

Abstract
Ischemic heart disease is a severe stress condition that causes extensive pathological alterations and triggers cardiac cell death. Accumulating evidence suggests that the unfolded protein response (UPR) is strongly induced by myocardial ischemia. The UPR is an evolutionarily conserved cellular response to cope with protein-folding stress, from yeast to mammals. Endoplasmic reticulum (ER) transmembrane sensors detect the accumulation of unfolded proteins and stimulate a signaling network to accommodate unfolded and misfolded proteins. Distinct mechanisms participate in the activation of three major signal pathways, viz. protein kinase RNA-like ER kinase, inositol-requiring protein 1, and activating transcription factor 6, to transiently suppress protein translation, enhance protein folding capacity of the ER, and augment ER-associated degradation to refold denatured proteins and restore cellular homeostasis. However, if the stress is severe and persistent, the UPR elicits inflammatory and apoptotic pathways to eliminate terminally affected cells. The ER is therefore recognized as a vitally important organelle that determines cell survival or death. Recent studies indicate the UPR plays critical roles in the pathophysiology of ischemic heart disease. The three signaling branches may elicit distinct but overlapping effects in cardiac response to ischemia. Here, we outline the findings and discuss the mechanisms of action and therapeutic potentials of the UPR in the treatment of ischemic heart disease.
Keywords: UPR, PERK, ATF6, IRE1, XBP1s, Ischemic heart disease, ER stress
1. Introduction
Ischemic heart disease is the leading cause of cardiovascular disease-related disability and death worldwide, which creates huge burden on the healthcare system and economy [1–3]. Despite extensive interests and mounting needs, our knowledge into the pathophysiology is still lagging behind. As a result, current care and cure are unable to arrest the progression and prevalence of ischemic heart disease.
Identification of better and more effective therapeutic approaches is not feasible without a deeper and more thorough understanding of the pathological mechanisms. Further, the present clinical situation of inability to treat ischemic heart disease suggests novel, additional pharmacological targets need to be discovered. Myocardial infarction creates sudden blockage of oxygen and nutrient supply to the myocardium. To survive this detrimental insult, cardiac myocytes undergo extensive remodeling, electrophysiologically, metabolically and structurally [4]. Persistent ischemia causes permanent damage in cardiac cells and renders them beyond rescue. On the other hand, timely and effective restoration of coronary blood flow by thrombolysis and/or percutaneous coronary intervention is the best approach to salvage myocardium from ischemia and improve clinical outcomes [5]. This so-called ischemia/reperfusion (I/R) however does not help without a price [6]. Numerous studies have shown that I/R per se leads to significant cardiac damage, in additional to ischemia [7,8], which may account for as much as 40% of the final infarction [6].
A growing body of evidence suggests that the unfolded protein response (UPR) is strongly activated in ischemic heart disease in humans and rodent models by various pathological events, such as overproduction of reactive oxygen species (ROS), inflammation, and metabolic derangement (Fig. 1) [9–13]. The UPR is an adaptive cellular process to accommodate protein-folding stress [14,15]. Upon activation, three signaling arms cooperate to restore cellular homeostasis, including protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6) (Fig. 2). However, during prolonged or overwhelming protein folding stress, the adaptation of UPR starts to fail and ER-initiated apoptosis ensues, which in turn, contributes to the development and progression of ischemic heart disease [16,17]. Here, we focus on the role and mechanisms of action of the UPR in myocardial infarction and I/R and explore potential therapeutic targets to tackle this devastating disease.
Fig. 1.
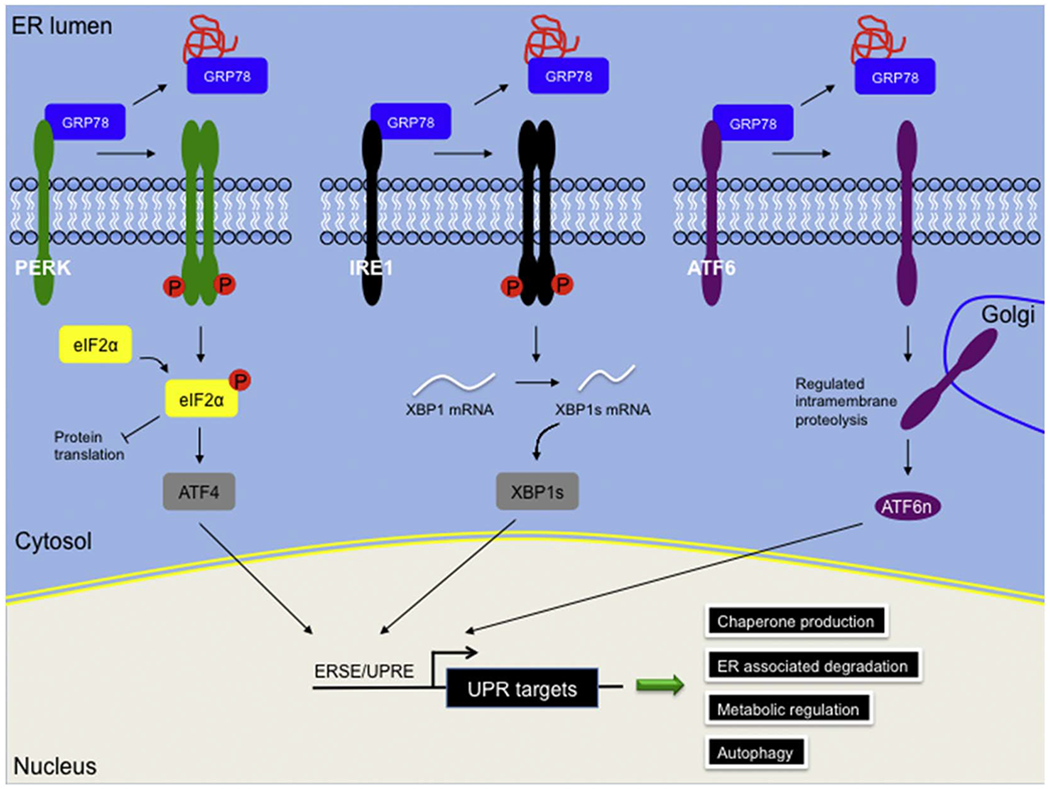
The mammalian unfolded protein response (UPR).
