain compartment.
DApred (distribution advantage), the ratio of brain clearance ratio in wild-type to brain clearance ratio in transgenic mice.
Based on the pharmacokinetic parameters and metrics estimated from the compartmental BBB model, the predicted partition coefficient of the brain, distribution advantage, and the ratio of clearance into the brain to clearance out of the brain were calculated and summarized in Table 7. The predicted partition coefficient of the brain and the ratio of clearances were closely matched with the observed Kp values calculated with the results from NCA (Tables 1 and 2). The ratios of the clearance into and out of the brain were calculated and compared with Kp values, and clearance ratios and predicted Kp from the models closely matched with each other in all genotypes. The agreement of the model-based predicted values to the observed values support the assumptions in the compartmental models and the model described the data well.
Discussion
Challenges in the successful treatment of primary and metastatic brain tumors include insufficient and heterogeneous distribution of therapeutics across an intact BBB, which can lead to lack of efficacy, as well as acquired drug resistance due to exposure to subtherapeutic concentrations (Lockman et al., 2010; Pafundi et al., 2013). Therefore, it is important when examining innovative therapeutic agents that target novel signaling pathways in brain tumors to understand the pharmacokinetic properties and distributional kinetics of these agents to the brain. SAR405838 was recently developed to target the p53 and MDM2 interaction, and it has advanced to clinical testing for the treatment of various solid tumors (Wang et al., 2014), but its efficacy in brain tumors was only recently addressed (Kim et al., 2018b). Given the general mechanism of action of p53 enhancement and the fact that some glioblastoma and other tumors of the brain overexpress MDM2, there is a great interest in exploring this target in brain tumors. Importantly, the distributional kinetics of this compound to, from, and in the brain is critical to its rational use in preclinical efficacy studies and in eventually informing the clinical use for brain tumors.
Recent studies from our group examined the potential efficacy of SAR405838 in a patient-derived xenograft model of primary brain tumor, glioblastoma (GBM). The overall conclusion of that study was that the limited brain distribution of SAR405838 diminishes its value as an effective treatment of brain tumor (Kim et al., 2018b). However, the specific mechanisms that influence the adequate delivery of an active concentration of SAR405838 to the brain or brain tumor were not examined. The current study shows that, of the most highly expressed efflux transporters in the BBB, SAR405838 is a substrate of p-glycoprotein (P-gp). P-gp and Bcrp are highly expressed in brain endothelial cells of human and mouse (Uchida et al., 2011; Agarwal et al., 2012), therefore it can be expected that P-gp may limit the distribution, hence the efficacy of SAR405838 in both the preclinical and clinical settings of both primary (e.g., GBM) and secondary tumors in the CNS.
In vitro cell accumulation experiments using MDCKII cells transfected with efflux transporter genes have confirmed that SAR405838 is a substrate of human P-gp, but it may not be a substrate of mouse Bcrp. LY335979 (zosuquidar), a selective competitive inhibitor of P-gp, increased the intracellular accumulation of SAR405838. Consistent with the in vitro study results, in vivo studies with wild-type and transporter knockout mice have confirmed that P-gp plays a crucial role in brain distribution of SAR405838. It is valuable to describe the distributional kinetics after an oral administration of SAR405838, because this drug is given by mouth in both preclinical efficacy studies and in clinical trials. Mdr1a/b–/–Bcrp1–/– FVB mice, after a single intravenous and oral administration of SAR405838, showed enhanced SAR405838 distribution to the brain. The AUCs in the plasma analyzed by NCA in wild-type and Mdr1a/b–/–Bcrp1–/– FVB mice were similar, even though the AUCs in the brain analyzed by NCA were over 30-fold higher in Mdr1a/b–/–Bcrp1–/– compared with wild-type mice after a tail vein injection. The results from oral dosing were consistent with the intravenous studies, where SAR405838 achieved significantly high brain distribution in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– compared with wild-type and Bcrp1–/– mice. Due to the expression of efflux transporters in the intestine, oral absorption and bioavailability can be influenced by the presence or absence of efflux transporters (Kruijtzer et al., 2002). In this regard, it is important to note that plasma concentration-time profile following the oral administration of SAR405838 in Mdr1a/b–/–Bcrp1–/– were no different than those of wild type and Bcrp1–/–. The oral bioavailability calculated in wild-type and Mdr1a/b–/–Bcrp1–/– mice also confirms that the role of efflux transporters, in the case of SAR405838, does not influence drug absorption in the intestine, unlike the brain. One of the reasons for a lack of effect in the intestine may be that concentrations (especially “free” concentrations that may interact with the transporters) of drug achieved in the intestinal lumen after an oral administration are much higher than in plasma that influence brain distribution, therefore, saturating intestinal transporters (Lin and Yamazaki, 2003). As such, the overall permeability of a drug in the intestine will be governed primarily by passive permeability for drugs that have a favorable intrinsic permeability due to its physicochemical properties, such as SAR405838 (Wang et al., 2014). The AUCbrain of SAR405838 in Mdr1a/b–/–Bcrp1–/– mice was comparable to that in Mdr1a/b–/– mice, indicative of, in this case, a lack of “compensation” of one efflux system (Bcrp) for the other (P-gp) (Kodaira et al., 2010; Agarwal et al., 2011a).
