

Abstract
The impressive clinical activity of small molecule receptor tyrosine kinase inhibitors (TKIs) for oncogene-addicted subgroups of non-small cell lung cancer (NSCLC) [for example those driven by activating mutations in the gene encoding epidermal growth factor receptor (EGFR) or rearrangements in the genes encoding the receptor tyrosine kinases anaplastic lymphoma kinase (ALK), ROS proto-oncogene 1 (ROS1), and rearranged during transfection (RET)] has established an oncogene-centric molecular classification paradigm in this disease. However, recent studies have revealed considerable phenotypic diversity downstream of tumor-initiating oncogenes. Co-occurring genomic alterations, particularly in tumor suppressor genes such as TP53 and LKB1 (also known as STK11), have emerged as core determinants of the molecular and clinical heterogeneity of oncogene-driven lung cancer subgroups through their effects on both tumor cell-intrinsic and non-cell-autonomous cancer hallmarks. In this review, we discuss the impact of co-mutations on the pathogenesis, biology, micro-environmental interactions, and therapeutic vulnerabilities of NSCLC and assess the challenges and opportunities that co-mutations present for personalized anti-cancer therapy, as well as the expanding field of precision immunotherapy.
Table of Contents Summary
Co-occurring genomic alterations contribute to the heterogeneity of driver oncogene-defined non-small cell lung cancer (NSCLC) subgroups. This Review discusses the effects of co-mutations on the pathogenesis, biology, microenvironmental interactions, and therapeutic vulnerabilities of NSCLC.
Introduction
The identification in 2004 of activating oncogenic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) in a subset of patients with non-small cell lung cancer (NSCLC) that exhibited dramatic clinical responses to the first-generation EGFR tyrosine kinase inhibitor (TKI) gefitinib launched the field of targeted therapy in NSCLC and reinforced the concept of oncogene addiction as a pillar of modern cancer therapeutics1, 2. Subsequent discovery of ALK re-arrangements in 20073 in 3–7% of NSCLC expanded the spectrum of targetable genomic alterations in this disease4. Since then, several additional driver events with robust transforming potential have been reported, including oncogenic ROS15, RET6, NTRK17 and NRG18 fusions, oncogenic somatic mutations in BRAF (V600E and non-V600E)9–11, intragenic insertions in ERBB2 (also known as HER2)12 and exon 14 skipping mutations in the MET proto-oncogene13–15. Pivotal clinical studies established the superiority of molecularly targeted therapy compared with platinum-doublet chemotherapy [G] for EGFR-mutant, ALK-rearranged and ROS1-rearranged NSCLC and led to the FDA approval of several first, second and third-generation small molecule inhibitors of mutant oncoproteins16–32. The robust clinical activity of these targeted agents, coupled with the apparent mutual exclusivity of strong oncogenic drivers in NSCLC, cemented a driver oncogene-centric paradigm in NSCLC oncogenesis and molecular classification. This prevailing model, commonly represented graphically as an “oncogenic pie chart”, constitutes the bedrock of NSCLC clinical practice and the framework that underpins the design and implementation of a generation of precision oncology clinical trials aimed at matching patients with available targeted therapies based on identification of a single genomic driver event (Figure 1). However, accumulating evidence points towards the existence of substantial clinical heterogeneity within oncogenic-driver defined NSCLC subgroups that is currently incompletely accounted for by the single oncogenic driver model. In this review, we discuss the emerging role of co-occurring genomic alterations as major determinants of both tumor cell-intrinsic as well as non-cell-autonomous cancer hallmark traits, including their impact on the composition of the tumor microenvironment and response to systemic anti-cancer therapies.
Figure 1. Single oncogenic driver paradigm of lung adenocarcinoma molecular classification.
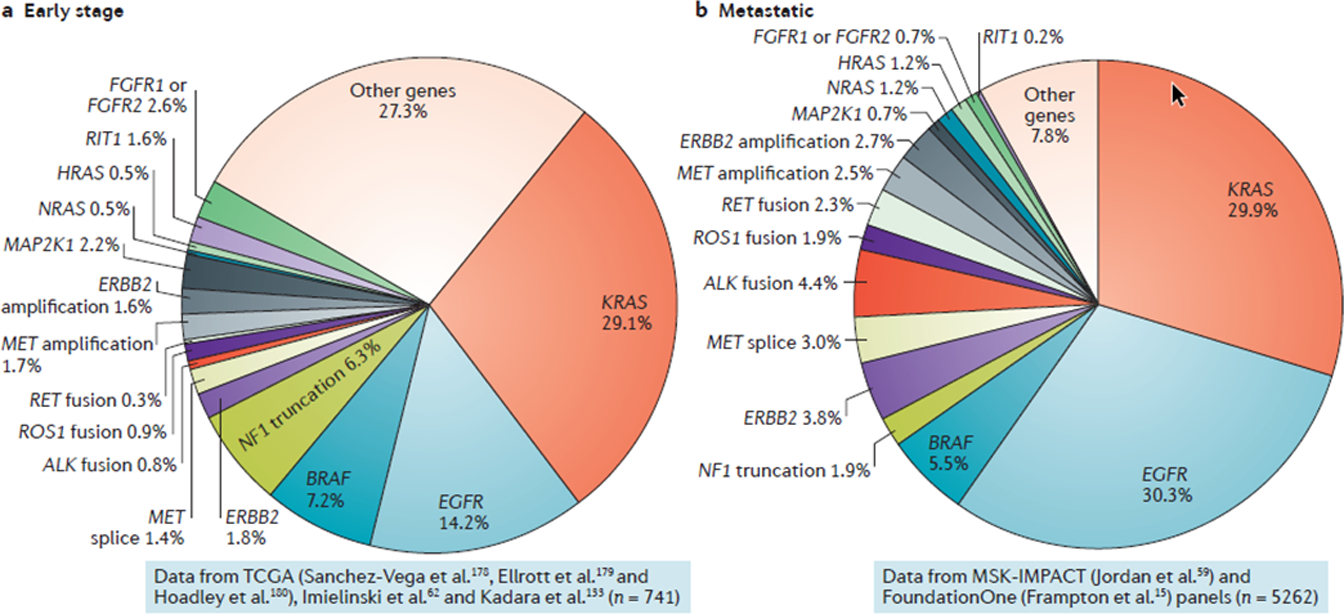
The dominant contemporary model of non-small cell lung cancer pathogenesis and molecular classification is based on identification of single and largely non-overlapping oncogenic driver events. Oncogenic pie charts are presented for early-stage (a) and metastatic (b) lung adenocarcinomas (LUADs). The prevalence of individual genomic alterations in early-stage disease is based on combined analysis of whole exome sequencing data from the PanCancer Atlas cohort of The Cancer Genome Atlas (TCGA) (n=785)177–179, as well as the cohorts reported by Imielinski et al (n=148)62 and Kadara et al (n=108)132, following exclusion of patients with stage 4 disease (n=741 patients in total). The prevalence of MET splice site alterations, MET amplification, ERBB2 amplification, HRAS and NRAS mutations as well as ALK, ROS1 and RET fusions was based on data from the TCGA and Imielinski cohorts only. Oncogenic driver alterations in advanced or metastatic LUAD (encompassing both treatment-naïve patients as well as patients that received prior anti-cancer therapies) are based on next-generation sequencing of pre-defined panels of cancer-relevant genes from patients treated at Memorial Sloan Kettering Cancer Center (N=860,MSK-IMPACT panel59) and samples referred to Foundation Medicine (n=4402,FoundationOne panel15) (n=5262 patients with advanced/metastatic LUAD in total). The prevalence of alterations in NF1, NRAS, HRAS, MAP2K1, FGFR1/2 and RIT1 is based on data from MSK-IMPACT only. It is notable that although the prevalence of oncogenic KRAS mutations is similar in both early and advanced stage LUADs the frequency of other driver alterations (for example truncating NF1 mutations) differs substantially depending on the disease stage. The increased prevalence of EGFR mutations in the metastatic dataset may partially reflect referral bias. Data were visualized and downloaded from the open source web program cBioPortal180, 181 or curated from the scientific literature.
Diversity in driver-defined subgroups.
There is mounting evidence that substantial molecular and clinical heterogeneity exists within oncogenic driver-defined subgroups of NSCLC (“intra-driver heterogeneity”). Despite known associations between certain NSCLC oncogenic subtypes and distinct tumor histopathologic features or growth patterns, NSCLCs driven by the same dominant oncogenic alteration can vary considerably in their histological appearance and immunohistochemical profile. For example, KRAS-mutant lung adenocarcinomas (LUADs) demonstrate dual propensity towards either solid growth pattern with positivity for the NKX2–1 homeobox transcription factor (also known as TTF1) or, alternatively, invasive mucinous adenocarcinoma histology and corresponding lack of NKX2–1 expression33. At the molecular level, considerable efforts have focused on segregating LUAD into molecular subtypes on the basis of multi-dimensional molecular profiling, coupled with unsupervised clustering computational approaches34. Enrichment for specific oncogenic drivers has been observed within distinct subtypes, however cluster membership typically transcends initiating oncogenes, thus providing further evidence for intra-driver molecular diversity34.
Most importantly, overwhelming evidence indicates that intra-driver molecular diversity translates into heterogeneous clinical behavior and variable sensitivity to anticancer therapies. Across clinical trials of first-line targeted therapy for oncogene-addicted subgroups of NSCLC, rates of objective response [G] typically range between 50% and 83% and complete responses [G] are rare; in addition, some patients exhibit de novo resistance16–26, 28–32, 35. Even more variable are duration of response to targeted therapy, progression-free survival and overall survival16–26, 28–32, 35. Phenotypic variability and therapeutic response heterogeneity are particularly evident within KRAS-mutant LUAD. The pervasive diversity of this oncogenotype was aptly demonstrated in a study that applied affinity propagation clustering analysis36 to mRNA expression data from 106 genomically-annotated NSCLC cell lines; strikingly, variation in mRNA expression within KRAS-mutant NSCLC cell lines was equivalent to that observed across the entire cell line panel37. Inter- and intra-driver heterogeneity are also evident following treatment with inhibitors of the immune checkpoint molecules PD-1 or PD-L1, with only ~20% of unselected NSCLC patients deriving durable clinical benefit38–43.
