

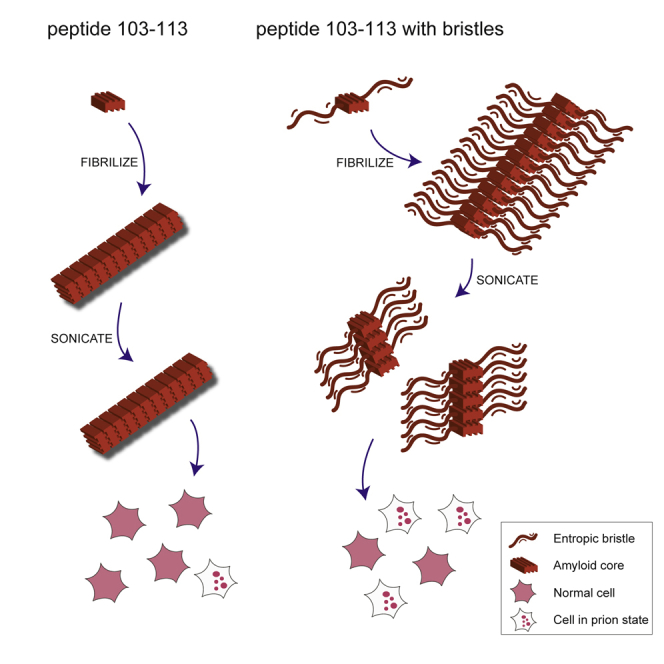
Summary
Prions of lower eukaryotes are self-templating protein aggregates with cores formed by parallel in-register beta strands. Short aggregation-prone glutamine (Q)- and asparagine (N)-rich regions embedded in longer disordered domains have been proposed to act as nucleation sites that initiate refolding of soluble prion proteins into highly ordered fibrils, termed amyloid. We demonstrate that a short Q/N-rich peptide corresponding to a proposed nucleation site in the prototype Saccharomyces cerevisiae prion protein Sup35 is sufficient to induce infectious cytosolic prions in mouse neuroblastoma cells ectopically expressing the soluble Sup35 NM prion domain. Embedding this nucleating core in a non-native N-rich sequence that does not form amyloid but acts as an entropic bristle quadruples seeding efficiency. Our data suggest that large disordered sequences flanking an aggregation core in prion proteins act as not only solubilizers of the monomeric protein but also breakers of the formed amyloid fibrils, enhancing infectivity of the prion seeds.
Keywords: prion, Sup35, entropic bristle, amyloid, fibril, brittle
Graphical Abstract
Highlights
-
•
A short peptide derived from Sup35 (p103–113) forms rigid amyloid fibrils
-
•
p103–113 fibrils can induce infectious Sup35 NM prions in mammalian cells
-
•
Embedding p103–113 in an N-rich sequence increases fibril brittleness
-
•
Increased fibril brittleness enhances prion-inducing capacity
A protein aggregate can contain infective properties, prions being the prime example. Michiels et al. show that these infective properties are encoded in the specific amino acid sequence flanking the aggregation core of a protein. These so-called entropic bristle sequences drive fiber brittleness, a crucial feature for amyloid infectivity.
Introduction
Protein aggregation propensity is a universal feature of polypeptides that is mediated by short linear aggregation-prone regions (APRs), generally between 5 and 15 residues in length, that have the intrinsic propensity to self-associate by beta strand interactions (Rousseau et al., 2006, Goldschmidt et al., 2010). These beta strand interactions form a nucleus that can grow into large amyloid-like beta sheeted protein inclusions by edge-strand addition of more APRs. Although recent cryoelectron microscopy high-resolution structures of mature amyloid fibrils demonstrated that the sequences surrounding the APRs are involved in stabilizing the final amyloid structure (Fitzpatrick et al., 2017), the necessary and sufficient role of APRs in driving protein aggregation has been amply demonstrated (Giasson et al., 2001, Krebs et al., 2004). Grafting APRs of known amyloidogenic proteins onto proteins that do not aggregate by themselves creates chimeras that recapitulate both the aggregation propensity and aggregate morphology of the donor protein (Ventura et al., 2004, Teng and Eisenberg, 2009). Conversely, it has also been shown that the introduction of point mutations that abolish the aggregation propensity of an APR is sufficient to reduce the aggregation propensity of the entire protein (Ganesan et al., 2016, Marshall et al., 2016). Finally, short peptides solely coding for such APRs are able to induce the aggregation of a full-length protein comprising that APR (Gallardo et al., 2016, Khodaparast et al., 2018, Betti et al., 2016). APRs generally only present a danger for aggregation in situations where proteins are partially or totally unfolded, such as during protein translation or translocation, under situations of physiological stress, or due to mutations that destabilize the native conformation. Indeed, APRs are generally buried in the hydrophobic core of globular proteins, where they are protected from aggregation by stabilizing native interactions. Solvent-exposed APRs in native proteins are generally APRs contributing to protein interaction interfaces (where again they are buried) or catalytic sites (where they are stabilized by ligand interaction).
The amyloid form of a very small subset of proteins, called prions (for protein infectious particle), also exhibits infectious properties, enabling it to impose its conformational state onto natively folded or unfolded copies of the same protein. Prions spread their amyloid state from cell to cell and even individual to individual. The only bona fide prion in mammals is the misfolded and aggregated prion protein PrP that causes transmissible spongiform encephalopathies. In lower eukaryotes, on the other hand, a growing number of diverse proteins can adopt self-templating protein conformations, thereby inducing heritable traits in their hosts (Crow and Li, 2011, Liebman and Chernoff, 2012, Wickner, 2016). The prototype of prion proteins in lower eukaryotes is the translation termination factor Sup35 of Saccharomyces cerevisiae (Uptain and Lindquist, 2002). Adoption of its prion conformation causes its loss of function, resulting in stop codon read-through and changes in the metabolic phenotype of the yeast host. Prion properties are governed by the glutamine (Q)- and asparagine (N)-rich amino-terminal prion domain N of Sup35 (DePace et al., 1998, Kushnirov et al., 2000). The middle domain (M) is highly charged and increases solubility of the protein, while translation termination is mediated by the carboxy-terminal (C) domain (Ter-Avanesyan et al., 1993, Glover et al., 1997, Liu et al., 2002). Recombinant NM protein rapidly forms amyloid fibrils in vitro, a process that can be greatly accelerated by addition of preformed NM amyloid seeds (Serio et al., 2000). Amyloid fibrils composed of recombinant NM are able to induce the prion state in Saccharomyces cerevisiae (Tanaka et al., 2004, King and Diaz-Avalos, 2004) and self-sustained NM aggregates when NM is ectopically expressed in mammalian cells (Krammer et al., 2009, Hofmann et al., 2013, Liu et al., 2016).
It remains poorly understood what it takes to render a universal property like amyloid aggregation into prion behavior, which requires the formation of brittle aggregates so that they can break into seeds that can spread and convert more native protein (Tanaka et al., 2006). This question is highly relevant, as the definition of a prion has recently been challenged by the discovery that several proteins that undergo pathogenic amyloid formation in human diseases display several characteristics that were thought to be the exclusive domain of prions. These so-called prionoids (Aguzzi and Lakkaraju, 2016), for example composed of tau (Polymenidou and Cleveland, 2012, Jucker and Walker, 2013, Holmes et al., 2014, Clavaguera et al., 2014, Spires-Jones and Hyman, 2014, Nisbet et al., 2015) or alpha-synuclein (Hansen et al., 2011, Polymenidou and Cleveland, 2012, Freundt et al., 2012, Herva and Spillantini, 2015, Reyes et al., 2015), have been shown to spread in a prion-like manner from cell to cell. Intercellular protein aggregate dissemination is thought to underlie the typical spreading pattern of protein deposition during disease progression in neurodegenerative diseases.