The results from compartmental BBB modeling agreed with the results from NCA, which indicated that the results were consistent regardless of the data analysis method. The transporter-mediated SAR405838 efflux at the BBB was characterized by the fact that the tissue transfer rate constant out of the brain (Kout) was considerably decreased in transgenic mice that lack P-gp. The simplified compartmental BBB model also gave an additional insight into brain distributional kinetics by calculating mean transit times (MTTs) and mean residence times of SAR405838 in the brain for in wild-type and transgenic mice that can be translated into a therapeutic exposure time in the brain. The mean residence time in the brain compartment is defined as the average number of times drug molecules visit the brain compartment (N) multiplied by the average time the molecule spends in the brain on one visit, the mean transit time (MTT) (Kong and Jusko, 1988). N is determined by the ratio of Kin and Ke, or CLin to CLsys, which are assumed to be the same across genotypes (Kong and Jusko, 1988). Therefore, the exposure of brain to drug, as exemplified by the mean residence time, will be much higher in the P-gp deficient genotypes than the P-gp intact genotypes.
Nonspecific drug binding to proteins in plasma and tissue is a critical factor to consider for CNS pharmacodynamics as well as distributional kinetics according to the “free drug hypothesis” (Trainor, 2007). This is especially true for the drugs targeting the CNS, where it is the unbound drug concentrations and unbound AUCs in the brain and plasma that indicate involvement of active efflux processes in CNS delivery of drugs (Kalvass and Maurer, 2002; Hammarlund-Udenaes et al., 2008). With the assumption that the free drug concentrations in the brain and in the plasma are in equilibrium, unbound (free) drug partition coefficient of brain (Kpuu, brain) is an informative parameter indicating the contribution of active transport (either influx or efflux) or metabolism in CNS drug distribution (Hammarlund-Udenaes et al., 2008).
There are several ways to experimentally determine the unbound drug concentration in the brain. Recently, the brain homogenate method using rapid equilibrium dialysis (RED) has been suggested as a valid way of determining brain unbound concentration (Waters et al., 2008; Liu et al., 2009). Even though the use of unbound fraction from equilibrium dialysis needs to be carefully evaluated, especially for lipophilic drugs that tend to be highly bound, the RED method is generally accepted as an efficient and practical way to understand tissue binding characteristics (Waters et al., 2008). Therefore, RED was used with brain homogenate and plasma to determine unbound brain and plasma concentrations in the current study. The results show that SAR405838 is more highly bound to components in the brain homogenate than in plasma, consistent with the current understanding about the correlation between lipophilicity and protein binding (Summerfield et al., 2007). As such, the Kpuu, brain was calculated as “low,” i.e., less than unity, in wild type and Bcrp1–/– due to the presence of active efflux transporter, and these values increased in Mdr1a/b–/– and Mdr1a/b–/–Bcrp1–/– mice to 0.5 and 0.3, respectively, when two major efflux transporters are absent. Interestingly, Kpuu, brain does not reach unity even without the major efflux transporter systems, which leads to the possibility of the presence of other efflux transporters that prevent SAR405838 from entering across the BBB.