What are the molecular underpinnings of this remarkable intra-driver heterogeneity in NSCLC? In many cases, divergent clinical behavior can be directly attributed to the distinct effects of individual oncogenic alleles. Multiple studies have affirmed the favorable prognostic impact of exon 19 EGFR deletions compared with exon 21 L858R amino acid substitution, although the molecular basis for this association has not been conclusively determined44, 45. Furthermore, EGFR exon 20 in-frame insertion mutants are recalcitrant to all currently FDA-approved EGFR TKIs due to insertion-imposed steric hindrance of the drug binding pocket, but exhibit sensitivity to poziotinib - a smaller and more flexible inhibitor - in vitro and in vivo46, 47. Among ALK-rearranged NSCLC, both the fusion partner as well as EML4-ALK fusion variantshave been considered candidate modifiers of transforming potential and response to ALK TKIs48, 49. For example, the PRKAR1A-ALK fusion was consistently demonstrated to be less sensitive to first, second and third generation ALK TKIs48. In addition, EML4-ALK variant 3 was associated with more frequent secondary resistance mutations (including the G1202R solvent front mutation) compared to EML4-ALK variant 1 and, consequently, longer progression-free survival with the 3rd generation ALK inhibitor lorlatinib, that is active against the EML4-ALKG1202R mutation49. Similarly, in RET-rearranged NSCLC, non-KIF5B-RET fusions have been associated with significantly higher response rates to RXDX-105 (a RET and BRAF inhibitor) but not to the potent and selective RET inhibitor LOXO-29250. Finally, distinct KRAS mutant alleles differentially engage downstream effectors with KRASG12C or KRASG12V preferentially activating RALA or RALB signaling and KRASG12D triggering increased PI3K–AKT and MAPK/ERKpathway activation51. Currently, the prognostic and predictive utility of KRAS alleles in NSCLC remains unclear but is likely to increase in view of the ongoing clinical development of covalent, direct KRASG12C inhibitors52–56. Nonetheless, distinct types of somatic mutations or gene rearrangements appear to only partially account for intra-driver heterogeneity because marked differences in biological behavior can also be observed between NSCLC that bear identical oncogenic alterations in driver genes. Taken together, these studies challenge the single-oncogene paradigm in NSCLC by unveiling multiple layers of heterogeneity within oncogenic subgroups that can only partially be attributed to the driver oncoprotein itself.
Co-occurring genomic alterations
The compendium of co-occurring genomic alterations in NSCLC are potentially more impactful than distinct mutations in oncogenic drivers with regard to determining tumor heterogeneity. LUADs and lung squamous cell carcinomas (LUSCs) are characterized by a high average number of somatic mutations per Mb in comparison to many other tumor types57. Although passenger mutations account for the largest fraction of this mutational burden, combinations of somatic mutations in bona fide cancer driver genes are identified in the majority of LUAD and a substantial fraction of LUSC, even when next generation sequencing platforms are limited to the evaluation of pre-defined sets of cancer-relevant genes. Importantly, large-scale profiling studies utilizing either whole exome sequencing or broad targeted sequencing panels in NSCLC tumors have revealed multiple non-random patterns of co-occurring or mutually exclusive mutations, which typically vary depending on the particular oncogenic driver mutation15, 34, 58–62. From an evolutionary standpoint, co-selection of oncogenic alterations implies functional co-operation that converges on improved fitness, whereas mutual exclusivity indicates redundancy (potentially manifesting as soft exclusivity) or antagonism (resulting in more strict patterns of mutual exclusivity due to deleterious effects of the combined alterations)63, 64. Thus, from its inception, NSCLC develops through a network of evolving genetic interactions that collectively determine cancer hallmark traits65, 66. This further suggests that early oncogenic events may channel tumor evolution towards distinct trajectories and influence the likelihood of positive or negative selection of subsequent genomic alterations. It is important to note, however, that even genomic alterations that do not show statistically significant patterns of co-occurrence may still have important interactions biologically. For example, mutations in TP53 (which encodes p53) are under-represented among KRAS-mutant LUAD compared to other oncogene-driven subgroups, yet p53 inactivation is common and impactful in KRAS-mutant LUAD67–71. The importance of co-mutations as mediators of diverse NSCLC phenotypes has only recently attracted focus and their functional impact remains largely uncaptured within current molecular stratification frameworks.
Knowledge of the clinical context is paramount when evaluating the functional importance as well as prevalence of co-occurring genomic alterations. In particular, it is critical to distinguish between early-stage, surgically resected tumors and locally advanced or metastatic disease because several distinct patterns of co-mutations are enriched in metastatic disease, likely reflecting acquisition of traits that promote tumor progression and metastatic dissemination72, 73. In addition, selective pressure imposed by previous anticancer therapy can substantially influence patterns of co-mutations; therefore, detailed knowledge of prior therapeutic exposures is critical for accurate interpretation and understanding of the functional effect of co-mutation patterns.
Determination of the clonal or sub-clonal nature as well as timing of individual co-alterations may also provide important information regarding their contributions to different stages of carcinogenesis and impact on therapeutic response. Early clonal events are more likely to impact core cancer hallmarks that are critical for tumor initiation. In addition, targeting clonal events is more likely to yield sustained responses; although both clonal and sub-clonal events can contribute to clinical resistance, clonal events are more likely to result in primary resistance. In the landmark TRACERx study, multiregion sequencing of 100 early-stage NSCLCs provided a measure of the extent of clonal driver events (1–18 in LUAD and 1–14 in LUSC) and sub-clonal driver events (0–10 in LUAD and 0–12 in LUSC) and established a catalog of clonal alterations74. Furthermore, evidence from other tumor types supports the notion that within a network of epistatic oncogenic interactions the chronology – or order – of individual genomic alterations can impart distinct phenotypic outcomes. For example in myeloproliferative disorders, the order in which mutations in JAK2 and TET2 arise in hematopoietic stem and progenitor cells can affect age of disease onset, influence the likelihood of the disease manifesting as polycythemia vera versus essential thrombocythemia and result in different propensities for development of thrombosis75, 76.
Finally, when assessing patterns of co-occurring events in human NSCLC it is important to also consider the impact of mutational processes and immune selection. Several distinct mutational signatures sculpt the genome of NSCLC – including signatures of tobacco exposure and APOBEC-mediated cytidine deamination57. Certain recurrent oncogenic mutations, for example classical mutations in PIK3CA (which encodes a catalytic subunit of PI3K), occur within APOBEC deaminase trinucleotide motifs and are enriched in tumors with a high APOBEC mutational footprint77. While the full extent to which mutational processes account for unique combinations of somatic genomic alterations in NSCLC is currently unknown, there is evidence that APOBEC-mediated mutagenesis fuels sub-clonal diversification and branched evolution78, 79. Furthermore, tumor genomes can be shaped by immunosurveillance through early elimination of clones that present strong antigenic neo-peptides. For example, the major histocompatibility complex (MHC) class I genotype of individual patients was demonstrated to impose restrictions on the tumor mutational landscape and predict for selection of distinct driver mutations80. Such immunoediting likely influences patterns of co-mutations in NSCLC and these associations warrant further study. On the other hand, imposition of a cold tumor immune microenvironment [G] as a result of tumor cell-intrinsic processes may relax immune selection and result in a more diverse spectrum of co-mutations.
Co-mutations within LUAD subgroups
KRAS-mutant LUAD
Activating mutations in KRAS are the most prevalent oncogenic driver event in both early-stage and metastatic LUAD, occurring in 25–32% of tumors34, 59, 60, 62, 67. As noted previously, KRAS-mutant LUADs are intrinsically heterogeneous in their biology and clinical behavior. We previously identified three robust and reproducible transcriptomic sub-groups of KRAS-mutant LUAD by applying non-negative matrix factorization consensus clustering81–83 to RNASeq data from 68 tumors from The Cancer Genome Atlas (TCGA) dataset67. Remarkably, superimposition of somatic genetic alterations of key tumor suppressor genes revealed non-overlapping patterns of co-occurring genomic alterations in the three subgroups: one subgroup was dominated by co-occurring TP53 alterations (thereafter referred to as KP), whereas co-mutations or genomic loss in LKB1 (also known as STK11) were a hallmark of the second cluster (referred to as KL), that was further enriched in somatic mutations in KEAP1 and ATM. Bi-allelic inactivation of the CDKN2A/CDKN2B locus was significantly enriched in the third cluster (referred to as KC), that was defined by lack of NKX2–1 expression. Notably, distinct KRAS alleles were not differentially distributed between the three clusters – with the exception of enrichment for KRASG12D in the KC subgroup in some cohorts. These findings established co-mutations as major determinants of the molecular diversity of KRAS-mutant LUAD.
Landmark large scale sequencing studies have established a census of major KRAS co-mutations in both early-stage and advanced LUAD15, 34, 59, 62. The significance of co-occurrence for individual pairs of genetic alterations varies depending on the size of the clinical cohort, the number of possible interactions that are surveyed and the sequencing platform. However, co-mutations in a set of core genes including LKB1, KEAP1, ATM and RBM10 are consistently enriched in KRAS-mutant LUAD (Figure 2). Additional significantly co-altered genes reported in some studies include PTPRD, U2AF1, POLE, NTRK3 and LRP1B. Mutations in TP53 and inactivation of CDKN2A, CDKN2B or combined CDKN2A/CDKN2B loss due to bi-allelic deletion, are common and functionally relevant co-alterations, although they are not enriched in KRAS-mutant compared to other oncogene-driven subgroups. Mutations in other established drivers within the receptor tyrosine kinase-RAS-RAF network including EGFR, ERBB2, BRAF, NF1, as well as ALK, ROS1 and RET rearrangementsare largely non-overlapping with KRAS, although the strength of their negative association varies depending on the individual gene.
Figure 2. Spectrum of major co-occurring genomic alterations in KRAS- and EGFR-mutant lung adenocarcinoma.