The peculiar sequence composition of several identified prion domains present in proteins of Saccharomyces cerevisiae has led to the proposal that the aggregation mechanism of prion(oid)s might fundamentally differ from the typical mechanism of nucleation by short APRs. Instead, prion formation has been proposed to be driven by a diffuse nucleus spanning long polypeptide segments up to 100 amino acids (Toombs et al., 2012, Lancaster et al., 2014). However, an alternative possibility is that, in addition to a nucleating core, the sequence context of these APRs in prion proteins somehow provides the additional properties required for the formation of a full-blown prion. As mentioned above, the prion domains found in Saccharomyces cerevisiae exhibit low sequence complexity, and their composition is heavily biased toward polar amino acids that strongly enhance protein solubility. In fact, it has been shown that such sequences can act as entropic bristles, thereby controlling the aggregation propensity and solubility of even highly aggregation-prone sequences such as the Alzheimer beta peptide (Finder et al., 2010).
We set out to create a synthetic prion seed coding for a short APR using the Sup35 model system. Our data indicate that aggregates formed by this 10-amino-acid peptide fragment are sufficient to induce the prion state in mammalian neuroblastoma cells ectopically expressing Sup35 NM. However, embedding this nucleating core in a non-native N-rich sequence, known to act as an entropic bristle, greatly enhances seeding efficiency. Aggregates formed by the bristled peptide resemble those formed by the full-length Sup35 NM protein and readily break into smaller seeds upon sonication, in contrast to peptides comprising the APR alone, which organize into rigid fibrils. Our bristled peptide mimics the Sup35 NM case and highlights the importance of a nucleating core embedded into a larger disordered region to obtain archetypical prion properties. It suggests that in addition to the reduced aggregation propensity of the monomeric protein, the large disordered regions in prions also increase the brittleness of the fibrils, leading to higher infectivity.
Results
Fibrils Composed of a Peptide Encompassing Amino Acid Residues 103–113 of Sup35 Accelerate NM Fibril Formation In Vitro
To test if amyloids formed by short peptides are able to seed Sup35 NM aggregation, 10 peptides were designed according to the Sup35 prion domain N. Peptides correspond to short sequences within the amino-terminal Q/N-rich region (QNR), the oligopeptide repeat region (OPR), and the carboxy-terminal N region (CTN) (Figure 1A; Table S1) and had previously been shown to form amyloid fibrils at pH 7.5 and 150 mM NaCl (Maurer-Stroh et al., 2010). Under our experimental conditions, all peptides except peptide p127–137 spanning the CTN formed amyloid fibrils, as confirmed by transmission electron microscopy (TEM) (Figure 1B) and thioflavin T (ThT) fluorescence (Figure 1C) after 6 days of rotation. To test if peptide fibrils decrease the lag time of NM fibril formation in vitro, Sup35 NM fibrillization assays were performed in the presence or absence of peptide seeds (Figure 1D). Unseeded Sup35 NM fibrillized with a lag time of ∼7 h, and addition of 2% (v/v) of preformed Sup35 NM fibrils abolished the lag time completely. Interestingly, most peptide fibrils did not affect Sup35 NM fibrillization kinetics. A significant decrease in lag time was observed for a peptide encompassing amino acid residues 103–113 of Sup35 (p103–113) (Figure 1E). Of note, p103–113 corresponds to a region in Sup35 previously identified to mediate first intermolecular contacts between Sup35 monomers during de novo amyloid assembly in vitro (Krishnan and Lindquist, 2005, Tessier and Lindquist, 2007).
Figure 1.

Sup35 Peptide p103–113 Fibrils Accelerate Sup35 NM Aggregation In Vitro
(A) Schematic diagram of functional domains N and M of Sup35. The regions corresponding to the synthetic peptides are marked. QNR, amino-terminal Q/N-rich region; OPR, oligopeptide repeat region; CTN carboxy-terminal N region.
(B) Synthetic peptides, rotated at 1 mM for 6 days, as observed by transmission electron microscopy (TEM). Indents represent higher magnification of the same image. p127–p137 aggregates were not observed by TEM and hence are not shown.
(C) ThT fluorescence of synthetic peptides rotated at 1 mM concentration for 6 days. Mean values and standard deviation (SD) of three independent replicates are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001, and ∗∗∗∗p < 0.00001 (unpaired t test [every condition compared to vehicle control]).
(D) ThT fluorescence of recombinant Sup35 NM (10 μM) incubated without (gray) or with 2% (v/v) preformed, sonicated fibrils (red). Three independent replicates are shown.
(E) Mean lag time and SD of three independent repeats of the data in (D). ∗p < 0.05 and ∗∗∗p < 0.0001 (unpaired t test [every condition compared to vehicle control]).
Induction of Cytosolic Sup35 NM Aggregation by Synthetic p103–113 Fibrils
Peptide fibrils produced in vitro were tested for their prion-inducing activity in mammalian cells. In the mouse neuroblastoma cell line N2a, the Sup35 NM domain fused to a carboxy-terminal GFP tag remains soluble (NM-GFPsol) when expressed in the cytosol (Hofmann et al., 2013). Upon addition of recombinant preformed Sup35 NM fibrils to the cell culture medium, fibrils gain access to the cytosol, where they induce self-perpetuating NM-GFP aggregates that vertically transmit to progeny cells and horizontally transmit to bystander cells (Krammer et al., 2009, Hofmann et al., 2013, Liu et al., 2016). Peptides fibrillized in vitro for 6 d or Sup35 NM fibrils were added to N2a NM-GFPsol cells for 24 h prior to image acquisition and analysis using an automated confocal microscope (Figure 2A). Interestingly, mainly fibrillized p103–113 induced NM-GFP aggregation in recipient cells, albeit at much lower rates compared to Sup35 NM fibrils (Figure 2B). Automated image analysis also demonstrated very low induction rates for some other peptides. Among them, p16–26 and p46–56 showed consistently positive induction rates. However, visual inspection of all captured images revealed that the few cells identified as positives exhibited condensed cytoplasm and thus represent false positives (Figure 2C). No induction was observed when cells were exposed to soluble peptides (Figure S1A). Altered parameters such as fibrillization time, concentrations during fibril assembly, and fibrillized peptide concentrations in the cell culture medium consistently led to highest induction rates for p103–113 (Figures S1B–S1D). Peptides p16–26 and p46–56 were tested together with all the altered parameters, and no induction was observed (Figures S1B–S1D). Thus, independent of the fibrillization protocol, only p103–113 repeatedly induced NM-GFPagg in recipient cells, consistent with the in vitro results described above. This is in line with our recent finding demonstrating that amino acid residues 98–123 serve as a nucleation core in cytosolically expressed Sup35 NM that drives prion induction in mammalian N2a cells (Duernberger et al., 2018). Importantly, none of the fibrillized peptides used in this study show cytotoxic effects in mouse neuroblastoma cells (Figure S2).
Figure 2.

