

Abstract
Background
Obstetric cholestasis has been linked to adverse maternal and fetal/neonatal outcomes. As the pathophysiology is poorly understood, therapies have been empiric. The first version of this review, published in 2001, and including nine randomised controlled trials involving 227 women, concluded that there was insufficient evidence to recommend any of the interventions alone or in combination. This is the first update.
Objectives
To evaluate the effectiveness and safety of therapeutic and delivery interventions in women with cholestasis of pregnancy.
Search methods
We searched the Cochrane Pregnancy and Childbirth Group's Trials Register (20 February 2013) and reference lists of identified studies.
Selection criteria
Randomised controlled trials that compared two intervention strategies for women with a clinical diagnosis of obstetric cholestasis.
Data collection and analysis
The review authors independently assessed trials for eligibility and risk of bias. We independently extracted data and checked these for accuracy.
Main results
We included 21 trials with a total of 1197 women. They were mostly at moderate to high risk of bias. They assessed 11 different interventions resulting in 15 different comparisons.
Compared with placebo, ursodeoxycholic acid (UDCA) showed improvement in pruritus in five (228 women) out of seven trials. There were no significant differences in instances of fetal distress in the UDCA groups compared with placebo (average risk ratio (RR) 0.67; 95% confidence interval (CI) 0.22 to 2.02; five trials, 304 women; random‐effects analysis: Tau² = 0.74; I² = 48%). There were significantly fewer total preterm births with UDCA (RR 0.46; 95% CI 0.28 to 0.73; two trials, 179 women). The difference for spontaneous preterm births was not significant (RR 0.99; 95% CI 0.41 to 2.36, two trials, 109 women).
Two trials (48 women) reported lower (better) pruritus scores for S‐adenosylmethionine (SAMe) compared with placebo, while two other trials of 34 women reported no significant differences between groups.
UDCA was more effective in improving pruritus than either SAMe (four trials; 133 women) or cholestyramine (one trial; 84 women), as was combined UDCA+SAMe when compared with placebo (one trial; 16 women) and SAMe alone (two trials; 68 women). However, combined UDCA+SAMe was no more effective than UDCA alone in regard to pruritus improvement (one trial; 53 women) and two trials (80 women) reported data were insufficient to draw any conclusions from. In one trial comparing UDCA and dexamethasone (83 women), a significant improvement with UDCA was seen only in a subgroup of women with severe obstetric cholestasis (23 women).
Danxiaoling significantly improved pruritus in comparison to Yiganling. No significant differences were seen in pruritus improvement with other interventions.
Eight trials reported fetal or neonatal deaths, with two deaths reported overall (both in the placebo groups).
Women receiving UDCA and cholestyramine experienced nausea, vomiting and diarrhoea. Guar gum caused mild abdominal distress, diarrhoea and flatulence during the first days of treatment. Women found charcoal suspension unpleasant to swallow. Dexamethasone caused nausea, dizziness and stomach pain in one woman.
One trial (62 women) looked at the timing of delivery intervention. There were no stillbirths or neonatal deaths in 'early delivery' or the 'await spontaneous labour' group. There were no significant differences in the rates of caesarean section, meconium passage or admission to neonatal intensive care unit between the two groups.
Authors' conclusions
Different approaches to assessing and reporting pruritus precluded pooling of trials comparing the effects of UDCA versus placebo on pruritus, but examination of individual trials suggests that UDCA significantly improves pruritus, albeit by a small amount. Fewer instances of fetal distress/asphyxial events were seen in the UDCA groups when compared with placebo but the difference was not statistically significant. Large trials of UDCA to determine fetal benefits or risks are needed.
A single trial was too small to rule in or out a clinically important effect of early term delivery on caesarean section.
There is insufficient evidence to indicate that SAMe, guar gum, activated charcoal, dexamethasone, cholestyramine, Salvia, Yinchenghao decoction (YCHD), Danxioling and Yiganling, or Yiganling alone or in combination are effective in treating women with cholestasis of pregnancy.
Plain language summary
Interventions for treating cholestasis in pregnancy
Obstetric cholestasis is a liver disorder in pregnancy that appears most often in the third trimester of pregnancy. The main symptom of this condition is itching (pruritus), which can be quite distressful to the pregnant woman. Bile acids accumulate within the liver and the blood level of bile acids are raised. The signs and symptoms spontaneously clear within the first few days after birth, or within two to three weeks. This condition is associated with preterm birth and is thought to be associated with complications in the unborn babies, including stillbirth. Most clinicians deliver babies early to reduce the risk of stillbirth. Therapies such as ursodeoxycholic acid (UDCA) and S‐adenosylmethionine (SAMe) seek to detoxify bile acids, or to change how they dissolve. Some agents (activated charcoal, guar gum, cholestyramine) have been used to bind bile acids in the intestine and thus get rid of them. Some of these agents have potential adverse effects for mothers due to the depletion of vitamin K, required for blood clotting.
We included 21 randomised controlled trials involving 1197 participants in this review. The trials were mostly at moderate to high risk of bias. Compared with placebo, UDCA showed improvement in itching in five trials (228 women), no benefit was observed in one trial (16 women) and one trial reported improvement only in women with severe disease (94 women). Distress in the unborn baby or symptoms of asphyxia were reported in five trials (304 women) and although there were fewer instances of fetal distress in the UDCA groups compared with placebo, the difference was not significant. The results from the four trials comparing SAMe and placebo were conflicting. Two trials (48 women) reported better pruritus scores for SAMe compared with placebo and two trials (34 women) reported no significant differences between groups for the disappearance of their pruritus.
Comparisons of guar gum, activated charcoal, dexamethasone, cholestyramine, Salvia, Yinchenghao decoction, Danxioling or Yiganling (used in Chinese medicine for their liver‐protective properties) with placebo or one with another was based on data from one trial. Further trials are required before any firm conclusions might be made about their effectiveness.
One trial (63 women) compared early delivery versus expectant management. There were no stillbirths or neonatal deaths in either group. No significant differences in caesarean section, passage of meconium‐stained liquor or admission to neonatal intensive care unit were observed.
Background
Description of the condition
Introduction and definition
Obstetric cholestasis (also known as intrahepatic cholestasis of pregnancy (ICP)) is an obstetric liver condition appearing most often in the third trimester of pregnancy. It is a relatively benign but often significantly distressing condition maternally, and may adversely affect fetal outcome as seen by associations with preterm labour, fetal distress and stillbirths. Significant features needed for a diagnosis of obstetric cholestasis are pruritus (itching), which classically affects palms and soles but may become generalised but without a rash apart from excoriations, together with increased concentrations of serum bile acids (fasting values usually at least 10 μmol/L) and/or increased concentrations of serum transaminases (e.g. alanine aminotransferase (ALT) greater than 50 U/L). Clinical pruritus may precede the development of abnormal biochemistry (Kenyon 2001). Following birth, there is usually spontaneous relief of signs and symptoms within the first few days, although occasionally resolution may take two to three weeks (Beuers 2006). Ongoing clinical symptoms and abnormal liver biochemical values within a month after birth is not consistent with a diagnosis of obstetric cholestasis. Histopathology of the liver of those affected by obstetric cholestasis shows non‐specific mild intrahepatic cholestasis with accumulation of bile pigments in hepatocytes and bile duct swelling (Heikkinen 1981). Accumulation of bile acids within the liver increases bile acid levels which may cause pruritus, perhaps due to increased availability of brain opiate receptors (Jones 1990), although the fact that pruritus may precede abnormal chemistry, including changes in bile acids, suggests that other mechanisms may be at work.
Epidemiology
The incidence of obstetric cholestasis varies across ethnic groups. It is observed in less than 1% of pregnancies in areas of Central and Western Europe and North America, and 1% to 2% in Scandinavia and the Baltic states, but can be as high as 5% to 15% in Araucanian Indians in Chile and Bolivia (Lammert 2000).
Pathophysiology
The exact pathophysiology of obstetric cholestasis is unknown but genetic, endocrine and environmental factors have been implicated. The role of genetics remains unsubstantiated but in high prevalence areas a strong family history is often present (Berg 1986; Eloranta 2001; Qui 1983; Reyes 1976; Shaw 1982). It is thought that mutations of bile acid transporter genes may impair maternal excretion and affect transplacental passage of bile acids (Milkiewicz 2002). Familial disorders such as progressive familial intrahepatic cholestasis and benign recurrent intrahepatic cholestasis may be linked to obstetric cholestasis via alterations in the binding domains of liver receptors for DNA and oestrogens (Leevy 1997). A higher than anticipated incidence of obstetric cholestasis has been found in mothers of patients with these two familial liver disorders (de Swiet 2002).
The precise role of oestrogens is unknown but their causal role is suggested by the appearance of obstetric cholestasis in the third trimester (when oestrogen concentrations are highest), the increased frequency of obstetric cholestasis in pregnancies with high oestrogen concentrations (e.g. multiple pregnancies) (Gonzalez 1989), and the resolution of symptoms following the cessation of pregnancy (Germain 2002). Women who develop obstetric cholestasis are at a higher risk of developing cholestasis with combined oral contraceptive pill use. This also suggests that oestrogen may be an aetiological factor (de Swiet 2002).
Similarly, the role of progesterone in obstetric cholestasis is unclear. While the total serum progesterone levels and the amount excreted in urine are similar to normal pregnancies, large amounts of sulphated progesterone have been detected in the plasma and urine of women with obstetric cholestasis (Meng 1997). In‐vitro animal studies suggest that high levels of progesterone metabolites induce trans‐inhibition of the bile salt export pump (BSEP), and consequently interfere with bile acid secretion into bile. This leads to intracellular accumulation of bile acids, which disrupts mitochondrial function and may explain the role of progesterone metabolites in the aetiopathogenesis of obstetric cholestasis (Vallejo 2006).
Seasonal variation in the prevalence of obstetric cholestasis indicates that environmental factors may have a role (Reyes 1997). It was also observed that only 60% of Chilean women who develop obstetric cholestasis have it in a subsequent pregnancy (Ribalta 1995). Pollutants in pesticides, erucic acid (a constituent of rape‐seed oil) and dietary deficiency of selenium have been suggested as possible environmental factors (Ribalta 1995).
Clinical features
Women present with pruritus without rash characteristically after 30 weeks' gestation (Kenyon 2002; Reyes 1992). Pruritus often worsens as the pregnancy progresses. Steatorrhoea and dark urine may occur. Jaundice is a rare symptom (de Swiet 2002). Increased rates of postpartum haemorrhage have been postulated to be due to vitamin K deficiency (Johnston 1979; Reid 1976; Reyes 1992). One study reported a higher rate of postpartum haemorrhage in women who had not taken vitamin K compared to those who had (Kenyon 2002). Gallstones may be present more often in these women (Kirkinen 1984; Ropponen 2006). Women with hepatitis C infection have a higher incidence of obstetric cholestasis (Locatelli 1999; Paternoster 2002).
Investigations
The most specific laboratory test for obstetric cholestasis is measurement of plasma or serum concentration of bile acids, such as cholic or chenodeoxycholic acid: values may be 10 to 100 times those found in healthy pregnant women (Bacq 1997; Heikkinen 1981). Increases of serum transaminases are also common (Reyes 1997). Unlike in other cholestatic diseases, increases of gamma glutamyl transferase (GGT) are less common (Walker 2002). Upper abdominal ultrasound should be performed to exclude gallbladder disease, duct dilatation and other liver pathology. Serology for hepatitis A, B, C, Epstein Barr virus (EBV) and cytomegalovirus (CMV) will help to exclude viral pathology, while an autoimmune screen including anti‐smooth muscle, liver‐kidney microsomal (LKM) and antimitochondrial antibodies will help to identify women with chronic active hepatitis or primary biliary cirrhosis (Bacq 1997; Heinonen 1999; Kenyon 2005).
Fetal effects
The implications of excess maternal serum bile acids on the fetus is not completely understood. Increased rates of fetal complications, perinatal mortality rates, stillbirths, low birthweight, preterm labour and birth, and fetal distress in labour have been linked with the condition (Alsulyman 1996; Davies 1995; Fisk 1988; Gaudet 2000; Jiang 1986; Johnston 1979; Laatikainen 1975; Reid 1976; Rioseco 1994; Roszkowski 1968; Williamson 2004; Wilson 1979; Ylostalo 1975). There is evidence to suggest an increased incidence of meconium‐stained liquor in women with obstetric cholestasis (RCOG 2011) and it is more common in those with serum bile acid levels over 40 µmol/L (Lee 2008). No specific fetal monitoring such as cardiotocography (CTG), ultrasound or amniocentesis for meconium presence has found to be beneficial or accurate in predicting an adverse outcome in obstetric cholestasis (RCOG 2011). Possible mechanisms that have been suggested include a toxic effect of bile acids on the fetal myocardium, leading to cardiac dysrhythmia and acute anoxia, as demonstrated in neonatal rat cardiomyocytes (Williamson 2001). It has been hypothesised that high bile acid concentration in the mother may cause bile acid pneumonia in the newborn (Zecca 2006; Zecca 2008).
Description of the intervention
Early delivery (e.g. around 37 weeks of pregnancy as discussed by RCOG) is widely practiced across the world on the assumption that it might pre‐empt stillbirths (Roncaglia 2002; RCOG 2011).
Topical emollients may provide temporary relief of pruritus for some women and antihistamines are also used to provide symptom relief. These are widely used (RCOG 2011). Chlorpheniramine is sometimes prescribed in obstetric cholestasis, although its role in reducing itching in obstetric cholestasis has not been substantiated. Other treatments aimed to decrease bile production (dexamethasone and phenobarbitone) are rarely used in UK practice. In the United States, hydroxyzine and diphenhydramine are commonly used as first‐line agents to treat pruritus in women with cholestasis.
Some agents have been used that bind bile acids in the intestine, facilitating their elimination and preventing enterohepatic recirculation (activated charcoal, guar gum, cholestyramine). Agents binding bile acids have potential adverse effects for mothers due to the depletion of vitamin K (Briggs 2001).
Other therapies such as ursodeoxycholic acid (UDCA) and S‐adenosylmethionine (SAMe) may detoxify bile acids, or change their solubility, thereby allowing increased choleresis and potentially reducing their adverse cellular effects.
Yinchenghao decoction (YCHD), and Danxioling and Yiganling are used in Chinese medicine for their hepato‐protective properties. There is little information available on these products.
Potential side effects for mother and fetus exist for dexamethasone, phenobarbitone, SAMe and UDCA since they all cross the placenta.
How the intervention might work
Early delivery pre‐empts stillbirth but can increase caesarean section rate and respiratory distress syndrome in neonates.
The efficacy of topical emollients has not been tested in clinical trials but they seem to provide temporary relief from pruritus in some women and are safe in pregnancy (RCOG 2011). Calamine lotion contains zinc oxide (ZnO) and 0.5% iron oxide (Fe2O3) and has antipruritic and antiseptic properties. One to two per cent menthol in aqueous cream affects A delta sensory nerve fibres and suppresses histamine‐induced itching (Bernhard 1994; Bromma 1995). Diprobase contains liquid paraffin, white soft paraffin, cetomacrogol and cetostearyl alcohol. The principle behind its use is to provide symptomatic relief from itching due to its moisturising properties. Balneum Plus cream contains urea and lauromacrogols; the hydrophilic properties of urea hydrate the skin and the local anaesthetic properties of lauromacrogols cause a soothing effect.
Chlorpheniramine is a first‐generation alkylamine antihistamine. Its use in obstetric cholestasis has not been tested in a clinical trial but it seems to provide symptomatic relief from itching in some women. It can cause sedation but is otherwise safe in pregnancy.
Dexamethasone decreases the synthesis of fetal and maternal adrenocorticotrophin hormone (ACTH). It also reduces production and secretion of oestrogen precursors dehydroepiandrosterone (DHEA) and DHEA sulphate from both maternal and fetal adrenal glands (Kauppila 1979; Simmer 1975). More than 50% of oestrogen in the maternal circulation is derived from the fetor‐placental unit. Reduction of maternal oestrogen levels may be the possible mechanism by which it may improve cholestasis (Diac 2006).
The role of phenobarbitone in cholestasis was first demonstrated by Cunningham in 1968 (Cunningham 1968). Animal models suggest that phenobarbitone increases the excretion of bile salts into the biliary tree and enhances bile flow (Klaasen 1970; Robinson 1971).
Activated charcoal is a highly porous carbon compound. It is widely used to treat acute poisoning following oral ingestion, where it binds to the toxin and prevents its absorption from the stomach and intestine. It can effectively adsorb bile salts in vitro (Krasopoulos 1980).
Guar gum is a viscous polysaccharide obtained from guar beans. It helps to hold plant cells together. Its main use is in the food industry where it is used to thicken or add texture to foods and drinks (Insel 2010). It is also used to add thickness in lotions and creams, to bind ingredients together in tablets and was widely used as an appetite suppresser in weight loss formulations in the past. Guar gums binds the bile acids to the intestinal contents, which are then expelled from the body (Morgan 1993).
Cholestyramine is a resin that binds to bile acids in the intestine and prevents their reabsorption. Consequently, it may interfere with the absorption of fat‐soluble vitamins, including vitamin K, which is essential for blood coagulation. This may increase the risk of postpartum haemorrhage in the mother and intracranial haemorrhage in the fetus (Sadler 1995).
S‐adenosylmethionine is produced from methionine and adenosine triphosphate (ATP) in all mammalian cells. Liver is the principal site where it is produced and metabolised (Cantoni 1952). It is an important methyl group donor and plays a crucial role in the biosynthesis of phospholipids, which are important for maintaining the fluidity of hepatic cell membranes and excretion of oestrogen metabolites (Boelsterli 1983). Interference with hepatic SAMe biosynthesis may cause and predispose hepatocytes to injury. Experiments on rat models indicate that SAMe can reverse cholestasis (Stramentinoli 1981). The exact mechanism of action remains unclear.
Ursodeoxycholic acid is a naturally occurring hydrophilic bile acid. Studies suggest that UDCA displaces endogenous hydrophobic, detergent‐like, toxic bile acids in cholestatic disorders without disrupting the bile acid pool (Stiehl 1999). UDCA has been attributed with cytoprotective and anti‐apoptotic properties (Mitsuyoshi 1999; Rodrigues 1998). Animal studies have shown that UDCA improves hepatocellular and cholangiocellular biliary secretion in cholestatic disorders by post‐transcriptional regulation of the apical transporters, BSEP and multidrug resistance protein 2 (MRP2) (Beuers 2001). Women with obstetric cholestasis treated with UDCA have reduced levels of cord blood bile acid levels. This may be due to up regulation of the expression of placental MRP2 (Azzaroli 2007).
Yinchenghao decoction (YCHD) is extracted from three different herbs:Artemisia capillaries, Gardenia jasminoides Ellis and Rheum officinale Baill. It was invented 2000 years ago and has been used in Chinese medicine to treat a wide range of liver disorders. Downregulation of the production of pro‐inflammatory cytokine tumour necrosis factor (TNF) by inhibition of NF‐kappaB activation (Cai 2006), an antifibrotic action, in part due to the inhibitory action on extracellular matrix (ECM) gene expression (Lee 2009), and decreased tumour growth factor 1 (TGF‐1) mRNA expression and inhibition of lipid peroxidation with reduced hepatic collagen accumulation (Lee 2007) have all been postulated as possible mechanisms for its hepato‐protective properties.
Why it is important to do this review
This is an update of a Cochrane review first published in 2001 (Burrows 2001), which concluded that there was insufficient evidence for any of the treatments for obstetric cholestasis so far evaluated in randomised controlled trials. None were found to be consistently effective in resolving maternal pruritus. It is therefore important to update this review to incorporate new evidence generated since 2001.
Objectives
To assess the effects of interventions to treat women with cholestasis of pregnancy, on maternal, fetal and neonatal outcomes.
Methods
Criteria for considering studies for this review
Types of studies
Randomised or quasi‐randomised controlled trials.
Types of participants
Women stated to have a diagnosis of intrahepatic cholestasis of pregnancy.
Types of interventions
Interventions used to treat obstetric cholestasis and its symptoms, compared with placebo, no treatment or another intervention.
Types of outcome measures
Primary outcomes
Maternal
Pruritus (scores, change in score, improvement)
Fetal/neonatal
Stillbirths and or neonatal deaths
Fetal distress/asphyxial events
Secondary outcomes
Maternal
Liver function as measured by bile acid and ALT
Caesarean section
Postpartum haemorrhage
Adverse effects of medication
Fetal/neonatal
Meconium‐stained liquor
Mean gestational age at birth
Spontaneous birth at less than 37 weeks
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
Admission to neonatal intensive care unit
Search methods for identification of studies
Electronic searches
We searched the Cochrane Pregnancy and Childbirth Group’s Trials Register by contacting the Trials Search Co‐ordinator (20 February 2013).
The Cochrane Pregnancy and Childbirth Group’s Trials Register is maintained by the Trials Search Co‐ordinator and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE;
weekly searches of EMBASE;
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Details of the search strategies for CENTRAL, MEDLINE and EMBASE, the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service can be found in the ‘Specialized Register’ section within the editorial information about the Cochrane Pregnancy and Childbirth Group.
Trials identified through the searching activities described above are each assigned to a review topic (or topics). The Trials Search Co‐ordinator searches the Register for each review using the topic list rather than keywords.
Searching other resources
We searched the reference lists of identified studies.
We did not apply any language restrictions.
Data collection and analysis
For the methods used when assessing the trials identified in the previous version of this review, seeAppendix 1. For this update we used the following methods when assessing the trials identified by the updated search.
Selection of studies
Two review authors (Vinita Gurung (VG), Michael Stokes (MS)) independently assessed for inclusion all the potential studies identified as a result of the search strategy. There were no disagreements. Studies presented only as abstracts were considered for inclusion on the same basis as studies published in full.
Data extraction and management
MS designed a form to extract data. For eligible studies, VG and MS extracted the data using the agreed form. We resolved discrepancies through discussion or, by consulting the other authors of the review (Philippa Middleton (PM), William Hague (WH), Jim Thornton (JT)). VG entered data into Review Manager software (RevMan 2012) and JT checked for accuracy.