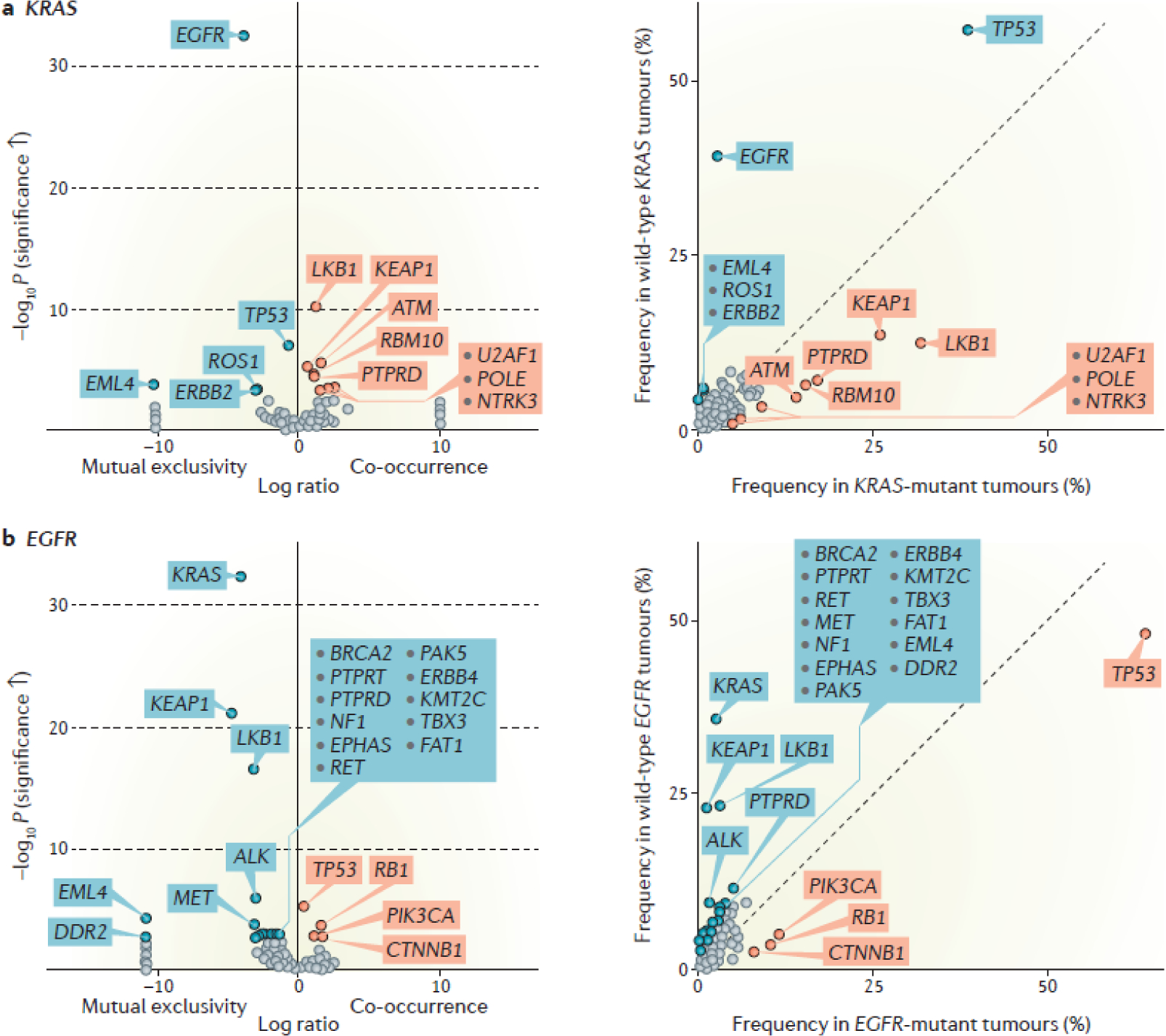
Volcano plots (left graphs) summarizing enrichment of individual co-alterations in: KRAS-mutant compared with KRAS-wild-type LUADs (a) and EGFR-mutant compared with EGFR-wild-type LUADs (b). The magnitude of co-mutation enrichment is indicated on the x-axis and is expressed as log2 (% in KRAS-mutant / % in KRAS-wild-type) or log2 (% in EGFR-mutant / % in EGFR-wild-type) respectively, whereas the statistical significance of the association is plotted on the y-axis and is expressed as –log10P value (derived from a Fisher’s exact test). Significantly enriched co-mutations based on a q value <0.05 (derived from Benjamini-Hochberg procedure182) are highlighted in red, whereas under-represented genomic events are highlighted in blue. The prevalence of each co-alteration in KRAS-mutant and KRAS-wild-type groups (or EGFR-mutant and EGFR-wild-type groups) is shown in the adjacent frequency plots (right graphs of parts a and b). Targeted next generation sequencing-based molecular profiling (MSK-IMPACT platform) from 860 patients with metastatic LUAD treated at Memorial Sloan Kettering Cancer Center were included in this enrichment analysis that was performed using the cBioPortal web program180, 181. Oncogene-driver specific, non-random patterns of co-occurring alterations in key tumor suppressor genes are evident for both KRAS-mutant and EGFR-mutant tumors.
Insights from genetically engineered mouse models and cell line studies have been pivotal towards elucidating the phenotypic sequelae of the most prominent KRAS co-mutations in NSCLC. Somatic deletion of Lkb1 is insufficient for initiation of lung carcinogenesis in mice as a singular event but dramatically accelerates KrasG12D-driven carcinogenesis and fosters early metastatic dissemination84. In addition, loss of Lkb1 results in epigenetic reprogramming and an expanded tumor histological repertoire, with high incidence of squamous or adenosquamous carcinomas, in agreement with data supporting enrichment of LKB1 mutations in human adenosquamous NSCLC84–86. Mechanistically, trans-differentiation [G] is mediated by LKB1 loss-triggered down-regulation of the Polycomb Repressive Complex 2 (PRC2) subunit EED and relief of PRC2-mediated repression of squamous differentiation genes86. Epigenetic reprogramming upon LKB1 loss in KRAS-mutant cells is further fueled by a metabolic network, that promotes increased flux of glucose-derived carbon towards serine biosynthesis and the methionine salvage pathway, bolstering synthesis of S-adenosyl methionine (SAM), a critical substrate for DNA methylation87. Generation of SAM via this pathway, coupled with up-regulation of DNA methyl-transferases, underpins an increase in global levels of CpG methylation in KL cells that is critical for tumor maintenance. Furthermore, KL NSCLC cells exhibit increased dependence on dTTP synthesis and rely on an unorthodox pathway of pyrimidine biosynthesis that utilizes mitochondrially generated carbamoyl phosphate88, 89. The unique metabolic phenotypes associated with combined expression of oncogenic KRAS and LKB1 inactivation – but not with mutations in either gene alone - may at least partially explain their preferential co-occurrence in human NSCLC.
Intricately linked with LKB1 inactivation is loss of KEAP1, an adaptor protein that mediates ubiquitination and proteasomal degradation of NRF2, a key transcription factor in cellular antioxidant, metabolic, cyto-protective, and anti-inflammatory pathways90. Somatic mutations in LKB1 and KEAP1 significantly co-occur with mutant KRAS and with each other in NSCLC15, 59, 63, 91. In the conditional KrasG12D/+;Trp53Fl/FL mouse model, loss of Keap1 increases both tumor burden and the percent of high-grade lesions, pointing towards roles in both tumor initiation and progression92. At the cellular level KEAP1 loss results in increased cellular proliferation in vivo and an altered metabolic profile characterized by increased glucose-derived carbon flux towards the pentose phosphate and serine–glycine biosynthetic pathways93–95 with enhanced dependence on glutaminolysis for tricarboxylic acid (TCA) cycle anaplerosis92, a dependence that is further enhanced by co-occurring LKB1 alterations96. Thus, NRF2- mediated metabolic reprogramming and regulation of redox homeostasis likely underpin the strong co-selection of KEAP1 with LKB1 and KRAS mutations in NSCLC at least partially because oncogenic KRAS itself promotes oxidative stress and anabolic metabolism and because KL NSCLC cells depend on the pentose phosphate pathway for NAPDH generation and for detoxification of reactive oxygen species because these cells have defective fatty acid oxidation94, 97, 98. This suggests that up-regulation of a NRF2-driven transcriptional program may represent a bottleneck in the evolution of LKB1-deficient NSCLC. Potentiation of cellular anabolic, antioxidant and detoxification pathways collectively support the aggressive clinical phenotype of KEAP1-mutant NSCLC that is concordant with its role as an independent negative prognostic indicator92, 99.
Loss of p53 or expression of either dominant negative or dominant gain-of-function Trp53 mutants also co-operate with oncogenic Kras to induce LUADs with shortened latency and increased metastatic proclivity, although these tumors are less aggressive than those with Lkb1 loss69–71, 84. Notably, the selective pressure for p53 inactivation is most critical in aggressive, high-grade lesions that exhibit high levels of ERK signaling, whereas engagement of p53-mediated signaling is minimal in low-grade adenomas, thus highlighting stage and signal intensity - dependent patterns of co-operativity100–102. This notion is further supported by identification of TP53 mutational inactivation as a clonal and predominantly early event in established NSCLC that precedes genome doubling and subsequent branched evolution74, 103.
Mutations in ATM, encoding an apical kinase in the DNA damage response pathway, also significantly co-occur with mutant KRAS. In murine models the impact of Atm inactivation on Kras-driven lung carcinogenesis is context-dependent and varies according to the functional status of p53104. In a p53-proficient setting, bi-allelic loss of Atm is tolerated but does not promote KrasG12D-initiated neoplasia. In contrast, complete Atm inactivation is incompatible with cellular viability in the context of KrasG12D expression and bi-allelic Trp53 inactivation, suggesting that excessive DNA damage in this context removes incipient cancer cells from the proliferative pool. Interestingly, Kras-driven lung carcinogenesis is accelerated by incomplete Atm loss in a p53 deficient settingThus, data from genetically engineered mouse models (GEMMs) point towards a context-dependent, conditional haplo-insufficient role for Atm loss in KrasG12D-driven lung tumorigenesis. Selection against complete ATM inactivation may explain the mutual exclusivity of ATM and TP53 mutations in human LUAD as well as the enrichment of ATM mutations in the KL subgroup67. However, ATM haplo-insufficiency has not been convincingly established yet in human LUAD, where there is evidence for complete lack of ATM expression by immunohistochemistry in a significant proportion of LUAD105.
Intriguingly, analysis of patterns of co-occurrence and mutual exclusivity between a set of 505 pre-selected candidate functional genomic events in 6456 tumors from the Pan-Cancer TCGA Dataset using a novel algorithmic approach (SELECT algorithm) identified somatic mutations in RBM10 as the top-scoring KRAS co-occurrence motif in both NSCLC and colorectal adenocarcinoma63. RBM10 encodes a splicing regulator that is involved in cellular growth control via regulation of NOTCH signaling106, 107. In vivo depletion of Rbm10 in mice using CRISPR/Cas9- mediated gene editing concurrently with activation of endogenous oncogenic KrasG12D confers a modest fitness advantage that is lost when Trp53 or Lkb1 are also inactivated108. The precise phenotypic consequences of RBM10 inactivation in NSCLC and the mechanisms that underpin its oncogenic cooperation with KRAS remain incompletely understood.
Somatic genomic alterations in CDKN2A [encoding the p16 and p14ARF (p19ARF in the mouse) tumor suppressors] and CDKN2B (encoding p15) are observed in ~20% and ~12% of metastatic KRAS-mutant NSCLC respectively and bi-allelic loss of the CDKN2A/CDKN2B locus is a hallmark of the KC subgroup15, 67. KC tumors are characterized by lack of NKX2–1 expression and frequent activation of a gastrointestinal transcriptional program (manifesting histologically as invasive mucinous carcinoma in some cases), enrichment for the KrasG12D mutation and poor prognosis. Several of these features are recapitulated in mice where endogenous expression of oncogenic KrasG12D is coupled with bi-allelic deletion of Cdkn2a/Cdkn2b, resulting in concurrent abrogation of p16, p19ARF and p15. Both isolated Cdkn2a loss (leading to inactivation of p16 and p19ARF) as well as combined Cdkn2a/Cdkn2b inactivation accelerate KrasG12D-driven lung carcinogenesis and promote loco-regional metastatic spread but combined loss of p16, p19ARF and p15 elicits a more marked phenotype than Cdkn2a loss alone with enhanced cellular proliferation, frequent loss of NKX2–1 expression and up-regulation of the embryonal protein HMGA2, increased burden of poorly differentiated, high-grade tumors, enhanced metastatic proclivity and curtailed survival109. Mechanistically, loss of NKX2–1 unleashes a hepatocyte nuclear factor 4-alpha (HNF4A)-driven gastric differentiation program whereas concomitant loss of HNF4A promotes de-repression of HMGA2110.