The mammalian UPR consists of three distinct but overlapping signaling branches. Under resting conditions, PERK, IRE1 and ATF6 are sequestered on the ER membrane by interacting with ER-resident chaperone GRP78. Accumulation of unfolded proteins leads to dissociation of GRP78 and activates the three downstream pathways via different mechanisms. Dimmerization and autophosphorylation of PERK stimulates phosphorylation of eIF2α, which on one hand transiently attenuates protein translation to create a “window-of-repair” for protein folding, and on the other hand, increases ATF4 translation. Dimerization and autophosphorylation of IRE1 enhances an endoribonuclease activity that cleaves a cryptic intron from downstream target XBP1. The spliced XBP1 (XBP1s) is a potent transcriptional factor targeting the ER stress responsive element (ERSE) / unfolded protein response element (UPRE) sites of numerous UPR genes. ATF6 is single transmembrane protein, which translocated to the Golgi from ER upon protein folding stress. ATF6 undergoes regulated intramembrane proteolysis on the Golgi membrane and the cytosolic N-terminus of nuclear ATF6 (ATF6n) acts as a strong transcriptional factor. Stimulation of the UPR leads to activation of various downstream signaling, such as chaperone production, ER-associated degradation, metabolic regulation and autophagy, which together aim to restore ER homeostasis.
Fig. 2.
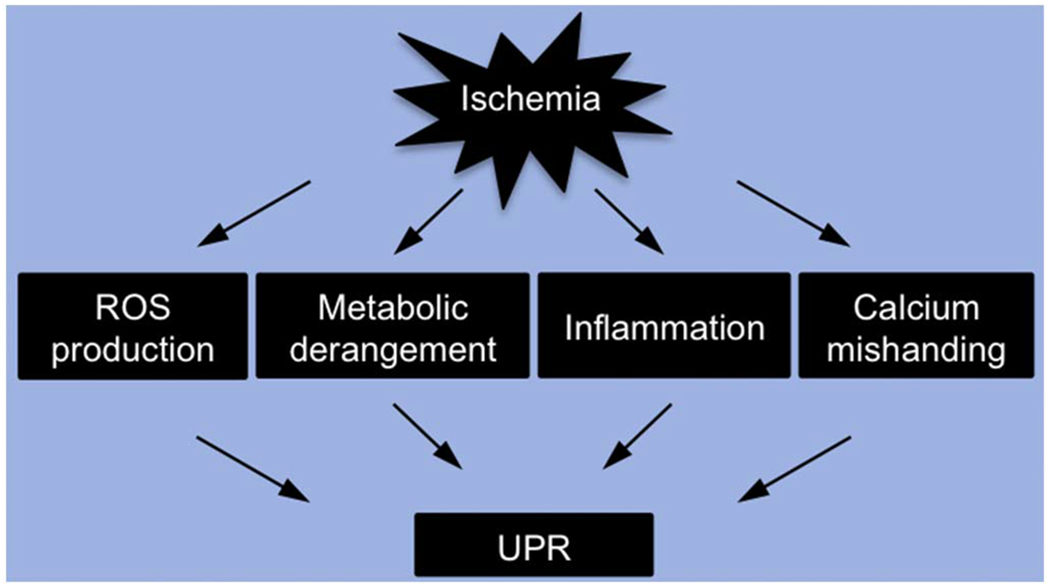
Activation of the UPR in ischemic heart disease.
Multiple events participate in the pathogenesis of ischemic heart disease, including overproduction of reactive oxygen species (ROS), metabolic derangement, inflammation, and calcium mishandling. Most, if not all, of these signaling pathways lead to perturbation of the ER homeostasis and induction of the UPR.
2. The UPR and ER molecular chaperones in ischemic heart disease
Glucose-regulated protein of 78 kDa (GRP78), also known as immunoglobulin heavy chain binding protein (BiP), is one of the most abundant molecular chaperones residing in the ER. GRP78 was originally discovered as a glucose-regulated target due to its upregulation by glucose deprivation in transformed cells [18]. Besides as a critical chaperone involved in protein folding, GRP78 also serves as a sensor and regulator of the UPR. At resting conditions, GRP78 binds the three signaling arms of the UPR and retains them on the ER membrane. In response to accumulation of misfolded proteins, however, GRP78 preferentially interacts with protein clients on the hydrophobic patches and releases the tethering with the UPR transducers. Liberated PERK undergoes autophosphorylation and activation, while ATF6 is translocated from ER to the Golgi for regulated intramembrane cleavage and maturation. As for IRE1, autophosphorylation leads to stimulation of an endoribonuclease activity, which acts on a downstream target mRNA X-box binding protein 1 (XBP1). After excision of a cryptic exon of 26 bp, the spliced XBP1 (XBP1s) becomes a potent transcriptional factor. The three signaling branches then transiently suppress protein synthesis, enhance ER protein-folding capacity, and augment ER-associated degradation, which together orchestrate to restore cellular homeostasis. Additionally, GRP78 is a bono fide target of the UPR [19]. XBP1s and activated ATF6 may form heterodimers and directly stimulate the transcription and translation of GRP78. In so doing, the UPR may be quenched after acute adaption, and chronic pathological activation of the UPR may be prevented.
Accumulating evidence has shown that GRP78 is upregulated in the heart under multiple cardiac pathological conditions [20] (Table 1). Using human heart samples, Ortega et al. found that GRP78 is induced at protein level in patients with either dilated or ischemic cardiomyopathy [21]. On the other hand, mouse hearts subjected to in vivo myocardial infarction exhibit increased GRP78 expression in cardiac myocytes near the infarct region but not in healthy cells in the remote area [10]. Additionally, Hardy and Raiter found that simulated ischemia for 4 h leads to upregulation of GRP78 in cultured cardiac myocytes [22]. Moreover, Shintani-Ishida et al. showed that early ischemic preconditioning increases myocardial GRP78 in a rat coronary artery occlusion model [23]. More importantly, transfection of GRP78 antisense oligonucleotides attenuates the preconditioning-mediated protection to ischemia, suggesting that GRP78 plays a critical role in ischemic preconditioning [23].
Table 1.