Fibrillized Peptide p103–113 Induces NM-GFP Aggregation in Mouse Neuroblastoma Cells
(A) Schematic diagram of the workflow for the peptide aggregate induction assay adapted to 96-well-plate format for automated microscopy image analysis with CellVoyager 6000. N2a cells stably expressing soluble NM-GFP (N2a NM-GFPsol) were incubated with fibrillized peptides or Sup35 NM fibrils for 24 h. Fixed cells were subjected to automated image acquisition and analysis.
(B) Induction of NM-GFP aggregation in recipient N2a NM-GFPsol cells. 1 mM peptides were fibrillized for 6 days, sonicated, and subsequently added to N2a NM-GFPsol cells at a final concentration of 20 μM (monomer concentration). Shown is the mean ± SD (n = 9, three independent experiments). ∗∗p < 0.01 and ∗∗∗p < 0.0001 (one-way ANOVA).
(C) Confocal microscopy images of N2a NM-GFPsol cells exposed to peptide fibrils. Arrow indicates NM-GFP aggregates. Scale bar, 5 μm.
See also Figures S1 and S2.
p103–113-Induced NM-GFP Aggregates Exhibit Prion Characteristics
The criteria for a bona fide prion in mammalian cells include inducible aggregation, self-perpetuation of the aggregated state, inheritance of aggregates by progeny, and dissemination of aggregates to bystander cells (Ghaemmaghami et al., 2007, Grassmann et al., 2013, Krauss and Vorberg, 2013). To test if the p103–113-induced NM-GFP aggregates were stably maintained upon cell division, recipient cells were exposed to 30 μM (monomer equivalent) sonicated p103–113 fibrils for 2 days, and NM-GFP cells with induced aggregates were cultured further to isolate cell clones persistently propagating NM-GFP aggregates. Confocal microscopy revealed that NM-GFP aggregates were still present after 1-week cultivation, suggesting that at least a subset of the induced NM-GFP aggregates was inherited by daughter cells (Figure 3A). Moreover, isolated clones (Figure 3B) of N2a NM-GFP cells previously exposed to p103–113 fibrils still maintained NM-GFP aggregates, confirming that p103–113 fibrils are able to seed self-propagating NM-GFP aggregates (Figure 3C). To assess the infectious properties of induced protein aggregates, we selected two clones containing several small punctate aggregates and cocultured them with recipient N2a cells expressing soluble NM carboxy-terminally tagged with a hemagglutinin (HA) antibody epitope (NM-HAsol) (Krammer et al., 2009) (Figure 3B). NM-HA aggregate induction was observed for both donor clones (Figure 3D). Live-cell imaging of N2a NM-GFP cell clone 2 harboring NM-GFP aggregates (NM-GFPagg) cocultured with N2a cells stably expressing soluble mCherry-tagged NM confirmed induction of aggregates in NM-mCherrysol cells and subsequent propagation of the induced NM-mCherry aggregates by progeny (Figure 3E; Video S1). We conclude that a short amyloidogenic peptide (p103–113) targeting a predicted nucleation region is able to induce self-perpetuating NM prions in N2a cells.
Figure 3.

NM-GFP Aggregates Induced by Fibrillized Peptide p103–113 Exhibit Prion Properties
(A) Confocal microscopy analysis of N2a NM-GFPsol cells 2 days after exposure to fibrillized peptide p103–113. Scale bar, 5 μm.
(B) Schematic diagram of the workflow for isolating N2a NM-GFP cell clones harboring NM-GFP aggregates induced by p103–113 and subsequent coculture. Isolated clones were cocultured with recipient N2a NM-HAsol cells for 24 h to study the intercellular aggregate transmission efficiency.
(C) Presence of NM-GFP aggregates in cell clones. The N2a NM-GFPagg cell clones 1 and 2 contain several small NM-GFP aggregates. Nuclei were stained with Hoechst (blue). Scale bar, 5 μm.
(D) N2a NM-GFPagg donor clones and recipient N2a NM-HAsol cells were cocultured for 24 h and subsequently assessed for the percentage of N2a NM-HA-expressing cells with induced NM-HA aggregates. Shown is the mean ± SD (n = 3). No aggregates were observed in NM-HAsol cells that were cultured alone.
(E) Donor N2a NM-GFPagg clone 2 was cocultured with recipient N2a NM-mCherrysol cells. Aggregate induction was monitored by time-lapse microscopy for 4 h after co-plating. Images from time points 20–160 min are shown. Arrows indicate aggregates. Scale bar, 5 μm.
See also Video S1.
Coculture of N2a NM-GFPagg clone 2 and N2a NM-mCherrysol. Cells were analyzed by time-lapse microscopy. Recording was initiated after 4 h in coculture.
Embedding p103–113 in a Non-native N-rich Sequence Affects Fibril Morphology, Structure, and Brittleness
The foregoing results demonstrated that synthetic p103–113 fibrils are able to induce cytosolic NM-GFP prions in mammalian cells. However, we observed striking differences in the induction rates using peptide fibrils compared to fibrils composed of full-length NM. Morphologically, p103–113 fibrils appear microcrystalline in nature compared to the elongated Sup35 NM fibrils (Figure 4A). Sonication of our Sup35 NM fibrils drastically increased the number of short aggregates, while the same treatment left p103–113 fibrils relatively unaffected (Figure 4A). This results in p103–113 seeds that are ∼10-fold larger than the small aggregates formed by Sup35 NM (Figure 4B). It has been shown that the size distribution and particle concentration of sonicated Sup35 NM fibrils is of pivotal importance for seeding efficiency (Marchante et al., 2017), hence likely explaining the limited seeding efficiency of p103–113 fibrils. A key feature of the native Sup35 prion domain is the high abundance of N and Q residues in an extended sequence with high disorder, with N being the most over-represented residue relative to the frequency in other proteins (Espinosa Angarica et al., 2013). To capture this feature in our design, we capped the nucleation core p103–113 by one or two repeats of a tetrapeptide sequence, (Asn-Ala-Asn-Pro [NANP]), previously shown to increase solubility of amyloidogenic peptides (Finder et al., 2010), yielding peptides p103–113_Bristle_I and p103–113_Bristle_II (Table S2). We used an N-rich sequence including a proline residue, a so-called aggregation gatekeeper (Rousseau et al., 2006), to avoid aggregation potential of the flanking regions themselves. Of note, this NANP repeat sequence is not present in the native Sup35 sequence. Following 6 days of rotation, p103–113 and p103–113_Bristle_I organized into microcrystalline aggregates (Figure 4A), and only a small fraction of total peptide remained soluble (Figure 4C). Adding two NANP repeats on both sides of p103–113, on the other hand, drastically increased solubility (Figure 4C) and slowed down aggregation (Figure 4D). Even though solubility increased, p103–113_Bristle_II still organized into fibrils (Figure 4A) and remarkably showed a higher ThT fluorescence signal compared to p103–113, indicating underlying structural differences (Figure 4E). To confirm the latter observation, we used another amyloid-specific dye, p-FTAA (Hammarström et al., 2010). The emission spectrum of p-FTAA allows us to deduct structural information and shows that p103–113_Bristle_II fibrils closely resemble NM fibrils, while p103–113 and p103–113_Bristle_I fibrils show a different p-FTAA emission spectrum compared to NM fibrils (Figures S3A and S3B). Moreover, NM and p103-113_Bristle_II fibrils disassemble at a lower urea concentration compared to p103-113 and p103-113_Bristle_I fibrils, showing that the structural differences also result in altered fibril stabilities (Figures S3C–S3D). Fibrils formed by p103-113_Bristle_II showed a similar morphology compared to full-length Sup35 NM fibrils (Figures 4A and S4) and showed increased sensitivity to sonication, generating a higher number of smaller seeds compared to p103–113 and p103–113_Bristle_I (Figures 4A–4B). The size distributions of sonicated NM and p103–113_Bristle_II fibrils were confirmed by atomic force microscopy (AFM), while the large p103–113 and p103–113_Bristle_I fibrils did not adsorb on the AFM surface (Figure S5).