EGFR-mutant LUAD
Although EGFR –mutant tumors represent the prototypical oncogene-addicted LUAD subgroup that spearheaded adoption of the single-driver model, the overwhelming majority of EGFR-mutant lung tumors harbor one or more co-mutations, even when the analysis is limited to pre-defined sets of cancer-relevant genes within established panels (FoundationOne, Guardant360, MSK-IMPACT)15, 59, 72, 73. The spectrum of enriched genomic co-alterations in advanced EGFR-mutant LUAD is dominated by recurrent mutations in a core set of genes including TP53 (54.6% % - 64.6%), RB1 (9.6%−10.33%), CTNNB1 (which encodes β-catenin; 5.3%−9.6%), and PIK3CA (9%−12.4%) as well as amplifications involving EGFR itself (22% - 25.5%), NKX2–1 (12.2% - 16.7%), CDK4 (7%−10%), CDK6 and CCNE115, 72, 73 (Figure 2). The spectrum and prevalence of co-mutations does not appear to vary depending on the specific initiating EGFR mutation and is similar across the three most common subtypes (EGFR exon 19 deletion, EGFRL858R and EGFR exon 20 insertions)47. Prior therapy is associated with increased average number of co-alterations. Mutations in PIK3CA and CTNNB1 are more frequent in advanced stage tumors compared with early stage LUADs, pointing towards functional roles in malignant progression and metastasis, whereas alterations in TP53 (62.5%), RB1 (9.5 −12.5%) and NKX2–1 (12.5%) appear to occur with comparable frequencies in early- and advanced-stage tumors15, 34, 72, 73.
Somatic mutations in TP53 represent by far the most prevalent co-alteration in EGFR-mutant LUAD (54.6%−64.5%) and their clinical significance has been evaluated in several studies. TP53 mutations are mostly truncal events (present in all geographically distinct segments of the tumor) that occur early during tumor evolution and prior to whole genome doubling, and are frequently accompanied by truncal loss of heterozygosity at the TP53 locus, indicating strong selective pressure for complete TP53 inactivation in early stage LUADs103. Furthermore, tumors bearing co-mutations in TP53 exhibit higher degrees of copy number genomic instability (aneuploidy), and a higher somatic mutation burden, both on the trunk and in the branches of the tumor phylogenetic tree103. Therefore, TP53 co-mutations impact the natural history of EGFR-mutant NSCLC at least partially by allowing tolerance of a greater degree of genomic instability that results in both larger numbers of co-occurring truncal drivers as well as late sub-clonal diversification with focal emergence of high amplitude amplifications and deletions in mediators of therapeutic resistance103. In keeping with a more complex genomic landscape and a larger burden of clonal or sub-clonal co-drivers, multiple clinical studies have identified TP53 co-alterations as a negative prognostic marker in EGFR-mutant LUAD and a consistent predictor of worse clinical outcomes following EGFR TKI therapy72, 73, 111–116.
Mutational inactivation of RB1 is a clonal and early genetic event in 9.5%−12.5% of EGFR-mutant LUAD72, 73, 103, 117. The majority of RB1-mutant tumors also harbor TP53 co-alterations, underscoring the critical contributions of these archetypal tumor suppressor genes to cell cycle control. TP53 and RB1 co-mutations mark the earliest ancestors of EGFR-mutant LUAD that transform to small cell carcinoma following exposure to EGFR TKIs and dramatically increase the risk of small cell transformation, although loss of RB1 is insufficient to directly induce neuroendocrine trans-differentiation.117–119. Alterations in other regulators of G1/S cell cycle transition including amplification of CDK4, CDK6 and CCNE1 are prevalent and appear to be enriched in tumors that express the EGFRT790M gatekeeper mutation that confers TKI resistance, although data regarding their preferential occurrence in EGFR-mutant compared to EGFR wild-type LUAD are less consistent between studies73. Similarly, genomic alterations –most commonly deletion events - in the CDKN2A and CDKN2B genes are observed in ~24.6% and 20.2% of EGFR-mutant tumors and these alterations are typically truncal, further underscoring the significance of G1/S checkpoint dysregulation in the early stages of lung carcinogenesis driven by mutant EGFR72, 73, 103.
Activating mutations in CTNNB1 represent one of the most consistently co-selected alterations in EGFR-mutant LUAD across different studies. CTNNB1 mutations are rare in early-stage EGFR-mutant LUAD (1.8% in the TCGA cohort) but their prevalence increases in late-stage tumors (5.3% - 9.6%), in agreement with earlier studies that identified a central role for WNT signaling in LUAD metastasis and experimental data demonstrating increased invasive potential of EGFR and CTNNB1 co-mutated NSCLC cells in vitro15, 34, 72, 73, 120, 121. However, in a mouse LUAD model driven by compound EgfrL858R/T790M mutations genetic deletion of Ctnnb1 reduced tumor burden indicating non-redundant functions in tumor initiation122. Interestingly, mutations in CTNNB1 have been reported to occur more frequently in LUAD with the EGFRT790M mutation following exposure to first or second generation EGFR TKIs suggesting enhanced genetic interaction in this setting73. Mutant EGFR has further been shown to directly tyrosine phosphorylate β-catenin resulting in its stabilization and nuclear accumulation120.
PIK3CA mutations, including classical kinase (H1074R and H1074L) and helical (E545K and E542K) domain mutations are observed in 9%−12.4% of advanced stage EGFR-mutant LUAD and, like CTNNB1 mutations, are encountered preferentially in advanced-stage tumors15, 34, 72, 73. In vitro, co-occurring PIK3CA mutations promote cellular invasion and migration whereas in vivo they are associated with worse overall survival in some studies but do not appear to impact response rates and progression-free survival with first or second line EGFR TKI therapy73, 115, 123.
NKX2–1 amplification is significantly enriched in EGFR-mutant LUAD and constitutes a classical example of a context-dependent genetic interaction. In mouse models of Kras-mutant LUAD, Nkx2–1 loss fosters metastasis and Nkx2–1 haplo-insufficiency promotes both initiation and progression of invasive mucinous adenocarcinomas124; therefore, in this genomic background Nkx2–1 functions as a tumor suppressor gene. In contrast, hemizygous Nkx2–1 loss suppresses EgfrL858R-driven lung carcinogenesis, indicating that sustained NKX2–1 expression is essential for tumor initiation downstream of mutant Egfr125. Mechanistically, NKX2–1 transactivates the receptor ROR1, which directly binds to EGFR and sustains EGFR–ERBB3 heterodimerization, ERBB3 phosphorylation and pro-survival PI3K–AKT signaling; in addition, ROR1 can interact with and phosphorylate SRC, providing a parallel pathway to AKT activation126. Thus, the function of NKX2–1 as a lineage survival oncogene in EGFR-mutant NSCLC provides a plausible explanation for its preferential amplification in this oncogenic subgroup.
ALK, ROS1, RET and other oncogenic fusion-driven molecular subgroups
Recent studies have also begun to shed light on the co-mutation landscape and genomic architecture of LUAD driven by oncogenic fusions15, 127, although the clinical significance of co-alterations in this setting is less well characterized. Interestingly, advanced-stage ALK-rearrangement -positive LUAD are enriched in somatic alterations in CDKN2A (32.5%) and CDKN2B (26.5%), but are less likely to harbor TP53 alterations (23.8%−26.5%) compared with other driver subgroups15, 128. TP53 co-mutations promote genomic instability and are an independent negative prognostic factor in ALK-re-arrangement-positive LUAD, regardless of the type of systemic therapy used128–130. The prevalence of additional co-mutations in this group is low,128 and both the rarity of co-drivers and strong addiction to the ALK fusion oncoprotein may account for the long progression-free survival observed in patients with ALK-rearrangements with the potent and selective second and third generation ALK inhibitors alectinib, brigatinib and lorlatinib23, 26, 27. Similarly to LUAD with ALK fusions, both RET and ROS1 fusion-positive LUAD are characterized by high rates of concurrent CDKN2A loss (29.8% and 30.4% for RET and ROS1-rearranged tumors respectively) and CDKN2B loss (25% and 17.7% respectively) and relative paucity of TP53 mutations, although the frequency of TP53 mutations appears to be somewhat higher compared to ALK-rearranged tumors (34.6%−45.5% for RET-and 45.6% for ROS1- rearranged tumors)15, 131. The key finding that TP53 somatic mutations are underrepresented across LUAD driven by different oncogenic fusions was validated in a subsequent study that further identified frequent bi-allelic SETD2 deletions in this group127. The functional consequence of these associations is currently incompletely understood. The co-alteration spectrum of LUAD driven by NRG1 or NTRK1 fusion events has not been elucidated to date.
Other oncogenic subgroups.
A distinct pattern of co-occurring alterations is observed in LUAD driven by MET exon 14 skipping mutations. Specifically, these tumors are characterized by highly significant enrichment of MDM2 and CDK4 amplification (41.6%) compared with other driver oncogenes, as well as amplification of MET itself15. In contrast, mutations in TP53 (33.57%) are under-represented, whereas loss of CDKN2A (24.1%) and CDKN2B (17.5%) occur with similar frequencies to that in the overall population of patients with NSCLC15. The spectrum of co-occurring alterations in BRAF-mutant NSCLC mirrors the background frequency of alterations in TP53 (53.3%), LKB1 (16.2%), ATM (5.8%), NF1 (6.9%), PIK3CA (6.6%), KEAP1 (6.6%), MYC (10.8%), NKX2–1 (7.3%), although alterations in RB1, MDM2, CDKN2A (16.6%) and CDKN2B (11.2%) are less frequent within this molecular subgroup15. Finally, patients with ERBB2-mutant NSCLC exhibit preferential amplification of NKX2–1 (19.4%) and ERBB2 itself (14.4%) as well as frequent mutations in RB1 (8.9%), but the frequencies of co-mutations in TP53 (51.7%), CDKN2A (27.2%), CDKN2B (17.2%), PIK3CA (5%), CTNNB1 (4.4%) and MDM2 amplification (7.2%) are similar to that observed in the overall population of patients with NSCLC15.