Diseases and defective genes.
| Pathway | Gene | Model/disease | Phenotype | Ref. |
|---|---|---|---|---|
| Protein folding | GRP78 | Cardiomyopathy, human | Upregulated | [21] |
| GRP78 | Myocardial infarction, mouse | Upregulated | [10] | |
| GRP78 | Simulated ischemia, NRVM | Upregulated | [22] | |
| GRP78 | Preconditioning, rat | Upregulated. Knockdown in NRVM dampens protection by preconditioning | [23] | |
| GRP94 | Knockout, mouse | Embryonic lethality; deficient ES cells do not different into cardiac cells | [39–40] | |
| Calreticulin | Knockout, mouse | Embryonic lethality; ventricular well thinning | [42] | |
| PERK | CHOP | Knockout, mouse | Reduced retinal ganglion cell death after retinal I/R | [46] |
| CHOP | Knockout, mouse | Reduced infarction in cardiac I/R | [17] | |
| CHOP | Atherosclerosis, human | Upregulated in unstable thin-cap atheroma | [47] | |
| CHOP | Knockout, mouse | Reduced proximal tubule damage in renal I/R | [48] | |
| CHOP | Cardiomyopathy, human | Upregulated | [50–52] | |
| CHOP | Knockout, mouse | Reduced pathological remodeling in pressure overload | [50] | |
| IRE | XBP1s | Cardiomyopathy, human | Reduced after implantation of left ventricular assist device | [58] |
| XBP1s | Cardiac-specific knockout, mouse | Elevated infarction in cardiac I/R | [58] | |
| XBP1s | Cardiac-specific overexpression, mouse | Reduced infarction in cardiac I/R | [58] | |
| XBP1s | Cardiac-specific overexpression, mouse | Reduced infarction in cerebral I/R | [59] | |
| IRE1 | Atherosclerosis, mouse | Inhibitor reduces plaque size | [65] | |
| ATF6 | ATF6 | Simulated ischemia, NRVM | Upregulated | [68] |
| ATF6 | Cardiac-specific overexpression, mouse | Reduced infarction in ex vivo cardiac I/R | [69] | |
| ATF6 | Cardiac-specific knockout, mouse | Elevated infarction in cardiac I/R | [70] | |
| ATF6 | Cardiac-specific overexpression, dominant negative mutant | Exacerbated response in mouse myocardial infarction | [71] | |
| ATF6 | ATF6 inhibitor, mouse | Exacerbated response in myocardial infarction | [71] |
The mechanisms of GRP78-mediated cardioprotection remain to be fully clarified. Most studies attribute this beneficial effect to the chaperone function, to ameliorate protein-folding stress and quench chronic pro-apoptotic signaling of the UPR [20,24]. Recently, emerging evidence suggests that upregulation of GRP78 in response to ER stress may trigger its translocation to cell surface and mediate a pro-survival signaling transduction pathway [25]. Studies have shown that GRP78 is localized on the cell surface, such as endothelial cells [26], macrophages [27], immortalized cell lines [28], and tumor cells [29–32]. Jacobsen et al. found that a human monoclonal antibody derived from the phage display library recognizes cell surface localized GRP78 in breast cancer cells [33]. Additionally, Pizzo and colleagues isolated autoantibodies from prostate cancer patients, which bind GRP78 on cell surface. Interestingly, the epitope on GRP78 for these autoantibodies is also recognized by activated α2-macroglobin (α2M*) [34]. Further studies show that the interaction between cell surface GRP78 and antibodies or α2M* elicits intracellular anti-apoptotic signaling [35]. In addition, using a phage display peptide library, Hardy et al. identified a 12-amino acids peptide that specifically binds cell surface GRP78 in endothelial cells [36]. This interaction leads to enhancement of angiogenesis and protection against limb ischemia [37]. Moreover, Hardy and Raiter show that AdoPep1, a GRP78-binding peptide of 12-amino acids derived from the metalloproteinase domain of ADAM15 (a disintegrin and metalloproteinase 15), can protect cardiac myocytes from ischemia-induced cell death at both in vitro and in vivo levels [22]. In aggregate, these results suggest that GRP78 may confer cardioprotective effects in the heart by both chaperone function and cell surface localization and pro-survival pathway activation.
ER is also the host of other protein folding chaperones and quality control system. Glucose-regulated protein of 94 kDa (GRP94) is the HSP90 (heat shock protein of 90 kDa) counterpart in the ER lumen. Like GRP78, GRP94 plays a critical role in ER protein folding [38]. Targeted disruption of GRP94 leads to embryonic lethality on day 7 of gestation [39]. While GRP94+/− embryonic stem (ES) cells does not affect the ER stress response, homozygous deletion in ES cells severely impairs differentiation to cardiac myocytes, highlighting an essential role in cardiogenesis [40]. Indeed, GRP94 is highly expressed in both atrial and ventricular myocytes in the developing heart [41]. However, the role of GRP94 in the heart under pathological conditions remains to be fully clarified. The other critical component of the protein quality control system in the ER is calnexin/calreticulin, which functions to ensure correct folding and assembly of glycoproteins. After N-glycan is transferred from donor to substrate glycoproteins, glucosidases I and II cooperate to remove the terminal two glucose molecules, and the exposed, third glucose is recognized by calnexin/calreticulin. ER protein of 57 kDa (ERp57), an integral component of the calnexin/calreticulin system and an oxidoreductase in the ER, catalyzes disulfide bond formation between ERp57 and substrates. When protein folding is complete, cleavage of the last glucose in N-glycan releases cargo proteins from calnexin/calreticulin. If folding is failed, UDP glucose: glycoprotein glucosyltransferase attaches a new glucose to the N-glycan and another round of calnexin/calreticulin cycle starts for further protein folding. Using a fluorescent reporter mouse model, Mesaeli et al. found that calreticulin is highly abundant in the developing heart [42]. Homozygous deletion of calreticulin shows marked decreases in ventricular wall thickness and defects in trabeculation, which may stem from impairments in calcium signaling [42].
2.1. The PERK pathway in ischemic heart disease
PERK is a transmembrane serine/threonine kinase activated by ER stress via dimerization and autophosphorylation, which leads to phosphorylation of downstream target eIF2α (eukaryotic initiation factor 2α) and global inhibition of translation [43]. The attenuation of translation causes decreases in protein synthesis and reduction in new ER folding clients, which provides additional time for repair in the ER. However, translation of ATF4 (activating transcription factor 4) is activated under this condition due to the existence of a positive-acting upstream open-reading frame (uORF) in the 5′ untranslated region (UTR) [44]. ATF4 in turn stimulates a downstream target CHOP (C/EBP homologous protein), which is expressed at a very low level under resting conditions. As a transcription factor, CHOP has been shown to regulate multiple apoptosis-related genes, including Bcl-2 (B cell lymphoma −2) and GADD34 (growth arrest and DNA damage inducible 34) [45].
Emerging evidence shows that CHOP exerts strong pro-apoptotic function in multiple ischemic conditions. Nashine et al. found that I/R in the eye leads to upregulation of the UPR in retinal ganglion cells. Consistently, deficiency of CHOP significantly improves cell survival and functional recovery in response to I/R [46]. Miyazaki et al. showed that cardiac I/R activates the phosphorylation of eIF2α and upregulates CHOP gene expression [17]. More importantly, CHOP knockout mice show reduction of myocardial inflammation and improvements in cardiac function against I/R. In addition, Myoishi et al. found CHOP-dependent pathway is activated in unstable plaques [47]. Knockdown of CHOP by siRNA decreases ER stress-dependent death of cultured coronary artery smooth muscle cells and THP-1 cells [47]. Similar findings have been observed in a renal I/R model [48]. On the other hand, pharmacological inhibition of the UPR by 4-PBA (4-phenylbutyric acid) confers strong cardioprotection against myocardial infarction, which is accompanied by significant reduction of CHOP expression [49]. Collectively, these results suggest that the pro-apoptotic role of CHOP under chronic ER stress is a universal phenomenon in response to various ischemic insults.