The concomitant use of elacridar, a potent dual inhibitor of P-gp and Bcrp, with SAR405838 significantly improves the drug exposure in the brain without increasing the plasma concentration. There have been concerns about using transporter inhibitors with anticancer agents due to possible toxicity related to increased drug systemic exposure, because of drug-drug interactions at the level of the systemic clearance. However, for drugs that do not rely on transporters for their systemic clearance, such as SAR405838, combination therapy with efflux transporter inhibitors may be considered as a potential therapeutic strategy to overcome the BBB, especially with molecularly targeted agents in the treatment of glioblastoma, where only limited therapeutic regimens are available. The dosage and the interlaced schedule of dosing of the two interacting compounds need to be carefully assessed when using such a therapeutic drug-drug interaction strategy in clinical setting. Interestingly, coadministration of LY335979 (zosuquidar), a selective P-gp inhibitor, with SAR405838 did not change the brain delivery of SAR405838 in mice. The inhibitory potencies of elacridar and LY335979 against P-gp are reported to be similar (Jabeen et al., 2012), so the similar in vivo inhibitory efficacy might be expected with the same dose (5 mg/kg) of inhibitors, given similar concentrations. The discrepancies in the results between elacridar and LY335979 might be explained in several ways. One possibility is that the availability of a drug at the site of action, BBB in this case, can be lower with LY335979, so LY335979 may need higher dose to have the similar efficacy as elacridar. Another interesting possibility is that the binding site of SAR405838 to P-gp is different from that of LY335979 in mice, since LY335979 has been shown to inhibit human P-gp potently in the in vitro study with MDCKII cells transfected with human MDR1 in this study (Fig. 3).
In conclusion, this study has showed that P-glycoprotein, of the major efflux transporters at the blood-brain barrier (P-gp and Bcrp), plays a key role in limiting the brain distribution of a novel MDM2 inhibitor, SAR405838. The distribution to the brain has been shown to be increased significantly in mice that are lacking P-glycoprotein compared with mice that have an intact P-gp at the BBB. Lack of P-gp did not influence the systemic disposition (clearance or volume of distribution) of SAR405838. Both NCA and compartmental analysis resulted in similar estimates of systemic pharmacokinetic parameters and metrics, and the compartmental BBB model provided additional insights into the rate and extent of the delivery of SAR405838 to the brain. The model-estimated tissue transfer rates out of the brain were significantly higher in the presence of P-gp than in the absence of P-gp, even though the tissue transfer rates into the brain were unchanged among genotypes. Based on our findings, it may still be of interest to examine the efficacy of brain penetrant MDM2 inhibitors in the GBM patient, as long as the limitations in delivery across an intact BBB can be overcome. Treatments for CNS tumors need to be able to penetrate the intact BBB to have maximal therapeutic efficacy especially for the treatment of infiltrative CNS tumors, such as GBM (Agarwal et al., 2011b). Even though targeting MDM2 is promising for the treatment of GBM (Wade et al., 2013), the ability of therapeutic agents to reach adequate concentration in CNS will limit the potential efficacy due to lack of BBB permeability (Kim et al., 2018b). Moreover, subtherapeutic concentrations in CNS due to heterogeneous BBB permeability may result in acquired drug resistance (Sacher et al., 2014; Jung et al., 2016; Kim et al., 2018a). Therefore, it is critical to understand the delivery of these agents to the brain and to find either a novel MDM2 inhibitor, which can penetrate the intact BBB, modify the structure of SAR405838 to avoid the active efflux by P-gp, or find an additional novel means to improve the delivery of MDM2 inhibitors through the BBB.
Acknowledgments
The authors thank Jim Fisher, Clinical Pharmacology Analytical Laboratory, University of Minnesota, for his support in the development of the LC-MS/MS assays.
Abbreviations
- AUC
area under the curve
- BBB
blood-brain barrier
- BCRP
breast cancer resistance protein
- CL
clearance
- CNS
central nervous system
- DA
distribution advantage
- fu
free (unbound) fraction
- FVB
Friend leukemia virus strain B
- GBM
glioblastoma
- Ke
elimination rate constant
- Kp
brain-to-plasma ratio
- Kpuu
brain unbound (free) brain-to-plasma ratio
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MDCKII
Madin-Darby canine kidney II
- MDM2
mouse double minute 2 homolog
- Mdr1
multi-drug resistance protein 1 (p-glycoprotein)
- MTT
mean transit time
- NCA
noncompartmental analysis
- N.S.
not significant
- P-gp
P-glycoprotein
- p53
tumor protein p53
- PKO
Mdr1a/b−/−
- RED
rapid equilibrium dialysis
- TKO
triple knockout
- WT
wild type
Authorship Contributions
Participated in research design: Kim, Sarkaria, Elmquist.
Conducted experiments: Kim, Laramy, Gampa, Parrish.
Performed data analysis: Kim, Laramy, Parrish, Brundage, Sarkaria, Elmquist.
Wrote or contributed to the writing of the manuscript: Kim, Brundage, Sarkaria, Elmquist.