Effects on the immune microenvironment
In addition to their impact on cell-autonomous cancer hallmarks, co-mutations can also shape the NSCLC microenvironment and determine its immune contexture (Figure 3). Inactivating LKB1 genomic alterations, present in ~25% of KRAS-mutant LUAD, have emerged as a major driver of the cold, non-T cell-inflamed microenvironment in NSCLC, characterized by paucity of infiltrating CD3+, CD4+ and CD8+ T-cells and low tumor cell expression of PD-L1, despite intermediate to high tumor mutational burden (TMB)67, 68, 132–134. These findings are recapitulated in the KrasLSL-G12D/+; Lkb1Fl/Fl GEMM, where Cre-mediated Lkb1 ablation triggers marked influx of tumor-associated neutrophils with T cell suppressive properties including increased expression of Arginase 1 (ARG1) and Interleukin 10 (IL-10)135. Mechanistically, Lkb1 loss in this model results in altered tumor cytokine milieu with increased expression of interleukin 1β (IL-1β), IL-6, CXCL7 and G-CSF that foster myeloid cell recruitment135. In addition, LKB1 inactivation induces epigenetic repression of STING (also known as TMEM173), thus promoting insensitivity to cytosolic dsDNA accumulation136. Silencing of STING in this context is triggered by enhanced activity of the EZH2 and DNMT1 methyltransferases due – at least in part- to increased production of SAM through diversion of glucose towards the serine biosynthetic pathway in LKB1-deficient cells87, 136. Increased expression of MYC has also been observed following LKB1 loss137 and may provide an additional mechanistic clue to the immune inert phenotype of LKB10-deficient NSCLC because IL-23 and CCL9-mediated inflammation and exclusion of B cells, T cells and NK cells have been reported to underpin the strong oncogenic cooperation between KRAS and MYC in lung cancer pathogenesis138. Finally, LKB1 inactivation has also been reported to impinge on non-immune components of the microenvironment of KrasG12D-mutant mouse tumors, including increased collagen deposition as a result of elevated lysyl oxidase (LOX) expression and effects on angiogenesis139, 140.
Figure 3. Impact of co-mutations on the microenvironment of KRAS-mutant lung adenocarcinoma.
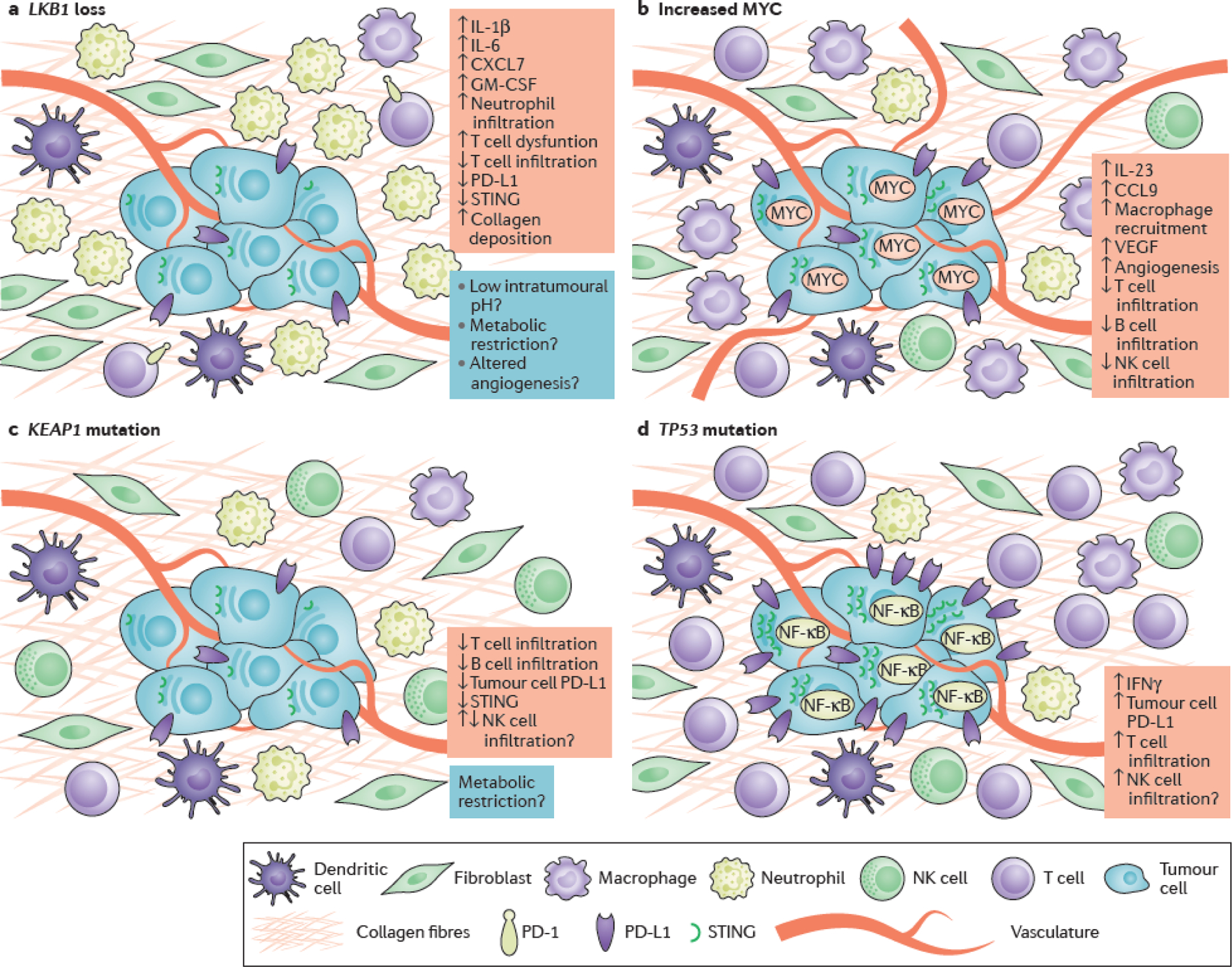
Schematic representation of co-mutation-associated changes in the immune and non-immune microenvironment of KRAS-mutant lung adenocarcinoma (LUAD). (a) LKB1 inactivation promotes epigenetic suppression of STING and insensitivity to cytosolic DNA that accumulates in the cytoplasm of KRAS- and LKB1-mutant (KL) cells due to dysfunctional mitochondria136. KL tumors are further characterized by a pro-inflammatory cytokine milieu with accumulation of immunosuppressive neutrophils, marked paucity of CD4+ and CD8+ T-cells and evidence of T-cell exhaustion68, 135. The potential contributions of immune cell metabolic restriction, altered angiogenesis and acidification of the tumor microenvironment (highlighted in blue) to the immune-inert phenotype of KL tumors remain as yet unexplored, but represent plausible directions for future study. (b) MYC fosters immune evasion of murine KrasG12D- driven LUADs through IL-23- mediated expulsion of T, B and NK cells and CCL9-mediated macrophage recruitment and secretion of immunosuppressive VEGF138. (c) KEAP1 mutations, which frequently co-occur with mutations in LKB1, particularly in the context of KRAS-mutant LUAD, have also been associated with low intra-tumoral density of infiltrating T- and B- lymphocytes, although the possible role of KEAP1 loss on NK cell infiltration remains unclear141. Stabilization of NRF2 as a result of KEAP1 inactivation may further promote reduced expression of STING through post-transcriptional regulation142. (d) Finally, somatic TP53 mutations have been shown to mediate NF-κB pathway activation in Kras-mutant murine models of LUAD146. Although TP53 mutations have been associated with reduced production of chemokines required for the recruitment of NK and T cells in some models and human tumors, in the context of KRAS-mutant LUAD TP53 co-alterations promote an inflamed tumor immune microenvironment with increased production of interferon γ (IFNγ) and increased expression of PD-L1 on the surface of tumor cells67, 68, 186.
Inactivating mutations in KEAP1 have also been associated with an altered NSCLC immune microenvironment134. In a conditional GEMM of LUAD (Keap1Fl/Fl;PtenFl/Fl), co-deletion of Keap1 and Pten resulted in immunologically cold tumors, akin to Lkb1-mutant NSCLC141. Interestingly, NRF2 was recently identified as a negative regulator of STING expression via effects on STING mRNA stability142 thus suggesting a tantalizing mechanistic connection between the effects of KEAP1 and LKB1 inactivation that warrants further study. Additional immune phenotypes may be uniquely associated with KEAP1 loss; for example, increased peri-tumoral accumulation of natural killer (NK) cells in KEAP1-mutant tumors was reported in a cohort of surgically resected early-stage LUAD132.
Finally, TP53 co-mutations are associated with an inflamed tumor immune microenvironment and increased tumor cell PD-L1 expression in KRAS-mutant NSCLC and GEMMs. This is at least in part due to activation of the nuclear factor κB (NF-κB) pathway driven by p53 loss, as well as increased tolerance of a higher mutational burden that may ostensibly result in enhanced immunogenicity due to increased neoantigen load143, 144, 145, 146.
The impact of co-mutations on other oncogene-driven subgroups of NSCLC, including those driven by EGFR mutations, ALK, ROS1 and RET translocations, as well as ERBB2 and MET exon 14 skipping mutations has not hitherto been determined and represents an area of active investigation. This will be particularly pertinent for BRAF-mutant NSCLCs, which are characterized by high tumor cell PD-L1 expression and more favorable clinical response to PD-1 and PD-L1 inhibitors147, 148.
Effects on drug sensitivity
Large-scale efforts aimed at linking tumor genomic alterations with sensitivity to cytotoxic and targeted therapies have uncovered a wealth of pharmacogenomic interactions in NSCLC and other cancer types149–152. These seminal high-throughput studies yielded multiple novel associations but also highlighted challenges in therapeutic response modeling that underscore the genomic complexity and biological heterogeneity of cancer. Interestingly, logic models – generated using the LOBICO (“Logic Optimization for Binary Input to Continuous Output”computational approach- that combine multiple input features such as mutations in cancer genes, gene fusions, recurrent copy number aberrations and binarized pathway activity scores (derived from gene expression profiling outperform single-gene models for prediction of drug sensitivity153, 154. Thus, co-occurring alterations can function as robust, and in many settings more precise, biomarkers of therapeutic response than single-gene predictors.