Consistently, CHOP expression is elevated in human heart failure of ischemic origin, along with other markers of the UPR [50,51]. Interestingly, CHOP is also stimulated in the heart of dilated cardiomyopathy in human [50–52]. In rodent models, thoracic aortic constriction (TAC) causes significant cardiac hypertrophy, pathological remodeling and heart failure, which are accompanied by augmentations of CHOP and other UPR markers [50,52]. Although the hypertensive cardiomyopathy is different compared with ischemic heart disease, hypoxia has been identified in pressure overload-induced heart failure [53]. It is therefore possible that the hypoxic response by TAC triggers the UPR and CHOP expression in the heart. In addition, cardiac myocyte hypertrophic growth involves new protein production, metabolic reprogramming, phospholipid biosynthesis and membrane expansion, most of which are potent inducers of the UPR [54–56]. Importantly, germline deletion of CHOP shows strong protection against heart failure progression in response to pressure overload [50]. Mechanistically, Caspase 12 expression is significantly diminished and pro-surviving proteins, including Bcl-2, are restored [50]. Moreover, expression of GADD34 is reduced in CHOP deficiency hearts after TAC. Since GADD34 is involved in translation stimulation and ER client expression [57], decrease of GADD34 may lead to a drop in ER load and improvements in cellular homeostasis. Collectively, these results indicate that chronic activation of the UPR under pathological conditions elicits cell death pathways and promotes progression of heart failure.
2.2. The IRE1/XBP1s pathway in ischemic heart disease
IRE1 is the most evolutionarily conserved ER stress transducer from yeast to mammals [14]. Accumulation of misfolded proteins in the ER stimulates dimerization and autophosphorylation of IRE1. The active IRE1 manifests an endoribonuclease activity toward multiple downstream targets. In mammalian cells, IRE1 recognizes and cleaves an atypical exon from XBP1, which creates frame-shift in the XBP1 mRNA. The resultant spliced XBP1 (XBP1s) is translated as a fully functional, larger basic leucine zipper transcriptional factor. Studies have shown that XBP1s stimulates genes involved in chaperone production, protein folding, ER-associated degradation and metabolic regulation.
Using a mouse I/R model, we have shown that reperfusion of as early as 5 minutes post coronary artery ligation stimulates XBP1s expression, which rises to approximately 6-fold by 4 h [58]. More importantly, XBP1s downstream target genes, such as GRP78, GRP94, start to increase 4 h after reperfusion. We have also examined the level of XBP1s in myocardial samples from patients with end-stage heart failure, and found that XBP1s mRNA level is reduced in human hearts following left ventricular assistant device mechanical support compared to patients prior to device implantation [58]. These results indicate that XBP1s is an acute, early response to I/R stress in the heart.
To further investigate the role of the IRE1/XBP1s pathway in the heart during I/R, we took advantage of the cardiac-specific XBP1 knockout (cKO) animal model [58]. We found that a significant increase in myocyte death and more profound pathological remodeling in cKO mice compared with either XBP1fl/fl or αMHC-Cre controls, suggesting that XBP1s induction is necessary to protect the heart from I/R injury in vivo. In addition, using a tetracycline inducible transgenic mouse model, we observed dramatic protection against reperfusion injury by XBP1s overexpression, with the infarct area of the transgenic group reduced by nearly 50%, suggesting that XBP1s expression is sufficient to protect the heart from I/R injury. Consistently, in a cerebral I/R injury model, Ibuki et al. found that overexpression of XBP1s in the brain suppresses cell death. Moreover, inhibition of XBP1s activation accelerates neuronal cell death in response to I/R [59]. Although the gain- and loss-of-function studies of XBP1s show consistent results in I/R, caution needs to be exercised when interpreting these results. In the transgenic mouse model, we only triggered short-term overexpression for 2 weeks [58]. However, prolonged induction of XBP1s in cardiac myocytes may cause persistent ER stress and detrimental consequences.
It has long been appreciated that chronic activation of the UPR leads to cell death, of which the IRE1 branch plays an essential role [60]. Pharmacological induction of the UPR by tunicamycin or thapsigargin stimulates the canonical UPR signaling, and also c-Jun N-terminal kinase (JNK) activation [61]. Importantly, the UPR-JNK phenotype is absent in IRE1α−/− mouse embryonic fibroblasts (MEFs), highlighting a critical role of IRE1 in this process. At the molecular level, the cytoplasmic domain of IRE1 recruits and interacts with tumor necrotic factor receptor associated factor 2 (TRAF2) and stimulates JNK phosphorylation and activation [61].
Further, emerging studies show that sustained IRE1 signaling under ER stress promotes cell death via other mechanisms [62]. Chronic activation of IRE1 triggers the regulated IRE1-dependent decay (RIDD) [63]. While early phase of RIDD may exert beneficial actions by reducing ER protein load, persistent RIDD targets ER chaperones for degradation, which contributes to impairment in ER folding capacity and cellular homeostasis [63]. In addition, prolonged activation of IRE1 may cause rapid decay of several microRNAs for Caspase 2 [64]. Therefore, Caspase 2 protein level is elevated and cell death ensues. To achieve these divergent actions, the cytoplasmic region of IRE1 may form distinct scaffold complexes with different cellular components, which are referred as UPRosome [60].
Along these lines, IRE1 has been implicated in pro-apoptotic and proinflammatory pathways in atherosclerosis models. A recent study by Tufanli et al. suggests that IRE1 regulates the expression of many proatherogenic genes, including several important cytokines and chemokines. This study reveals that at the in vivo level, IRE1 inhibitors lead to a significant decrease in hyperlipidemia-induced IL-1 (interleukin -1) and IL-18 (interleukin -18) production, lower T-helper type-1 immune responses, and reduced atherosclerotic plaque size without altering the plasma lipid profiles in ApoE−/− mice [65].