When information regarding any of the above was unclear, we attempted to contact authors of the original reports to provide further details.
Assessment of risk of bias in included studies
VG and PM independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreement by discussion or by consulting the other assessors.
(1) Random sequence generation (checking for possible selection bias)
We describe for each included study whether the method used to generate the allocation sequence was described in sufficient detail to allow an assessment of whether it produced comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We describe for each included study whether the method used to conceal the allocation sequence and determine whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We describe for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered studies to be at low risk of bias if they were blinded, or if we judged that the lack of blinding could not have affected the results. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods as:
low, high or unclear risk of bias participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We describe for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We describe for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We state whether attrition and exclusions were reported. We also mention the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we re‐included missing data in the analyses.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated” analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting bias
We describe for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (where it is clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the study’s pre‐specified outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest are reported incompletely and so cannot be used; study fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other sources of bias
We describe for each included study any important concerns we had about other possible sources of bias.
We assessed whether each study was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Cochrane Handbook (Higgins 2011). With reference to (1) to (6) above, we assessed the likely magnitude and direction of the bias and whether they were likely to impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analyses ‐ seeSensitivity analysis.
Measures of treatment effect
Dichotomous data
For dichotomous data, we presented results as summary risk ratio with 95% confidence intervals.
Continuous data
For continuous data, we used the mean difference if outcomes were measured in the same way between trials. We planned to use the standardised mean difference to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
If cluster‐randomised trials had been available they would have been included. We planned to adjust their sample sizes using the methods described in the Cochrane Handbook Section 16.3.4 using an estimate of the intra‐cluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial or from a study of a similar population. If we had used ICCs from other sources, we would have reported this and conducted sensitivity analyses to investigate the effect of variation in the ICC. If we had identified both cluster‐randomised trials and individually‐randomised trials, we planned to synthesise the relevant information. We would have considered it reasonable to combine the results from both if there was little heterogeneity between the study designs and the interaction between the effect of intervention and the choice of randomisation unit was considered to be unlikely.
We also planned to acknowledge heterogeneity in the randomisation unit and to perform a sensitivity analysis to investigate the effects of the randomisation unit (Higgins 2011)
Cross‐over trials
This is not an appropriate study design for the topic of this review.
Dealing with missing data
For included studies we noted levels of attrition. We explored the impact of included studies with high levels of missing data in the overall assessment of treatment effect by sensitivity analysis.
For all outcomes, we analysed the data, as far as possible, on an intention‐to‐treat (ITT) basis, i.e. we made an attempt to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the Tau², I² and Chi² statistics. We regarded heterogeneity as substantial if the Tau² is greater than zero and either I² is greater than 30% or there is a low P value (less than 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
If there were 10 or more studies for any outcome in the meta‐analysis, we had planned to investigate reporting biases (such as publication bias) using funnel plots. We planned to assess funnel plot asymmetry visually. If asymmetry was suggested by a visual assessment, we planned to perform exploratory analyses to investigate it.
Data synthesis
We carried out statistical analysis using the Review Manager software (RevMan 2012). We used fixed‐effect meta‐analysis for combining data where it was reasonable to assume that studies were estimating the same underlying treatment effect: i.e. where trials were examining the same intervention, and the trials’ populations and methods were judged sufficiently similar. If there was clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if substantial statistical heterogeneity was detected, we used random‐effects meta‐analysis to produce an overall summary if an average treatment effect across trials was considered clinically meaningful. We treated the random‐effects summary as the average of the range of possible treatment effects and we discussed the clinical implications of treatment effects differing between trials. If the average treatment effect was not clinically meaningful, we did not combine trials.
Where we used random‐effects analyses, we presented the results as the average treatment effect with its 95% confidence interval, and the estimates of Tau² and I².
Subgroup analysis and investigation of heterogeneity
If we had identified substantial heterogeneity, we planned to investigate it using subgroup analyses and sensitivity analyses. We planned to consider whether an overall summary was meaningful, and if it was, use random‐effects analysis to produce it.
We carried out the following subgroup analyses.
Bile acids levels ≥ 40 µmol/L versus bile acid levels less than 40 µmol/L.
We used primary outcomes only for the subgroup analysis.
We assessed subgroup differences by interaction tests available within RevMan (RevMan 2012). We reported the results of subgroup analyses quoting the Chi² statistic and P value, and the interaction test I² value.
Sensitivity analysis
When appropriate, in future updates, we will carry out sensitivity analysis to explore the effect of trial quality based on concealment of allocation, by excluding studies with unclear or high risk of bias for allocation concealment.
Results
Description of studies
SeeCharacteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
We included 21 trials, and excluded two studies. We were unable to trace one study published in Chinese. It is classified under studies awaiting classification.
Included studies
The original review included nine randomised controlled trials (Diaferia 1996; Floreani 1996; Frezza 1984; Frezza 1990; Kaaja 1994; Nicastri 1998; Palma 1997; Ribalta 1991; Riikonen 2000). The updated search identified 11 new studies and all were judged to be eligible for inclusion (Binder 2006; Fang 2009; Glantz 2005; Huang 2004; Kondrackiene 2005; Liu 2006; Luo 2008; PITCH 2012; Roncaglia 2004; Shi 2002; Zhang 2012). In addition, one study (Leino 1998) was a conference abstract and excluded from the original review (Burrows 2001). This has also been included in the update.
Thus 21 trials involving 1197 women are now included in this review. For a full description of the characteristics of included studies, see table of Characteristics of included studies.
Participants
All women had a diagnosis of obstetric cholestasis based on the presence of pruritus in pregnancy and abnormalities of liver function. The onset of pruritus varied among the studies, occurring before week 19 (Frezza 1984), week 28 (Nicastri 1998), week 29 (Diaferia 1996), week 32 (Ribalta 1991), week 33 (Palma 1997), week 35 (Zhang 2012), in the second half of pregnancy (Huang 2004), the last trimester (Floreani 1996) or the second or third trimester (Binder 2006; Kondrackiene 2005; Roncaglia 2004). In one study (PITCH 2012), women were randomised after week 24, irrespective of the time of onset of gestational pruritus. Nine studies did not specify a time for onset of pruritus (Frezza 1990; Fang 2009; Glantz 2005; Kaaja 1994; Leino 1998; Liu 2006; Luo 2008; Riikonen 2000; Shi 2002). Generally the inclusion criteria stipulated the severity and duration of pruritus, elevated levels of bile acids/salts and/or other liver function assays, consent to remain in hospital until the birth or undergo extensive fetal monitoring and the exclusion criteria stipulated absence of skin disease, chronic liver disease or other abnormalities unrelated to pregnancy. One study (Riikonen 2000) reported that one woman was in the study twice, during successive pregnancies.
Interventions
Eleven different interventions were compared with placebo, no treatment or another intervention in the included trials. One trial compared the timing of delivery in obstetric cholestasis. We grouped them into the following 15 comparisons (with some trials appearing in more than one comparison):
UDCA versus placebo ‐ seven studies (Diaferia 1996; Glantz 2005; Leino 1998; Liu 2006; Nicastri 1998; Palma 1997; PITCH 2012)
SAMe versus placebo ‐ four studies (Frezza 1984; Frezza 1990; Nicastri 1998; Ribalta 1991)
Guar gum versus placebo ‐ one study (Riikonen 2000)
Activated charcoal versus no treatment ‐ one study (Kaaja 1994)
Dexamethasone versus placebo ‐ one study (Glantz 2005)
UDCA versus SAMe ‐ five studies (Binder 2006; Floreani 1996; Nicastri 1998; Roncaglia 2004; Zhang 2012)
UDCA versus dexamethasone ‐ one study (Glantz 2005)
UDCA versus cholestyramine ‐ one study (Kondrackiene 2005)
UDCA+SAMe versus placebo ‐ one study (Nicastri 1998)
UDCA+SAMe versus SAMe ‐ three studies (Binder 2006; Nicastri 1998; Zhang 2012)
UDCA+SAMe versus UDCA ‐ four studies (Binder 2006; Luo 2008; Nicastri 1998; Zhang 2012)
UDCA+Salvia versus UDCA ‐ one study (Fang 2009)
Yinchenghao decoction (YCHD) versus SAMe ‐ one study (Huang 2004)
Danxiaoling Pill (DXLP) versus Yiganling ‐ one study (Shi 2002)
Early term delivery versus expectant management ‐ one study (PITCH 2012)
Ursodeoxycholic acid (UDCA) versus placebo (Diaferia 1996; Glantz 2005; Leino 1996; Liu 2006; Nicastri 1998; Palma 1997, PITCH 2012)
Participants in Leino 1998 received UDCA 450 mg/day in two doses for 14 days.The treatment and control interventions were identical in two studies (Diaferia 1996 and relevant arms of Nicastri 1998): 600 mg/day UDCA, or placebo (vitamin) given in two oral doses for 20 days (given after 30 weeks' gestation in Diaferia 1996). Participants in Glantz 2005 and Palma 1997 received a higher dose of UDCA or placebo over a longer period of time. UDCA 1000 mg/day or placebo was given as a single daily dose for three weeks in Glantz 2005 and as three divided doses or placebo (starch) in Palma 1997. In Liu 2006, women received UDCA (18 mg/kg body weight) three times a day for two weeks. The control group received a combination of 10% glucose, vitamin C and Inosine for two weeks. It is unclear whether the interventions were administered orally or by parenteral route. Participants in PITCH 2012 received UDCA 1 g daily increased in increments of 500 mg daily every three to 14 days up to a maximum UDCA dose 2 g/day if no biochemical or clinical improvement was observed.
S‐adenosylmethionine (SAMe) versus placebo (Frezza 1984; Frezza 1990; Nicastri 1998; Ribalta 1991)
In these studies 800 mg/day of SAMe dissolved in a 500 mL solution of saline (Frezza 1984), 5% dextrose (Frezza 1990; Nicastri 1998) or 5% glucose (Ribalta 1991) were administered intravenously (IV) over the course of three (Ribalta 1991) or four hours (Frezza 1984). The duration of administration was not reported in two studies (Frezza 1990; Nicastri 1998). A lower dose of SAMe 200 mg/day with placebo was also compared (Frezza 1984). The intervention was administered up to the day of delivery (Frezza 1984; Frezza 1990) or for a maximum of 20 days (Nicastri 1998; Ribalta 1991). Placebo treatment was either 5% dextrose solution (Frezza 1990), mannitol (800 mg) in a 5% glucose solution (Ribalta 1991), saline solution (Frezza 1984) or a vitamin solution (Nicastri 1998).
Guar gum versus placebo (Riikonen 2000)
Guar gum or placebo (wheat flour) at doses from 5 to 15 g/day (increases in dosage occurring at three‐day intervals) were given in three intermittent doses up until delivery. For the participants to be included in the intervention analysis, they had to take guar‐gum or placebo for at least 10 days.
Activated charcoal versus no treatment (Kaaja 1994)
Activated charcoal as a water suspension was given in a dose of 50 g three times a day for eight days.
Dexamethasone versus placebo (Glantz 2005)
Dexamethasone 12 mg/day was administered as a single daily oral dose for a week, followed by placebo for two weeks. Women in the control group took a single dose of placebo every day for three weeks.
Ursodeoxycholic acid (UDCA) versus S‐adenosylmethionine (SAMe) (Binder 2006; Floreani 1996; Nicastri 1998; Roncaglia 2004; Zhang 2012)
These studies differed with regards to dose, administration and duration of intervention. Binder 2006 used the highest dose of UDCA (750 mg/day) and this was administered orally three times a day until birth. In Nicastri 1998 and Roncaglia 2004, 600 mg/day of UDCA was administered as two oral daily doses for 20 days or until delivery respectively whereas in Floreani 1996, UDCA was given as a single oral dose of 450 mg/day until delivery.
Binder 2006, Floreani 1996,and Roncaglia 2004 administered 1000 mg/day of SAMe but the routes of administration and duration of intervention were different. In Binder 2006, SAMe 500 mg was administered IV twice daily for 12 days and subsequently as 500 mg twice daily oral dose until delivery. In Floreani 1996, SAMe was administered as a single intramuscular (IM) injection daily until birth whereas in Roncaglia 2004, it was given in two doses by oral route until delivery. In Nicastri 1998, 800 mg/day of SAMe was administered daily in two doses as IV infusions. These were given for a maximum of 20 days.
In Zhang 2012 UDCA (250 mg given orally four times per day) was compared with SAMe (1000 mg IV four times daily) alone.
Ursodeoxycholic acid (UDCA) versus dexamethasone (Glantz 2005)
In Glantz 2005, UDCA 1000 mg was administered as a daily single daily oral dose for three weeks. This was compared with dexamethasone 12 mg/day given as a single oral dose for one week and placebo during weeks two and three.
Ursodeoxycholic acid (UDCA) versus cholestyramine (Kondrackiene 2005)
UDCA (8 to 10 mg/kg body weight per day) was compared with cholestyramine (8 g/day). They were administered orally for two weeks.
Yinchenghao decoction (YCHD) versus S‐adenosylmethionine (SAMe) (Huang 2004)
YCHD given twice daily orally for three weeks was compared with SAMe IV infusion of 2 x 500 mg daily for three weeks.
Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus placebo (Nicastri 1998)
UDCA (600 mg/day, in two oral doses) plus SAMe (800 mg sulphate‐P‐toluenesulphatonate diluted in 500 mL 5% dextrose, in two IV infusions) were compared with placebo (vitamin) administered for a maximum of 20 days.
Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus S‐adenosylmethionine (SAMe) (Binder 2006; Nicastri 1998; Zhang 2012)
In Nicastri 1998, UDCA (600 mg/day, in two oral doses) plus SAMe (800 mg sulphate‐P‐toluenesulphatonate diluted in 500 mL 5% dextrose, in two IV infusions) were compared with SAMe 800 mg/day administered for a maximum of 20 days. In Binder 2006, UDCA (3 X 250 mg/day oral doses until delivery) plus SAMe (2 X 500 mg/day given by slow infusion for 14 days) was compared with SAMe (2 X 500 mg/day given by slow infusion for 14 days) alone. In Zhang 2012 UDCA plus SAMEe (dose not stated) was compared with SAMe (1000 mg IV four times daily) alone.
Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus Ursodeoxycholic acid (UDCA) (Binder 2006; Luo 2008; Nicastri 1998; Zhang 2012)
In Binder 2006, UDCA (3 X 250 mg/day oral doses until delivery) plus SAMe (2 X 500 mg/day given by slow infusion for 14 days) was compared with UDCA (3 X 250 mg/day oral doses until delivery) alone. In Zhang 2012, UDCA plus SAMEe (dose not stated) was compared with UDCA (250 mg given orally four times daily) alone. In Nicastri 1998, UDCA (600 mg/day, in two oral doses) plus SAMe (800 mg sulphate‐P‐toluenesulphatonate diluted in 500 mL 5% dextrose, in two IV infusions) was compared with UDCA (600 mg/day, in two oral doses) alone administered for a maximum of 20 days. In Luo 2008, SAMe (Transmetil 1 g added to 250 mL 5% glucose administered as an IV infusion once daily) plus UDCA (250 mg oral pill twice daily) were compared with UDCA pill alone (250 mg oral pill twice daily) for 10 days. Participants in both groups received dexamethasone (10 mg once a day orally) for three days before commencing the study drugs.
Ursodeoxycholic acid (UDCA)+Salvia versus Ursodeoxycholic acid (UDCA) (Fang 2009)
Salvia (10 mL in 10% 500 mL dextrose IV injection) and ursodeoxycholic acid (15 mg/kg/day divided into three oral doses per day) was compared with UDCA (same dose as above) only. Both were used for 14 days.
Danxiaoling Pill (DXLP) versus Yiganling (Shi 2002)
DXLP 9 g/day given three times a day orally for seven days was compared with Yiganling tablets given as four tablets three times a day for seven days.
Excluded studies
Two studies were excluded as they are not randomised controlled trials. For further details, seeCharacteristics of excluded studies.
Risk of bias in included studies
A summary of the risk of bias for the included studies is provided in the following figures: Figure 1; Figure 2.
1.
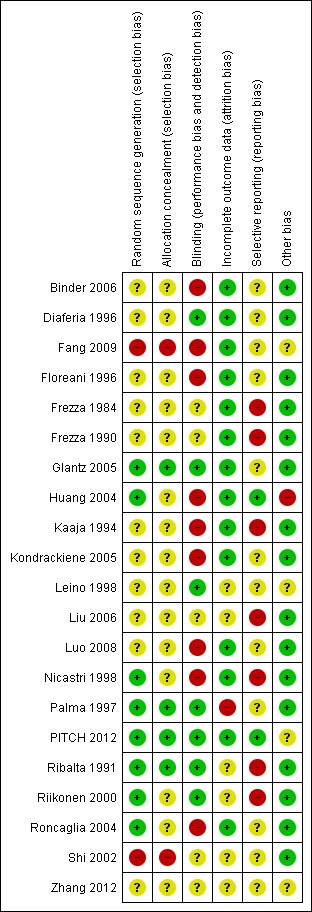
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
2.
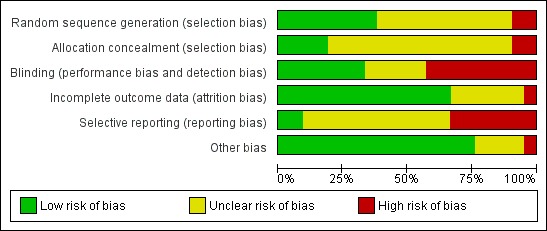
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Apart from two studies (Fang 2009; Shi 2002), which were quasi‐randomised controlled trials, all other studies were randomised controlled trials. Eight trials reported adequate methods for sequence generation (Glantz 2005; Huang 2004; Nicastri 1998; Palma 1997; PITCH 2012; Ribalta 1991; Riikonen 2000; Roncaglia 2004). Shi 2002 used alternation according to hospital admission for to generate random sequence. In Fang 2009, patients were divided into two groups based on the date of hospital admission. Floreani 1996 and Luo 2008 mentioned that the study participants were 'randomly assigned' to the two interventions, but it is unclear how this random sequence was generated. In Frezza 1990, participants were randomised according to a pre‐established code, but it is unclear how this code was derived. It is unclear whether the remaining studies had used a random sequence for intervention allocation.
Allocation concealment was adequate for four trials (Glantz 2005; Palma 1997; PITCH 2012; Ribalta 1991). There was a high risk of possible selection bias in two (Fang 2009; Shi 2002) and it was unclear in the remaining trials.
Blinding
Blinding of both participants and/or investigators was reported in seven studies (Diaferia 1996; Glantz 2005; Leino 1998; Palma 1997; PITCH 2012; Ribalta 1991; Riikonen 2000). Two studies were single blinded where only the investigators were informed of which treatment participants were receiving (Frezza 1984; Frezza 1990), and in nine studies no blinding occurred (Binder 2006; Fang 2009; Floreani 1996; Huang 2004; Kaaja 1994; Kondrackiene 2005; Luo 2008; Nicastri 1998; Roncaglia 2004). In the three remaining studies it is unclear whether the participants and/or investigators were blinded to trial allocation (Liu 2006; Shi 2002; Zhang 2012).
Incomplete outcome data
Fourteen of the 20 studies either experienced no dropouts or did not report losses to follow‐up and therefore we presume all women were included in the analysis (Binder 2006; Diaferia 1996; Fang 2009; Floreani 1996; Frezza 1984; Frezza 1990; Glantz 2005; Huang 2004; Kaaja 1994; Kondrackiene 2005; Luo 2008; Nicastri 1998; PITCH 2012; Roncaglia 2004). In Shi 2002 outcomes were reported for 25 (86%) participants for ALT and aspartate transaminase (AST), 27 (93%) for ALP and for 21 (72%) women for bilirubin levels out of 29 participants receiving Danxiaoling and for 16 of 29 (55%) participants for bilirubin in theYiganling group. In studies in which patients withdrew, no ITT analysis was conducted. Outcomes were reported in 15 of 25 (63%) participants randomised in Palma 1997, 39 of 48 (81%) in Riikonen 2000, and 18 of 20 (90%) in Ribalta 1991. The number of participants analysed in the results was unclear in one study (Leino 1998). In one study (Zhang 2012), 20 cases were reported to have been eliminated and not included in the analysis. However, It was unclear how many of these from each randomised group were lost to follow‐up (Zhang 2012), as only the total number of cases eliminated from the analysis was reported.
Palma 1997 excluded nine women who delivered before completion of two weeks of treatment from the analysis.
Selective reporting
While most trials reported maternal pruritus after treatment, variable and incomplete reporting precluded pooling of data for this outcome.
The other primary outcomes of perinatal mortality were not reported in all of the trials. In addition, several trials reported some outcomes only in graphical form.
Other potential sources of bias
Most trials appeared to have no other potential sources of bias, except Huang 2004 where there was an imbalance in numbers of women randomised to each group.
Effects of interventions
1. Ursodeoxycholic acid (UDCA) versus placebo
Seven trials (Diaferia 1996; Glantz 2005; Leino 1998; Liu 2006; Nicastri 1998; Palma 1997; PITCH 2012) involving 338 women looked at this comparison.
Primary outcomes (maternal)
Pruritus
All seven trials (338 women) comparing UDCA and placebo reported this outcome. Two studies (205 women) used a 100 mm visual analogue scale (VAS), four studies (105 women) evaluated itching on a 0‐4 categorical scale and one study (18 women) did not elaborate on the methods used to assess pruritus. Studies that used a 0‐4 scale (0 = absence of pruritus, 1 = occasional pruritus, 2 = discontinuous pruritus every day, with prevailing asymptomatic lapses, 3 = discontinuous pruritus with prevailing symptomatic lapses and, 4 = constant pruritus) analysed the data as a continuous outcome, which is not ideal as the assumption of normality on a short scale will not be met. We therefore planned to dichotomise data by classifying a pruritus score of 0‐2 as mild pruritus, and 3‐4 as severe pruritus. We also planned to dichotomise pruritus outcome after the end of intervention as "improvers" and "non‐improvers". Only Palma 1997 allowed dichotomisation of data. We could not pool results from any of these trials due to the differing methods of measuring and reporting pruritus.