Chemical and genetic screens in panels of molecularly annotated NSCLC cell lines as well as candidate target approaches have uncovered several KRAS co-mutation-driven molecular dependencies and collateral vulnerabilities. KL NSCLC cell lines are characterized by unique sensitivity to depletion of multiple components of the coatomer 1 (COPI) complex and pharmacological inhibition of lysosomal acidification (for example by exposure to bafilomycin A) as a result of critical dependence on lysosomal macromolecule degradation for supply of TCA cycle substrates155. Other studies have linked LKB1 loss with enhanced sensitivity to energetic stress triggered by the biguanides metformin and phenformin156 or the combination of phenformin with the mTOR inhibitor MLN0128157, as well as to endoplasmic reticulum stress induced by 2-deoxy-D-glucose158. Enhanced dependence on nucleotide (and especially dTTP) synthesis further underpins the selective sensitivity of KL cells to deoxythymidylate kinase (DTYMK) depletion and to combined treatment with gemcitabine - a deoxycytidine analog that inhibits DNA synthesis and further depletes dNTP pools by targeting ribonucleotide reductase159- and CHK1 inhibitors, that abrogate the CHK1-mediated checkpoint response to replicative stress160, 161. Furthermore, LKB1 deficient cells are selectively vulnerable to inhibition of the ATP1A1 Na+/K+-ATPase by cardiac glycosides162 and to several structurally distinct inhibitors of the HSP90 family of molecular chaperones67. While some of these vulnerabilities are associated with LKB1 inactivation irrespective of concurrent KRAS mutations, others, such as addiction to lysosomal enzymatic degradation, appear to be specific to the KL oncogenotype and thus represent de facto co-mutation-dependent vulnerabilities. In contrast, KL lung tumors exhibit resistance to MEK inhibitors in mouse models and LKB1 deficiency by immunohistochemistry is associated with lack of benefit from the addition of the vascular endothelial growth factor A (VEGFA) inhibitor bevacizumab to platinum doublet chemotherapy163, 164. Beyond the KL genotype, KRAS and KEAP1 co-altered NSCLC cells display chemically tractable selective dependence on GLUT8-mediated uptake for effective diversion of glucose towards the serine biosynthetic pathway165 and rely on glutaminolysis for TCA cycle anaplerosis; thus, they are selectively sensitive to glutaminase inhibition in both cell line and mouse models92. In agreement with the role of NRF2 as a transactivator of antioxidant as well as phase II detoxifying and cytoprotective enzymes that mediate resistance to cytotoxic chemotherapy, KEAP1 co-mutations are associated with resistance to multiple inhibitors of oncogenic kinases within the receptor tyrosine kinase–MAPK pathway in vitro166 and significantly worse clinical outcomes with platinum-doublet chemotherapy in KRAS-mutant LUAD99 or, as shown in preliminary data, with chemo-immunotherapy using pemetrexed-carboplatin (or cisplatin) plus the PD-1 antibody pembrolizumab in non-squamous NSCLC167. Finally, ATM co-alterations increase the sensitivity of KRAS and BRAF-mutant NSCLC cell lines to MEK inhibitor-induced apoptosis and genetic deletion of Atm is associated with increased sensitivity to poly(ADP-ribose) polymerase (PARP) and ATR inhibitors as well as to radiation therapy in mouse models of Kras-mutant LUAD168, 169. Notably, despite apparent mutual exclusivity between classical activating mutations in RAS pathway genes, oncogenic co-operativity has been observed between atypical, weakly activating mutations170. In this setting signaling inputs from multiple co-altered RAS pathway genes coordinately contribute towards thresholds of oncogenic activity that are critical for transformation and tumor maintenance but also bestow therapeutic vulnerabilities; for example, co-mutations in NF1 and RASA1, encoding two critical RAS pathway GTPase-activating proteins (GAPs), drive addiction to the MEK–ERK signaling axis and confer enhanced sensitivity to MEK inhibitors in a subset of both LUAD and LUSC171, 172.
Of particular relevance is the impact of co-occurring alterations on clinical outcomes with EGFR and ALK TKIs as well as other targeted therapies. TP53 co-mutations have consistently been associated with shorter progression-free survival following upfront treatment with 1st or 2nd generation EGFR TKIs and there is further evidence that they adversely impact clinical outcomes with the third generation, mutant-selective EGFR TKIs, in patients whose tumors have acquired the EGFRT790M gatekeeper mutation72, 115. In a study of 200 EGFR-mutant patients with extensive molecular profiling at baseline, pre-existing MET (present in 2% of cases) or ERBB2 (4% prevalence) amplification were also associated with significantly shorter progression-free survival with first-line 1st or 2nd generation EGFR TKI therapy, whereas among patients with acquired EGFRT790M mutation from a distinct cohort, co-mutations in RB1 and PTEN and amplification of MDM2 were independently associated with worse progression-free survival following treatment with 3rd generation EGFR TKIs72, 173. Co-alterations in BRAF, CDKN2A, CDKN2B, fibroblast growth factor receptor 3 (FGFR3) and amplification of MET and EGFR itself were all enriched in patients with acquired resistance to EGFR TKIs compared to pre-treatment tumors, indicating roles in mediating the drug resistant phenotype. Interestingly, co-mutations in PIK3CA don’t impact response to first, second or third generation EGFR TKIs113, 123, 174. Importantly, co-occurring clonal alterations in both TP53 and RB1, present in ~9 % of EGFR-mutant LUAD at baseline, substantially increase the risk of transformation to small cell carcinoma upon treatment with EGFR TKI; therefore co-occurring alterations can affect not only the likelihood and duration of response to targeted therapy but also impact mechanisms of acquired resistance117. It is currently unknown whether the likelihood of acquisition of an EGFRT790M secondary resistance mutation can also be influenced by the co-mutation status of the tumor.
In keeping with their prominent role in shaping tumor immunobiology and immune contexture, co-occurring genomic alterations can further impact clinical response to immune checkpoint inhibitors. This is particularly evident in KRAS-mutant LUAD. Inactivating somatic mutations in LKB1, present in ~25% of KRAS-mutant LUAD, have emerged as a major genomic driver of primary resistance to PD-1 and PD-L1 inhibition, despite KL LUAD harboring intermediate to high TMB68. Importantly, the negative impact of LKB1 genomic alterations on clinical outcomes with anti-PD1 or anti-PD-L1 therapy extends to PD-L1 positive tumors68. Therefore, somatic genomic alterations may represent independent predictors of clinical outcomes with immune checkpoint inhibitors, in addition to previously established markers such as PD-L1 expression and TMB. De novo resistance to immune checkpoint blockade following LKB1 loss is further associated with primary resistance to combined anti-PD-1 and anti-CTLA4 therapy with nivolumab and ipilimumab175. In contrast, KRAS-mutant tumors bearing co-mutations in TP53 exhibit high rates of clinical response to PD-1 axis immunotherapy and markedly improved progression-free and overall survival compared to KL68. In addition to LKB1, co-mutations in KEAP1 have also been implicated in de novo resistance to PD-1 blockade99 and both LKB1 and KEAP1 are associated with inferior clinical outcomes with chemo-immunotherapy with pemetrexed-carboplatin (or cisplatin)-pembrolizumab, particularly among PD-L1-positive and TMB-high tumors167. In this context, double LKB1;KEAP1 mutant tumors exhibita particularly recalcitrant clinical response phenotype167. Finally, mutations in PTEN have also been nominated as a candidate driver of primary resistance to immune checkpoint inhibition in NSCLC, in agreement with similar reports in melanoma175, 176.
Conclusions and perspectives.
As our understanding of the genomic landscape of NSCLC deepens, broad tumor genomic profiling becomes increasingly accessible and our therapeutic armamentarium continues to evolve, there is growing appreciation that the current single oncogenic driver model fails to adequately capture the clinical complexity of NSCLC and warrants revision. Co-occurring genomic alterations in oncogenic drivers and tumor suppressor genes have emerged as major tenets of the molecular diversity of NSCLC. Antecedent knowledge of a key set of major co-mutations may therefore allow more granular insights into NSCLC biology; facilitate development of improved clinical response prediction algorithms; anticipate and forestall the emergence of acquired resistance; and enable development of novel, highly personalized therapeutic approaches in the next wave of precision oncology clinical trials. Based on accumulated and emerging evidence we propose a next-generation, dynamic model for the molecular classification of NSCLC that encompasses the molecular and clinical diversity affected by co-mutations (Figure 4). Immediate priorities and challenges for the future are to catalog, functionalize and systematically evaluate the therapeutic utility of the full spectrum of co-occurring alterations in NSCLC and, simultaneously, to expeditiously translate the most robust and critical insights into more precise therapeutic strategies that yield improved clinical outcomes for NSCLC patients. These tasks will require novel computational tools and high throughput in vivo platforms as well as large, prospectively assembled collaborative clinical datasets and efficient and flexible umbrella clinical trial [G] designs.
Figure 4. Next-generation model for the molecular stratification of lung adenocarcinoma.