Figure 4.

Fibril Brittleness Increases Seeding Efficiency
(A) TEM images of preformed fibrils of Sup35 NM (10 μM monomer concentration), p103–113 (1 mM monomer concentration), p103–113_Bristle_I (1 mM monomer concentration), and p103–113_Bristle_II (1 mM monomer concentration) before and after sonication.
(B) Quantification of the length of individual aggregates (seeds) after sonication based on four independent TEM experiments, visualized as violin plots. Numbers above violin symbols represent average size of seeds. ∗∗∗∗p < 0.00001 (one-way ANOVA).
(C) Concentrations of the soluble fractions of peptides rotated for 6 days at 1 mM. ∗∗∗∗p < 0.00001 (one-way ANOVA).
(D) ThT kinetic assay of all peptides at 1 mM with continuous shaking. Three independent replicates are shown. Quantification of the lag time of each of the sigmoidal curves is shown on the bottom right. Mean values and SD of three independent replicates are shown. ∗∗p < 0.01 (one-way ANOVA).
(E) ThT fluorescence of synthetic peptides rotated at 1 mM concentration for 6 days. Mean values and SD of three independent replicates are shown. ∗∗∗p < 0.0001 (one-way ANOVA).
(F) ThT kinetic assay of recombinant Sup35 NM (10 μM) incubated without (gray) or with 2% (v/v) preformed, sonicated fibrils (red). Three independent replicates are shown.
(G) Mean lag time and SD of three independent repeats of the data in (F). ∗∗p < 0.01 and ∗∗∗p < 0.0001 (one-way ANOVA).
(H) Induction of NM-GFP aggregates in recipient N2a NM-GFPsol cells. 1 mM peptides were fibrillized for 6 days, sonicated, and subsequently added to NM-GFPsol cells at a final concentration of 20 μM (monomer concentration). Shown is the mean ± SD (n > 4). ∗∗p < 0.01.
See also Figures S3–S6.
Embedding p103–113 in a Non-native N-rich Sequence Enhances Prion Seeding Efficiency
Adding two NANP repeats to p103–113 significantly increases seeding efficiency of Sup35 NM in vitro, represented by a reduction of the lag phase of Sup35 NM aggregation (Figures 4F–4G). TEM images taken at different time points after seed addition confirm the latter observation (Figure S6). Finally, sonicated p103–113_Bristle_II fibrils show a 4-fold increase in seeding efficiency in mammalian N2a NM-GFPsol cells compared to the more rigid p103–113 fibrils, suggesting that the aggregates formed by p103–113_Bristle_II have an improved prion seeding capacity in the cell (Figure 4H). Moreover, when we inserted proline mutations in the 10-amino-acid core of p103–113 and p103–113_Bristle_II, this completely abrogated seeding potential, showing that the entropic bristles are not sufficient for prion induction (Figure 4H; Table S2). Interestingly, by using fluorescently labeled peptides, we show that uptake efficiency of p103–113_Bristle_II fibrils is doubled compared to p103–113 fibrils, while seeding efficiency is quadrupled (Figure 5). This indicates that increased uptake of smaller seeds is only part of the effect and that the number of seeds or intrinsic seeding capacity also accounts for the enhanced prion seeding. Finally, to confirm the specificity of our design, we capped p28–38, whose fibrils showed a minor but nonsignificant effect on Sup35 NM aggregation (Figures 1D and 1E), with the same NANP sequences (Tables S1 and S2). Fibrils formed by p28–38 and its bristled variants do not seed Sup35 NM aggregation in vitro or in mammalian N2a cells, confirming the importance of the p103–113 aggregation core (Figure 6). In conclusion, flanking an aggregation core of Sup35 with non-native N-rich sequences increases solubility of the APR, allowing it to organize into elongated fibrils structurally similar to full-length Sup35 NM aggregates that show increased brittleness to form more effective prion seeds in a mammalian cellular system.
Figure 5.

Differences in Uptake and In Cellula Prion Seeding Capacity of Fluorescently Labeled p103–113 and p103–113_Bristle_II Seeds in Mouse Neuroblastoma Cells
(A) Confocal microscopy images of N2a NM-HAsol cells exposed to sonicated fibrils formed by p103–113 and p103–113_Bristle_II, fluorescently labeled with fluorescein isothiocyanate (FITC). 1 mM peptides were fibrillized for 6 days, sonicated, and subsequently added to N2a NM-HAsol cells at a final concentration of 20 μM. Arrows indicate aggregates. Scale bar, 5 μm.
(B) Percentage of cells positive for FITC-labeled peptide seeds. Shown is the mean ± SD (n = 3, three independent experiments). ∗∗∗p < 0.0001 (unpaired t test).
(C) Percentage of recipient cells with NM-HA aggregates. Shown is the mean ± SD (n = 3, three independent experiments). ∗∗∗p < 0.0001 (unpaired t test).
Figure 6.

Adding NANP Sequences to p28–38 Does Not Result in Improved Seeding Capacity In Vitro and in Mouse Neuroblastoma Cells
(A) ThT kinetic assay of recombinant Sup35 NM (10 μM) incubated without (gray) or with 2% (v/v) preformed, sonicated fibrils (red). At least three independent replicates are shown.
(B) Mean lag time and SD of at least three independent repeats of the data in (A). ∗∗∗∗p < 0.00001 (one-way ANOVA).
(C) Confocal microscopy images of N2a NM-GFPsol cells exposed to sonicated peptide fibrils. 1 mM peptides were fibrillized for 6 days, sonicated, and subsequently added to N2a NM-GFPsol cells at a final concentration of 20 μM. Scale bar, 5 μm.
(D) Percentage of recipient cells with NM-GFP aggregates. Shown is the mean ± SD (n = 3, independent experiments).
Discussion
The ordered assembly of proteins into amyloid is a key feature of many neurodegenerative diseases and functional processes (Chiti and Dobson, 2017, Blanco et al., 2012, Fowler et al., 2006). Amyloid formation is driven by short hydrophobic stretches, called aggregation-prone regions (APRs), that organize into an ordered intermolecular cross-beta structure. Homotypic interactions are preferred between these APRs, resulting in a highly sequence-specific process (Krebs et al., 2004, O’Nuallain et al., 2004, Wright et al., 2005).