Four studies (158 women) reported a significant improvement in the pruritus score with both UDCA and placebo. Of these, two studies (127 women) reported a statistically significant reduction in pruritus score with UDCA compared with placebo (Diaferia 1996; PITCH 2012). One study (70 women) reported significant improvements in pruritus score with UDCA only and one study reported no difference in pruritus score on ITT analysis but significant improvement with UDCA was seen in a subgroup of women with bile acids ≥ 40 µmol/L.
In Diaferia 1996, pruritus was assessed before treatment (day 0) and at five‐day intervals thereafter, on a 0‐4 scale. Pruritus score was reported as mean and standard deviation (SD) at day 0 and day 20. The difference in pruritus score was statistically significant both in UDCA (reported P value < 0.001) and placebo (reported P value < 0.01), but favoured UDCA over placebo.
In Glantz 2005, no significant difference in pruritus score (100 mm VAS) was seen between the UDCA and placebo groups after three weeks treatment (94 women; no P value reported). However, in the 23 women with severe cholestasis (at least 40 μmol/L bile acids), the pruritus score fell to a mean of about 15 in the UDCA group compared with a mean of about 52 in the placebo group (reported P value 0.001). These data are not reported graphically in this review.
Leino 1998 reported a significant improvement in pruritus scores within two weeks in the UDCA group. However, they did not report numerical or graphical data.
In Liu 2006, pruritus was evaluated on a 0‐4 scale. Results were reported as mean and SD at trial entry and two weeks later. After 14 days treatment, a significant reduction in the pruritus scores was observed in the UDCA group (reported P value < 0.05) compared with the placebo group (reported P value > 0.05).
In Nicastri 1998, pruritus was evaluated by the participant every three days up to 24 hours after delivery using the 0‐4 scoring system. The change in pruritus score after 20 days treatment was analysed as a continuous outcome and reported as mean and SD. A significant reduction in pruritus score was observed with both UDCA (reported P value < 0.01; 8 women) and placebo (reported P value < 0.01; 8 women).
In Palma 1997, a weekly assessment of pruritus was performed in all the study participants by the same clinician using the 0‐4 scoring system. They reported a significant improvement in pruritus score after two weeks (P < 0.01; 15 women) and three weeks (P = 0.02; 15 women) treatment with UDCA compared with placebo. Data for improvement in pruritus score were presented as a graph. Similar numbers of women (seven of the eight women in the UDCA group and five of the seven women in the placebo group) showed a reduction in pruritus score after three weeks (risk ratio (RR) 1.23; 95% confidence interval (CI) 0.72 to 2.10 (Analysis 1.1)); all seven 'improvers' in the UDCA group had low scores (under 1.5) compared with two of the five 'improvers' in the placebo group, which was also a non‐significant difference.
PITCH 2012 prespecified in their trial protocol, published before data unblinding, that their primary outcome was to be the mean of all worst itching scores in the preceding 24 hours (100 mm VAS) measured between randomisation and delivery. The authors of this trial surveyed patients and obstetricians who indicated that the average minimum worthwhile improvement was 30 mm. The results (Analysis 1.2; Analysis 1.3) showed a statistically significant reduction but the 95% CI around the effect was only 22 mm, i.e. smaller than the minimum worthwhile treatment effect for most women and doctors.
1.1. Analysis.

Comparison 1 UDCA versus placebo, Outcome 1 Pruritus improvement.
1.2. Analysis.
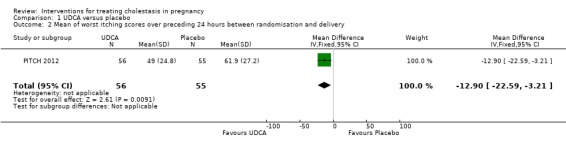
Comparison 1 UDCA versus placebo, Outcome 2 Mean of worst itching scores over preceding 24 hours between randomisation and delivery.
1.3. Analysis.
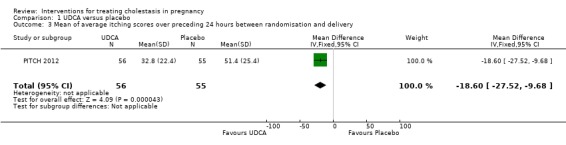
Comparison 1 UDCA versus placebo, Outcome 3 Mean of average itching scores over preceding 24 hours between randomisation and delivery.
Primary outcomes (fetal/neonatal)
Stillbirth
Two out of the three studies reported one stillbirth each, both in the placebo group (RR 0.31; 95% CI 0.03 to 2.84; 233 women (Analysis 1.4). In Glantz 2005, a woman on clomipramine for long‐term depressive disorder experienced itching from 33 weeks' gestation. After going into spontaneous labour at week 38, intrauterine death was diagnosed. Her serum bile acid concentrations were 16 µmol/L at trial inclusion and at two weeks later. In Palma 1997, the woman with a stillbirth had received placebo for two weeks and fetal death occurred after minor signs of fetal distress were noted.
1.4. Analysis.
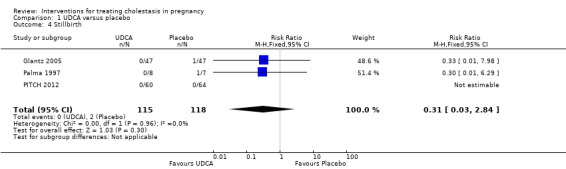
Comparison 1 UDCA versus placebo, Outcome 4 Stillbirth.
No neonatal deaths were reported.
Fetal distress/asphyxial events
Five of the seven trials comparing UDCA with placebo reported fetal distress and/or asphyxial events in some form but the difference was not statistically significant (average RR 0.67 95% CI 0.22 to 2.02; random‐effects analysis: Tau² = 0.74; I² = 48%) 304 women (Analysis 1.5)).
1.5. Analysis.
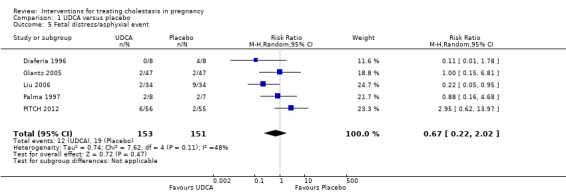
Comparison 1 UDCA versus placebo, Outcome 5 Fetal distress/asphyxial event.
In Diaferia 1996 and Palma 1997, this outcome included women who had operative births for fetal distress and in Liu 2006 it was defined as abnormal results of antepartum testing prompting delivery. Glantz 2005 defined asphyxial events as all operative births due to asphyxia, umbilical arterial pH less than 7.05 or Apgar score less than seven at five minutes. In Liu 2006 one baby from the UDCA group and seven babies from the placebo group were reported to have asphyxia neonatorum (which was not clearly defined in the paper). PITCH 2012 reported asphyxial events defined as induction or caesarean section for fetal compromise.
Subgroup analysis (bile acid levels ≥ 40 µmol/L versus bile acid levels < 40 µmol/L)
One study presented data for the subgroups of bile acid levels ≥ 40 µmol/L (RR 0.31 95% CI 0.01 to 6.85; 23 women) versus bile acids < 40 µmol/L (RR 1.03 95% CI 0.15 to 6.90; 71 women) for one of the review's primary outcomes (asphyxial events). There were no differences between these subgroups (Analysis 1.6), (test for subgroup differences: Chi² = 0.42, df = 1 (P = 0.52), I² = 0%).
1.6. Analysis.
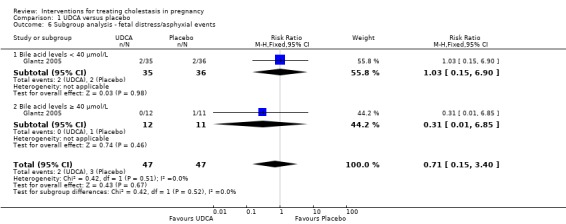
Comparison 1 UDCA versus placebo, Outcome 6 Subgroup analysis ‐ fetal distress/asphyxial events.
Secondary outcomes (maternal)
Liver function
Bile acid concentrations appeared significantly lower after treatment with UDCA compared with placebo (three trials). However, due to extreme heterogeneity and large differences in SDs, we have not presented the data for analysis.
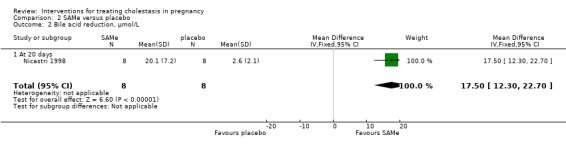
In Nicastri 1998, a significant reduction in bile acids after treatment with UDCA compared with placebo was reported (mean difference (MD) 30.40 µmol/L lower; 95% CI 23.32 to 37.48, 16 women (Analysis 1.7)).
1.7. Analysis.
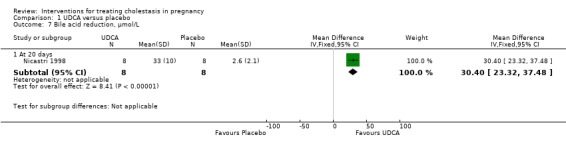
Comparison 1 UDCA versus placebo, Outcome 7 Bile acid reduction, µmol/L.
In two trials, alanine aminotransferase (ALT) concentrations were significantly lower after treatment with UDCA compared with placebo (average MD ‐111.0 IU/L; 95% CI ‐182.48 to ‐39.51; random‐effects analysis: Tau² = 1726.45; I² = 48%; 83 women (Analysis 1.8)). In one trial, ALT concentrations were significantly lower after treatment with UDCA compared with placebo (MD ‐50.88 IU/L; 95% CI ‐75.14 to ‐26.62, 16 women (Analysis 1.9)). In one trial a significant reduction in ALT after treatment with UDCA compared with placebo was seen (MD 121.00 IU/L lower; 95% CI 100.93 to 141.07, 16 women (Analysis 1.10)). Analysis 1.10 is presented as change data.
1.8. Analysis.
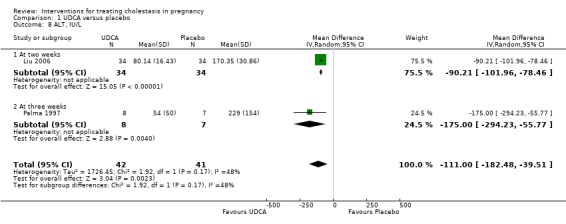
Comparison 1 UDCA versus placebo, Outcome 8 ALT, IU/L.
1.9. Analysis.
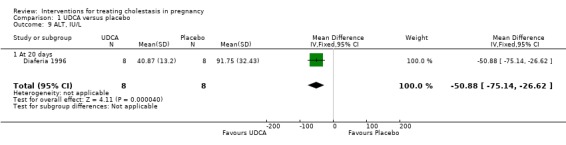
Comparison 1 UDCA versus placebo, Outcome 9 ALT, IU/L.
1.10. Analysis.
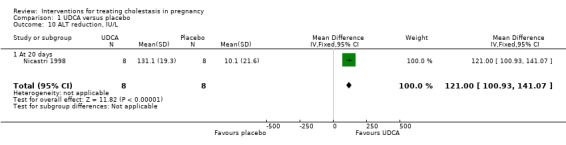
Comparison 1 UDCA versus placebo, Outcome 10 ALT reduction, IU/L.
In Glantz 2005, liver function tests were reported only graphically as medians and P values. The final bile acid concentrations were significantly lower after treatment in the UDCA group compared with the placebo group (P = 0.001). For ALT, there was a significantly greater reduction in the UDCA group compared with the placebo group overall (P = 0.01). Leino 1998 reported a reduction of ALT and bile acid levels to the upper limit of normal pregnancy values in the UDCA group but did not report numerical or graphical data by randomisation group.
Caesarean section (and mode of birth)
In four trials, no significant differences were seen between UDCA and placebo for rates of caesarean section (RR 1.00 95% CI 0.82 to 1.23; 210 women (Analysis 1.11)). Glantz 2005 did not report caesarean births but did indicate that rates of elective birth (both caesarean and vaginal) were not significantly different between the two groups (32% for UDCA and 38% for placebo).
1.11. Analysis.
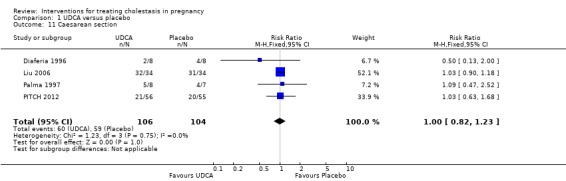
Comparison 1 UDCA versus placebo, Outcome 11 Caesarean section.
Postpartum haemorrhage
There was no significant difference in the rates of postpartum haemorrhage in the two trials (RR 0.77 95% CI 0.20 to 2.98; 127 women (Analysis 1.12)).
1.12. Analysis.
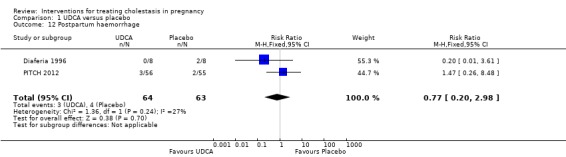
Comparison 1 UDCA versus placebo, Outcome 12 Postpartum haemorrhage.
Adverse effects of medication
No adverse effects for mothers or babies in either group were reported in three trials (Leino 1998; Liu 2006 and Nicastri 1998); and Diaferia 1996 reported that there were no important adverse effects in either the mothers or babies during or after the administration of UDCA.
In Glantz 2005, one participant in the UDCA group experienced diarrhoea and one in the placebo group suffered a severe headache. Palma 1997 reported that one woman in the UDCA group experienced transient morning nausea and mild vomiting, which resolved after changing the time of UDCA intake. PITCH 2012 reported 13 adverse events (seven mild, six moderate) in the treatment group and 10 in placebo group (eight mild, two moderate). The drug was stopped due to adverse events in one participant in the treatment group and one in the placebo group. The difference between the two groups was not significant (RR 1.32 95% CI 0.66 to 2.63; 220 women (Analysis 1.13)).
1.13. Analysis.
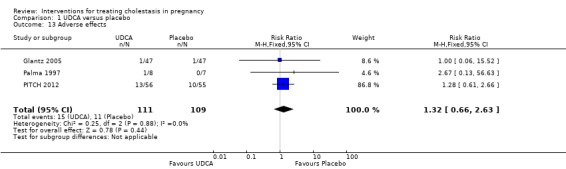
Comparison 1 UDCA versus placebo, Outcome 13 Adverse effects.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
No statistically significant differences in meconium‐stained liquor were seen between the UDCA and placebo groups in three trials (average RR 0.56 95% CI 0.24 to 1.30; random‐effects analysis: Tau² = 0.36; I² = 67% 274 women (Analysis 1.14)).
1.14. Analysis.
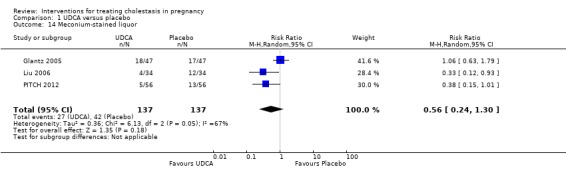
Comparison 1 UDCA versus placebo, Outcome 14 Meconium‐stained liquor.
Mean gestational age at birth
In three trials there was a non‐significant increase in gestational age at birth in the UDCA group (average MD 2.68 weeks 95% CI ‐0.13 to 5.48; random‐effects analysis: Tau² = 4.81; I² = 96%; 142 women (Analysis 1.15)). Leino 1998 reported a higher birthweight in the UDCA group coinciding with advanced gestation at birth in this group, but did not report any numerical data by comparison group.
1.15. Analysis.
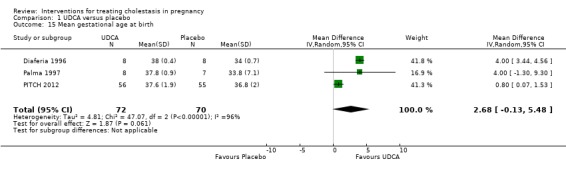
Comparison 1 UDCA versus placebo, Outcome 15 Mean gestational age at birth.
Spontaneous birth at less than 37 weeks
In two trials, no significant differences in rates of spontaneous preterm birth at less than 37 weeks were seen between the UDCA and placebo groups (RR 0.99; 95% CI 0.41 to 2.36; 109 women (Analysis 1.16)). Nicastri 1998 reported that two women in the UDCA group had spontaneous preterm labour but did not report this outcome for the women in the placebo group.
1.16. Analysis.
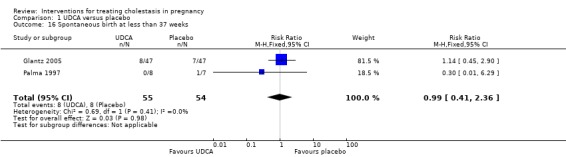
Comparison 1 UDCA versus placebo, Outcome 16 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
Two trials (different from the two reporting spontaneous preterm birth above) reported the total number of preterm births at less than 37 weeks of gestation. There were significantly fewer total preterm births in the UDCA group compared with placebo (RR 0.46 95% CI 0.28 to 0.73; 179 women (Analysis 1.17)).
1.17. Analysis.
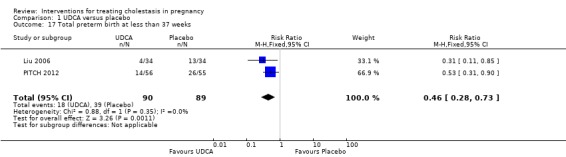
Comparison 1 UDCA versus placebo, Outcome 17 Total preterm birth at less than 37 weeks.
Admission to neonatal unit
One trial reported no significant difference in admission rates to the neonatal intensive care unit between the UDCA and the placebo groups (RR 0.48, 95% CI 0.18 to 1.31; 124 women (Analysis 1.18)).
1.18. Analysis.
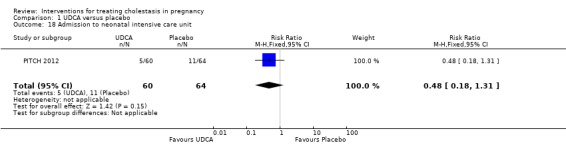
Comparison 1 UDCA versus placebo, Outcome 18 Admission to neonatal intensive care unit.
2. S‐adenosylmethionine (SAMe) versus placebo
Four trials (Frezza 1984; Frezza 1990; Nicastri 1998; Ribalta 1991) involving 82 women looked at this comparison.
Primary outcomes (maternal)
Pruritus
Four trials (82 women) reported this outcome. One trial (30 women) reported significant improvements in pruritus score with SAMe, whereas another trial (18 women) reported reduction in pruritus with 800 g daily dose of SAMe but not with 200 g daily dose. Two studies (34 women) reported a significant improvement in the pruritus score with both SAMe and placebo. None of these studies performed a subgroup analysis for improvement in pruritus in women with bile acids ≥ 40 µmol/L.
Three studies (52 women) evaluated itching on a 0‐4 scale. Data were reported as mean and SD. We planned to dichotomise and re‐analyse data but this was not possible because pruritus scores at trial entry and after intervention were not reported.
Frezza 1984 assessed pruritus on day 0 (before entering the study), and at day 10 and day 20 of treatment. Pruritus was graded from 0 to 4. The reductions in mean grade of pruritus score after 10 and 20 days of treatment were analysed and presented as a continuous outcome. A significant reduction in pruritus grade was reported with 800 g daily dose of SAMe (reported P value < 0.02 after day 10 and < 0.01 after day 20) compared with placebo but not for the 200 g daily dose.
In Frezza 1990, pruritus was assessed on a 10 cm analogue scale every three days up to 24 hours after delivery. The authors reported the mean pruritus scores after treatment as significantly lower (better) in the SAMe group compared with the placebo group (reported P value < 0.01; 30 women), but gave no numerical data.
Nicastri 1998 evaluated pruritus on a 0‐4 scale every three days. The mean changes in pruritus score in the two groups were reported as a continuous outcome. A significant reduction in mean pruritus score was observed both in the SAMe group (reported P value < 0.01; 8 women) and the placebo group (P < 0.01; 8 women).
Ribalta 1991 assessed the severity of pruritus on a 0‐4 scale immediately before treatment and every five days until delivery, one to three days after delivery and one to three months afterwards. They were analysed as a continuous outcome. The severity of pruritus reduced in both groups. The mean pruritus score decreased more in the placebo group but this difference was not significant.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
One trial reported this outcome and there were no stillbirths or neonatal deaths (Analysis 2.1).
2.1. Analysis.
Comparison 2 SAMe versus placebo, Outcome 1 Stillbirth/neonatal death.
Fetal distress/asphyxial events
In Frezza 1984, all the infants born to women in the SAMe group had Apgar scores of seven or above at five minutes. They did not report these figures for the placebo group. Comparisons were therefore not possible. In Ribalta 1991, all the newborns had Apgar scores of seven or above in both the groups. In Ribalta 1991, caesarean sections were performed for various indications, including fetal distress, but the actual number of caesarean sections for this indication was not specified.
Secondary outcomes (maternal)
Liver function
In one trial (16 women), reductions in bile acid, and ALT were significantly greater in the SAMe group compared with placebo (Analysis 2.2; Analysis 2.3).
2.2. Analysis.
Comparison 2 SAMe versus placebo, Outcome 2 Bile acid reduction, µmol/L.
2.3. Analysis.
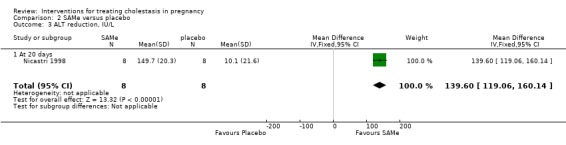
Comparison 2 SAMe versus placebo, Outcome 3 ALT reduction, IU/L.
In Frezza 1984, the final values of serum transaminases, conjugated bilirubin and total bile acids were reported to be lower in women treated with 800 mg per day SAMe than women who received placebo (total of 12 women for this comparison). In Frezza 1990 (30 women), after a mean 18 days of treatment with SAMe, total bile acids, ALT and AST were all reported to be significantly lower than for the placebo group (P = 0.01 for all four comparisons). Ribalta 1991 (18 women) reported no significant differences in results of the various liver function tests, but these were only presented in graphical form.
