

Abstract
To report a rare case of Muller cell sheen dystrophy and to describe its clinical and diagnostic aspects. A 42-year-old woman presented with unilateral defective vision. Fundus evaluation revealed bilateral glistening retinal reflexes throughout the posterior pole with a wrinkled appearance in the right. Spectral Domain-OCT in the right showed abnormal internal limiting membrane, intraretinal schisis with serous detachment at macula. Angiography revealed staining along vessels. Electroretinogram in the affected eye was negative. At 4 months of follow up, vision dropped and intraretinal schisis increased. Family screening was negative.
Keywords: Cellophane, glistening retinal reflexes, internal limiting membrane, internal limiting membrane dystrophy, macular edema, muller cell, muller cell sheen dystrophy, negative electroretinogram, schisis
Muller cell sheen dystrophy, otherwise known as Familial Internal limiting membrane dystrophy has been described in literature to be a rare, heritable dystrophy. Dalma et al., in 1991, first described this abnormal cellophane like sheen of retina in ten patients belonging to a single family and termed it as “autosomal dominant, late-onset, cellophane-like sheen vitreomacular dystrophy”.[1] Later Polk and Gass et al. in 1997, described the clinical course and pedigree of five affected members of a family and suggested an autosomal dominant mode of inheritance and named it as “Familial Internal limiting membrane dystrophy”.[2] In 1998, Kellner et al. described the electrophysiological data of a single affected patient and named it as “Muller cell sheen dystrophy” highlighting Muller cell involvement in this disorder.[3] In 2013, Dalma et al. described SD-OCT alterations in the 10 patients they had described earlier.[4]
Case Report
A 42-year-old woman presented with diminution in vision in the right eye since 4 days. Snellen's best corrected visual acuity was 20/40 in the right and 20/20 in the left. She denied of any systemic conditions or any preceding illness. Anterior segment was quiet. Intraocular pressure was normal. Color vision was normal. Fundus examination [Fig. 1a] revealed bilateral glistening retinal reflexes throughout the posterior pole up to equator with a wrinkled cellophane appearance in the right. Spectral Domain-OCT in the right eye [Fig. 1b] showed abnormal internal limiting membrane (ILM) in folds, schitic spaces between ILM and nerve fiber layer, intraretinal schisis with serous detachment at macula. Left eye OCT revealed a normal fovea with an epiretinal membrane along inferior macula [Fig. 2c]. Ultrasonography demonstrated retinochoroidal complex measuring 1.86 mm. Axial length measured 22.41 mm. Fundus fluorescein angiography [Fig. 1c] in the right revealed normal early phase with hyperfluorescence and staining along vessels in late phases. It was normal in the left. Electroretinogram in the affected right eye had reduced b wave and left was normal [Fig. 3]. Systemic investigations demonstrated no inflammatory cause or any systemic illness. However, an empirical course of oral prednisolone was administered in tapering doses for two months. At 4 months of follow up, vision dropped to 20/120 in right. Fundus [Fig. 2a] revealed radiating cellophane reflexes with subretinal exudation at macula. Left eye remained the same. SD-OCT [Fig. 2b] demonstrated increased intraretinal schisis with subretinal exudation at macula with a new area of serous detachment temporal to macula.
Figure 1.
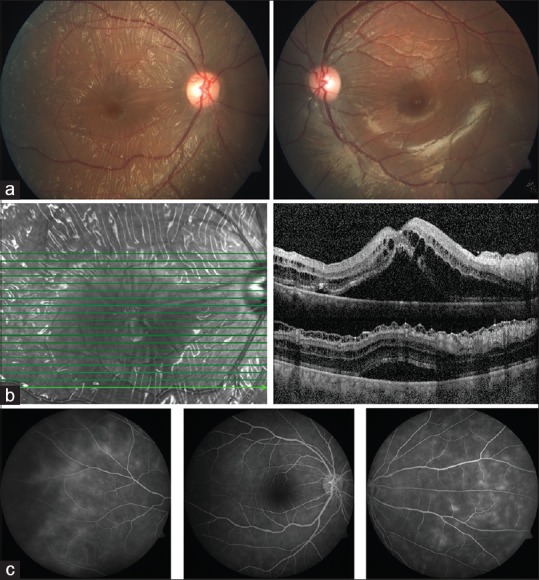
(At Presentation) (a) Fundus photograph in OD shows glistening cellophane retinal reflexes with folds in posterior pole up to equator. OS has retinal sheen. (b) SD-OCT in OD shows abnormal internal limiting membrane, schitic spaces between ILM, and Nerve fiber Layer, intraretinal schisis with serous detachment at macula (c) Fluorescein angiography in OD reveals small vessel leak in late phases
Figure 2.