Prions are considered a subclass of amyloids in the sense that they organize into very similar supramolecular fibrillar structures but additionally are characterized by infectivity and cell-to-cell propagation (Tuite and Cox, 2003). In mammals, the only so far bona fide prions composed of the host protein PrP cause neurodegenerative diseases. In lower eukaryotes, by contrast, proteins with prion activities can also fulfill physiological functions. Several prion domains of proteins from Saccharomyces cerevisiae contain long intrinsically disordered domains enriched in Q and N residues that drive the protein’s conversion into the prion state. Remarkably, compositionally similar prion-forming domains have been identified in over 250 mammalian proteins, some of which are associated with neurodegenerative diseases (King et al., 2012, Lancaster et al., 2014). Of note, multiple non-prion amyloids composed, for example, of tau (Polymenidou and Cleveland, 2012, Jucker and Walker, 2013, Holmes et al., 2014, Clavaguera et al., 2014, Spires-Jones and Hyman, 2014, Nisbet et al., 2015), alpha-synuclein (Hansen et al., 2011, Polymenidou and Cleveland, 2012, Freundt et al., 2012, Herva and Spillantini, 2015, Reyes et al., 2015), or amyloid-beta (Bahr et al., 1998, Kane et al., 2000, Meyer-Luehmann et al., 2006, Eisele et al., 2010, Nath et al., 2012, Stöhr et al., 2012) have been shown to share some prion properties on a molecular and cellular level. Thus, amyloids should be classified according to their prion properties rather than by the classical “prion versus non-prion” classification (Aguzzi and Lakkaraju, 2016). Whether typical aggregation-nucleating cores reside in prion domains, as they do in other amyloids, and whether the long sequences with high disorder flanking these potential cores encode a functional role is still a matter of debate.
Using the budding yeast Saccharomyces cerevisiae Sup35 protein, we here provide evidence that prion induction indeed can be driven by a short amino acid stretch, comparable to the mechanism underlying non-prion amyloid formation. More specifically, we found that fibrils formed by a short stretch (amino acids 103–113) originating from the N domain of Sup35 are able to induce the aggregation of Sup35 NM in vitro and in mammalian cells. These p103–113-induced aggregates indeed possess archetypal prion properties in mammalian cells, including inheritance of aggregates by progeny and cell-to-cell spreading propensity. Interestingly, deletion studies of Sup35 NM expressed in N2a cells previously demonstrated that a region comprising amino acid residues 98–123 is critically involved in prion induction and maintenance, arguing that this region represents the nucleation core of the NM prion state in a mammalian cell environment (Duernberger et al., 2018). The fact that other studies (Osherovich et al., 2004, Shkundina et al., 2006) showed that this region is dispensable for prion activity in yeast cells highlights that, depending on the host, different regions can nucleate and/or seed Sup35 NM prions.
Importantly, we observed much lower prion induction rates for p103–113 compared to Sup35 NM self-seeding, indicating that additional properties are needed to reach the full prion seeding potential. Taking into consideration the N-rich disordered sequence background of the Sup35 prion domain, we designed variants of p103–113 by flanking it with multiple non-native N-rich (NANP) sequences (Finder et al., 2010). The effect of these modifications is twofold. First, solubility of the peptide increased, as seen from aggregation kinetics and critical concentration calculations. Second, the aggregated fraction showed an elongated morphology and lower physical stability, reminiscent of full-length Sup35 fibrils, that readily break into a high number of small seeds upon sonication, in contrast to the rigid microcrystalline-like aggregates formed by non-flanked p103–113. This dual effect resulted in an improved seeding capacity of the peptide fibrils in vitro and in cellula.
Although we cannot exclude the possibility of additional aggregation cores in the Sup35 NM sequence, this work provides evidence that, in a mammalian cellular context, prion induction can be driven by at least one short aggregation nucleating core (p103–113). Even more important, in contrast to non-prion amyloids, this work also highlights the importance of the N-rich disordered amino acid background. On the one hand, this background ensures solubility of the prion protein to avoid oversensitive aggregation induction (Sabate et al., 2015), while on the other hand, it also drives the brittleness of prion fibrils once they form. Brittleness has been put forward as a crucial prion property (Tanaka et al., 2006), and Marchante and colleagues (Marchante et al., 2017) highlighted the importance of aggregate size and concentration in terms of prion induction in the Sup35 case. Our data confirm these findings by the design of a synthetic prion seed consisting of an amyloidogenic core of Sup35, flanked by non-native N-rich sequences. Reversibly, our data show that rigid fibrils are less infectious than more brittle fibrils, suggesting that trapping amyloids (prion and non-prion) in end-stage fibrils would make them more inert to propagation and could be a valid therapeutic strategy to combat amyloid-associated diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Peptides (custom made) | Genscript | N/A |
| ProLong Gold Antifade with DAPI | ThermoFisher scientific | Cat# P36935 |
| Experimental Models: Cell Lines | ||
| N2a NM-GFPsol | In-house | Described previously (Hofmann et al., 2013, Liu et al., 2016) |
| N2a NM-HAsol | In-house | Described previously (Krammer et al., 2009) |
| N2a NM-mCherrysol | In-house | Described previously (Hofmann et al., 2013) |
| Software and Algorithms | ||
| Graphpad Prism (Version 7.0) | Graphpad software | https://www.graphpad.com/RRID:SCR_002798 |
| R-Studio | R | https://www.rstudio.com/RRID:SCR_000432 |
| ImageJ (Version 1.51h) | NIH Image | https://imagej.nih.gov/ij/RRID: SCR_003070 |
| Yokogawa analysis software | Yokogawa Inc. | N/A |
| Other | ||
| ClarioStar plate reader | BMG labtech | N/A |
| PolarStar Optima plate reader | BMG labtech | N/A |
| DynaPro DLS plate reader | Wyatt | N/A |
| JEM-1400 transmission electron microscope | Jeol | N/A |
| CellVoyager CV6000 | Yokogawa Inc. | CV6000 |
Lead Contact and Materials Availability
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Frederic Rousseau (Frederic.rousseau@kuleuven.vib.be). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Cell Lines
N2a cells were cultured in Opti-MEM (GIBCO) supplemented with glutamine, 10% (v/v) fetal bovine serum (FCS) (PAN-Biotech GmbH) and antibiotics. All cells were incubated at 37°C and 5% CO2. N2a cells stably producing soluble NM-GFP (N2a NM-GFPsol), NM-HA (N2a NM-HAsol) or NM-mCherry (NM-mCherrysol) have been described previously (Hofmann et al., 2013, Liu et al., 2016, Krammer et al., 2009). All experiments were performed with cells that were passaged less than 10 times past defrosting. The total numbers cells and the viability of cells were determined using the Vi-VELLTMXR Cell Viability Analyzer (Beckman Coulter).
Method Details
Peptide synthesis
Peptides were synthesized by FMOC solid phase peptide synthesis. An Intavis Multipep RSi synthesis robot was used for all peptides that were synthetized in-house. After synthesis, crude peptides were stored as dry ether precipitates at –20°C. Crude peptides were tested for purity and all peptides used were > 90% pure. All peptides were aminoterminally acetylated and carboxyterminally amidated. FITC-labeled peptides were custom made by Genscript.
Fibrillization of peptides
Peptides were diluted to 1 mM or 2 mM starting concentration as follows. First, 50 μl of 2 M HCl were added dropwise to solve the peptides. After 5 min incubation at room temperature, the pH was neutralized with 50 μl 2 M NaOH and the peptides were diluted in 200 μl double distilled water. Peptides were loaded onto a SpinX 0.2 μm filter columns (Costar) to remove preformed fibrils and aggregates. Peptides were snap frozen and stored at −20°C. For fibrillization, 300 μl peptide aliquots were shaken at 50 rpm with concomitant rotation (NeoLab). Before addition to cells, fibrillized peptides were sonicated, applying 10% power for 3 min (Branson Digital Sonifier).