Caesarean section
In one trial, no significant differences were seen between the SAMe and placebo groups for caesarean section (RR 1.14; 95% CI 0.75 to 1.74; 18 women (Analysis 2.4)).
2.4. Analysis.
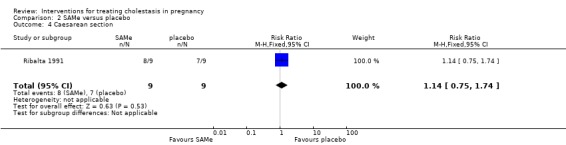
Comparison 2 SAMe versus placebo, Outcome 4 Caesarean section.
Adverse effects
Frezza 1984 reported that SAMe was well tolerated by women and no adverse effects were seen and in Frezza 1990 no adverse effects were recorded for women or their children. Ribalta 1991 reported that one woman experienced problems in peripheral veins due to prolonged daily IV infusions.
Secondary outcomes (fetal/neonatal)
Spontaneous labour/birth at less than 37 weeks
In one trial, two women in the SAMe group and five in the placebo group had preterm labour before 37 weeks (RR 0.40; 95% CI 0.09 to 1.75; 30 women (Analysis 2.5)). Nicastri 1998 reported three preterm births in the SAMe group but did not state how many there were in the placebo group. Ribalta 1991 reported the total preterm births (see below) but did not specify the number of spontaneous preterm births.
2.5. Analysis.
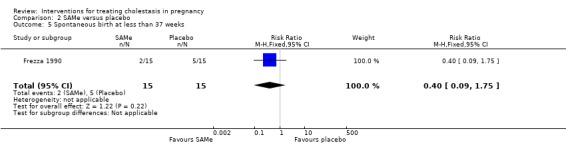
Comparison 2 SAMe versus placebo, Outcome 5 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
In one study, six women in the SAMe group versus eight in the placebo group had preterm births (RR 0.75; 95% CI 0.45 to 1.26; 18 women (Analysis 2.6)).
2.6. Analysis.
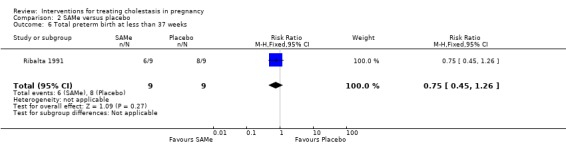
Comparison 2 SAMe versus placebo, Outcome 6 Total preterm birth at less than 37 weeks.
The following secondary outcomes were not reported for this comparison: postpartum haemorrhage, meconium‐stained liquor, mean gestational age at birth, or admission to neonatal unit.
3. Guar gum versus placebo
One trial (Riikonen 2000) involving 39 women studied this comparison.
Primary outcomes (maternal)
Pruritus
In one trial both investigators and participants assessed change in pruritus following treatment. From the women's perspective, nine (48%) women receiving guar gum and five (25%) receiving placebo experienced a reduction in pruritus (RR 1.89; 95% CI 0.77 to 4.64 (Analysis 3.1)). From the investigator's perspective, six (32%) women receiving guar gum and five (25%) receiving placebo had a reduction in pruritus (RR 1.26; 95% CI 0.46 to 3.46 (Analysis 3.1)). The difference was not significant for either group.
3.1. Analysis.
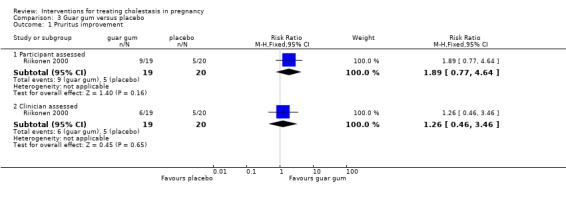
Comparison 3 Guar gum versus placebo, Outcome 1 Pruritus improvement.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
No neonatal or infant deaths were reported.
Fetal distress/asphyxial events
This outcome was not reported.
Secondary outcomes (maternal)
Liver function
In one trial, there were no significant differences seen between guar gum and placebo in reducing the levels of bile acids (MD ‐7.40; 95% CI ‐24.22 to 9.42; 39 women (Analysis 3.2)) or ALT (MD ‐37.50; 95% CI ‐137.33 to 62.33; 39 women (Analysis 3.3)).
3.2. Analysis.
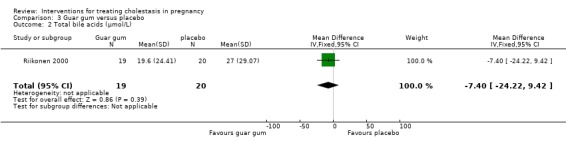
Comparison 3 Guar gum versus placebo, Outcome 2 Total bile acids (µmol/L).
3.3. Analysis.
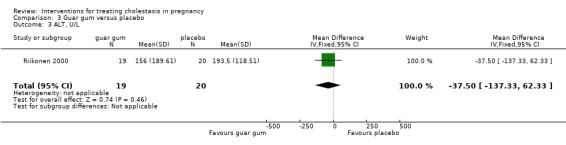
Comparison 3 Guar gum versus placebo, Outcome 3 ALT, U/L.
Adverse effects of medication
Eight women (42%) in the guar gum group and six (30%) in the placebo group reported mild abdominal distress, diarrhoea and flatulence during the first days of treatment, showing no significant difference overall (RR 1.40 95% CI 0.60 to 3.29) (Analysis 3.4)). None of the participants discontinued the study.
3.4. Analysis.
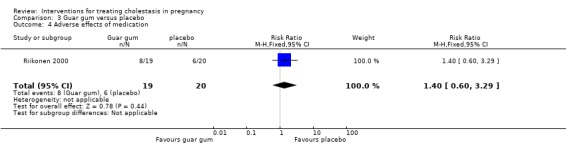
Comparison 3 Guar gum versus placebo, Outcome 4 Adverse effects of medication.
Secondary outcomes (fetal/neonatal)
Mean gestational age at birth
The mean gestational age for women in the guar gum group was 38.40 weeks and 38.30 weeks for placebo (MD 0.10 weeks; 95% CI ‐0.73 to 0.93 (Analysis 3.5).
3.5. Analysis.

Comparison 3 Guar gum versus placebo, Outcome 5 Mean gestational age at birth.
The following secondary outcomes were not reported for this comparison: caesarean section, postpartum haemorrhage, meconium‐stained liquor, spontaneous or total preterm birth, or admission to neonatal unit.
4. Activated charcoal versus no treatment
One trial (Kaaja 1994) involving 20 women looked at this comparison.
Primary outcomes (maternal)
Pruritus
Participants maintained a daily written record of pruritus using four‐point scale. Four (40%) women taking activated charcoal compared to none in the no treatment group reported relief of itching after eight days follow‐up. This difference was not significant (RR 9.00; 95% CI 0.55 to 147.95; 20 women (Analysis 4.1)).
4.1. Analysis.
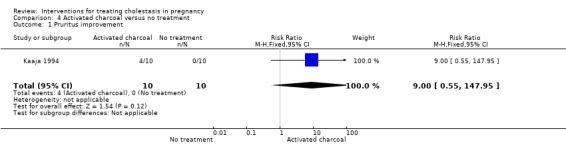
Comparison 4 Activated charcoal versus no treatment, Outcome 1 Pruritus improvement.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
No details were provided.
Fetal distress/asphyxial events
Outcome not reported.
Secondary outcomes (maternal)
Liver function
After eight days treatment, seven (70%) women taking activated charcoal compared with one (10%) woman in the no treatment group had decreased bile acid concentrations. This was a significant difference (MD ‐45.20 µmol/L; 95% CI ‐74.31 to ‐16.09; 20 women (Analysis 4.2)). However, there were no significant differences between charcoal and no treatment in final ALT concentrations (MD 74.60; 95% CI ‐141.33 to 290.53; 20 women (Analysis 4.3)).
4.2. Analysis.

Comparison 4 Activated charcoal versus no treatment, Outcome 2 Bile acids after 8 days treatment, µmol/L.
4.3. Analysis.
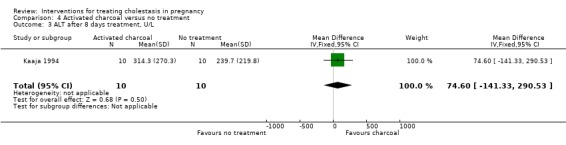
Comparison 4 Activated charcoal versus no treatment, Outcome 3 ALT after 8 days treatment, U/L.
Adverse effects of medication
Some participants reported that they found the charcoal suspension unpleasant to swallow; and some noted that their stools were black.
Secondary outcomes (fetal/neonatal)
Mean gestational age at birth
There was no significant difference in mean gestation at birth between the two groups (MD ‐1.00 week; 95% CI ‐2.77 to 0.77 (Analysis 4.4)).
4.4. Analysis.
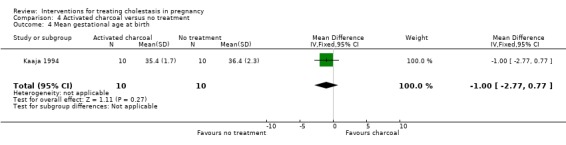
Comparison 4 Activated charcoal versus no treatment, Outcome 4 Mean gestational age at birth.
The following secondary outcomes were not reported for this comparison: caesarean section, postpartum haemorrhage, meconium‐stained liquor, spontaneous or total preterm birth, or admission to neonatal unit.
5. Dexamethasone versus placebo
One trial (Glantz 2005) involving 83 women studied this comparison.
Primary outcomes (maternal)
Pruritus
No significant difference in pruritus score (100‐mm VAS) was seen between the dexamethasone and placebo groups after three weeks treatment (83 women; no P value reported).
Primary outcomes (fetal/neonatal)
Stillbirths
One stillbirth was reported in the placebo group and none in the dexamethasone group (RR 0.43; 95% CI 0.02 to 10.31; 83 women (Analysis 5.1)).
5.1. Analysis.
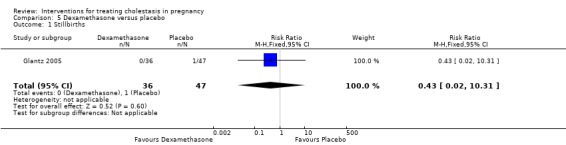
Comparison 5 Dexamethasone versus placebo, Outcome 1 Stillbirths.
Fetal distress/asphyxial events
Asphyxial events included operative birth due to asphyxia, arterial umbilical pH less than 7.05 and Apgar score of less than seven at five minutes. Four (11%) babies born to women receiving dexamethasone suffered asphyxial events compared with two (4%) babies born to women who received placebo (RR 2.61; 95% CI 0.51 to 13.47; 83 women (Analysis 5.2)).
5.2. Analysis.
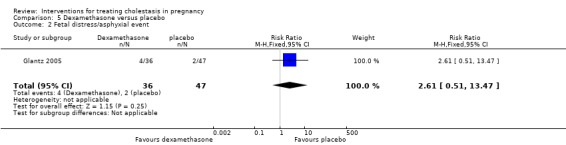
Comparison 5 Dexamethasone versus placebo, Outcome 2 Fetal distress/asphyxial event.
Subgroup analysis (bile acid levels ≥ 40 µmol/L versus bile acid levels < 40 µmol/L)
Glantz 2005 presented data for the subgroups of bile acids ≥ 40 µmol/L versus bile acids < 40 µmol/L for one of the review's primary outcomes (fetal distress/asphyxial events) (Analysis 5.3). There were no differences between subgroups, (test for subgroup differences: Chi² = 0.69, df = 1 (P = 0.40), I² = 0%).
5.3. Analysis.
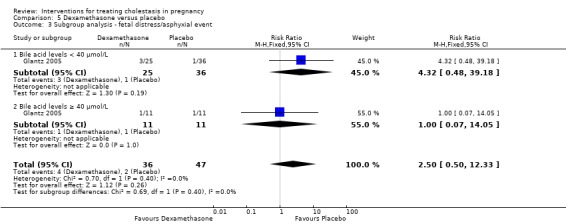
Comparison 5 Dexamethasone versus placebo, Outcome 3 Subgroup analysis ‐ fetal distress/asphyxial event.
Secondary outcomes (maternal)
Liver function
In one study, liver function tests were reported only graphically as medians and P values. The final bile acid concentrations were significantly reduced in the dexamethasone group compared with placebo overall (P = 0.01); and also in the women with severe cholestasis (P = 0.01). For ALT, there was not a significantly greater reduction in the dexamethasone group compared with the placebo group overall.
Caesarean section
Glantz 2005 did not report caesarean births but did indicate that rates of elective birth (both caesarean and vaginal) were not significantly different between the two groups (33% for dexamethasone and 38% for placebo).
Adverse effects of medication
One woman on dexamethasone suffered nausea, dizziness and stomach pain and one woman receiving placebo complained of severe headache.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
No significant differences for meconium‐stained liquor were found between dexamethasone and placebo (RR 1.00; 95% CI 0.56 to 1.78; 83 women (Analysis 5.4)). Similarly, the results were not significant in the severe subgroup, with five out of 11 women receiving dexamethasone having meconium‐stained liquor compared with six out of 11 women receiving placebo (RR 0.83; 95% CI 0.36 to 1.94 (Analysis 5.4)).
5.4. Analysis.
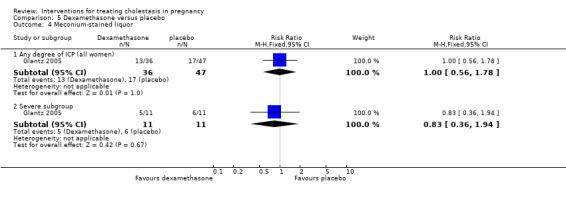
Comparison 5 Dexamethasone versus placebo, Outcome 4 Meconium‐stained liquor.
Spontaneous birth at less than 37 weeks
No significant differences between dexamethasone and placebo were seen for spontaneous preterm birth at less than 37 weeks' gestation (RR 1.52; 95% CI 0.21 to 10.90; random‐effects analysis: Tau² = 1.56; I² = 77%) 83 women (Analysis 5.5) or for the subgroup of women with severe cholestasis (RR 0.60; 95% CI 0.19 to 1.92; 22 women (Analysis 5.5)). There was evidence of a difference between subgroups, (test for subgroup differences: Chi² = 4.10, df = 1 (P = 0.04), I² = 75.6%).
5.5. Analysis.
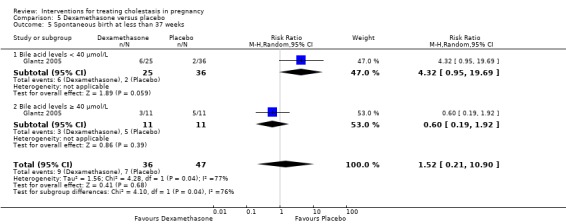
Comparison 5 Dexamethasone versus placebo, Outcome 5 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
No statistically significant differences were found between dexamethasone and placebo (RR 1.16; 95% CI 0.26 to 5.10; random‐effects analysis: Tau² = 0.88; I² = 77%: 83 women (Analysis 5.6)). 4/11 women receiving dexamethasone versus 7/11 women receiving placebo in the severe subgroup with bile acid levels ≥ 40µmol/L had a preterm birth (RR 0.57; CI 0.23 to 1.41; 22 women (Analysis 5.6). There was evidence of a difference between subgroups, (test for subgroup differences: Chi² = 4.11, df = 1 (P = 0.04), I² = 75.7%).
5.6. Analysis.
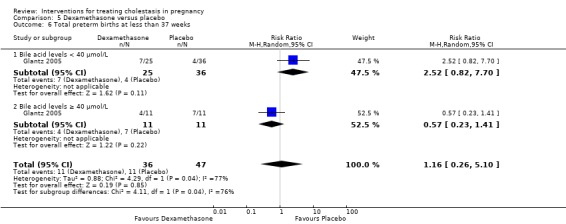
Comparison 5 Dexamethasone versus placebo, Outcome 6 Total preterm births at less than 37 weeks.
For this comparison the following secondary outcomes were not reported: postpartum haemorrhage, mean gestational age at birth or admission to neonatal unit.
6. Ursodeoxycholic acid (UDCA) versus S‐adenosylmethionine (SAMe)
Five trials (Binder 2006; Floreani 1996; Nicastri 1998; Roncaglia 2004; Zhang 2012) involving 212 women compared these two interventions.
Primary outcomes (maternal)
Pruritus
Nicastri 1998 reported a significantly greater fall in pruritus score on a 0‐4 scale with both interventions (reported P value < 0.01). Results were analysed as a continuous outcome. Dichotomisation of data for re‐analysis was not possible because results were presented as mean and SD. Zhang 2012 reported improvements in pruritus symptoms in both groups, but did not report the actual scores and stated that the differences were not statistically significant. The other three trials reported number of women with improved pruritus after treatment ‐ Binder 2006 on a 10‐point scale, and Floreani 1996 and Roncaglia 2004 on four‐point scales. Improvements were seen favouring the UDCA group in comparison to the SAMe group in the following categories: any improvement (average RR 1.46, 95% CI 0.83 to 2.59; 117 women; 3 studies; I² = 67%); marked improvement (RR 1.73, 95% CI 1.00 to 2.98; 51 women; 1 study); complete resolution (RR 21.00, 95% CI 1.40 to 315.98; 20 women; 1 study); and complete resolution or marked improvent (average RR 4.68, 95% CI 0.26 to 83.44; 71 women; 2 studies; I² = 78%). However in the analyses for any improvement and complete or marked improvement, substantial statistical heterogeneity was observed, and so a random‐effects model was used in these analyses. SeeAnalysis 6.1.
6.1. Analysis.
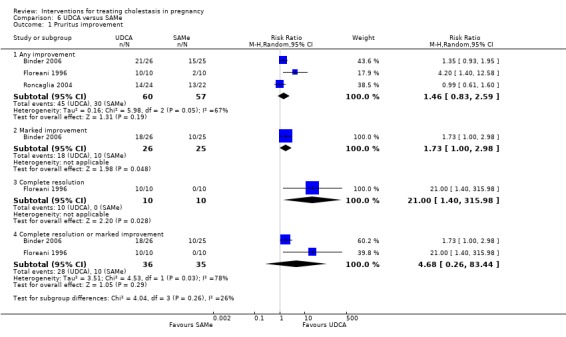
Comparison 6 UDCA versus SAMe, Outcome 1 Pruritus improvement.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
Binder 2006 reported zero stillbirths in either group and Zhang 2012 reported zero perinatal deaths in either group. Three trials did not comment on this outcome.
Fetal distress/asphyxial events
For Binder 2006, we included those women who delivered by caesarean section for fetal asphyxia to avoid duplication and overestimation of rates of fetal distress. For Roncaglia 2004, we included women with babies who had an Apgar score of less than seven at five minutes in our analysis. Floreani 1996 reported that none of the babies had Apgar scores less than seven at five minutes. Overall, there were no significant differences in fetal distress between the two groups (RR 0.94; 95% CI 0.25 to 3.58; 117 women (Analysis 6.2)).
6.2. Analysis.
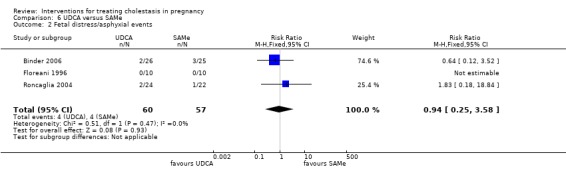
Comparison 6 UDCA versus SAMe, Outcome 2 Fetal distress/asphyxial events.
Secondary outcomes (maternal)
Liver function
In one trial, women on UDCA had a greater fall in bile acid concentrations compared with SAMe (MD 12.90 µmol/L; 95% CI 4.36 to 21.44; 16 women (Analysis 6.3)). Another trial reported a significantly lower bile acid concentration in the UDCA group compared with the SAMe group after treatment (MD ‐27.00 µmol/L; 95% CI ‐43.67 to ‐10.33; 51 women (Analysis 6.3)) while ALT concentrations were lower with SAMe (MD ‐2.20 µkat/L; 95% CI ‐3.55 to ‐0.85; 51 women (Analysis 6.4)).
6.3. Analysis.
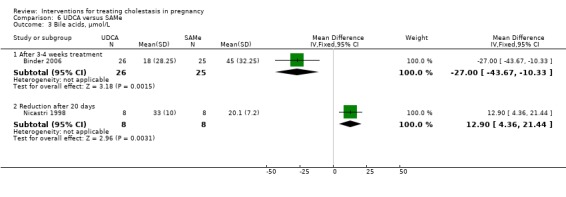
Comparison 6 UDCA versus SAMe, Outcome 3 Bile acids, µmol/L.
6.4. Analysis.
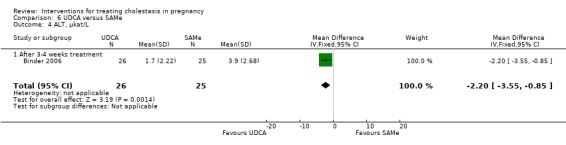
Comparison 6 UDCA versus SAMe, Outcome 4 ALT, µkat/L.
Roncaglia 2004 reported differences in laboratory variables as median and P values in relation to treatment. A significant reduction was reported in bile acids (reported P value = 0.001), and ALT (reported P value = 0.001) in the group receiving UDCA, whereas the changes from baseline were not significant in the group receiving SAMe.
All liver function results in Floreani 1996 were presented graphically ‐ after 15 days treatment; women in the UDCA group showed significantly lower total bile acid concentrations compared with women in the SAMe group (reported P value < 0.05) and there were no significant differences seen for ALT concentrations after 15 days treatment with either UDCA or SAMe (20 women in total).
Caesarean section
Three trials studies reported caesarean sections with no overall difference seen between UDCA and SAMe (RR 0.90; 95% CI 0.52 to 1.58; 117 women (Analysis 6.5)).
6.5. Analysis.
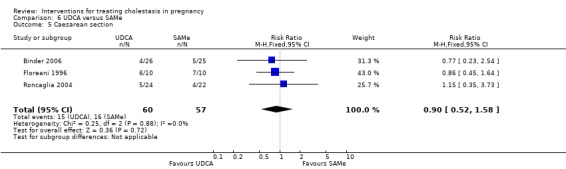
Comparison 6 UDCA versus SAMe, Outcome 5 Caesarean section.