2.3. The ATF6 pathway in ischemic heart disease
ATF6 is a 670 amino acids single pass type 2 transmembrane protein. At basal conditions, ATF6 is localized on the ER membrane via interaction with GRP78. Upon induction of the UPR, dissociation of GRP78 leads to liberation of ATF6 and consequent translocation from ER to the Golgi, which requires a conserved region of amino acids 468–500 in the ER luminal domain [66]. In the Golgi, ATF6 is subjected to regulated intramembrane proteolysis by site-1 and site-2 proteases (S1P and S2P), which resembles the posttranslational processing of sterol regulatory element binding proteins (SREBPs) [67]. The other conserved domain of amino acids 550–640 is indispensible for S1P recognition and initiation of intramembrane cleavage. The soluble cytoplasmic region of 400 amino acids is then translocated to the nucleus. This nuclear ATF6 (ATF6n) possesses both DNA-binding and transactivation domains, which contributes to the upregulation of a host of ER chaperones to enhance the folding capacity of the ER and restore cellular homeostasis.
Ischemia in the heart leads to potent activation of the UPR at both in vitro [10,68] and in vivo levels [69,70]. Doroudgar et al. show that simulated ischemia for 20 h using cultured myocytes stimulates the ER-resident chaperone GRP78 expression and this effect is largely diminished when ATF6 is reduced by siRNA [68]. At the functional level, ATF6 knockdown causes more severe cell death upon ischemia in neonatal rat ventricular myocytes (NRVMs). These in vitro findings are further confirmed in an in vivo setting. Martindale et al. took a transgenic approach to overexpress ATF6n in an inducible manner in cardiac myocytes [69]. Upon induction by tamoxifen injection, the transgenic hearts show significant protection against global I/R as assessed by increases in recovery of ventricular developed pressure, decreases in cardiomyocyte apoptosis, and reduction in necrotic release of lactate dehydrogenase. Consistently, ATF6 inhibition by a specific inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride leads to deterioration of cardiac function after myocardial infarction, which is similar to a transgenic mouse model expressing the dominant negative mutant of ATF6 [71]. In aggregate, these results suggest that the ATF6 branch of the UPR confers strong cardioprotection against ischemic heart disease.
Multiple mechanisms have been proposed to explain the pro-surviving effects of ATF6. The active ATF6n may form a heterodimer with XBP1s and bind the ER stress responsive element (ERSE) in promoters of ER chaperones, including GRP78, GRP84, ERp72, etc. In addition, ATF6n may directly stimulate sarco/endoplasmic reticulum calcium ATPase 2 (SERCA2) and promote the restoration of calcium homeostasis [72]. Indeed, dominant negative mutation of ATF6 strongly diminishes calcium depletion-mediated upregulation of SERCA2, suggesting that ATF6 is required for maximal, optimal induction of SERCA2. Not surprisingly, a conserved ERSE site has been discovered in the SERCA2 promoter, indicating that SERCA2 is a direct transcriptional target of ATF6. Moreover, a recent study highlights a novel role of ATF6 in protecting the heart against I/R. Overproduction of ROS is at the central stage of reperfusion injury. Studies by Jin et al. show that overexpression of ATF6n is sufficient to protect NRVMs from H2O2-induced cell damage [70]. Knockdown of ATF6 leads to exacerbation of ROS production by simulated I/R and more profound cell death. Moreover, a comprehensive survey shows that ATF6 directly stimulates a group of antioxidant genes, including catalase and peroxiredoxin 5. Further analysis discovers 2 conserved ERSE sites in the catalase promoter, suggesting that catalase is a direct transcriptional target of ATF6. In addition, catalase induction by I/R is significantly diminished by ATF6 knockout. Ex vivo studies show that deficiency of ATF6 exacerbates recovery from I/R, and overexpression of either ATF6 or catalase leads to a strong rescue. Collectively, these findings suggest that ATF6 confers strong cardioprotection against I/R, which is mediated by multiple pro-surviving mechanisms.
2.4. The UPR and inflammation
Inflammation is a collection of inflammatory responses to tissue injury or infection, which plays critical roles in maintaining homeostasis at both cellular and organism levels. Numerous epidemiological, clinical, and experimental evidence has firmly established a causal effect of inflammation in disease initiation and progression, including cardiovascular disease [73]. Like the UPR, acute inflammatory response aims to repair and protect physiological function, whereas chronic stimulation of inflammation is implicated in pathogenesis under various conditions. Importantly, multiple stimuli of the UPR are potent inducers of inflammation, such as overproduction of ROS, calcium derangements, and metabolic dysregulation [74]. Indeed, previous studies have shown that the UPR may directly elicit inflammatory response [75]. ER is an oxidative environment for disulfide bond formation. The oxidative folding machinery, consisting of protein disulfide isomerase and ER oxidoreductase, catalyzes disulfide bond formation in client proteins and transmits electrons to oxygen, which is a major source of ER-derived ROS. Moreover, PERK phosphorylation and activation leads to global translational attenuation. As a consequence, the short-lived IκB kinase may not be efficiently regenerated, the inhibition of NFκB pathway is therefore diminished, and inflammatory response ensues [76]. Further, chronic activation of the UPR leads to formation of IRE1-TRAF2 complex in cytosol that induces JNK phosphorylation and inflammation [61]. On the other hand, multiple cytokines from the inflammatory response may directly stimulate the UPR [77,78]. Taken these findings together, the UPR and inflammation are intimately intercalated with prominent crosstalk, which may play critical roles in the pathogenesis of ischemic heart disease.
2.5. Temporal dynamics of the UPR
Activation of the UPR may activate both cytoprotective and pro-apoptotic signaling pathways, which together determine the final fate of the cell (Fig. 3). Studies have shown that the three branches are stimulated with different temporal dynamics. Administration of tunicamycin or thapsigargin in HEK293 cells leads to activation of all three signaling branches, albeit at different time course [79]. The IRE1 pathway is quickly stimulated, peaks at 4 h and then diminishes after 12 h of tunicamycin treatment. ATF6 follows a similar pattern. PERK, however, manifests persistent activation throughout the treatment time. Lin et al. propose that the temporal regulation may determine cell fate. Forced activation of IRE1 to prevent the quenching improves cell survival, which is associated with induction of XBP1s, not JNK. In contrast, chemical-genetic induction of PERK in cultured cells, without activating either IRE1 or ATF6, leads to exacerbation of cell death even in the absence of UPR stimuli [80]. Using a mouse I/R model, we have shown that the induction of XBP1s is acute, potent and transient, which is consistent with a temporal cytoprotective role of XBP1s in I/R injury [58]. Whereas most studies with acute induction of the UPR show beneficial effects in cell survival, sustained stimulation of all three branches may disrupt the feedback regulation, and lead to impairments in cellular homeostasis.
Fig. 3.