Recombinant expression and purification of Sup35 NM
Competent E. coli cells, grown in LB supplemented with 75 μg/ml ampicillin, were transformed with a pJCSUP35 plasmid coding for Sup35 NM. Transformants were selected by picking colonies grown on LB plates supplemented with 75 μg/ml ampicillin. Transformants were grown by shaking at 37°C in LB supplemented with 75 μg/ml ampicillin. Expression was induced with 1 mM IPTG for 2 h when cells reached log phase growth (OD600 0.3). Cells were harvested by centrifugation (6,238 g for 15 min). Purification was carried out under denaturing conditions and at room temperature. First, cells were lysed by shaking in buffer A (50 mM Tris [pH 8.0] with 8 M urea) for 30 min. Lysed cells were centrifuged (39,191 g for 30 min) and supernatant was applied to a HiTrap Fast Flow column, 5 mL column volume (GE Healthcare Life Sciences). The column was first washed with 5 column volumes of buffer A with 40 mM imidazole and eluted with 2 column volumes of buffer A with 400 mM imidazole. Eluate was applied to a HiTrap Q Sepharose Fast Flow column, 5 mL column volume (GE Healthcare Life Sciences). The column was first washed with 5 column volumes of buffer A with and eluted with a gradient of 0-500 mM NaCl in buffer A. A final purification step was performed by adding the eluate to another HiTrap FF column as described above. Finally, the purified Sup35 NM protein was dialyzed into a 50 mM Tris buffer (pH 8) with 8 M urea.
Transmission electron microscopy (TEM)
Formvar film coated 400-mesh copper grids (Agar Scientific Ltd., England) were first glow-discharged. 10 μl of each sample was adsorbed for 10 min and the grids were washed by contact with one drop of ultrapure water. Finally, negative staining was performed by contact with one drop of uranyl acetate (2% w/v) for 1 min. The grids were examined using a JEM-1400 transmission electron microscope (Jeol, Japan) at accelerating voltage 80 keV.
Fibril- and seed dimension measurements
Using ImageJ, we manually selected all fibrils or seeds. Only fibrils and seeds that were clearly distinguishable from a larger bundle were used for the analysis. The latter was done to avoid artifacts due to drying on a TEM grid.
Thioflavin-T kinetic assay
Aggregation kinetics were obtained by placing 100 μl of the peptide solution with a final concentration of 25 μM Thioflavin-T (ThT) into a flat-bottom 96-well microclear plate. Fluorescence emission was monitored at 480-10 nm after excitation at 440-10 nm. Every 2-5 min ThT fluorescence was measured. Endpoint ThT fluorescence values were also measured by adding 25 μM (final concentration) of ThT to aged/ rotated peptide solutions.
In vitro seeding assay
A Sup35 NM stock solution of 500 μM (in 50 mM Tris [pH 8.0] with 8 M urea) was diluted 50x in 5 mM potassium dihydrogen phosphate (pH 7.4) to allow aggregation. Samples of 100 μl were placed into a flat-bottom 96-well microclear plate with or without mature Sup35 NM fibrils or peptide fibrils (2% molar ratio). ThT was added to a final concentration of 25 μM and fluorescence was measured with a FLUOstar OPTIMA multidetection microplate reader. Peptides were allowed to fibrilize by continous shaking/rotating for 6 d. Aggregate seeds were then prepared by sonication for 3 min (10% intensity, Branson Digital Sonifier). A four-parameter dose-response curve, or four-parameter logistic curve (4PL) was used to fit all sigmoidal curves (Graphpad Prism).
P-FTAA assay
Fibrils formed from peptides or Sup35 NM by rotation for 6 d were incubated with a final concentration of 0.5 μM p-FTAA and incubated for at least 1 h. Samples were transferred to a 96-well plate (black, non-treated, half area with transparent bottom, Corning) and the emission spectra of p-FTAA after excitation at 440-10 nm was measured using a ClarioStar plate reader (BMG labtech, Germany).
Urea denaturation assay
Fibrils formed from peptides or Sup35 NM by rotation for 6 d were incubated with the corresponding concentration of urea (a stock of 10 M urea was prepared) and incubated for at least 1 h. Samples were transferred to an UNcle machine (Unchained Labs) that allows to measure scattering at 266 nm. Each sample was measured 3 times and the experiment was repeated with 3 different batches of fibrils. A four-parameter dose-response curve, or four-parameter logistic curve (4PL) was used to fit all sigmoidal curves and deduce EC50 values (Graphpad Prism).
Aggregate induction assay
N2a NM-GFPsol or N2a NM-HAsol cells were cultured on 96-well μ-clear black plates (Greiner) at a density of 4x104 cells per well for 1 h. Peptides (20-40 μM, monomer equivalent) were added to the cells. After 16 h of incubation, the cells were fixed in 4% paraformaldehyde and nuclei were counterstained with Hoechst. Cells were imaged with an automated confocal microscope (CellVoyager CV6000; Yokogawa Inc.) using a 20x objective. Maximum intensity projections were generated from Z stacks. Images from 16 fields per well were taken. On average, a total of 3-4x104 cells/well and at least two wells per treatment were analyzed. To assess toxicity, NM-GFPsol cells were cultured as described above with peptides (20 μM, monomer concentration). Cells were counted based on Hoechst staining and compared to vehicle-treated cells.
Time Lapse Analysis
For the coculture of donor N2a NM-GFPagg clone 2 and recipient NM-mCherrysol, a total of 4x104 cells/well were seeded on 96-well μ-clear black plates (Greiner) at a ratio of 1.5:1 donor to recipient cells. Cells were maintained in a humidified 5% CO2 atmosphere at 37°C. Time lapse analysis was performed using CellVoyager CV6000 (Yokogawa Inc.). Recordings started 4 h post seeding. Images were acquired every 10 min for 6 h with a 40x water objective (N.A. = 1.15).
Isolation of single cell clones by limiting dilution
Cells were diluted into medium to a final concentration of 10 cells/ ml. A total of 100 μl cell suspension was transferred to individual wells of a 96-well plate. Wells containing only single cell colonies were selected, and clones with NM-GFP aggregates were expanded.
Immunofluorescence Staining and Confocal Microscopy Analysis
Cells were fixed in 4% paraformaldehyde and permeabilized in 0.5% Triton X-100. For antibody staining, cells were incubated for 1 h with Alexa Fluor 647 conjugated mouse anti-HA TANA2 (1:500; MBL) in 5% (v/v) ChemiBLOCKER (Millipore) at room temperature. Nuclei were counterstained for 15 min with 4 μg/ml Hoechst 33342 (Molecular Probes). 96-well plates were scanned using CellVoyager CV6000 (Yokogawa Inc.). Confocal laser scanning microscopy was performed on a Zeiss LSM 700 laser-scanning microscope (Carl Zeiss). Maximum intensity projections were generated from Z stacks.
Determination of soluble fraction
Peptides rotated for at least 6 d at 1 mM were ultracentrifuged at 250,000 g for 1 h at 4°C. Soluble fractions were isolated, and concentrations were determined by using tyrosine absorbance at 280 nm.
Atomic force microscopy
AFM images of sonicated fibrils (prepared as described above) were recorded at room temperature using a NanoWizard III AFM (JPK Instruments). 50 μl of the seeds were dropped on mica, left to incubate for 5 min and washed 3 times with buffer (5 mM potassium dihydrogen phosphate, pH 7.4) before any AFM measurements. Imaging was performed in the tapping mode with DNP cantilevers (k∼0.06 N/m from thermal noise method and f∼40kHz). To minimize the tip-sample loading force, the free oscillation A0 of the cantilever was set to 10 nm and the amplitude set point between 0.7 and 0.9A0. Data were analyzed offline using the JPK data processing software.