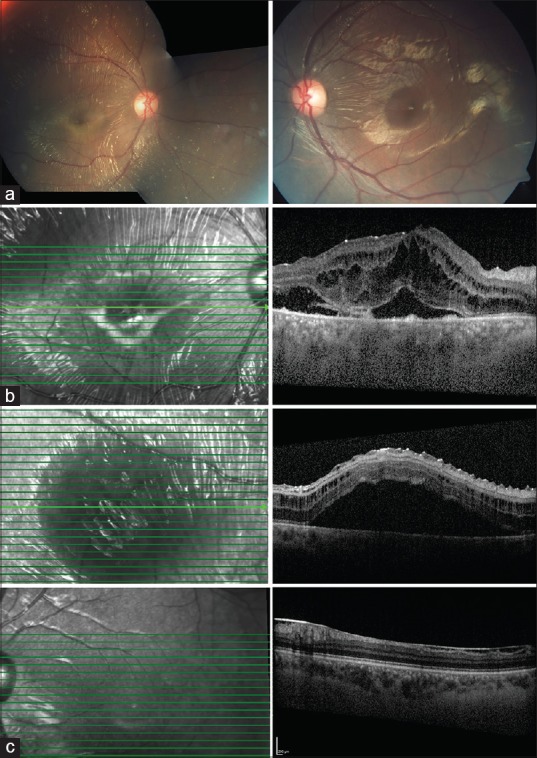
(At 4 months follow up) (a) OD has retinal reflexes, folds with subretinal exudation (b) SD-OCT in OD has increased Intraretinal schisis with a new area of serous detachment temporal to macula (c) OS has epiretinal membrane in inferior macula
Figure 3.
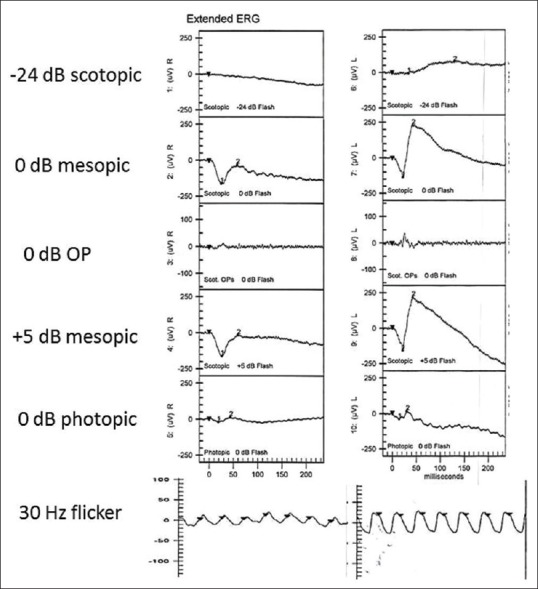
(ERG): OD negative ERG, OS normal
Discussion
We present here a case of a woman in her forties with bilateral retinal sheen with unilateral retinal folds, intraretinal schisis, small vessel leak with negative electroretinogram. All patients reported in literature had a late onset of presentation with an asymmetric clinical course. Our patient presented in her fourth decade with the right eye much more affected than the other. Left eye would probably undergo the same natural course as the right. Gass et al. in their pedigree analysis hypothesized an autosomal dominant pattern of inheritance.[2] No genetic locus has been defined yet. Family members could not be evaluated in our case.