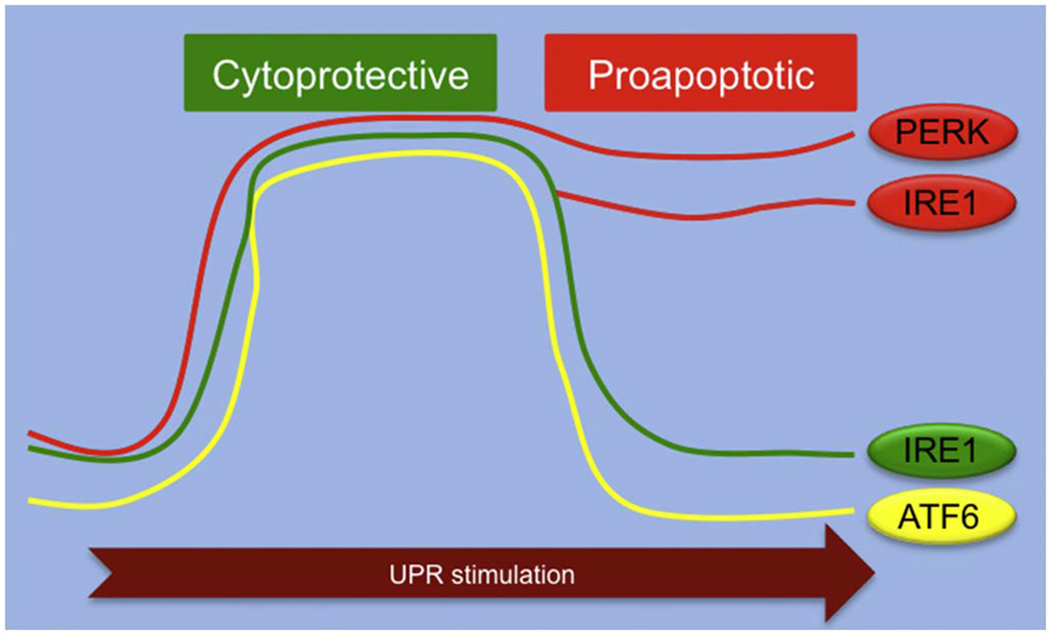
The temporal dynamics of the UPR.
The UPR stimuli typically activate all three branches. Both IRE1 and ATF6 are stimulated in an acute, transient manner, which is largely cytoprotective. The diminishment of IRE1 and ATF6 when facing long-lasting stress, combined with persistent activation of PERK, leads to augmentation of the pro-apoptotic signaling and cell death. Note that the pro-apoptotic property of the IRE1 signaling may be induced by irremediable protein folding stress, which contributes to cellular demise under conditions of pathological chronic activation of the UPR. The temporal dynamics of the UPR therefore plays a pivotal role in determining cell fate in response to various stresses.
Emerging evidence suggests that the IRE1 branch acts as a unique cell fate executor [62]. Whereas acute UPR induction stimulates the pro-surviving XBP1s signaling via IRE1 autophosphorylation and oligomerization, chronic protein-folding stress leads to the pro-apoptotic action of IRE1. Activation of IRE1 stimulates RIDD toward multiple RNA substrates, including ER folding cargos [63]. This action, together with PERK-mediated translation attenuation, contributes to reduction of ER folding load and helps restore cell homeostasis. Sustained activation of RIDD however degrades transcripts of ER chaperones, which may impair the ER folding capacity [63]. Moreover, recent studies indicate that RIDD targets multiple microRNAs for pro-apoptotic Caspase 2 [64]. As a consequence, sustained activation of IRE1 leads to upregulation of Caspase 2 and following cell death. Indeed, allosteric inhibition of the RIDD activity of IRE1 preserves ER stress-induced retinal degeneration and pancreatic β cell loss [81]. Taken together, these results suggest that IRE1 may function as a cell fate executor, controlled by dynamic cytosolic scaffolds with different components [60].
3. Conclusions and future perspectives
Despite extensive interests and mounting needs, ischemic heart disease remains a leading cause of morbidity and mortality {Benjamin, 2018 #1331}. Current therapies are insufficient to arrest disease initiation and progression. Discovering new treatment approaches requires a better and further understanding of the underlying pathophysiology. Molecular alterations occurring in cardiac ischemia, such as ROS overproduction, metabolic derangements, inflammation, etc., may lead to potent, acute induction of the UPR in the heart. Indeed, accumulating evidence points to a critical role of the UPR in the etiology of ischemic heart disease. Whereas the pro-surviving effects of the UPR may dominate in the early phase of ischemia, persistent activation of the UPR could cause adverse consequences. While we continue to gain insights about the versatile roles of the UPR in ischemic heart disease over the past decades, future work may be focused on dissecting the temporal dynamics of the UPR and fine-tuning individual signaling branches for therapeutic gain.
Acknowledgements
We thank members from the Wang lab for valuable discussions. This work was supported by grants from American Heart Association (14SDG18440002 and 17IRG33460191 to ZVW), American Diabetes Association (1-17-IBS-120 to ZVW), NIH (HL137723 to ZVW), and China Scholarship Council (201606270140 to XDW).
Footnotes
Disclosures
None.