Postpartum haemorrhage
Binder 2006 and Roncaglia 2004 reported estimated blood loss (mL) at birth rather than the incidence of postpartum haemorrhage. The differences were not significant.
Adverse effects of medication
Binder 2006, Nicastri 1998, Roncaglia 2004 and Zhang 2012 noted no adverse effects on women or the fetuses with either therapy. Floreani 1996 noted that both drugs were "well tolerated".
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
Two trials compared meconium‐stained liquor at birth, with no significant difference seen between UDCA and SAMe (RR 0.47; 95% CI 0.17 to 1.27; 97 women (Analysis 6.6)).
6.6. Analysis.
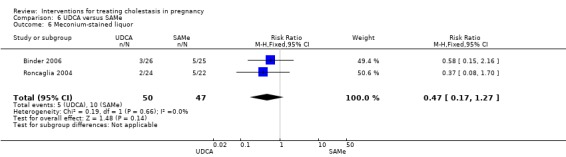
Comparison 6 UDCA versus SAMe, Outcome 6 Meconium‐stained liquor.
Mean gestational age at birth
No significant difference in gestational age at birth between UDCA and SAMe was seen in two trials (MD ‐0.04 weeks; 95% CI ‐0.84 to 0.76; 66 women (Analysis 6.7)).
6.7. Analysis.
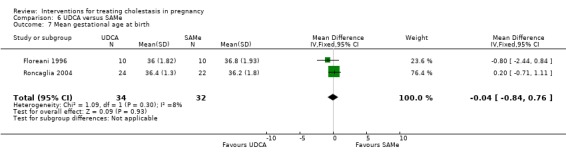
Comparison 6 UDCA versus SAMe, Outcome 7 Mean gestational age at birth.
Binder 2006 only reported ranges and not SD (no significant differences were seen between UDCA and SAMe).
Spontaneous birth at less than 37 weeks
In two trials, no significant difference between UDCA and SAMe was seen for spontaneous births less than 37 weeks (RR 0.59; 95% 0.22 to 1.59; 62 women (Analysis 6.8)).
6.8. Analysis.
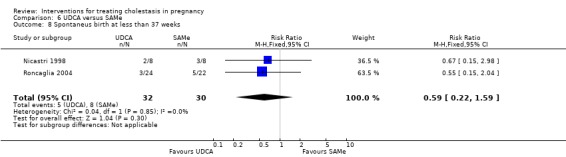
Comparison 6 UDCA versus SAMe, Outcome 8 Spontaneus birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
Two other trials reported the total number of births at less than 37 weeks of gestation for the two groups, but did not specify how many of them were spontaneous preterm births. There was no significant difference between groups (RR 0.71, 95% CI 0.33 to 1.54; 71 women (Analysis 6.9)).
6.9. Analysis.
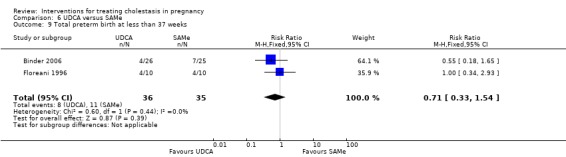
Comparison 6 UDCA versus SAMe, Outcome 9 Total preterm birth at less than 37 weeks.
Admission to neonatal unit
Two trials reported the number of babies that were admitted to the neonatal unit and the difference was not significant (RR 0.51; 95% CI 0.21 to 1.27; 97 babies (Analysis 6.10)).
6.10. Analysis.
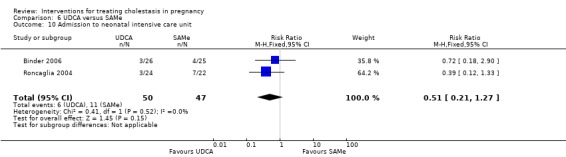
Comparison 6 UDCA versus SAMe, Outcome 10 Admission to neonatal intensive care unit.
7. Ursodeoxycholic acid (UDCA) versus dexamethasone
One study (Glantz 2005) involving 83 women compared these two interventions.
Primary outcomes (maternal)
Pruritus
Improvement in pruritus after three weeks of treatment was reported graphically. No significant differences were seen overall, although in the subgroup with severe obstetric cholestasis, UDCA was significantly more effective than dexamethasone (P = 0.01).
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
There were no stillbirths or neonatal deaths in either group.
Fetal distress/asphyxial events
No statistically significant difference in asphyxial events results were found between UDCA and dexamethasone (RR 0.34; 95% CI 0.08 to 1.45; 83 women (Analysis 7.1)).
7.1. Analysis.
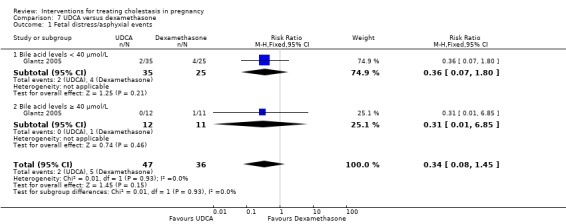
Comparison 7 UDCA versus dexamethasone, Outcome 1 Fetal distress/asphyxial events.
Subgroup analysis (bile acid levels ≥ 40 µmol/L versus bile acid levels < 40 µmol/L)
Glantz 2005 presented data for the subgroups of bile acids ≥ 40 µmol/L versus bile acids < 40 µmol/L for fetal distress/asphyxial events.
In the severe subgroup (bile acids ≥ 40 µmol/L), 0/12 in the UDCA group and 1/11 in the dexamethasone group were reported to have asphyxial events (RR 0.31; 95% CI 0.01 to 6.85; 23 women (Analysis 7.1). There were no differences between subgroups (Analysis 7.1), (test for subgroup differences: Chi² = 0.01, df = 1 (P = 0.93), I² = 0%).
Secondary outcomes (maternal)
Liver function
UDCA was significantly better than dexamethasone in reducing serum bile acid concentrations (P = 0.001) and ALT (P = 0.01). In the subgroup of women with severe cholestasis, these measures of liver function showed significantly greater reductions for UDCA compared with dexamethasone. These results were reported as graphs and P values.
Caesarean section
Glantz 2005 did not report caesarean births but did indicate that rates of elective birth (both caesarean and vaginal) were not significantly different between the two groups (32% for UDCA and 33% for dexamethasone).
Adverse effects of medication
In one study, one woman on UDCA complained of diarrhoea while one woman receiving dexamethasone suffered from nausea, dizziness and stomach pain. This was a non‐significant difference (RR 0.77 95% CI 0.05 to 11.83; 83 women (Analysis 7.2)).
7.2. Analysis.

Comparison 7 UDCA versus dexamethasone, Outcome 2 Adverse effects of medication.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
The differences between UDCA and dexamethasone for passage of meconium‐stained liquor was not statistically significant (RR 1.06; 95% CI 0.60 to 1.87; 83 women (Analysis 7.3)). In the severe subgroup, 6/12 women in the UDCA group and 5/11 in the dexamethasone group had meconium‐stained liquor.
7.3. Analysis.
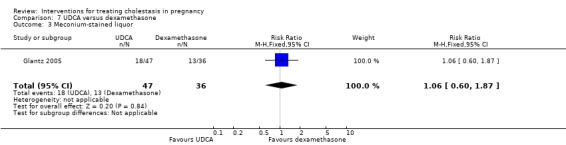
Comparison 7 UDCA versus dexamethasone, Outcome 3 Meconium‐stained liquor.
Spontaneous birth at less than 37 weeks
The results for spontaneous preterm birth between UDCA and dexamethasone were not significantly different (RR 0.68; 95% CI 0.29 to 1.59; 83 women (Analysis 7.4)). In the severe subgroup, 4/12 women in the UDCA group and 4/11 in the dexamethasone group had a spontaneous preterm birth.
7.4. Analysis.
Comparison 7 UDCA versus dexamethasone, Outcome 4 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
There were no significant differences seen between groups (RR 0.87; 95% CI 0.44 to 1.71; 83 women (Analysis 7.5)).
7.5. Analysis.
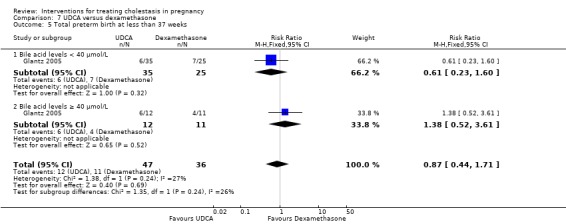
Comparison 7 UDCA versus dexamethasone, Outcome 5 Total preterm birth at less than 37 weeks.
The following secondary outcomes were not reported: postpartum haemorrhage, mean gestational age at birth or admission to neonatal unit.
8. Ursodeoxycholic acid (UDCA) versus cholestyramine
One trial (Kondrackiene 2005) involving 84 women compared these two interventions.
Primary outcomes (maternal)
Pruritus
Self‐assessment of pruritus was performed by participants on a 0‐4 scale. Pruritus was relieved after three to four days in the UDCA group compared to seven to 10 days cholestyramine group. UDCA was found to result in a lower mean pruritus score compared with cholestyramine. After four days, the pruritus score was significantly lower in the group receiving UDCA compared with the group receiving cholestyramine (reported P value < 0.05 after four days; P < 0.001 after 14 days). Results were presented as mean and SD and dichotomization was not possible. Also, a significantly higher number of women in the UDCA group reported a reduction of pruritus score by more than 50% (RR 3.50; 95% CI 1.81 to 6.77; 84 women (Analysis 8.1).
8.1. Analysis.
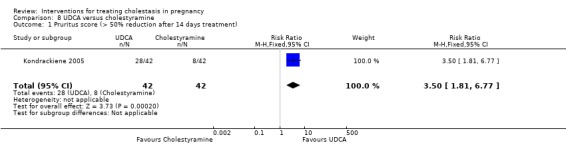
Comparison 8 UDCA versus cholestyramine, Outcome 1 Pruritus score (> 50% reduction after 14 days treatment).
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
In the only trial there were no stillbirths or neonatal deaths in either group.
Fetal distress/asphyxial events
One out of 42 women in each group suffered morbidity associated with fetal distress (RR 1.00; 95% CI 0.06 to 15.47; 84 women (Analysis 8.2)).
8.2. Analysis.
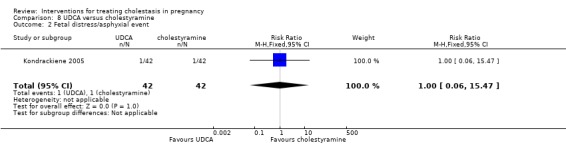
Comparison 8 UDCA versus cholestyramine, Outcome 2 Fetal distress/asphyxial event.
Secondary outcomes (maternal)
Liver function
The trial did not observe significant differences in bile acid concentrations between the two groups after treatment (MD ‐1.80 µmol/L; 95% CI ‐13.10 to 9.50; 84 women (Analysis 8.3)). For ALT, women in the UDCA group had much lower concentrations after treatment than women in the cholestyramine group (MD ‐144.20 U/L; 95% CI ‐186.63 to ‐101.77; 84 women (Analysis 8.4)).
8.3. Analysis.
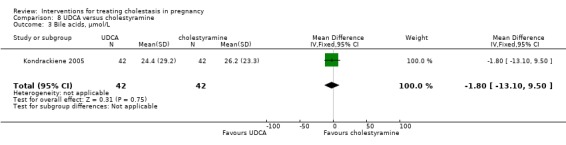
Comparison 8 UDCA versus cholestyramine, Outcome 3 Bile acids, µmol/L.
8.4. Analysis.
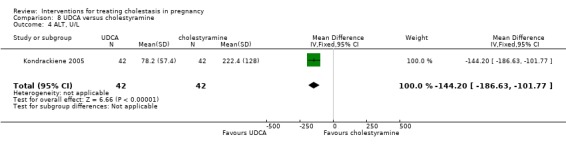
Comparison 8 UDCA versus cholestyramine, Outcome 4 ALT, U/L.
Caesarean section
There were no statistically significant differences between the two groups in rates of caesarean section (one trial; RR 2.33; 95% CI 0.65 to 8.42; 84 women (Analysis 8.5)). Reasons for the seven caesareans in the UDCA group were three multiple pregnancies, one placenta praevia, one cephalo‐pelvic disproportion, one fetal distress and one advanced maternal age. The three caesareans in the cholestyramine group were performed for fetal distress, twin pregnancy and cephalo‐pelvic disproportion (one case each).
8.5. Analysis.
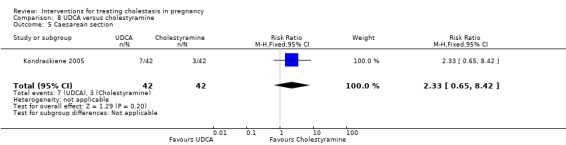
Comparison 8 UDCA versus cholestyramine, Outcome 5 Caesarean section.
Adverse effects of medication
Cholestyramine use was found to have a greater number of adverse effects with 12 out of 42 women suffering adverse effects (11 women suffering nausea, five women suffering vomiting and one woman suffering diarrhoea) compared with no adverse events reported for women in the UDCA group (RR 0.04; 95% CI 0.00 to 0.65, (Analysis 8.6)).
8.6. Analysis.
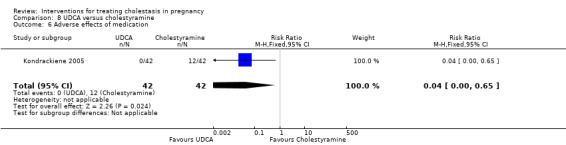
Comparison 8 UDCA versus cholestyramine, Outcome 6 Adverse effects of medication.
Secondary outcomes (fetal/neonatal)
Mean gestational age at birth
Women receiving UDCA had a significantly shorter gestational length than women in the cholestyramine group (MD ‐1.30 weeks; 95% CI ‐1.99 to ‐0.61: one trial; 84 women (Analysis 8.7)).
8.7. Analysis.
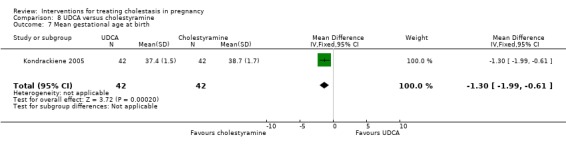
Comparison 8 UDCA versus cholestyramine, Outcome 7 Mean gestational age at birth.
Spontaneous birth at less than 37 weeks
The study did not report spontaneous preterm births separately for the two interventions.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
No significant difference was seen for preterm births between the two groups ((RR 0.60; 95% CI 0.15 to 2.35; 84 women (Analysis 8.8)).
8.8. Analysis.

Comparison 8 UDCA versus cholestyramine, Outcome 8 Total preterm birth at less than 37 weeks.
The following secondary outcomes were not reported: postpartum haemorrhage, meconium‐stained liquor or admission to neonatal unit.
9. Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus placebo
One trial (16 women) contributed data to this comparison (Nicastri 1998).
Primary outcomes (maternal)
Pruritus
Pruritus was assessed on a 0‐4 scale and results were analysed as a continuous outcome. Dichotomisation of data for reanalysis was not possible because results were presented as mean and SD. Significant change in pruritus score from the baseline was reported after treatment in the two groups (reported P value < 0.01).
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
This outcome was not reported.
Fetal distress/asphyxial events
This outcome was not reported.
Secondary outcomes (maternal)
Liver function
Compared with women given placebo, women given UDCA + SAMe had significantly greater decreases in bile acids (MD 41.70 µmol/L; 95% CI 35.57 to 47.83; 16 women (Analysis 9.1)).
9.1. Analysis.
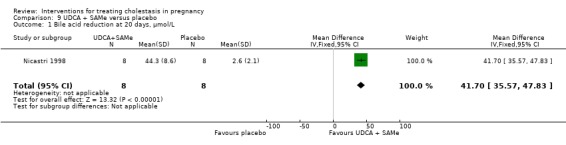
Comparison 9 UDCA + SAMe versus placebo, Outcome 1 Bile acid reduction at 20 days, µmol/L.
Adverse effects of medication
No adverse effects were observed in the mother or the babies in either group.
Secondary outcomes (fetal/neonatal)
Spontaneous birth at less than 37 weeks
One case of spontaneous preterm birth was reported in the UDCA + SAMe group but this outcome was not reported for the placebo group in Nicastri 1998.
The following secondary outcomes were not reported: caesarean section, postpartum haemorrhage, meconium‐stained liquor, mean gestational age at birth, total preterm birth or admission to neonatal unit.
10. Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus S‐adenosylmethionine (SAMe)
Three trials (147 women) contributed data to this comparison (Binder 2006; Nicastri 1998; Zhang 2012).
Primary outcomes (maternal)
Pruritus
One study reported no significant differences for any improvement in pruritus on a 10‐point scale between the two groups (RR 1.42; 95% CI 0.99 to 2.03; 52 women). However when restricted only to women with marked improvement, significantly more in the UDCA + SAMe group reported marked improvements in their pruritus compared with those in the other SAMe alone group (RR 1.85; 95% CI 1.09 to 3.14; 52 women) Analysis 10.1.
10.1. Analysis.
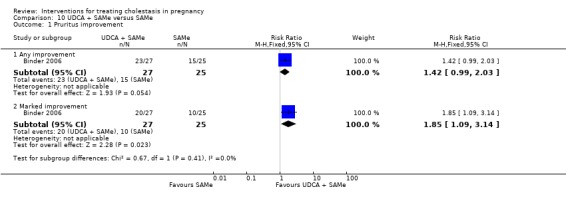
Comparison 10 UDCA + SAMe versus SAMe, Outcome 1 Pruritus improvement.
Nicastri 1998 reported a significant reduction in pruritus after treatment with UDCA + SAMe compared with SAMe alone. They used 0‐4 scale for assessing pruritus but analysed results as a continuous outcome. Dichotomisation of data and reanalysis was not possible because results were reported as mean and SD. Zhang 2012 reported improvements in pruritus symptoms in both groups, but did not report the actual scores and stated that the differences were not statistically significant.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
There were no stillbirths or neonatal deaths in two trials (Analysis 10.2). This outcome was not reported in Nicastri 1998.
10.2. Analysis.
Comparison 10 UDCA + SAMe versus SAMe, Outcome 2 Stillbirths/neonatal deaths.
Fetal distress/asphyxial events
In one trial, one woman (4%) in the UDCA + SAMe group and three (12%) in the SAMe group had an operative birth for fetal asphyxia (RR 0.31 95% CI 0.03 to 2.78; 52 women (Analysis 10.3)).
10.3. Analysis.
Comparison 10 UDCA + SAMe versus SAMe, Outcome 3 Fetal distress/asphyxial event.
Secondary outcomes (maternal)
Liver function
The two trials reported contrasting results for improvement in bile acid levels. In one trial bile acid after 3‐4 weeks were significantly lower in the UDCA + SAMe group compared with SAMe alone (MD ‐25.00 µmol/L; 95% CI ‐40.16 to ‐9.84; 52 women) and in the other trial, reduction in bile acid levels were significantly lower in the SAMe only group compared to UDCA + SAMe group after 20 days (MD 24.20 µmol/L 95% CI 16.43 to 31.97; 16 women) ‐ Analysis 10.4.
10.4. Analysis.
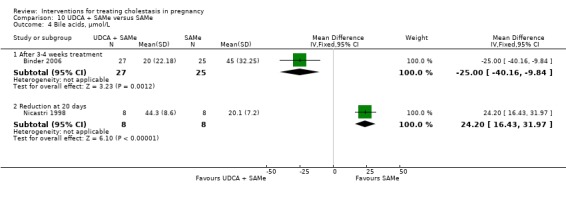
Comparison 10 UDCA + SAMe versus SAMe, Outcome 4 Bile acids, µmol/L.
Only one trial reported ALT concentrations after treatment ‐ these were significantly lower after treatment with UDCA + SAMe compared with SAMe alone (MD ‐2.40 µkat/L; 95% CI ‐3.59 to ‐1.21; 52 women (Analysis 10.5)).
10.5. Analysis.
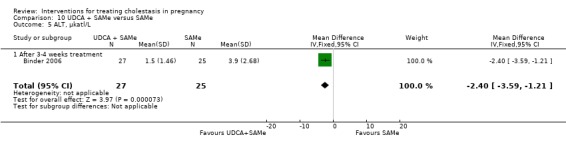
Comparison 10 UDCA + SAMe versus SAMe, Outcome 5 ALT, µkatl/L.
Caesarean section
In one trial no significant difference was seen between the UDCA + SAMe group compared with the SAMe group for caesareans (RR 0.37; 95% CI 0.08 to 1.74, 52 women (Analysis 10.6)).
10.6. Analysis.
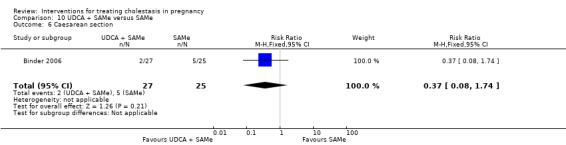
Comparison 10 UDCA + SAMe versus SAMe, Outcome 6 Caesarean section.
Postpartum haemorrhage
The three trials did not report the incidence of postpartum haemorrhage. Binder 2006 compared the estimated blood loss at delivery, which was 296 mL in the UDCA+SAMe group compared with 295 mL in the SAMe only group (MD 1.00; 95% CI ‐76.75 to 78.75; 52 women (Analysis 10.7)).
10.7. Analysis.
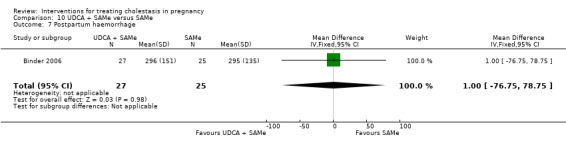
Comparison 10 UDCA + SAMe versus SAMe, Outcome 7 Postpartum haemorrhage.
Adverse effects of medication
No adverse effects were reported in two trials. Zhang 2012 reported that no adverse drug reactions were observed.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
In one trial there were no significant differences between the UDCA + SAMe and SAMe only groups for passage of meconium‐stained liquor (RR 0.46; 95% CI 0.09 to 2.31; 52 women (Analysis 10.8)).