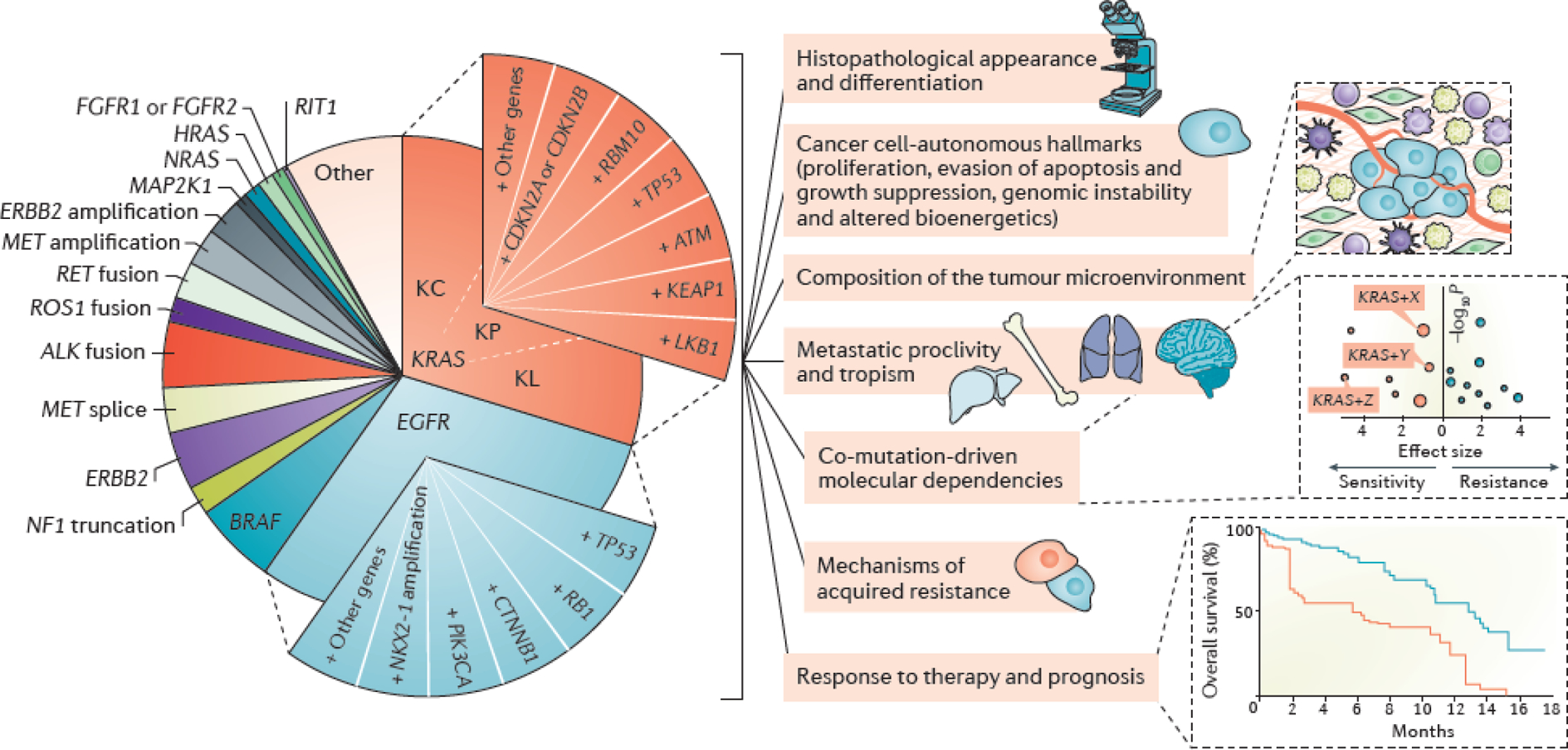
Oncogenic subgroups of lung adenocarcinoma (LUAD) are divided into smaller subsets on the basis of key co-occurring genomic alterations. Co-mutations constitute major determinants of tumor molecular diversity and can impact both tumor cell-autonomous and non-cell-autonomous cancer hallmarks; determine prognosis; predict response to systemic therapies and influence mechanisms of innate and acquired resistance. For simplicity, only KRAS and EGFR co-alterations are depicted graphically. For KRAS-mutant LUADs the previously identified KL, KP, and KC transcriptome-based subgroups are also indicated67; co-mutations in LKB1, KEAP1 and ATM are significantly enriched in the KL subgroup, whereas co-occurring alterations in TP53 and bi-allelic inactivation of CDKN2A/CDKN2B are hallmarks of the KP and KC subgroups respectively. Co-mutations in RBM10 don’t appear to exhibit predilection for any of the three KRAS transcriptomic subgroups. It should therefore be noted that several of the reported co-alterations within oncogene-defined groups are not mutually exclusive. Although co-mutation-defined cohorts are represented as slices of equal size, both the spectrum and prevalence of individual co-mutations evolve according to disease stage, prior treatment exposures, immune editing and the mutational processes that are operational at each stage of carcinogenesis.
Acknowledgements
We acknowledge support from a Department of Defense Lung Cancer Research Program Career Development Award (to F.S.); RP160652 Cancer Prevention Research Institute of Texas Multi-Investigator Research Award (to J.V.H.); 1R01 CA205150 (to J.V.H.); Lung SPORE grant 5 P50 CA070907; a Stand Up To Cancer-American Cancer Society Lung Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT17-15). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. In addition, we would like to acknowledge generous philanthropic contributions to the University of Texas MD Anderson Lung Cancer Moonshots Program.
Glossary
- Platinum-doublet chemotherapy
Cisplatin or carboplatin-based combinations with a second chemotherapeutic agent, most commonly pemetrexed (LUAD), taxanes (LUAD or LUSC) and gemcitabine (LUSC)
- Objective response
Measurable decrease in tumor burden of a predefined amount in response to therapy
- Complete responses
The disappearance of all signs of cancer in response to treatment, including both target and non-target lesions (with reduction of all lymph nodes to <10mm in short axis), without emergence of any new lesions
- A less strict pattern of mutual exclusivity in which combinations of somatic mutations in different genes occur less frequently than expected but do not result in toxic functional effects and can therefore rarely co-occur in the same cell
- Cold tumor immune microenvironment
Tumor microenvironment characterized by lack or paucity of infiltrating T cells
- Trans-differentiation
Conversion of one differentiated somatic cell type to another without passage through an intermediate pluripotent or progenitor cell state
- Umbrella clinical trial
A clinical trial that assesses multiple targeted therapeutic strategies in a single cancer type
Footnotes
Competing interests
J.V.H. reports royalties and licensing fees from Spectrum Pharmaceuticals and Biotree; research support from AstraZeneca, Bayer, GlaxoSmithKline and Spectrum Pharmaceuticals; Advisory Committee membership from AstraZeneca, Boehringer-Ingelheim, Exelixis, Genentech, GlaxoSmithKline, Guardant Health, Hengrui Therapeutics, Eli Lilly, Novartis, Spectrum Pharmaceuticals, EMD Serono and Synta Pharmaceuticals. F.S. reports honoraria from Bristol-Myers Squibb, outside the scope of this work and consultancy fees from Tango Therapeutics.
Peer review information
Nature Reviews Cancer thanks F. Hirsch, M. Ladanyi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Lynch TJ et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350, 2129–39 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Paez JG et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–500 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Soda M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–6 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kwak EL et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363, 1693–703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergethon K et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30, 863–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohno T et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18, 375–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaishnavi A et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med 19, 1469–1472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Cuesta L et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 4, 415–22 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Marchetti A et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 29, 3574–9 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Paik PK et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 29, 2046–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nieto P et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature 548, 239–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens P et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–6 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Ma PC et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res 65, 1479–88 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Kong-Beltran M et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 66, 283–9 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Frampton GM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 5, 850–9 (2015). [DOI] [PubMed] [Google Scholar]; This paper and Jordan et al. (2017) are the largest reported studies of comprehensive genomic profiling in patients with advanced lung adenocarcinoma. [Au: Edited to avoid use of reference numbers in case these need to be renumbered at proof for any reason. I have also put the annotation under reference 59.]
- 16.Mok TS et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361, 947–57 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Zhou C et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12, 735–42 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Rosell R et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13, 239–46 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Yang JC et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16, 141–51 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Soria JC et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 378, 113–125 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Mok TS et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 376, 629–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok TS et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J Clin Oncol 36, 2244–2250 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Peters S et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med 377, 829–838 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Solomon BJ et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371, 2167–77 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Soria JC et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 389, 917–929 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Camidge DR et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med 379, 2027–2039 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Solomon BJ et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol 19, 1654–1667 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Planchard D et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 18, 1307–1316 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Mitsudomi T et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 11, 121–8 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Maemondo M et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362, 2380–8 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Sequist LV et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31, 3327–34 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Ramalingam SS et al. Dacomitinib versus erlotinib in patients with advanced-stage, previously treated non-small-cell lung cancer (ARCHER 1009): a randomised, double-blind, phase 3 trial. Lancet Oncol 15, 1369–78 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Rekhtman N, Ang DC, Riely GJ, Ladanyi M & Moreira AL KRAS mutations are associated with solid growth pattern and tumor-infiltrating leukocytes in lung adenocarcinoma. Mod Pathol 26, 1307–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw AT et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 371, 1963–71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dueck D & Frey BJ Non-metric affinity propagation for unsupervised image categorization. 2007 Ieee 11th International Conference on Computer Vision, Vols 1–6, 198–205 (2007). [Google Scholar]
- 37.Kim J et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 538, 114–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borghaei H et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 373, 1627–39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carbone DP et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med 376, 2415–2426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fehrenbacher L et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–46 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Herbst RS et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–50 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Horn L et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J Clin Oncol, JCO2017743062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rittmeyer A et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackman DM et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res 12, 3908–14 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Choi YW et al. EGFR Exon 19 Deletion is Associated With Favorable Overall Survival After First-line Gefitinib Therapy in Advanced Non-Small Cell Lung Cancer Patients. Am J Clin Oncol 41, 385–390 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Robichaux JP et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 24, 638–646 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riess JW et al. Diverse EGFR Exon 20 Insertions and Co-Occurring Molecular Alterations Identified by Comprehensive Genomic Profiling of NSCLC. J Thorac Oncol 13, 1560–1568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Childress MA et al. ALK Fusion Partners Impact Response to ALK Inhibition: Differential Effects on Sensitivity, Cellular Phenotypes, and Biochemical Properties. Mol Cancer Res 16, 1724–1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin JJ et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J Clin Oncol 36, 1199–1206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drilon A et al. A Phase I/Ib Trial of the VEGFR-Sparing Multikinase RET Inhibitor RXDX-105. Cancer Discov 9, 384–395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ihle NT et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 104, 228–39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haigis KM KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shepherd FA et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 31, 2173–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu HA et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol 10, 431–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ostrem JM & Shokat KM Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 15, 771–785 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jordan EJ et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov 7, 596–609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper and Frampton et al. (2015) are the largest reported studies of comprehensive genomic profiling in patients with advanced lung adenocarcinoma.
- 60.Ding L et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campbell JD et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48, 607–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imielinski M et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mina M et al. Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer Cell 32, 155–168 e6 (2017). [DOI] [PubMed] [Google Scholar]; This study descibes a novel algorithmic approach for assessment of cancer evolutionary dependencies and represents the most comprehensive pan-cancer analysis of co-alteration patterns reported to date.
- 64.Campbell PJ Cliques and Schisms of Cancer Genes. Cancer Cell 32, 129–130 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–74 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Skoulidis F et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 5, 860–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identified co-alterations in LKB1, TP53 and CDKN2A/CDKN2B as key determinants of the molecular diversity of KRAS-mutant LUAD and was the first report linking LKB1 co-mutations with a non- T cell-inflamed tumor immune microenvironment, in LUAD.
- 68.Skoulidis F et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified inactivating LKB1 genomic alterations as a major driver of de novo resistance to PD-1 axis blockade in KRAS-mutant LUAD.
- 69.Johnson L et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–6 (2001). [DOI] [PubMed] [Google Scholar]
- 70.Jackson EL et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 65, 10280–8 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Fisher GH et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 15, 3249–62 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu HA et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin Cancer Res 24, 3108–3118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the clinical impact of co-occurring alterations in a large cohort of treatment-naïve patients with advanced EGFR-mutant LUAD.
- 73.Blakely CM et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 49, 1693–1704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper examines the clinical impact of co-occurring alterations in a large cohort of patients with EGFR-mutant LUAD and available ctDNA-based molecular profiling.
- 74.Jamal-Hanjani M et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]; This landmark study established a census of clonal and subclonal alterations in early stage NSCLC and identified copy number heterogeneity as an independent predictor of short relapse-free survival following surgical resection.