References
- [1].Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Flaxman A, Murray CJ, Naghavi M, The global burden of ischemic heart disease in 1990 and 2010: the Global Burden of Disease 2010 study, Circulation 129 (14) (2014) 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Murray CJ, Naghavi M, Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: the Global Burden of Disease 2010 study, Circulation 129 (14) (2014) 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, Heart disease and stroke statistics-2017 update: a report from the American Heart Association, Circulation 135 (10) (2017) e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sobel BE, Salient biochemical features in ischemic myocardium, Circ. Res 35 (Suppl. 3) (1974) 173–181. [PubMed] [Google Scholar]
- [5].Lefer DJ, Marban E, Is Cardioprotection Dead, ? Circulation 136 (1) (2017) 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yellon DM, Hausenloy DJ, Myocardial reperfusion injury, N. Engl. J. Med 357 (11) (2007) 1121–1135. [DOI] [PubMed] [Google Scholar]
- [7].Hausenloy DJ, Yellon DM, Myocardial ischemia-reperfusion injury: a neglected therapeutic target, J. Clin. Invest 123 (1) (2013) 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Turer AT, Hill JA, Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy, Am. J. Cardiol 106 (3) (2010) 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani R, Silvestri F, Piro M, Liuzzo G, Biasucci LM, Mellone P, Feroce F, Capogrossi M, Baldi F, Fandrey J, Ehrmann M, Crea F, Abbate A, Baldi A, Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy, J. Am. Coll. Cardiol 50 (11) (2007) 1029–1037. [DOI] [PubMed] [Google Scholar]
- [10].Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC, Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes, Circ. Res 99 (3) (2006) 275–282. [DOI] [PubMed] [Google Scholar]
- [11].Szegezdi E, Duffy A, O’Mahoney ME, Logue SE, Mylotte LA, O’Brien T, Samali A, ER stress contributes to ischemia-induced cardiomyocyte apoptosis, Biochem. Biophys. Res. Commun 349 (4) (2006) 1406–1411. [DOI] [PubMed] [Google Scholar]
- [12].Nickson P, Toth A, Erhardt P, PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress, Cardiovasc. Res 73 (1) (2007) 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE, Activation of endoplasmic reticulum stress response during the development of ischemic heart disease, Am. J. Physiol. Heart Circ. Physiol 291 (3) (2006) H1411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Walter P, Ron D, The unfolded protein response: from stress pathway to homeostatic regulation, Science 334 (6059) (2011) 1081–1086. [DOI] [PubMed] [Google Scholar]
- [15].Wang M, Kaufman RJ, Protein misfolding in the endoplasmic reticulum as a conduit to human disease, Nature 529 (7586) (2016) 326–335. [DOI] [PubMed] [Google Scholar]
- [16].Rao RV, Ellerby HM, Bredesen DE, Coupling endoplasmic reticulum stress to the cell death program, Cell Death Differ. 11 (4) (2004) 372–380. [DOI] [PubMed] [Google Scholar]
- [17].Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y, C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation, Arterioscler. Thromb. Vasc. Biol 31 (5) (2011) 1124–1132. [DOI] [PubMed] [Google Scholar]
- [18].Lee AS, Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian-cells, Trends Biochem. Sci 12 (1) (1987) 20–23. [Google Scholar]
- [19].Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K, Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II, J. Biochem 136 (3) (2004) 343–350. [DOI] [PubMed] [Google Scholar]
- [20].Glembotski CC, Endoplasmic reticulum stress in the heart, Circ. Res 101 (10) (2007) 975–984. [DOI] [PubMed] [Google Scholar]
- [21].Ortega A, Rosello-Lleti E, Tarazon E, Molina-Navarro MM, Martinez-Dolz L, Gonzalez-Juanatey JR, Lago F, Montoro-Mateos JD, Salvador A, Rivera M, Portoles M, Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy, PLoS One 9 (9) (2014) e107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hardy B, Raiter A, Peptide-binding heat shock protein GRP78 protects cardiomyocytes from hypoxia-induced apoptosis, J. Mol. Med. (Berl.) 88 (11) (2010) 1157–1167. [DOI] [PubMed] [Google Scholar]
- [23].Shintani-Ishida K, Nakajima M, Uemura K, Yoshida K, Ischemic preconditioning protects cardiomyocytes against ischemic injury by inducing GRP78, Biochem. Biophys. Res. Commun 345 (4) (2006) 1600–1605. [DOI] [PubMed] [Google Scholar]
- [24].Ni M, Lee AS, ER chaperones in mammalian development and human diseases, FEBS Lett. 581 (19) (2007) 3641–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ni M, Zhang Y, Lee AS, Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting, Biochem. J 434 (2) (2011) 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Raiter A, Weiss C, Bechor Z, Ben-Dor I, Battler A, Kaplan B, Hardy B, Activation of GRP78 on endothelial cell membranes by an ADAM15-derived peptide induces angiogenesis, J. Vasc. Res 47 (5) (2010) 399–411. [DOI] [PubMed] [Google Scholar]
- [27].Misra UK, Sharma T, Pizzo SV, Ligation of cell surface-associated glucose-regulated protein 78 by receptor-recognized forms of alpha 2-macroglobulin: activation of p21-activated protein kinase-2-dependent signaling in murine peritoneal macrophages, J. Immunol 175 (4) (2005) 2525–2533. [DOI] [PubMed] [Google Scholar]
- [28].Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC, GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth, Mol. Cell. Biol 28 (2) (2008) 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, Pasqualini R, Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands, Cancer Cell 6 (3) (2004) 275–284. [DOI] [PubMed] [Google Scholar]
- [30].Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, Schneider A, Gubbins EF, Solomon L, Chen Z, Lesniewski R, Henkin J, Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78, Cancer Res. 65 (11) (2005) 4663–4672. [DOI] [PubMed] [Google Scholar]
- [31].Kim Y, Lillo AM, Steiniger SC, Liu Y, Ballatore C, Anichini A, Mortarini R, Kaufmann GF, Zhou B, Felding-Habermann B, Janda KD, Targeting heat shock proteins on cancer cells: selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand, Biochemistry 45 (31) (2006) 9434–9444. [DOI] [PubMed] [Google Scholar]
- [32].Misra UK, Deedwania R, Pizzo SV, Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK, J. Biol. Chem 280 (28) (2005) 26278–26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jakobsen CG, Rasmussen N, Laenkholm AV, Ditzel HJ, Phage display derived human monoclonal antibodies isolated by binding to the surface of live primary breast cancer cells recognize GRP78, Cancer Res. 67 (19) (2007) 9507–9517. [DOI] [PubMed] [Google Scholar]
- [34].Gonzalez-Gronow M, Cuchacovich M, Llanos C, Urzua C, Gawdi G, Pizzo SV, Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum, Cancer Res. 66 (23) (2006) 11424–11431. [DOI] [PubMed] [Google Scholar]
- [35].Misra UK, Payne S, Pizzo SV, Ligation of prostate cancer cell surface GRP78 activates a proproliferative and antiapoptotic feedback loop: a role for secreted prostate-specific antigen, J. Biol. Chem 286 (2) (2011) 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hardy B, Raiter A, Weiss C, Kaplan B, Tenenbaum A, Battler A, Angiogenesis induced by novel peptides selected from a phage display library by screening human vascular endothelial cells under different physiological conditions, Peptides 28 (3) (2007) 691–701. [DOI] [PubMed] [Google Scholar]
- [37].Hardy B, Battler A, Weiss C, Kudasi O, Raiter A, Therapeutic angiogenesis of mouse hind limb ischemia by novel peptide activating GRP78 receptor on endothelial cells, Biochem. Pharmacol 75 (4) (2008) 891–899. [DOI] [PubMed] [Google Scholar]
- [38].Zhu G, Lee AS, Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis, J. Cell. Physiol 230 (7) (2015) 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wanderling S, Simen BB, Ostrovsky O, Ahmed NT, Vogen SM, Gidalevitz T, Argon Y, GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion, Mol. Biol. Cell 18 (10) (2007) 3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mao C, Wang M, Luo B, Wey S, Dong D, Wesselschmidt R, Rawlings S, Lee AS, Targeted mutation of the mouse Grp94 gene disrupts development and perturbs endoplasmic reticulum stress signaling, PLoS One 5 (5) (2010) e10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Barnes JA, Smoak IW, Immunolocalization and heart levels of GRP94 in the mouse during post-implantation development, Anat. Embryol. (Berl.) 196 (4) (1997) 335–341. [DOI] [PubMed] [Google Scholar]