10.8. Analysis.
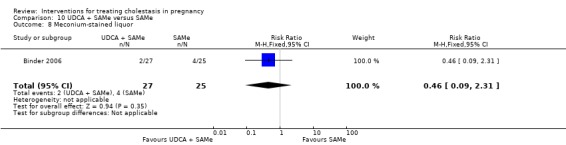
Comparison 10 UDCA + SAMe versus SAMe, Outcome 8 Meconium‐stained liquor.
Mean gestational age at birth
Binder 2006 indicated that this outcome did not differ significantly between the UDCA + SAMe and SAMe only groups but did not report mean gestational age at birth with SD.
Spontaneous birth at less than 37 weeks
One trial reported three cases of spontaneous preterm labour in the SAMe group compared with one in the UDCA + SAMe group (RR 0.33; 95% CI 0.04 to 2.56; 16 women (Analysis 10.9)).
10.9. Analysis.
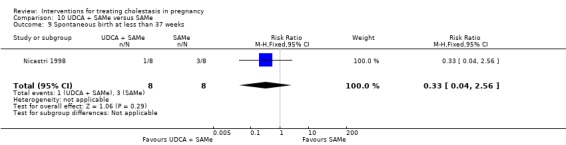
Comparison 10 UDCA + SAMe versus SAMe, Outcome 9 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic) ‐ not a pre‐specified outcome
In Binder 2006, the rate of preterm birth at less than 36 weeks was 28% (7/25) in the SAMe group compared with 15% in the combined therapy group (4/27) for less than 36 weeks (RR 0.53; 95% CI 0.18 to 1.59; 52 women (Analysis 10.10)).
10.10. Analysis.
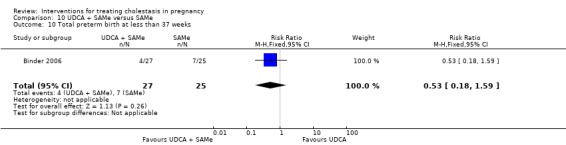
Comparison 10 UDCA + SAMe versus SAMe, Outcome 10 Total preterm birth at less than 37 weeks.
Admission to neonatal unit
No significant differences between UDCA + SAMe and SAMe only group were seen for the outcome of admission to the neonatal unit (RR 0.46; 95% CI 0.09 to 2.31; 52 women (Analysis 10.11)).
10.11. Analysis.
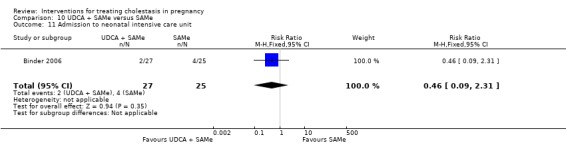
Comparison 10 UDCA + SAMe versus SAMe, Outcome 11 Admission to neonatal intensive care unit.
11. Ursodeoxycholic acid and S‐adenosylmethionine (UDCA+SAMe) versus Ursodeoxycholic acid (UDCA)
Four trials (215 women) contributed data to this comparison (Binder 2006; Luo 2008; Nicastri 1998; Zhang 2012).
Primary outcomes (maternal)
Pruritus
One study reported the effect of treatment on pruritus as deterioration, not affected, mild improvement and marked improvement. No significant differences were seen between UDCA + SAMe and UDCA alone for improvement in pruritus, either for any improvement (RR 1.05; 95% CI 0.83 to 1.35; 53 women) or marked improvement (RR 1.07; 95% CI 0.76 to 1.50; 53 women) ‐ see Analysis 11.1.
11.1. Analysis.
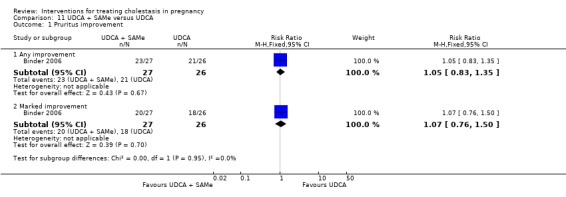
Comparison 11 UDCA + SAMe versus UDCA, Outcome 1 Pruritus improvement.
Nicastri 1998 used a 0‐4 scale for assessing pruritus and analysed results as a continuous outcome, which may not be the appropriate analysis. They found a significant reduction in pruritus score for UDCA + SAMe compared with UDCA alone. Luo 2008 reported mean itching score (0‐4 scale) as mean and SD before and after treatment and we were therefore unable to include this in the meta‐analysis. The results reported were: UDCA+SAMe 'before treatment' 3.89 ± 1.52, 'after treatment' 1.12 ± 0.63; UDCA 'before treatment' 3.90 ± 1.43, 'after treatment' 2.78 ± 0.79. Zhang 2012 reported improvements in pruritus symptoms in both groups, but did not report the actual scores and stated that the differences were not statistically significant.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
There were no stillbirths or neonatal deaths in two trials (Analysis 11.2). The other two trials did not report this outcome.
11.2. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 2 Stillbirths/neonatal deaths.
Fetal distress/asphyxial events
In one trial, two women in the UDCA group and one in the UDCA + SAMe group had an operative birth for fetal asphyxia(RR 0.48; 95% CI 0.05 to 4.99; 53 women (Analysis 11.3)).Luo 2008 pre‐specified an Apgar score of ≤ 7 as one of the fetal outcomes, but these data were either not reported or not translated.
11.3. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 3 Fetal distress/asphyxial event.
Secondary outcomes (maternal)
Liver function
One study found no significant differences between UDCA + SAMe and UDCA alone for bile acid concentrations (MD 2.00 µmol/L; 95% CI ‐11.71 to 15.71; 53 women) after treatment, whereas another study found a significant reduction in bile acid concentrations with UDCA (MD 11.30 µmol/L; 95% CI 2.16 to 20.44; 16 women) (Analysis 11.4)).
11.4. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 4 Bile acids, µmol/L.
Binder 2006 found significantly lower concentrations after treatment with combined therapy (MD ‐2.40 µkat/L; 95% CI ‐3.59 to ‐1.21; 53 women (Analysis 11.5)) and Luo 2008 reported a greater reduction with combined therapy (MD 1.28 IU/L; 95% CI 1.15 to 1.41; 64 women) (Analysis 11.6)).
11.5. Analysis.

Comparison 11 UDCA + SAMe versus UDCA, Outcome 5 ALT, µkatl/L.
11.6. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 6 Reduction in ALT (IU/L) after treatment.
Caesarean section
Two trials reported this outcome. The rates of caesarean section in the two groups were not statistically significant (RR 0.59; 95% CI 0.35 to 1.02; 116 women) (Analysis 11.7)).
11.7. Analysis.
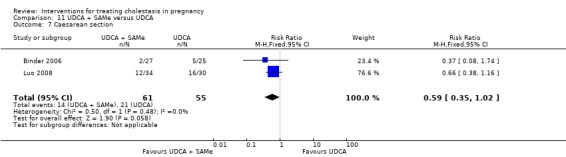
Comparison 11 UDCA + SAMe versus UDCA, Outcome 7 Caesarean section.
Postpartum haemorrhage
None of the three studies reported the incidence of postpartum haemorrhage.
Adverse effects of medication
There were no adverse effects reported in the three studies. Luo 2008 did not report this outcome.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
In one trial, no significant differences between UDCA + SAMe and UDCA alone were seen for the outcome of meconium‐stained liquor (RR 0.64; 95 CI 0.12 to 3.54; 53 women (Analysis 11.8)). Luo 2008 prespecified this outcome but data were not reported or translated.
11.8. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 8 Meconium‐stained liquor.
Mean gestational age at birth
Three trials did not report mean gestation at birth with SDs. Binder 2006 indicated that this outcome did not differ significantly between the two groups.
Spontaneous birth at less than 37 weeks
In one study, one woman who received UDCA + SAMe and two women who received UDCA only went into spontaneous labour at less than 37 weeks (RR 0.50; 95% CI 0.06 to 4.47; 16 women (Analysis 11.9)).
11.9. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 9 Spontaneous birth at less than 37 weeks.
Total preterm birth at less than 37 weeks (spontaneous and iatrogenic)
In one study, no significant difference was noted for the outcome of total preterm births at less than 37 weeks' gestation in the two groups (RR 0.69; 95% CI 0.29 to 1.62; 64 women) (Analysis 11.10)). In Binder 2006, the total preterm birth rate (< 36 weeks) was 15% in both groups.
11.10. Analysis.
Comparison 11 UDCA + SAMe versus UDCA, Outcome 10 Total preterm births at less than 37 weeks.
Admission to neonatal unit
One trial reported that two babies in the UDCA + SAMe group were admitted to neonatal intensive care unit for moderate respiratory distress syndrome (RDS) and that three babies in the UDCA group (severe prematurity in one baby and for RDS in two babies) were admitted to the neonatal unit (RR 0.64; 95% CI 0.12 to 3.54; 53 babies (Analysis 11.11)).
11.11. Analysis.
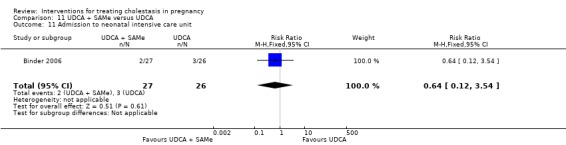
Comparison 11 UDCA + SAMe versus UDCA, Outcome 11 Admission to neonatal intensive care unit.
12. Ursodeoxycholic acid (UDCA) and Salvia versus UDCA (Ursodeoxycholic acid)
One trial (128 women) contributed data to this comparison (Fang 2009).
Primary outcomes (maternal)
Pruritus
Reduction in pruritus on a 0‐4 scale from moderate/severe to mild pruritus (3.6 to 1.4) was reported in 58/72 (80.5%) women in the UDCA + Salvia group compared with 43/56 (76.7%) in the UDCA group. The difference was not significant (RR 1.05; 95% CI 0.87 to 1.26; 128 women) (Analysis 12.1)). These effects were seen within four to six days in Salvia + UDCA group and eight to 10 days in the UDCA group.
12.1. Analysis.
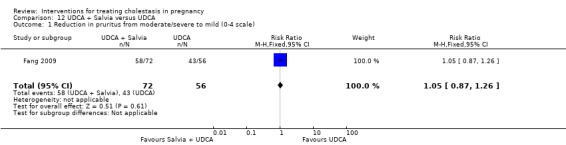
Comparison 12 UDCA + Salvia versus UDCA, Outcome 1 Reduction in pruritus from moderate/severe to mild (0‐4 scale).
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
The study did not report this outcome.
Fetal distress/asphyxial events
Thirteen women in the combination group and 11 women in the UDCA only group had caesarean births due to fetal distress. The difference was not statistically significant(RR 0.92; 95% CI 0.45 to 1.89; 128 women (Analysis 12.2)).
12.2. Analysis.
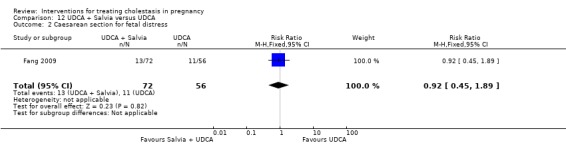
Comparison 12 UDCA + Salvia versus UDCA, Outcome 2 Caesarean section for fetal distress.
Secondary outcomes (maternal)
Liver function
Fang 2009 found a significant reduction in the levels of ALT after treatment with UDCA + Salvia compared with UDCA alone ((MD ‐14.90 µmol/L; 95% CI ‐24.42 to ‐5.38; 128 women) (Analysis 12.3)). Data on bile acids were not available.
12.3. Analysis.
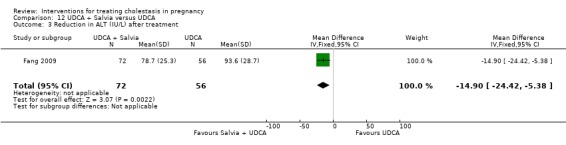
Comparison 12 UDCA + Salvia versus UDCA, Outcome 3 Reduction in ALT (IU/L) after treatment.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
No significant differences between UDCA + Salvia and UDCA alone were seen for the outcome of meconium‐stained liquor (RR 0.86; 95 CI 0.38 to 1.98; 128 women (Analysis 12.4)).
12.4. Analysis.
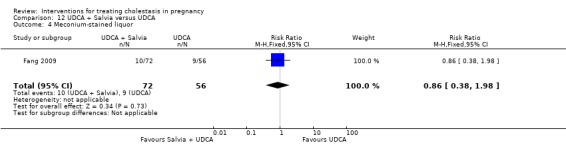
Comparison 12 UDCA + Salvia versus UDCA, Outcome 4 Meconium‐stained liquor.
The following secondary outcomes were not reported: caesarean section, postpartum haemorrhage, adverse effects of medication, mean gestational age at birth, spontaneous preterm birth, total preterm birth or admission to neonatal unit.
13. Yinchenghao decoction (YCHD) versus S‐adenosylmethionine (SAMe)
One trial (60 women) contributed data to this comparison (Huang 2004).
Primary outcomes (maternal)
Pruritus
One trial demonstrated that there were no significant differences between YCHD and SAMe in improving degree of pruritus after treatment (RR 1.00; 95% CI 0.77 to 1.29; 60 women (Analysis 13.1)).
13.1. Analysis.
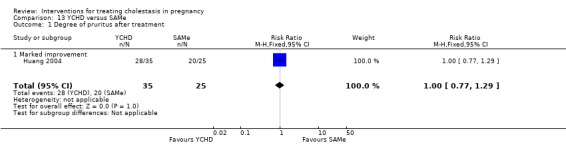
Comparison 13 YCHD versus SAMe, Outcome 1 Degree of pruritus after treatment.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
There were no stillbirths or neonatal deaths in either group.
Fetal distress/asphyxial events
No statistically significant difference in asphyxial events was found between the two groups (RR 0.86; 95% CI 0.29 to 2.50; 60 women (Analysis 13.3)).
13.3. Analysis.
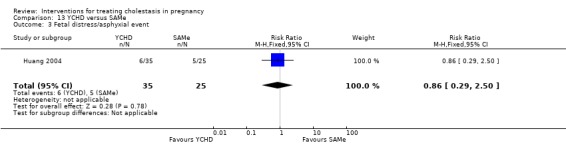
Comparison 13 YCHD versus SAMe, Outcome 3 Fetal distress/asphyxial event.
Secondary outcomes (maternal)
Liver function
There was no significant reduction in the levels of bile salt (CGA) (MD ‐1.50; 95% CI ‐6.12 to 3.12; 60 women (Analysis 13.4)) or ALT (MD 3.40; 95% CI ‐12.37 to 19.17; 60 women (Analysis 13.5)) when comparing the two intervention groups.
13.4. Analysis.
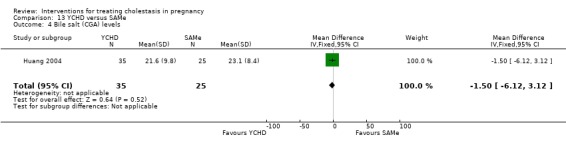
Comparison 13 YCHD versus SAMe, Outcome 4 Bile salt (CGA) levels.
13.5. Analysis.
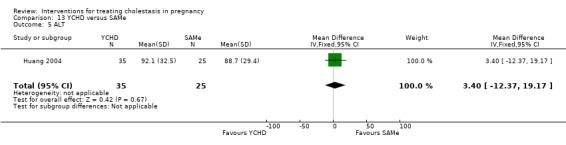
Comparison 13 YCHD versus SAMe, Outcome 5 ALT.
Caesarean section
No significant differences were seen between the YCHD and SAMe group for caesarean section (RR 0.93; 95% CI 0.56 to 1.55; 60 women (Analysis 13.6)).
13.6. Analysis.
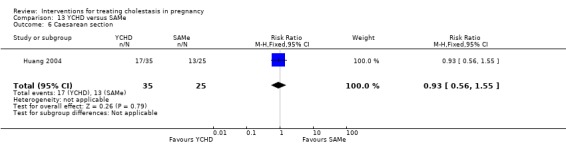
Comparison 13 YCHD versus SAMe, Outcome 6 Caesarean section.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
In one trial, no significant differences between YCHD and SAMe were seen for the outcome of meconium‐stained liquor (RR 0.86; 95% CI 0.29 to 2.50; 60 women (Analysis 13.7)).
13.7. Analysis.
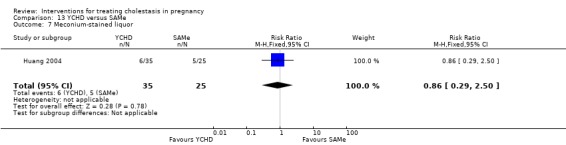
Comparison 13 YCHD versus SAMe, Outcome 7 Meconium‐stained liquor.
Mean gestational age at birth
Mean gestational age at birth in the YCHD group was 38.1 and 37.4 weeks in the SAMe group. The difference between the two groups was not significant (MD 0.70 weeks; 95% CI ‐0.35 to 1.75 (Analysis 13.8).
13.8. Analysis.
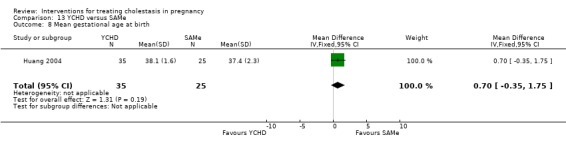
Comparison 13 YCHD versus SAMe, Outcome 8 Mean gestational age at birth.
The following secondary outcomes were not reported: postpartum haemorrhage, adverse effects of medication, spontaneous preterm birth, total preterm birth or admission to neonatal unit.
14. Danxiaoling versus yiganling
One trial (58 women) contributed data to this comparison (Shi 2002).
Primary outcomes (maternal)
Pruritus
In one trial, all 58 women, 29 women in each group, noticed improvement in pruritus after treatment (MD 1.00; 95% CI 0.94 to 1.07; 58 women). More women receiving Danxiaoling experienced marked improvement in pruritus in comparison to the Yiganling group and this difference was statistically significant (MD 1.67; 95% CI 1.14 to 2.44; 58 women). SeeAnalysis 14.1.
14.1. Analysis.
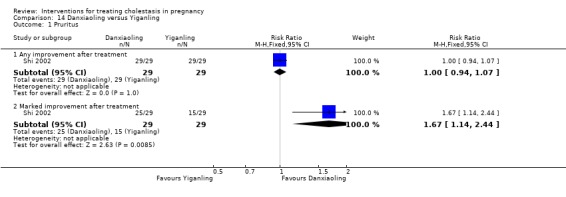
Comparison 14 Danxiaoling versus Yiganling, Outcome 1 Pruritus.
Primary outcomes (fetal/neonatal)
Stillbirth/neonatal death
There were no stillbirths or neonatal deaths in either group (Analysis 14.2).
14.2. Analysis.
Comparison 14 Danxiaoling versus Yiganling, Outcome 2 Stillbirths/neonatal deaths.
Fetal distress/asphyxial events
Shi 2002 did not report this outcome.
Secondary outcomes (maternal)
Liver function
Shi 2002 found no significant difference in the levels of bile acids (CGA) (MD ‐3.83; 95% CI ‐22.59 to 14.93; 58 women (Analysis 14.3)), or ALT (MD 5.20; 95% CI ‐36.90 to 47.30; 54 women (Analysis 14.4)).
14.3. Analysis.
Comparison 14 Danxiaoling versus Yiganling, Outcome 3 Bile acid levels (CGA).
14.4. Analysis.
Comparison 14 Danxiaoling versus Yiganling, Outcome 4 ALT.
Caesarean section
In one trial, no significant differences were seen between the Danxiaoling and Yiganling groups for caesarean section (RR 0.60; 95% CI 0.16 to 2.28; 58 women (Analysis 14.5)).
14.5. Analysis.
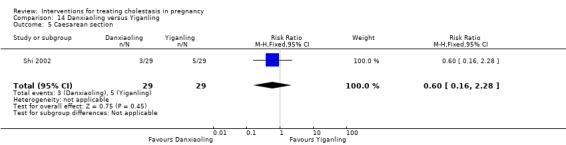
Comparison 14 Danxiaoling versus Yiganling, Outcome 5 Caesarean section.
Secondary outcomes (fetal/neonatal)
Meconium‐stained liquor
Significantly lower incidence of meconium‐stained liquor was observed in the group receiving Danxioling in comparison to the group receiving Yiganling (RR 0.40; 95% CI 0.18 to 0.89; 58 women (Analysis 14.6)).
14.6. Analysis.

Comparison 14 Danxiaoling versus Yiganling, Outcome 6 Meconium‐stained liquor.
Spontaneous birth at less than 37 weeks
There was no significant difference in the rates of spontaneous preterm births in the two groups (RR 0.33; 95% CI 0.04 to 3.02; 58 women (Analysis 14.7)).
14.7. Analysis.
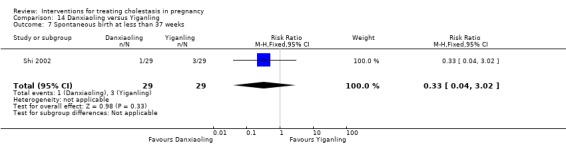
Comparison 14 Danxiaoling versus Yiganling, Outcome 7 Spontaneous birth at less than 37 weeks.
The following secondary outcomes were not reported: postpartum haemorrhage, adverse effects of medication, mean gestational age at birth, total preterm birth or admission to neonatal unit.
15. Early term delivery v expectant management
One trial (62 women) contributed data to this comparison (PITCH 2012).
There were no stillbirths or neonatal deaths in either group (Analysis 15.1) and no significant differences in caesarean section (RR 0.68; 95% CI 0.30 to 1.52; 62 women (Analysis 15.2)), passage of meconium‐stained liquor (RR 0.55; 95% CI 0.15 to 2.01; 63 women (Analysis 15.3)) or admission to neonatal intensive care unit (RR 0.55; 95% CI 0.05 to 5.76; 63 women (Analysis 15.4)).
15.1. Analysis.
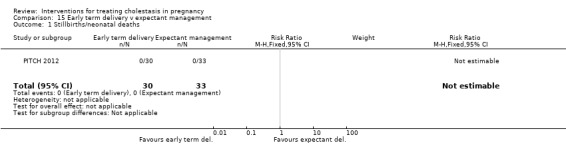
Comparison 15 Early term delivery v expectant management, Outcome 1 Stillbirths/neonatal deaths.