Quantification and Statistical Analysis
Image Analysis
The image analysis was performed using the CellVoyager Analysis support software. An image analysis routine was developed for single cell segmentation and aggregate identification (Yokogawa Inc.). The total number of cells was determined based on the Hoechst signal, and recipient cells were detected by their GFP signal. Green aggregates were identified via morphology and intensity characteristics. The percentage of recipient cells with NM-GFPagg was calculated as the number of aggregate-positive cells per total recipient cells set to 100%. A small percentage (below 1%) of false-positive induced recipient cells was detected due to the heterogeneity of GFP expression levels in individual cells. The mean percentage of false-positives determined in control NM-GFPsol cells was subtracted from all samples. Please note that negative values were sometimes obtained when no induction was observed.
Statistics
All experiments were repeated at least 3 times. Statistical analysis was performed with unpaired t test for comparison of 2 groups or one-way ANOVA with multiple comparisons (after verifying normality) to compare multiple groups. GraphPad Prism was used to perform all statistical analyses. All P values, exact values of n and specific tests used can be found in each figure legend. Significance was assigned only if P values were lower than 0.05.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
Acknowledgments
E.M. was supported by a PhD Fellowship from the Funds for Scientific Research Flanders (FWO). N.L. was supported by the Fund for Scientific Research Flanders Post-Doctoral Fellowship (FWO 12P0919N). The Switch Laboratory was supported by grants from the European Research Council under the European Union’s Horizon 2020 Framework Programme ERC grant agreement 647458 (MANGO) to J.S., the Flanders Institute for Biotechnology (VIB), KU Leuven, the Industrial Research Council of KU Leuven, the FWO, and the Hercules Stichting. Electron microscopy was performed at the electron microscopy platform of VIB-KULeuven.
Author Contributions
Conceptualization, E.M., S.L., R.G., J.S., I.V., and F.R.; Methodology, E.M., S.L., J.S., I.V., and F.R; Validation, E.M. and S.L.; Formal Analysis, E.M. and S.L.; Investigation, E.M., S.L., R.G., N.L., M.M.-G., and Y.D.; Visualization, E.M. and S.L.; Writing – Original Draft, E.M., S.L., and I.V.; Writing – Review & Editing, R.G., J.S., I.V., and F.R.; Funding Acquisition, J.S., I.V., and F.R.; Resources, J.S., I.V., and F.R.; Supervision, R.G., J.S., I.V., and F.R.
Declaration of Interests
The authors declare no competing interests.
Published: February 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.098.
Contributor Information
Joost Schymkowitz, Email: joost.schymkowitz@kuleuven.vib.be.
Ina Vorberg, Email: ina.vorberg@dzne.de.
Frederic Rousseau, Email: frederic.rousseau@kuleuven.vib.be.
Supplemental Information
References
- Aguzzi A., Lakkaraju A.K.K. Cell biology of prions and prionoids: a status report. Trends Cell Biol. 2016;26:40–51. doi: 10.1016/j.tcb.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Bahr B.A., Hoffman K.B., Yang A.J., Hess U.S., Glabe C.G., Lynch G. Amyloid beta protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J. Comp. Neurol. 1998;397:139–147. [PubMed] [Google Scholar]
- Betti C., Vanhoutte I., Coutuer S., De Rycke R., Mishev K., Vuylsteke M., Aesaert S., Rombaut D., Gallardo R., De Smet F. Sequence-specific protein aggregation generates defined protein knockdowns in plants. Plant Physiol. 2016;171:773–787. doi: 10.1104/pp.16.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco L.P., Evans M.L., Smith D.R., Badtke M.P., Chapman M.R. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. 2012;20:66–73. doi: 10.1016/j.tim.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 2017;86:27–68. doi: 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Clavaguera F., Hench J., Lavenir I., Schweighauser G., Frank S., Goedert M., Tolnay M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127:299–301. doi: 10.1007/s00401-013-1231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow E.T., Li L. Newly identified prions in budding yeast, and their possible functions. Semin. Cell Dev. Biol. 2011;22:452–459. doi: 10.1016/j.semcdb.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePace A.H., Santoso A., Hillner P., Weissman J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- Duernberger Y., Liu S., Riemschoss K., Paulsen L., Bester R., Kuhn P.H., Schölling M., Lichtenthaler S.F., Vorberg I. Prion replication in the mammalian cytosol: functional regions within a prion domain driving induction, propagation, and inheritance. Mol. Cell. Biol. 2018;38:136. doi: 10.1128/MCB.00111-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y.S., Obermüller U., Heilbronner G., Baumann F., Kaeser S.A., Wolburg H., Walker L.C., Staufenbiel M., Heikenwalder M., Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa Angarica V., Ventura S., Sancho J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genomics. 2013;14:316–317. doi: 10.1186/1471-2164-14-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finder V.H., Vodopivec I., Nitsch R.M., Glockshuber R. The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42. J. Mol. Biol. 2010;396:9–18. doi: 10.1016/j.jmb.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A.W.P., Falcon B., He S., Murzin A.G., Murshudov G., Garringer H.J., Crowther R.A., Ghetti B., Goedert M., Scheres S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D.M., Koulov A.V., Alory-Jost C., Marks M.S., Balch W.E., Kelly J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:E6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt E.C., Maynard N., Clancy E.K., Roy S., Bousset L., Sourigues Y., Covert M., Melki R., Kirkegaard K., Brahic M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 2012;72:517–524. doi: 10.1002/ana.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo R., Ramakers M., De Smet F., Claes F., Khodaparast L., Khodaparast L., Couceiro J.R., Langenberg T., Siemons M., Nyström S. De novo design of a biologically active amyloid. Science. 2016;354:aah4949. doi: 10.1126/science.aah4949. [DOI] [PubMed] [Google Scholar]
- Ganesan A., Siekierska A., Beerten J., Brams M., Van Durme J., De Baets G., Van der Kant R., Gallardo R., Ramakers M., Langenberg T. Structural hot spots for the solubility of globular proteins. Nat. Commun. 2016;7:10816. doi: 10.1038/ncomms10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S., Phuan P.W., Perkins B., Ullman J., May B.C., Cohen F.E., Prusiner S.B. Cell division modulates prion accumulation in cultured cells. Proc. Natl. Acad. Sci. USA. 2007;104:17971–17976. doi: 10.1073/pnas.0708372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson B.I., Murray I.V., Trojanowski J.Q., Lee V.M. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Glover J.R., Kowal A.S., Schirmer E.C., Patino M.M., Liu J.J., Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L., Teng P.K., Riek R., Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassmann A., Wolf H., Hofmann J., Graham J., Vorberg I. Cellular aspects of prion replication in vitro. Viruses. 2013;5:374–405. doi: 10.3390/v5010374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström P., Simon R., Nyström S., Konradsson P., Aslund A., Nilsson K.P. A fluorescent pentameric thiophene derivative detects in vitro-formed prefibrillar protein aggregates. Biochemistry. 2010;49:6838–6845. doi: 10.1021/bi100922r. [DOI] [PubMed] [Google Scholar]
- Hansen C., Angot E., Bergström A.L., Steiner J.A., Pieri L., Paul G., Outeiro T.F., Melki R., Kallunki P., Fog K. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herva M.E., Spillantini M.G. Parkinson’s disease as a member of prion-like disorders. Virus Res. 2015;207:38–46. doi: 10.1016/j.virusres.2014.10.016. [DOI] [PubMed] [Google Scholar]