In the posterior retina, ILM attains a thickness of 0.5-2.0 μm and it's thickest at fovea. Defective Muller cells cause abnormal basement membrane production in the outer aspect of ILM causing its thickening and undulation, more pronounced in the posterior pole due to thicker ILM.[2,3] Muller cells form the structural framework, so their derangement causes schitic spaces, which have been described to initially start in the inner layers of retina around the arcades and slowly progress towards the outer layers, finally causing serous detachment.[4] Dalma et al. described that only when cystic spaces reached outer nuclear layer, fovea was involved and macular edema appeared.[4] Visual loss was predominantly due to serous detachment at macula. In later stages, it progresses to IS/OS disruption and foveal atrophy. Our case presented with sudden onset diminution in vision which was due to occurrence of subretinal fluid at macula, schitic changes would probably have been persisting since a long time. Muller cells basement membrane surrounds the capillaries which explains small vessel leak on angiography and subsequent schisis formation.[2]
Electroretinogram demonstrates b wave reduction and delayed implicit time which is explained by widespread muller cell dysfunction. The asymmetry in ERG also correlates with the clinical picture. Kellner et al. in his article on electrophysiological evaluation of visual loss demonstrated progressive reduction in ERG in both eyes over 1 year, in which one eye ERG was normal at first.[3]
Differential diagnosis includes Fenestrated sheen macular dystrophy, idiopathic epiretinal membranes and cellophane reflex in certain retinal dystrophies. Polk and Gass et al. mentioned the use of nonsteroidal anti-inflammatory agents, steroids, acetazolamide and grid laser photocoagulation in their patients, but did not have any significant improvement.[2] Vitrectomy with internal limiting membrane peeling were performed in 3 eyes but failed to demonstrate a significant response.[2] Renner et al. in 2014 demonstrated successful outcome following vitrectomy with internal limiting membrane peeling in a patient, though schisis reduced following surgery, the schitic spaces returned at one year follow up.[5] No effective treatment is proven for this condition.
Muller cell sheen dystrophy or Internal limiting membrane dystrophy is a rare heritable dystrophy, characterized by late onset, retinal sheen with retinal folds throughout fundus, intraretinal schisis cavities, progressing to serous detachment at macula, and later atrophy.[2] A primary defect in Muller cells causing abnormal basement membrane production in the outer aspect of internal limiting membrane is the most probable cause.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Dr Emmett Cunningham, MD, PhD, MPH, California, Stanford University school of Medicine.
References
- 1.Dalma A, Dalma-Weiszhausz J, Jimenez-Sierra JM. Autosomal dominant late onset celophane-like sheen vitreomacular dystrophy: A new entity. Invest Ophthalmol Vis Sci. 1991;32:1228. [Google Scholar]
- 2.Polk TD, Gass JDM, Green WR, Novak MA, Johnson MW. Familial internal limiting membrane dystrophy. A new sheen retinal dystrophy. Arch Ophthalmol. 1997;115:878–85. doi: 10.1001/archopht.1997.01100160048007. [DOI] [PubMed] [Google Scholar]
- 3.Kellner U, Kraus H, Heimann H, Helbig H, Bornfeld N, Foerster M. Electrophysiological evaluation of visual loss in Muller cell sheen dystrophy. Br J Ophthalmol. 1998;82:650–4. doi: 10.1136/bjo.82.6.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cardenas VF, Weiszhausz JD, Munoz RM, Dalma A. SD-OCT progressive alterations in a family affected with muller cell sheen dystrophy. Invest Ophthalmol Vis Sci. 2013;54 ARVO abstract 5921. [Google Scholar]
- 5.Renner AB, Radeck V, Kellner U, Jagle H, Helbig H. Ten-year follow-up of two unrelated patients with Muller cell sheen dystrophy and first report of successful vitrectomy. Doc Ophthalmol. 2014;129:191–202. doi: 10.1007/s10633-014-9463-9. [DOI] [PubMed] [Google Scholar]