15.2. Analysis.
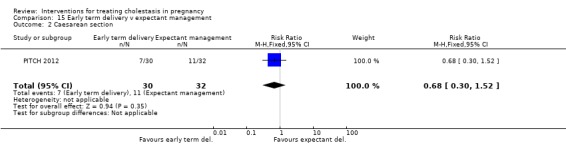
Comparison 15 Early term delivery v expectant management, Outcome 2 Caesarean section.
15.3. Analysis.
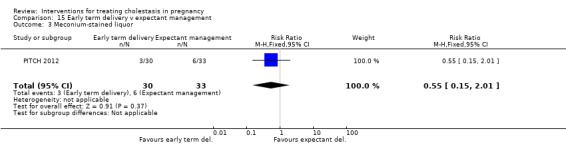
Comparison 15 Early term delivery v expectant management, Outcome 3 Meconium‐stained liquor.
15.4. Analysis.
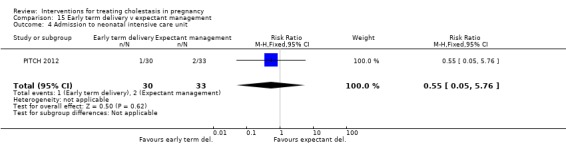
Comparison 15 Early term delivery v expectant management, Outcome 4 Admission to neonatal intensive care unit.
Discussion
Summary of main results
Although we found 21 trials with a total of 1197 women, in general the quality of evidence is low.
Only one trial reported a quantified reduction in itching as a prespecified primary outcome across all women with obstetric cholestasis. The different approaches to measuring and reporting pruritus and the very high level of statistical heterogeneity precluded any aggregation of UDCA versus placebo trials on this outcome. Individual assessment of these trials for maternal pruritus, assessed either through a categorical 0‐4 scale or on a 100 mm visual analogue (VAS) scale, demonstrated a greater support for UDCA compared with placebo and most other treatments in improving pruritus scores. One study reported no significant difference in pruritus reduction when comparing UDCA with placebo and dexamethasone, except in the subgroup of women with severe obstetric cholestasis (bile acids ≥ 40 µmol/L), where a significant improvement in pruritus was observed with UDCA. UDCA was also more effective in improving pruritus than either SAMe or cholestyramine. A combination of UDCA and SAMe was more effective than SAMe and placebo in improving pruritus. Pruritus was significantly reduced with Danxiaoling when compared to Yiganling, but the use of these medicines are currently limited to East Asia. Information on safety and efficacy and further evidence from well designed randomised controlled trials are needed before they can be adopted globally. The results from trials comparing other interventions in regards to pruritus improvement were either inconsistent or not significant.
Eight trials reported fetal or neonatal deaths, with two deaths reported overall (both in the placebo groups). There were fewer instances of fetal distress in the UDCA groups compared with placebo (RR 0.67; 95% CI 0.22 to 2.02; 304 women; I² = 48% (Analysis 1.5)) but the difference was not significant. In the UDCA group, the rates of passage of meconium‐stained liquor were lower and the mean gestational age at birth was higher but neither reached statistical significance. While the rate of total preterm births was significantly lower in the UDCA group when compared to placebo, there was no significant difference for the outcome of spontaneous preterm birth between the two groups. This could be interpreted as the rates of iatrogenic preterm births at less than 37 weeks of gestation being lower in women treated with UDCA, but evidence from larger trials is required before any robust interpretation can be attempted. The rates of fetal distress were similar when UDCA was compared with SAMe, cholestyramine and UDCA + Salvia. The group receiving combined UDCA + SAMe had fewer instances of fetal distress/asphyxial events when compared to the group randomised to UDCA or SAMe monotherapy. The rates of fetal distress were higher in the group receiving dexamethasone when compared to UDCA and placebo.
One trial compared early term delivery versus expectant management. There were no stillbirths or neonatal deaths in the two groups. There were no significant differences in the rates of caesarean section, meconium passage or admission to neonatal intensive care unit.
Overall completeness and applicability of evidence
The 21 studies included in this review are spread thinly over 15 comparisons. In only three comparisons was it possible to include more than two trials (with seven studies comparing UDCA versus placebo, four comparing SAMe versus placebo, and four trials comparing UDCA versus SAMe). In the remaining trials, it was not possible to answer with adequate levels of reliability how beneficial the relative merits of the interventions considered in this review are owing to the general paucity of data.
Quality of the evidence
The quality of the studies included in this review ranged from excellent to poor. Two large studies comparing UDCA with placebo were of high quality (Glantz 2005; PITCH 2012). Two (Fang 2009; Shi 2002) were quasi‐randomised controlled trials and two trials (Leino 1998; Zhang 2012) were available as conference abstracts only. The methods used for random sequence generation were described in eight trials (38%), allocation concealment was judged as adequate in four trials (19%) and there was no blinding in nine trials (43%). There were no dropouts reported in 10 studies (48%) and therefore all women were included in the analysis of those trials.
Due to the varying methods of measuring and reporting pruritus, pooling of data for this outcome was not possible. Only one trial reported a quantified reduction in itching as a prespecified primary outcome across all women with obstetric cholestasis. Eight trials (40%) used 0‐4 scale for pruritus assessment and analysed them as a continuous outcome, which may not be an appropriate method of analysis for such a short scale. Dichotomisation of data and re‐analysis was possible in one trial only. One trial did not specify the methods used for assessing pruritus.
The definition of fetal distress/asphyxial event was clearly predefined in one trial only. The definition of fetal distress/asphyxial events varied across the studies. Four trials did not report this outcome and it was unclear in three trials.
Potential biases in the review process
The evidence for this review is derived from studies identified in a detailed search process. Three studies (Elias 2001; Mazzella 2010; Wang 2003) were identified but Elias 2001 is no longer on www.controlled‐trials.com (where it was originally identified from) and we could not find any published randomised controlled trial by this author. Mazzella 2010 was registered on October 2010 and is not open for participant recruitment yet and Wang 2003 is published in Chinese and we were not able to locate this article. Should any of these studies be published or traced, we will include them in future updates of this review.
While we endeavoured to use a systematic process for including and excluding studies in this review and adhered to the criteria defined in our protocol, the final selection is of course open to interpretation or criticism. For further details please see Characteristics of included studies; Characteristics of excluded studies.
Agreements and disagreements with other studies or reviews
This previous version of this review Burrows 2001 included just nine randomised controlled trials with data from 227 women. In summary, the authors found insufficient evidence to recommend any of the interventions alone or in combination in treating women with obstetric cholestasis. In this update of the review there is, in general, slightly stronger support for the comparison between UDCA versus placebo (with the addition of Glantz 2005; Liu 2006 and PITCH 2012), although it is noted that the overview for this comparison should be treated conservatively until greater clarity is available from further research.
Authors' conclusions
Implications for practice.
There is support for use of UDCA in improving maternal pruritus in obstetric cholestasis although the size of the benefit is small.
Women should be informed that there is insufficient evidence to recommend UDCA to improve fetal outcome. There is some apparent decrease in some measures of fetal/neonatal morbidity associated with UDCA, including lower rates of meconium passage and higher mean gestational age at birth, but we cannot conclude this reliably due to the high level of statistical heterogeneity. Definitive evidence for fetal benefit with any intervention is still lacking.
There is insufficient evidence to indicate that SAMe, guar gum, activated charcoal, dexamethasone, cholestyramine, YCHD, DXLP, Yiganling alone or in combination are effective in treating women with cholestasis of pregnancy.
There is insufficient evidence to recommend early term delivery in obstetric cholestasis.
Implications for research.
UDCA and early term delivery are the most widely used treatments for obstetric cholestasis. Further trials of UDCA are justified. Such trials should be sufficiently large to test its effectiveness in reducing adverse fetal outcomes, and to confirm its fetal safety. Large trials of UDCA versus no treatment are feasible, but will probably have to rely on surrogate fetal outcomes such as admission to intensive care, and birth asphyxia, which are susceptible to bias. They should therefore be double blind.
It is unlikely that sufficiently large trials of near term early delivery are feasible, and there is increasing evidence from trials in other high risk pregnancies that this type of induction has few maternal side effects, so further near term induction trials are not a priority. However, if preterm induction prior to say 37 weeks is considered to pre‐empt stillbirth this should be evaluated in a trial first, before implementation.
Feedback
Gludd, July 2007
Summary
Could you explain why you chose to exclude trials published in abstract form only. Due to publication bias, trials are more likely to be published if they report statistically significant results. Excluding abstracts may therefore lead to an overestimate of treatment effects.
(Summary of comment from Lise Lotte Gluud, July 2007)
Reply
This review has been recently updated by a new review team and we have now included a randomised controlled trial published in abstract form (Leino 1998). However, it was not reported in a way that enabled the results to be included in RevMan 2012 and so is included in the text of the UCDA versus placebo results.
Contributors
Feedback: Lise Lotte Gluud
Reply to feedback: Vinita Gurung, Philippa Middleton, and Jim G Thornton
What's new
| Date | Event | Description |
|---|---|---|
| 21 November 2016 | Amended | Results in analysis 6.1 for Floreani 1996 corrected. |
History
Protocol first published: Issue 4, 1997 Review first published: Issue 4, 2001
| Date | Event | Description |
|---|---|---|
| 6 May 2014 | Amended | Michael Stokes added on the byline as an author and his contribution specified. |
| 1 March 2013 | New citation required and conclusions have changed | In this update, there is now support for a modest beneficial effect of ursodeoxycholic acid (UDCA) on pruritus, in the UDCA versus placebo comparison. |
| 20 February 2013 | New search has been performed | Search updated. Twelve studies have been included (Binder 2006; Fang 2009; Glantz 2005; Huang 2004; Kondrackiene 2005; Leino 1998; Liu 2006; Luo 2008; Roncaglia 2004; Shi 2002; PITCH 2012; Zhang 2012). A new team of review authors prepared this review update. The methods have also been updated. |
| 6 June 2011 | Feedback has been incorporated | The authors have replied to the feedback by Gludd from July 2007. SeeFeedback. |
| 30 November 2009 | Amended | Search updated. Nineteen reports added to Studies awaiting classification. |
| 30 October 2008 | Amended | Converted to new review format. |
| 13 November 2007 | Feedback has been incorporated | Feedback added. |
Acknowledgements
Bob Burrows, Ornella Clavisi and Elizabeth Burrows for preparing the first version of this review.
We acknowledge Michael Stoke's contribution to the update of this review.
We acknowledge Leanne Jones, Research Associate, Cochrane Pregnancy and Childbirth Group, for her support in the 2013 update.
This project was supported by the National Institute for Health Research, via Cochrane programme Grant funding to Cochrane Pregnancy and Childbirth. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
As part of the pre‐publication editorial process, this review has been commented on by three peers (an editor and two referees who are external to the editorial team) and the Group's Statistical Adviser.
Appendices
Appendix 1. Methods used to assess trials included in previous versions of this review
Data collection and analysis
Three reviewers (R Burrows, O Clavisi, E Burrows) independently searched for, assessed and selected trials for inclusion in this review. The decision to include trials was made without knowledge of authors, institutional affiliations, journal of publication and results. Any disagreements about inclusion of studies were resolved through discussion.
Two reviewers (O Clavisi, E Burrows) independently appraised included trials for their methodological quality using the validity criteria for intervention studies developed by Sackett et al (Sackett 2000). Individual appraisals were then compared and agreement reached. The validity criteria used are: 1. adequate method of randomisation (e.g. random number tables); 2. concealment of allocation (e.g. opaque envelopes); 3. blinding of study participants, caregivers and assessors; 4. inclusion of all randomised patients in the analysis of results; 5. adequate follow‐up of study participants (> 80%); 6. study patients have similar prognostic factors at the start of the trial; 7. study patients treated equally during the trial.
If any of this information was unavailable in the publication, we contacted the authors. However, no additional information was obtained.
Data and analyses
Comparison 1. UDCA versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.72, 2.10] |
| 2 Mean of worst itching scores over preceding 24 hours between randomisation and delivery | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | ‐12.90 [‐22.59, ‐3.21] |
| 3 Mean of average itching scores over preceding 24 hours between randomisation and delivery | 1 | 111 | Mean Difference (IV, Fixed, 95% CI) | ‐18.6 [‐27.52, ‐9.68] |
| 4 Stillbirth | 3 | 233 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.03, 2.84] |
| 5 Fetal distress/asphyxial event | 5 | 304 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.22, 2.02] |
| 6 Subgroup analysis ‐ fetal distress/asphyxial events | 1 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.15, 3.40] |
| 6.1 Bile acid levels < 40 µmol/L | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.15, 6.90] |
| 6.2 Bile acid levels ≥ 40 µmol/L | 1 | 23 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 6.85] |
| 7 Bile acid reduction, µmol/L | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 At 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 30.4 [23.32, 37.48] |
| 8 ALT, IU/L | 2 | 83 | Mean Difference (IV, Random, 95% CI) | ‐109.00 [‐182.48, ‐39.51] |
| 8.1 At two weeks | 1 | 68 | Mean Difference (IV, Random, 95% CI) | ‐90.21 [‐101.96, ‐78.46] |
| 8.2 At three weeks | 1 | 15 | Mean Difference (IV, Random, 95% CI) | ‐175.0 [‐294.23, ‐55.77] |
| 9 ALT, IU/L | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐50.88 [‐75.14, ‐26.62] |
| 9.1 At 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐50.88 [‐75.14, ‐26.62] |
| 10 ALT reduction, IU/L | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 121.0 [100.93, 141.07] |
| 10.1 At 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 121.0 [100.93, 141.07] |
| 11 Caesarean section | 4 | 210 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.82, 1.23] |
| 12 Postpartum haemorrhage | 2 | 127 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.20, 2.98] |
| 13 Adverse effects | 3 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.66, 2.63] |
| 14 Meconium‐stained liquor | 3 | 274 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.24, 1.30] |
| 15 Mean gestational age at birth | 3 | 142 | Mean Difference (IV, Random, 95% CI) | 2.68 [‐0.13, 5.48] |
| 16 Spontaneous birth at less than 37 weeks | 2 | 109 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.41, 2.36] |
| 17 Total preterm birth at less than 37 weeks | 2 | 179 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.28, 0.73] |
| 18 Admission to neonatal intensive care unit | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.18, 1.31] |
Comparison 2. SAMe versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Stillbirth/neonatal death | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Bile acid reduction, µmol/L | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 17.5 [12.30, 22.70] |
| 2.1 At 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 17.5 [12.30, 22.70] |
| 3 ALT reduction, IU/L | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 139.6 [119.06, 160.14] |
| 3.1 At 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 139.6 [119.06, 160.14] |
| 4 Caesarean section | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.75, 1.74] |
| 5 Spontaneous birth at less than 37 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.09, 1.75] |
| 6 Total preterm birth at less than 37 weeks | 1 | 18 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.45, 1.26] |
Comparison 3. Guar gum versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Participant assessed | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.89 [0.77, 4.64] |
| 1.2 Clinician assessed | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.46, 3.46] |
| 2 Total bile acids (µmol/L) | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | ‐7.40 [‐24.22, 9.42] |
| 3 ALT, U/L | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | ‐37.5 [‐137.33, 62.33] |
| 4 Adverse effects of medication | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.60, 3.29] |
| 5 Mean gestational age at birth | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.73, 0.93] |
Comparison 4. Activated charcoal versus no treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.55, 147.95] |
| 2 Bile acids after 8 days treatment, µmol/L | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐45.20 [‐74.31, ‐16.09] |
| 3 ALT after 8 days treatment, U/L | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 74.60 [‐141.33, 290.53] |
| 4 Mean gestational age at birth | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐1.0 [‐2.77, 0.77] |
Comparison 5. Dexamethasone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Stillbirths | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.02, 10.31] |
| 2 Fetal distress/asphyxial event | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.61 [0.51, 13.47] |
| 3 Subgroup analysis ‐ fetal distress/asphyxial event | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.50 [0.50, 12.33] |
| 3.1 Bile acid levels < 40 µmol/L | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.32 [0.48, 39.18] |
| 3.2 Bile acid levels ≥ 40 µmol/L | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.05] |
| 4 Meconium‐stained liquor | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Any degree of ICP (all women) | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.56, 1.78] |
| 4.2 Severe subgroup | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.36, 1.94] |
| 5 Spontaneous birth at less than 37 weeks | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.21, 10.90] |
| 5.1 Bile acid levels < 40 µmol/L | 1 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 4.32 [0.95, 19.69] |
| 5.2 Bile acid levels ≥ 40 µmol/L | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.19, 1.92] |
| 6 Total preterm births at less than 37 weeks | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.26, 5.10] |
| 6.1 Bile acid levels < 40 µmol/L | 1 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 2.52 [0.82, 7.70] |
| 6.2 Bile acid levels ≥ 40 µmol/L | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.23, 1.41] |
Comparison 6. UDCA versus SAMe.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Any improvement | 3 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.83, 2.59] |
| 1.2 Marked improvement | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [1.00, 2.98] |
| 1.3 Complete resolution | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 21.0 [1.40, 315.98] |
| 1.4 Complete resolution or marked improvement | 2 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 4.68 [0.26, 83.44] |
| 2 Fetal distress/asphyxial events | 3 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.25, 3.58] |
| 3 Bile acids, µmol/L | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 After 3‐4 weeks treatment | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐27.0 [‐43.67, ‐10.33] |
| 3.2 Reduction after 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 12.90 [4.36, 21.44] |
| 4 ALT, µkat/L | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐2.2 [‐3.55, ‐0.85] |
| 4.1 After 3‐4 weeks treatment | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐2.2 [‐3.55, ‐0.85] |
| 5 Caesarean section | 3 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.52, 1.58] |
| 6 Meconium‐stained liquor | 2 | 97 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.17, 1.27] |
| 7 Mean gestational age at birth | 2 | 66 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.84, 0.76] |
| 8 Spontaneus birth at less than 37 weeks | 2 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.22, 1.59] |
| 9 Total preterm birth at less than 37 weeks | 2 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.33, 1.54] |
| 10 Admission to neonatal intensive care unit | 2 | 97 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.21, 1.27] |
Comparison 7. UDCA versus dexamethasone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fetal distress/asphyxial events | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.08, 1.45] |
| 1.1 Bile acid levels < 40 µmol/L | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.07, 1.80] |
| 1.2 Bile acid levels ≥ 40 µmol/L | 1 | 23 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 6.85] |
| 2 Adverse effects of medication | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.05, 11.83] |
| 3 Meconium‐stained liquor | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.60, 1.87] |
| 4 Spontaneous birth at less than 37 weeks | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.29, 1.59] |
| 5 Total preterm birth at less than 37 weeks | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.44, 1.71] |
| 5.1 Bile acid levels < 40 µmol/L | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.23, 1.60] |
| 5.2 Bile acid levels ≥ 40 µmol/L | 1 | 23 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.52, 3.61] |
Comparison 8. UDCA versus cholestyramine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus score (> 50% reduction after 14 days treatment) | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.5 [1.81, 6.77] |
| 2 Fetal distress/asphyxial event | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.06, 15.47] |
| 3 Bile acids, µmol/L | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐13.10, 9.50] |
| 4 ALT, U/L | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | ‐144.2 [‐186.63, ‐101.77] |
| 5 Caesarean section | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.65, 8.42] |
| 6 Adverse effects of medication | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.04 [0.00, 0.65] |
| 7 Mean gestational age at birth | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | ‐1.30 [‐1.99, ‐0.61] |
| 8 Total preterm birth at less than 37 weeks | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.15, 2.35] |
Comparison 9. UDCA + SAMe versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Bile acid reduction at 20 days, µmol/L | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 41.70 [35.57, 47.83] |
Comparison 10. UDCA + SAMe versus SAMe.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Any improvement | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.99, 2.03] |
| 1.2 Marked improvement | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.85 [1.09, 3.14] |
| 2 Stillbirths/neonatal deaths | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Fetal distress/asphyxial event | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.03, 2.78] |
| 4 Bile acids, µmol/L | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 After 3‐4 weeks treatment | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | ‐25.0 [‐40.16, ‐9.84] |
| 4.2 Reduction at 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 24.20 [16.43, 31.97] |
| 5 ALT, µkatl/L | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | ‐2.4 [‐3.59, ‐1.21] |
| 5.1 After 3‐4 weeks treatment | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | ‐2.4 [‐3.59, ‐1.21] |
| 6 Caesarean section | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.08, 1.74] |
| 7 Postpartum haemorrhage | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | 1.0 [‐76.75, 78.75] |
| 8 Meconium‐stained liquor | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.09, 2.31] |
| 9 Spontaneous birth at less than 37 weeks | 1 | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.56] |
| 10 Total preterm birth at less than 37 weeks | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.18, 1.59] |
| 11 Admission to neonatal intensive care unit | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.09, 2.31] |
Comparison 11. UDCA + SAMe versus UDCA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Any improvement | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.83, 1.35] |
| 1.2 Marked improvement | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.76, 1.50] |
| 2 Stillbirths/neonatal deaths | 2 | 135 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Fetal distress/asphyxial event | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.05, 4.99] |
| 4 Bile acids, µmol/L | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 After 3‐4 weeks treatment | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 2.0 [‐11.71, 15.71] |
| 4.2 Reduction at 20 days | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 11.30 [2.16, 20.44] |
| 5 ALT, µkatl/L | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | ‐2.4 [‐3.59, ‐1.21] |
| 5.1 After 3‐4 weeks treatment | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | ‐2.4 [‐3.59, ‐1.21] |
| 6 Reduction in ALT (IU/L) after treatment | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 1.28 [1.15, 1.41] |
| 7 Caesarean section | 2 | 116 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.35, 1.02] |
| 8 Meconium‐stained liquor | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.12, 3.54] |
| 9 Spontaneous birth at less than 37 weeks | 1 | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.06, 4.47] |
| 10 Total preterm births at less than 37 weeks | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.29, 1.62] |
| 11 Admission to neonatal intensive care unit | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.12, 3.54] |
Comparison 12. UDCA + Salvia versus UDCA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Reduction in pruritus from moderate/severe to mild (0‐4 scale) | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.87, 1.26] |
| 2 Caesarean section for fetal distress | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.45, 1.89] |
| 3 Reduction in ALT (IU/L) after treatment | 1 | 128 | Mean Difference (IV, Fixed, 95% CI) | ‐14.90 [‐24.42, ‐5.38] |
| 4 Meconium‐stained liquor | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.38, 1.98] |
Comparison 13. YCHD versus SAMe.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Degree of pruritus after treatment | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.77, 1.29] |
| 1.1 Marked improvement | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.77, 1.29] |
| 2 Stillbirths/neonatal deaths | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Fetal distress/asphyxial event | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.29, 2.50] |
| 4 Bile salt (CGA) levels | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐1.5 [‐6.12, 3.12] |
| 5 ALT | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 3.40 [‐12.37, 19.17] |
| 6 Caesarean section | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.56, 1.55] |
| 7 Meconium‐stained liquor | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.29, 2.50] |
| 8 Mean gestational age at birth | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐0.35, 1.75] |
13.2. Analysis.

Comparison 13 YCHD versus SAMe, Outcome 2 Stillbirths/neonatal deaths.