Footnotes
This work was supported by the National Institutes of Health [Grants R01 CA138437, R01 NS077921, U54 CA210181, U01 CA227954, and P50 CA108960].
References
- Agarwal S, Hartz AM, Elmquist WF, Bauer B. (2011a) Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des 17:2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. (2011b) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. (2012) Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos 40:1164–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay CA. (1993) The mdm-2 oncogene can overcome wild-type p53 suppression of transformed cell growth. Mol Cell Biol 13:301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridén M, Ljungqvist H, Middleton B, Bredberg U, Hammarlund-Udenaes M. (2010) Improved measurement of drug exposure in the brain using drug-specific correction for residual blood. J Cereb Blood Flow Metab 30:150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainaut P, Hollstein M. (2000) p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 77:81–137. [DOI] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25:1737–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabeen I, Pleban K, Rinner U, Chiba P, Ecker GF. (2012) Structure-activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J Med Chem 55:3261–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, Bahleda R, Wagner AJ, Choy E, de Jonge MJ, et al. (2016) TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 7:12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS. (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338. [DOI] [PubMed] [Google Scholar]
- Kim M, Kizilbash SH, Laramy JK, Gampa G, Parrish KE, Sarkaria JN, Elmquist WF. (2018a) Barriers to effective drug treatment for brain metastases: a multifactorial problem in the delivery of precision medicine. Pharm Res 35:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ma DJ, Calligaris D, Zhang S, Feathers RW, Vaubel RA, Meaux I, Mladek AC, Parrish KE, Jin F, et al. (2018b) Efficacy of the MDM2 inhibitor SAR405838 in glioblastoma is limited by poor distribution across the blood-brain barrier. Mol Cancer Ther 17:1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. (2010) Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther 333:788–796. [DOI] [PubMed] [Google Scholar]
- Kong AN, Jusko WJ. (1988) Definitions and applications of mean transit and residence times in reference to the two-compartment mammillary plasma clearance model. J Pharm Sci 77:157–165. [DOI] [PubMed] [Google Scholar]
- Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. (2002) Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol 20:2943–2950. [DOI] [PubMed] [Google Scholar]
- Laramy JK, Kim M, Parrish KE, Sarkaria JN, Elmquist WF. (2018) Pharmacokinetic assessment of cooperative efflux of the multitargeted kinase inhibitor ponatinib across the blood-brain barrier. J Pharmacol Exp Ther 365:249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Yamazaki M. (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98. [DOI] [PubMed] [Google Scholar]
- Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, et al. (2005) Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther 313:1254–1262. [DOI] [PubMed] [Google Scholar]
- Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, Monshouwer M. (2009) Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos 37:787–793. [DOI] [PubMed] [Google Scholar]
- Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, et al. (2010) Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res 16:5664–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Wu HH, Dasgupta G. (2000) MDM2--master regulator of the p53 tumor suppressor protein. Gene 242:15–29. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245. [DOI] [PubMed] [Google Scholar]
- Pafundi DH, Laack NN, Youland RS, Parney IF, Lowe VJ, Giannini C, Kemp BJ, Grams MP, Morris JM, Hoover JM, et al. (2013) Biopsy validation of 18F-DOPA PET and biodistribution in gliomas for neurosurgical planning and radiotherapy target delineation: results of a prospective pilot study. Neuro-oncol 15:1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher AG, Jänne PA, Oxnard GR. (2014) Management of acquired resistance to epidermal growth factor receptor kinase inhibitors in patients with advanced non-small cell lung cancer. Cancer 120:2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane R, Mittapalli RK, Elmquist WF. (2013) Development and evaluation of a novel microemulsion formulation of elacridar to improve its bioavailability. J Pharm Sci 102:1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy JK, et al. (2018) Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-oncol 20:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. (2007) Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther 322:205–213. [DOI] [PubMed] [Google Scholar]
- Trainor GL. (2007) The importance of plasma protein binding in drug discovery. Expert Opin Drug Discov 2:51–64. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, Terasaki T. (2011) Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 117:333–345. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. (2000) Surfing the p53 network. Nature 408:307–310. [DOI] [PubMed] [Google Scholar]
- Wade M, Li YC, Wahl GM. (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barrière C, Stuckey JA, Meagher JL, Bai L, Liu L, et al. (2014) SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res 74:5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters NJ, Jones R, Williams G, Sohal B. (2008) Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci 97:4586–4595. [DOI] [PubMed] [Google Scholar]