- [42].Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH, Michalak M, Calreticulin is essential for cardiac development, J. Cell Biol 144 (5) (1999) 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC, Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control, Mol. Cell. Biol 18 (12) (1998) 7499–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vattem KM, Wek RC, Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells, Proc. Natl. Acad. Sci. U. S. A 101 (31) (2004) 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li Y, Guo Y, Tang J, Jiang J, Chen Z, New insights into the roles of CHOP-induced apoptosis in ER stress, Acta Biochim. Biophys. Sin 46 (8) (2014) 629–640. [DOI] [PubMed] [Google Scholar]
- [46].Nashine S, Liu Y, Kim BJ, Clark AF, Pang IH, Role of C/EBP homologous protein in retinal ganglion cell death after ischemia/reperfusion injury, Invest. Ophthalmol. Vis. Sci 56 (1) (2014) 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M, Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome, Circulation 116 (11) (2007) 1226–1233. [DOI] [PubMed] [Google Scholar]
- [48].Chen BL, Sheu ML, Tsai KS, Lan KC, Guan SS, Wu CT, Chen LP, Hung KY, Huang JW, Chiang CK, Liu SH, CCAAT-enhancer-binding protein homologous protein deficiency attenuates oxidative stress and renal ischemia-reperfusion injury, Antioxid. Redox Signal 23 (15) (2015) 1233–1245. [DOI] [PubMed] [Google Scholar]
- [49].Luo T, Kim JK, Chen B, Abdel-Latif A, Kitakaze M, Yan L, Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis, Chem. Biol. Interact 225 (2015) 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, Asanuma H, Asakura M, Takashima S, Komuro I, Kitakaze M, Minamino T, Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload, Circulation 122 (4) (2010) 361–369. [DOI] [PubMed] [Google Scholar]
- [51].Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, Yamazaki S, Asano Y, Okada K, Tsukamoto O, Sanada S, Asanuma H, Asakura M, Takashima S, Kitakaze M, Komuro I, X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes, J. Mol. Cell. Cardiol 48 (6) (2010) 1280–1289. [DOI] [PubMed] [Google Scholar]
- [52].Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M, Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis, Circulation 110 (6) (2004) 705–712. [DOI] [PubMed] [Google Scholar]
- [53].Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W, Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy, Cell Metab. 9 (6) (2009) 512–524. [DOI] [PubMed] [Google Scholar]
- [54].Heineke J, Molkentin JD, Regulation of cardiac hypertrophy by intracellular signalling pathways, Nat. Rev. Mol. Cell Biol 7 (8) (2006) 589–600. [DOI] [PubMed] [Google Scholar]
- [55].Taegtmeyer H, Stanley WC, Too much or not enough of a good thing? Cardiac glucolipotoxicity versus lipoprotection, J. Mol. Cell. Cardiol 50 (1) (2011) 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kolwicz SC Jr., Purohit S, Tian R, Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes, Circ. Res 113 (5) (2013) 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D, CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum, Genes Dev. 18 (24) (2004) 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE, Hill JA, Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway, Cell 156 (6) (2014) 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ibuki T, Yamasaki Y, Mizuguchi H, Sokabe M, Protective effects of XBP1 against oxygen and glucose deprivation/reoxygenation injury in rat primary hippocampal neurons, Neurosci. Lett 518 (1) (2012) 45–48. [DOI] [PubMed] [Google Scholar]
- [60].Hetz C, Martinon F, Rodriguez D, Glimcher LH, The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha, Physiol. Rev 91 (4) (2011) 1219–1243. [DOI] [PubMed] [Google Scholar]
- [61].Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D, Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1, Science 287 (5453) (2000) 664–666. [DOI] [PubMed] [Google Scholar]
- [62].Chen Y, Brandizzi F, IRE1: ER stress sensor and cell fate executor, Trends Cell Biol. 23 (11) (2013) 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR, IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates, Cell 138 (3) (2009) 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, Oakes SA, IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2, Science 338 (6108) (2012) 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tufanli O, Telkoparan Akillilar P, Acosta-Alvear D, Kocaturk B, Onat UI, Hamid SM, Cimen I, Walter P, Weber C, Erbay E, Targeting IRE1 with small molecules counteracts progression of atherosclerosis, Proc. Natl. Acad. Sci. U. S. A 114 (8) (2017) E1395–e1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen X, Shen J, Prywes R, The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi, J. Biol. Chem 277 (15) (2002) 13045–13052. [DOI] [PubMed] [Google Scholar]
- [67].Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL, ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs, Mol. Cell 6 (6) (2000) 1355–1364. [DOI] [PubMed] [Google Scholar]
- [68].Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC, Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response, J. Biol. Chem 284 (43) (2009) 29735–29745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC, Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6, Circ. Res 98 (9) (2006) 1186–1193. [DOI] [PubMed] [Google Scholar]
- [70].Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC, ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart, Circ. Res 120 (5) (2017) 862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Toko H, Takahashi H, Kayama Y, Okada S, Minamino T, Terasaki F, Kitaura Y, Komuro I, ATF6 is important under both pathological and physiological states in the heart, J. Mol. Cell. Cardiol 49 (1) (2010) 113–120. [DOI] [PubMed] [Google Scholar]
- [72].Thuerauf DJ, Hoover H, Meller J, Hernandez J, Su L, Andrews C, Dillmann WH, McDonough PM, Glembotski CC, Sarco/endoplasmic reticulum calcium ATPase-2 expression is regulated by ATF6 during the endoplasmic reticulum stress response: intracellular signaling of calcium stress in a cardiac myocyte model system, J. Biol. Chem 276 (51) (2001) 48309–48317. [DOI] [PubMed] [Google Scholar]
- [73].Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, Heart disease and stroke statistics-2018 update: a report from the American Heart Association, Circulation (2018), 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- [74].Zhang K, Kaufman RJ, From endoplasmic-reticulum stress to the inflammatory response, Nature 454 (7203) (2008) 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang C, Syed TW, Liu R, Yu J, Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease, Front. Cardiovasc. Med 4 (2017) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D, Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2, Mol. Cell. Biol 24 (23) (2004) 10161–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ, Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response, Cell 124 (3) (2006) 587–599. [DOI] [PubMed] [Google Scholar]
- [78].Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H, Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha, J. Biol. Chem 280 (40) (2005) 33917–33925. [DOI] [PubMed] [Google Scholar]
- [79].Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P, IRE1 signaling affects cell fate during the unfolded protein response, Science 318 (5852) (2007) 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lin JH, Li H, Zhang Y, Ron D, Walter P, Divergent effects of PERK and IRE1 signaling on cell viability, PLoS One 4 (1) (2009) e4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schurer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR, Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress, Cell 158 (3) (2014) 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]