- Hofmann J.P., Denner P., Nussbaum-Krammer C., Kuhn P.H., Suhre M.H., Scheibel T., Lichtenthaler S.F., Schätzl H.M., Bano D., Vorberg I.M. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc. Natl. Acad. Sci. USA. 2013;110:5951–5956. doi: 10.1073/pnas.1217321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B.B., Furman J.L., Mahan T.E., Yamasaki T.R., Mirbaha H., Eades W.C., Belaygorod L., Cairns N.J., Holtzman D.M., Diamond M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA. 2014;111:E4376–E4385. doi: 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M., Walker L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M.D., Lipinski W.J., Callahan M.J., Bian F., Durham R.A., Schwarz R.D., Roher A.E., Walker L.C. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J. Neurosci. 2000;20:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodaparast L., Khodaparast L., Gallardo R., Louros N.N., Michiels E., Ramakrishnan R., Ramakers M., Claes F., Young L., Shahrooei M. Aggregating sequences that occur in many proteins constitute weak spots of bacterial proteostasis. Nat. Commun. 2018;9:866. doi: 10.1038/s41467-018-03131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C.-Y., Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- King O.D., Gitler A.D., Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer C., Kryndushkin D., Suhre M.H., Kremmer E., Hofmann A., Pfeifer A., Scheibel T., Wickner R.B., Schätzl H.M., Vorberg I. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc. Natl. Acad. Sci. USA. 2009;106:462–467. doi: 10.1073/pnas.0811571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S., Vorberg I. Prions ex vivo: what cell culture models tell us about infectious proteins. Int. J. Cell Biol. 2013;2013 doi: 10.1155/2013/704546. 704546–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs M.R.H., Morozova-Roche L.A., Daniel K., Robinson C.V., Dobson C.M. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004;13:1933–1938. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R., Lindquist S.L. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov V.V., Kochneva-Pervukhova N.V., Chechenova M.B., Frolova N.S., Ter-Avanesyan M.D. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000;19:324–331. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster A.K., Nutter-Upham A., Lindquist S., King O.D. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics. 2014;30:2501–2502. doi: 10.1093/bioinformatics/btu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebman S.W., Chernoff Y.O. Prions in yeast. Genetics. 2012;191:1041–1072. doi: 10.1534/genetics.111.137760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.J., Sondheimer N., Lindquist S.L. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+] Proc. Natl. Acad. Sci. U S A. 2002;99:16446–16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Hossinger A., Hofmann J.P., Denner P., Vorberg I.M. Horizontal transmission of cytosolic Sup35 prions by extracellular vesicles. MBio. 2016;7:136. doi: 10.1128/mBio.00915-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchante R., Beal D.M., Koloteva-Levine N., Purton T.J., Tuite M.F., Xue W.F. The physical dimensions of amyloid aggregates control their infective potential as prion particles. eLife. 2017;6:552. doi: 10.7554/eLife.27109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall K.E., Vadukul D.M., Dahal L., Theisen A., Fowler M.W., Al-Hilaly Y., Ford L., Kemenes G., Day I.J., Staras K., Serpell L.C. A critical role for the self-assembly of amyloid-β1-42 in neurodegeneration. Sci. Rep. 2016;6:30182. doi: 10.1038/srep30182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S., Debulpaep M., Kuemmerer N., Lopez de la Paz M., Martins I.C., Reumers J., Morris K.L., Copland A., Serpell L., Serrano L. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods. 2010;7:237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A.L. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Nath S., Agholme L., Kurudenkandy F.R., Granseth B., Marcusson J., Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of β-amyloid. J. Neurosci. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisbet R.M., Polanco J.C., Ittner L.M., Götz J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015;129:207–220. doi: 10.1007/s00401-014-1371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B., Williams A.D., Westermark P., Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- Osherovich L.Z., Cox B.S., Tuite M.F., Weissman J.S. Dissection and design of yeast prions. PLoS Biol. 2004;2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M., Cleveland D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 2012;209:889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J.F., Olsson T.T., Lamberts J.T., Devine M.J., Kunath T., Brundin P. A cell culture model for monitoring α-synuclein cell-to-cell transfer. Neurobiol. Dis. 2015;77:266–275. doi: 10.1016/j.nbd.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Rousseau F., Serrano L., Schymkowitz J.W.H. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 2006;355:1037–1047. doi: 10.1016/j.jmb.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Sabate R., Rousseau F., Schymkowitz J., Batlle C., Ventura S. Amyloids or prions? That is the question. Prion. 2015;9:200–206. doi: 10.1080/19336896.2015.1053685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio T.R., Cashikar A.G., Kowal A.S., Sawicki G.J., Moslehi J.J., Serpell L., Arnsdorf M.F., Lindquist S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- Shkundina I.S., Kushnirov V.V., Tuite M.F., Ter-Avanesyan M.D. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics. 2006;172:827–835. doi: 10.1534/genetics.105.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T.L., Hyman B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J., Watts J.C., Mensinger Z.L., Oehler A., Grillo S.K., DeArmond S.J., Prusiner S.B., Giles K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Chien P., Naber N., Cooke R., Weissman J.S. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Collins S.R., Toyama B.H., Weissman J.S. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- Teng P.K., Eisenberg D. Short protein segments can drive a non-fibrillizing protein into the amyloid state. Protein Eng. Des. Sel. 2009;22:531–536. doi: 10.1093/protein/gzp037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Avanesyan M.D., Kushnirov V.V., Dagkesamanskaya A.R., Didichenko S.A., Chernoff Y.O., Inge-Vechtomov S.G., Smirnov V.N. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Tessier P.M., Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–561. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toombs J.A., Petri M., Paul K.R., Kan G.Y., Ben-Hur A., Ross E.D. De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. USA. 2012;109:6519–6524. doi: 10.1073/pnas.1119366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite M.F., Cox B.S. Propagation of yeast prions. Nat. Rev. Mol. Cell Biol. 2003;4:878–890. doi: 10.1038/nrm1247. [DOI] [PubMed] [Google Scholar]
- Uptain S.M., Lindquist S. Prions as protein-based genetic elements. Annu. Rev. Microbiol. 2002;56:703–741. doi: 10.1146/annurev.micro.56.013002.100603. [DOI] [PubMed] [Google Scholar]
- Ventura S., Zurdo J., Narayanan S., Parreño M., Mangues R., Reif B., Chiti F., Giannoni E., Dobson C.M., Aviles F.X., Serrano L. Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA. 2004;101:7258–7263. doi: 10.1073/pnas.0308249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner R.B. Yeast and fungal prions. Cold Spring Harb. Perspect. Biol. 2016;8:a023531. doi: 10.1101/cshperspect.a023531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.F., Teichmann S.A., Clarke J., Dobson C.M. The importance of sequence diversity in the aggregation and evolution of proteins. Nature. 2005;438:878–881. doi: 10.1038/nature04195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coculture of N2a NM-GFPagg clone 2 and N2a NM-mCherrysol. Cells were analyzed by time-lapse microscopy. Recording was initiated after 4 h in coculture.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.