- 75.Ortmann CA et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 372, 601–612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swanton C Cancer evolution constrained by mutation order. N Engl J Med 372, 661–3 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Henderson S, Chakravarthy A, Su X, Boshoff C & Fenton TR APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep 7, 1833–41 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Zhang J et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Bruin EC et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marty R et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 171, 1272–1283 e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brunet JP, Tamayo P, Golub TR & Mesirov JP Metagenes and molecular pattern discovery using matrix factorization. Proceedings of the National Academy of Sciences of the United States of America 101, 4164–4169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Devarajan K Nonnegative Matrix Factorization: An Analytical and Interpretive Tool in Computational Biology. Plos Computational Biology 4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Monti S, Tamayo P, Mesirov J & Golub T Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning 52, 91–118 (2003). [Google Scholar]
- 84.Ji H et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–10 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Li F et al. LKB1 Inactivation Elicits a Redox Imbalance to Modulate Non-small Cell Lung Cancer Plasticity and Therapeutic Response. Cancer Cell 27, 698–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang H et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun 8, 14922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kottakis F et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov 3, 870–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim J et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identifies a co-mutation-specific metabolic dependence of KRAS;LKB1 co-altered cells on the urea cycle enzyme CPS1 in order to maintain pyrimidine pools.
- 90.Jaramillo MC & Zhang DD The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27, 2179–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Babur O et al. Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol 16, 45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Romero R et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Singh A et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest 123, 2921–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeNicola GM et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeNicola GM et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galan-Cobo A et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma Cancer Research In press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ying H et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jeon SM, Chandel NS & Hay N AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arbour KC et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 24, 334–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feldser DM et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468, 572–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Junttila MR et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468, 567–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Muzumdar MD et al. Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nat Commun 7, 12685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nahar R et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat Commun 9, 216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a comprehensive evaluation of the clonal and subclonal genomic landscape of early-stage EGFR-mutant LUAD in patients of Asian descent [Au:OK?].
- 104.Schmitt A et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res 77, 3040–3056 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Petersen LF et al. Loss of tumour-specific ATM protein expression is an independent prognostic factor in early resected NSCLC. Oncotarget 8, 38326–38336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bechara EG, Sebestyen E, Bernardis I, Eyras E & Valcarcel J RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 52, 720–33 (2013). [DOI] [PubMed] [Google Scholar]
- 107.Zhao J et al. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci Rep 7, 40488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rogers ZN et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet 50, 483–486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a novel in vivo platform for the rapid evaluation of the functional impact of co-occurring alterations in GEMMs of KRAS-mutant LUAD.
- 109.Schuster K et al. Nullifying the CDKN2AB locus promotes mutant K-ras lung tumorigenesis. Mol Cancer Res 12, 912–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Canale M et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res 23, 2195–2202 (2017). [DOI] [PubMed] [Google Scholar]
- 112.Sato S et al. Impact of Concurrent Genomic Alterations Detected by Comprehensive Genomic Sequencing on Clinical Outcomes in East-Asian Patients with EGFR-Mutated Lung Adenocarcinoma. Sci Rep 8, 1005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.VanderLaan PA et al. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer 106, 17–21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Labbe C et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 111, 23–29 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Kim Y et al. Concurrent Genetic Alterations Predict the Progression to Target Therapy in EGFR-Mutated Advanced NSCLC. J Thorac Oncol 14, 193–202 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Jakobsen JN, Santoni-Rugiu E, Grauslund M, Melchior L & Sorensen JB Concomitant driver mutations in advanced EGFR-mutated non-small-cell lung cancer and their impact on erlotinib treatment. Oncotarget 9, 26195–26208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee JK et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J Clin Oncol 35, 3065–3074 (2017). [DOI] [PubMed] [Google Scholar]; This paper reported that pre-existing co-alterations in TP53 and RB1 can predict transformation to small cell carcinoma as a mechanism of acquired resistance to EGFR TKI therapy in EGFR-mutant LUAD.
- 118.Niederst MJ et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6, 6377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marcoux N et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J Clin Oncol 37, 278–285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nakayama S et al. beta-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res 74, 5891–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Malladi S et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pacheco-Pinedo EC et al. Wnt/beta-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J Clin Invest 121, 1935–45 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Eng J et al. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J Thorac Oncol 10, 1713–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Winslow MM et al. Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 473, 101–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maeda Y et al. Kras(G12D) and Nkx2–1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest 122, 4388–400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamaguchi T et al. NKX2–1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 21, 348–61 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Lee JJ et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 177, 1842–1857 e21 (2019). [DOI] [PubMed] [Google Scholar]
- 128.Kron A et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol 29, 2068–2075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alidousty C et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: a central role of TP53 mutations. J Pathol 246, 67–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aisner DL et al. The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations-The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res 24, 1038–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Michels S et al. Clinicopathological Characteristics of RET Rearranged Lung Cancer in European Patients. J Thorac Oncol 11, 122–7 (2016). [DOI] [PubMed] [Google Scholar]
- 132.Kadara H et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol 28, 75–82 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scheel AH et al. PD-L1 expression in non-small cell lung cancer: Correlations with genetic alterations. Oncoimmunology 5, e1131379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cristescu R et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koyama S et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res 76, 999–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kitajima S et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 9, 34–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first report that LKB1 loss results in impaired innate immune sensing of cytosolic DNA due to epigenetic repression of STING.
- 137.Partanen JI, Nieminen AI, Makela TP & Klefstrom J Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci U S A 104, 14694–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kortlever RM et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315 e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gao Y et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc Natl Acad Sci U S A 107, 18892–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Okon IS et al. Protein kinase LKB1 promotes RAB7-mediated neuropilin-1 degradation to inhibit angiogenesis. J Clin Invest 124, 4590–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Best SA et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab 27, 935–943 e4 (2018). [DOI] [PubMed] [Google Scholar]
- 142.Olagnier D et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 9, 3506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Petitjean A et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28, 622–9 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cha YJ, Kim HR, Lee CY, Cho BC & Shim HS Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer 97, 73–80 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Meylan E et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 462, 104–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dudnik E et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J Thorac Oncol 13, 1128–1137 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Negrao M et al. Driver Mutations are Associated with Distinct Patterns of Response to Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Journal of Thoracic Oncology 13, S733–S734 (2018). [Google Scholar]
- 149.Garnett MJ et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Seashore-Ludlow B et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov 5, 1210–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Iorio F et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reported that combinations of oncogenic alterations are better predictors of drug sensitivity than individual alterations.
- 154.Knijnenburg TA et al. Logic models to predict continuous outputs based on binary inputs with an application to personalized cancer therapy. Sci Rep 6, 36812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim HS et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 155, 552–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with McMillan et al. (2018), [Au:OK; I think this is about ref 155 and 165, and not 154 and 164?] this paper provides compelling evidence that co-occurring genomic alterations confer unique molecular dependencies and therapeutic vulnerabilities based on large scale chemical and genetic screens in panels of molecularly annotated NSCLC cell lines.
- 156.Shackelford DB et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Momcilovic M et al. Heightening Energetic Stress Selectively Targets LKB1-Deficient Non-Small Cell Lung Cancers. Cancer Res 75, 4910–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Inge LJ et al. LKB1 inactivation sensitizes non-small cell lung cancer to pharmacological aggravation of ER stress. Cancer Lett 352, 187–95 (2014). [DOI] [PubMed] [Google Scholar]
- 159.Wang J, Lohman GJ & Stubbe J Enhanced subunit interactions with gemcitabine-5’-diphosphate inhibit ribonucleotide reductases. Proc Natl Acad Sci U S A 104, 14324–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zegerman P & Diffley JF DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst) 8, 1077–88 (2009). [DOI] [PubMed] [Google Scholar]
- 161.Liu Y et al. Gemcitabine and Chk1 Inhibitor AZD7762 Synergistically Suppress the Growth of Lkb1- Deficient Lung Adenocarcinoma. Cancer Res 77, 5068–5076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kim N et al. Cardiac glycosides display selective efficacy for STK11 mutant lung cancer. Sci Rep 6, 29721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen Z et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper first established the concept of the co-clinical trial and identified that Lkb1 and Trp53 co-alterations can influence the clinical response of Kras-mutant lung cancer to chemotherapy and targeted therapy in mice [Au:OK?].
- 164.Bonanno L et al. LKB1 Expression Correlates with Increased Survival in Patients with Advanced Non-Small Cell Lung Cancer Treated with Chemotherapy and Bevacizumab. Clin Cancer Res 23, 3316–3324 (2017). [DOI] [PubMed] [Google Scholar]
- 165.McMillan EA et al. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 173, 864–878 e29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Kim et al. (2013), [Au:OK? Is this about ref 155 and 165, and not 154 and 164?] this paper provides compelling evidence that co-occurring genomic alterations confer unique molecular dependencies and therapeutic vulnerabilities based on large scale chemical and genetic screens in panels of molecularly annotated NSCLC cell lines.
- 166.Krall EB et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Skoulidis F e.a. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J Clin Oncol (2019). [Google Scholar]
- 168.Smida M et al. MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated. Nat Commun 7, 13701 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Torok JA et al. Deletion of Atm in Tumor but not Endothelial Cells Improves Radiation Response in a Primary Mouse Model of Lung Adenocarcinoma. Cancer Res 79, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stites EC, Trampont PC, Haney LB, Walk SF & Ravichandran KS Cooperation between Noncanonical Ras Network Mutations. Cell Rep 10, 840 (2015). [DOI] [PubMed] [Google Scholar]
- 171.Hayashi T et al. RASA1 and NF1 are Preferentially Co-Mutated and Define A Distinct Genetic Subset of Smoking-Associated Non-Small Cell Lung Carcinomas Sensitive to MEK Inhibition. Clin Cancer Res 24, 1436–1447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kitajima S & Barbie DA RASA1/NF1-Mutant Lung Cancer: Racing to the Clinic? Clin Cancer Res 24, 1243–1245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lai GGY et al. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor-Mutant Non-Small-Cell Lung Cancer. J Clin Oncol, JCO1800177 (2019). [DOI] [PubMed] [Google Scholar]
- 174.Wu SG, Chang YL, Yu CJ, Yang PC & Shih JY The Role of PIK3CA Mutations among Lung Adenocarcinoma Patients with Primary and Acquired Resistance to EGFR Tyrosine Kinase Inhibition. Sci Rep 6, 35249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hellmann MD et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 33, 843–852 e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Peng W et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 6, 202–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sanchez-Vega F et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ellrott K et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281 e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hoadley KA et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Benjamini Y & Hochberg Y Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Statistical Methodology 57, 289–300 (1995). [Google Scholar]
- 183.Sweis RF et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res 4, 563–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Seiwert TY et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res 21, 632–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–5 (2015). [DOI] [PubMed] [Google Scholar]; This seminal study hightlighted tumor-cell instrinsic oncogenic signaling - in this case activation of the WNT–β-catenin pathway - as a driver of immune evasion via establishment of a non-T cell-inflamed tumor immune microenvironment.
- 186.Spranger S & Gajewski TF Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 18, 139–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]