Comparison 14. Danxiaoling versus Yiganling.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Any improvement after treatment | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.94, 1.07] |
| 1.2 Marked improvement after treatment | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [1.14, 2.44] |
| 2 Stillbirths/neonatal deaths | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Bile acid levels (CGA) | 1 | 58 | Mean Difference (IV, Fixed, 95% CI) | ‐3.83 [‐22.59, 14.93] |
| 4 ALT | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | 5.20 [‐36.90, 47.30] |
| 5 Caesarean section | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.16, 2.28] |
| 6 Meconium‐stained liquor | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.18, 0.89] |
| 7 Spontaneous birth at less than 37 weeks | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 3.02] |
Comparison 15. Early term delivery v expectant management.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Stillbirths/neonatal deaths | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Caesarean section | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.30, 1.52] |
| 3 Meconium‐stained liquor | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.15, 2.01] |
| 4 Admission to neonatal intensive care unit | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.05, 5.76] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Binder 2006.
| Methods | Randomised controlled trial. | |
| Participants | 78 women randomised. Setting: Prague, Czech Republic. Recruitment: January 1999 to March 2005. Inclusion criteria: women with singleton pregnancies at < 36 weeks' gestation with generalised itching starting in the second half of pregnancy, serum liver enzymes > 1 μkat/L and bile acid levels > 6 μmol/L. Exclusion criteria: hepatitis A, B, C, acute CMV, herpes virus infection, gallbladder stones. |
|
| Interventions |
SAMe (n = 25). 2 x 500 mg/day given by slow infusion for 14 days and subsequently 2 x 500 mg/day orally until birth (median treatment duration 3 weeks, range 1 to 10). UDCA (n = 26). 3 x 250 mg/day orally until birth (median treatment duration 4 weeks, range 2 to 8). SAMe+UDCA (n = 27). Dosages as above (median treatment duration 3 weeks, range 1 to 12). All the participants were admitted to prenatal intensive care unit, but were discharged and followed up in the outpatient clinic in cases of remarkable clinical and biochemical improvement. A 10‐point score was used by the participants to determine itching, where score 1 indicated isolated episodes of pruritus and score 10 indicated a continuous pruritus with impairment of the sleeping rhythm, and described the impact of the mode of treatment on the severity of itching as 'deterioration', 'not affected', 'mild improvement' and 'marked improvement'. Blood samples were collected for LFTs and bile acids on alternate days in the most severe cases and weekly during remission. The fetus was monitored by CTG and ultrasound scans. Amnioscopy was performed when possible. Corticosteroids were not given for fetal lung maturity. Pregnancy was terminated in case the symptoms endangered the fetus and no later than 1 week if the disease progressed despite intervention. In the case of marked clinical and biochemical improvement, women were allowed to progress to term. |
|
| Outcomes |
Maternal: status of pruritus; biochemical parameters; adverse effects. Fetal/neonatal: perinatal outcomes; adverse effects. |
|
| Notes | Medications that could affect pruritus, transaminases and bile acid concentrations were not used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote "Randomisation into three groups was carried out by means of sealed envelopes". No further description of randomisation. |
| Allocation concealment (selection bias) | Unclear risk | "The women were divided to treatment group with the envelope method." No other details on whether envelopes were sequentially numbered, opaque or sealed. |
| Blinding (performance bias and detection bias) All outcomes | High risk | The route and the duration of interventions were different in each of the 3 groups and therefore blinding would not have been possible. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Some outcomes were not reported in a way that could be used in this review (e.g. gestational age at birth not reported with SDs, preterm birth reported but not spontaneous preterm birth). |
| Other bias | Low risk | No other additional bias noted |
Diaferia 1996.
| Methods | Randomised controlled trial. | |
| Participants | 16 women randomised. Setting: Bari, Italy. Inclusion criteria: women aged between 20‐39 with ICP in the third trimester of pregnancy, where pruritus appeared after week 29 of pregnancy. Exclusion criteria: hepatitis A, B, C, CMV and HSV; chronic liver disease; urinary tract infection; gestational diabetes; hypertension. |
|
| Interventions |
UDCA (n = 8). 600 mg/day of UDCA in 2 oral doses for 20 days after week 30 of gestation. Placebo (n = 8). Placebo (vitamin‐supradyn) in 2 oral doses for 20 days. Participants were admitted in the hospital during the duration of the study. No other drug was used to improve pruritus and LFTs. The severity of pruritus was assessed before randomisation and repeated every 5 days using the following score: 0 = absence of pruritus; 1 = occasional pruritus; 2 = discontinuous pruritus every day, with prevailing asymptomatic lapses; 3 = discontinuous pruritus with prevailing symptomatic lapses; 4 = constant pruritus, day and night. Blood samples were collected weekly for assays of liver function and bile acids. Ultrasound examinations and CTGs were performed to assess the fetus. |
|
| Outcomes |
Maternal: pruritus; liver function and bile acid assays; mode of birth; PPH; adverse effects. Fetal/neonatal: fetal distress; gestation at birth; birthweight; Apgar score at 1 and 5 minutes; adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described only as "randomised". |
| Allocation concealment (selection bias) | Unclear risk | Described only as "randomised". |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "double‐blind, placebo‐controlled" ‐ the investigators and the participants were blinded to the treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up were reported. |
| Selective reporting (reporting bias) | Unclear risk | Perinatal death not reported. |
| Other bias | Low risk | No other additional bias noted. |
Fang 2009.
| Methods | Quasi‐randomised controlled trial. | |
| Participants | 128 women randomised. Setting: First Affiliated Hospital of Xi’an Jiaotong University (Obstetrics Department) Inclusion criteria: women with singleton pregnancy presenting with antepartum itching and abnormal ALT and AST which resolved postpartum. Exclusion criteria: antenatal problems such as vomiting, loss of appetite, lethargy or any medical problems, known liver disease or hepatitis prior to pregnancy. |
|
| Interventions |
Salvia+UDCA (N = 72). Salvia injection IV (10 mL in 10% 500 mL Dextrose) and UDCA 15 mg TDS PO for 14 days. UDCA (N = 56). UDCA 15 mg TDS PO for 14 days. |
|
| Outcomes |
Maternal: reduction in pruritus score; monitoring of CG, TB, ALT and AST levels. Fetal/neonatal: CS for fetal distress; meconium‐stained liquor; Apgar score and birthweight. |
|
| Notes | Article in Chinese. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quasi‐randomised controlled trial. "A total of 128 patients were divided into two groups based on the date of admission into the First Affiliated Hospital of Xi'An Jiaotong University." |
| Allocation concealment (selection bias) | High risk | Quasi‐randomised controlled trial. "A total of 128 patients were divided into two groups based on the date of admission into the First Affiliated Hospital of Xi'An Jiaotong University." |
| Blinding (performance bias and detection bias) All outcomes | High risk | The route of administration of the interventions being compared were different and therefore blinding would not have been possible. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There appear to be no losses to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Results for outcomes described in the abstract and methods are reported. However, stillbirth, neonatal death or preterm birth are not reported. |
| Other bias | Unclear risk | It is unclear why there are 72 women in the experimental group and 58 women in the control group. |
Floreani 1996.
| Methods | Randomised controlled trial. | |
| Participants | 20 women randomised. Setting: Padova, Italy. Inclusion criteria: skin pruritus due to ICP during the last trimester of pregnancy , total serum bile acids ≽ 2 μmol/L and ALT ≽ 40 U/L. Exclusion criteria: dermatological or other causes of pruritus; abnormalities unrelated to pregnancy (acute hepatitis A, hepatitis B and C were excluded). |
|
| Interventions |
UDCA (n = 10). 450 mg/day oral until birth. SAMe (n = 10). 1000 mg/day IM until birth. Participants were admitted to the obstetrics ward before 34 weeks' gestation for strict fetal monitoring. They were examined by the same hepatologist. The severity of pruritus was assessed before treatment and subsequently every 3 days using the following score: 0 = absence of pruritus; 1 = occasional pruritus; 2 = discontinuous pruritus every day, with prevailing relapses at night; 3 = permanent pruritus during day and night. Fasting blood samples were obtained immediately before treatment, every 3 days until birth and then 5 days later. All fetal monitoring and delivery decisions were made by the treating obstetrician. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acids; mode of birth. Fetal/neonatal: gestation at birth; birthweight; Apgar score at 5 minutes. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly assigned" ‐ no further details given. |
| Allocation concealment (selection bias) | Unclear risk | "randomised by closed envelope system", no other details on whether envelopes were sequentially numbered, opaque or sealed. |
| Blinding (performance bias and detection bias) All outcomes | High risk | It is apparent from the study that blinding was not possible because the route of administration of the 2 interventions (oral vs injection) were different. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Liver function outcomes were reported only as graphs. |
| Other bias | Low risk | No other additional bias noted. |
Frezza 1984.
| Methods | Randomised controlled trial. | |
| Participants | 18 women randomised. Setting: Milan, Italy. Recruitment: between 1979 to 1982. Inclusion criteria: women between 28 and 32 weeks of pregnancy with history of gestational pruritus starting after 19th week of gestation and elevated serum bile acids, bilirubin and transaminases. Normalisation of biochemical parameters and resolution of itching after birth. Exclusion criteria: acute hepatitis A, hepatitis B, dermatological diseases. |
|
| Interventions |
SAMe (n = 6). Daily IV dose of 200 mg of SAMe (as disulphate‐p‐toluenesulfonate stable salt) dissolved in 500 mL of saline solution over 4 hours beginning at 8 am for 20 days. SAMe (n = 6). Daily IV dose of 800 mg of SAMe (as disulphate‐p‐toluenesulfonate stable salt) dissolved in 500 mL of saline solution over 4 hours beginning at 8 am for 20 days. Placebo (n = 6). Daily IV dose of 500 mL of saline solution over 4 hours beginning at 8 am for 20 days. Pruritus was assessed before randomisation and were repeated 10 and 20 days after treatment. It was graded as: 0, no pruritus; Grade 1+, rare; Grade 2+, occasional; Grade 3+, frequent; Grade 4+, almost continuous. Fasting blood samples were obtained for ALT, AST, ALP, bilirubin and total bile acid levels before randomisation and at 10‐day intervals. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acids, maternal adverse events. Fetal/neonatal: Apgar scores. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Unclear risk | Quote ‐ "Women were randomly allocated to three groups of six". No other details provided. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Single blinded. Participants were blinded. The medical staff were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | High risk | Fetal outcomes, mean length of gestation, preterm birth rates, mode of birth and blood loss at birth were not reported. Some outcomes were only presented as graphs. |
| Other bias | Low risk | No other additional bias noted. |
Frezza 1990.
| Methods | Randomised placebo‐controlled trial‐ using a pre‐established code, single‐blinded. | |
| Participants | 30 women randomised. Setting: Milan, Italy. Inclusion criteria: pruritus, with or without jaundice, and elevated levels of serum bile acids, bilirubin, ALT and AST during the last trimester of pregnancy. Exclusion criteria: acute hepatitis A, hepatitis B, dermatological conditions. |
|
| Interventions |
SAMe (n = 15). Daily IV dose of 800 mg of SAMe diluted in 500 mL of 5% dextrose. Half of the dosage was infused in the morning, and half in the afternoon. It was administered up to the day of birth and was withdrawn 12 hours after birth. Placebo (n = 15). Daily IV dose of 500 mL of 5% dextrose. Half of the dosage was infused in the morning, and half in the afternoon. It was administered up to the day of birth and was withdrawn 12 hours after birth. Pruritus was a scored on a 10 cm analogue scale every 3 days up to 24 hours after birth. LFTs were measured before randomisation and 24 hours after birth. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acid; adverse effects. Fetal/neonatal: preterm birth at < 37 weeks; birthweight < 2500 g. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ "According to pre‐established code, consecutive patients were randomised to receive either SAMe or placebo". It is unclear how this code was generated. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Single‐blind. Participants were blinded. The medical staff were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | High risk | Fetal mortality (stillbirth and neonatal deaths), mean length of gestation, mode of birth and blood loss at birth were not reported. Some outcomes were only presented as graphs. |
| Other bias | Low risk | No other bias apparent. |
Glantz 2005.
| Methods | Randomised controlled trial. | |
| Participants | 130 women randomised. Setting: 106 antenatal clinics and all 6 departments of obstetrics in the Västra Götaland region, Sweden. Recruitment: February 1999 ‐ January 2002. Inclusion criteria: women at < 37 weeks' gestation with gestational pruritus and fasting serum bile acid levels > 10 μmol/L. Exclusion criteria: diabetes pre‐eclampsia, intrauterine growth restriction, liver disease (including viral hepatitis), history of manic disorders, bleeding peptic ulcer. |
|
| Interventions |
UDCA (n = 47). 1 g/day as a single oral dose, for 3 weeks. Dexamethasone (n = 36). 12 mg/day as a single oral dose for 1 week, and placebo during weeks 2 and 3. Placebo (n = 47). Given daily as a single oral dose for 3 weeks. A 100 mm long VAS was used to score itching: no pruritus at all at 0 mm; worst possible pruritus at 100 mm. Blood samples were collected at entry for bile acids, ALT and bilirubin. They were repeated after 2‐3 days, after 4‐5 days and after 1, 2 and 3 weeks of treatment. If the pregnancy continued after 3 weeks of treatment, the above biochemical parameters were measured weekly until birth. CTG monitoring was done each time the samples were taken. |
|
| Outcomes |
Primary outcomes: spontaneous preterm birth (< 37 weeks) in singleton pregnancies, asphyxial events (operative delivery due to asphyxia, postpartum pH < 7.05 in umbilical arterial blood or Apgar score < 7 at 5 minutes), and meconium staining of amniotic fluid, placenta, and membranes. Secondary outcomes: changes in biochemical markers (bile acids, ALT and bilirubin), status of pruritus, total prematurity rate, total elective birth rate, maternal blood loss during vaginal birth. |
|
| Notes | Severe obstetric cholestasis was defined as serum bile acids ≥ 40 µmol/L. Subgroup analysis was done for this group. Funding: FoU, Västra Götaland. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study drugs were randomised in blocks of 6 (2 each of UDCA, dexamethasone and placebo). Method of sequence generation not specifically described but likely to be adequate. |
| Allocation concealment (selection bias) | Low risk | The hospital pharmacy was responsible for randomisation. Study drugs were provided in tins with a study code, each containing 6 treatments: 2 each of UDCA, dexamethasone, and placebo. Staff at the site were instructed to hand out the treatments consecutively, starting with the lowest study code number. The lower numbers randomised in the dexamethasone group may be due to only small numbers of women being randomised before study drugs were changed, with a large amount of study medication not being used. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Dr Falk Pharma supplied identical looking UDCA, placebo and empty capsules. The empty capsules were filled with dexamethasone at the hospital pharmacy. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up. 80/130 women completed the full 3‐week treatment period (31 in the UDCA group, 19 in the dexamethasone group and 30 in the placebo group). 3 women, 1 from each group, discontinued due to side effects. 1 woman in each group did not take the medication after being randomised due to fear of side effects. The remaining 44 women discontinued their treatment because of spontaneous or planned birth. |
| Selective reporting (reporting bias) | Unclear risk | Pruritus and liver function were reported only graphically as medians (with some P values reported). |
| Other bias | Low risk | No other additional bias noted. |
Huang 2004.
| Methods | Randomised controlled trial. | |
| Participants | 60 women randomised. Recruitment: July ‐ October 2002. Inclusion criteria: primigravidae, singleton pregnancies, pruritus in the second half of pregnancy, raised serum CG (> 10 UNL) and ALT. Exclusion criteria: PIH; gestational diabetes; anaemia; other liver (hepatitis A, B, C, D) and gallbladder diseases. |
|
| Interventions |
YCHD (n = 35). BD orally for 3 weeks. SAMe (n = 25). IV infusion of 2 x 500 mg daily for 3 weeks. Pruritus, serum bile acids and LFTs were assessed after 3 weeks treatment. |
|
| Outcomes |
Maternal: improvement in pruritus; serum CG; ALT; bilirubin; length of gestation; delivery by CS. Fetal/neonatal: mortality; Apgar score < 7; meconium‐stained liquor; preterm birth at < 37 weeks; birthweight; asphyxial events; umbilical cord artery pH, PO2, PCO2. |
|
| Notes | Full article in Chinese, abstract published in English. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not mentioned. Unlikely to be blinded because these 2 drugs have different modes of administration. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up. |
| Selective reporting (reporting bias) | Low risk | All the pre‐specified outcomes reported. |
| Other bias | High risk | Imbalance in numbers randomised to each group (35 versus 25) which may indicate a failure of proper randomisation. |
Kaaja 1994.
| Methods | Randomised controlled trial. | |
| Participants | 19 women randomised (1 woman entered trial in 2 successive pregnancies). Setting: Helsinki, Finland. Inclusion criteria: women with pruritus and abnormalities of liver function. Exclusion criteria: hepatitis A and B, gallbladder pathology. |
|
| Interventions |
Activated charcoal (n = 10). Activated charcoal as a water suspension, 50 g 3 times a day for 8 days. vs No treatment (n = 10). Normal follow‐up of ICP with no charcoal administration. Participants maintained a daily record of pruritus: 0 = no itching; 1 = mild itching; 2 = moderate itching, does not disturb sleep; 3 = intense itching, disturbs sleep; 4 = very intense (intolerable) itching, forces participant to scratch continuously. Fasting blood samples were collected for total bile acids and LFTs at the start of the study and were repeated on day 4 and 8. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acids. Fetal/neonatal: gestation at delivery, birthweight. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up reported. |
| Selective reporting (reporting bias) | High risk | Few fetal/neonatal outcomes not reported. |
| Other bias | Low risk | No other additional bias noted. |
Kondrackiene 2005.
| Methods | Randomised controlled trial. | |
| Participants | 84 women randomised. Setting: Lithuania. Recruitment: between October 1999 and September 2002. Inclusion criteria: women at 25 ‐ 39 weeks of gestation with pruritus starting in the second or third trimester of pregnancy and elevation of at least 1 of the following biochemical markers: ALT > 45 U/L, AST > 40 U/L, fasting serum bile acids > 10 μmol/L. Exclusion criteria: chronic liver disease; viral infections (hep A, B, C, CMV, HSV, EBV); skin disease; allergies; symptomatic cholelithiasis. |
|
| Interventions |
UDCA (n = 42). 8‐10 mg/kg body weight orally daily for 14 days. Cholestyramine (n = 42). 8 g orally daily for 14 days. Daily self‐assessment of pruritus by the participants using the following score: 0 = no pruritus; 1 = occasional; 2 = intermittent pruritus everyday with asymptomatic periods prevailing; 3 = intermittent pruritus everyday with symptomatic periods prevailing; 4 = constant pruritus day and night. Fasting blood samples were collected at entry and on the day after the completion of treatment for the analysis of LFTs and bile acid assay. Delivery decisions were made by managing obstetricians independent of the study. |
|
| Outcomes |
Primary end point: reduction in the severity of pruritus by more than 50% after 14 days of treatment. Secondary end points: reduction of ALT and serum bile acid levels; mode of birth; drug safety, gestation at birth, Apgar score at 1 and 5 minutes, birthweight. |
|
| Notes | Cholestyramine may cause PPH in mother and intracranial haemorrhage in fetus due to the malabsorption of vitamin K. These outcomes were not analysed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote ‐ 'All patients gave written informed consent before inclusion into the study and were randomised to receive either UDCA or cholestyramine'. No description of random sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | "Sealed envelopes." Unclear as not specified as consecutive and opaque |
| Blinding (performance bias and detection bias) All outcomes | High risk | "Open" trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All 84 women were included in analyses although 10/42 women in the UDCA group and 4/42 in the cholestyramine group either did not complete the study or were protocol violations. In the UDCA group, 4 women discontinued treatment and 6 women were protocol violations (apparently 6 women took UDCA before inclusion in the trial). In the cholestyramine group, 3 women experienced adverse events (nausea and vomiting) and 1 women discontinued treatment. |
| Selective reporting (reporting bias) | Unclear risk | Most expected outcomes were reported although outcomes related to bleeding were not reported (see Notes above). |
| Other bias | Low risk | No other additional bias noted. |
Leino 1998.
| Methods | Randomised controlled trial. | |
| Participants | 18 women with ICP were included in analyses: 10 in the UDCA group and 8 in the placebo group. | |
| Interventions | 450 mg of UDCA in 2 doses for 14 days vs placebo. | |
| Outcomes | Daily assessment of pruritus, diverse reactions, itching. The following were assessed at before treatment and at 7 days: fasting serum levels of total bile salts, ALAT, ALP, estradiol, progesterone, prolactin, cholesterol, triglycerides, APTT and thrombocytes. | |
| Notes | Conference abstract. Very limited information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Very limited information. |
| Allocation concealment (selection bias) | Unclear risk | Very limited information. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Reported as double‐blind but no further information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Very limited information. |
| Selective reporting (reporting bias) | Unclear risk | Very limited information. Serum ALP was assessed but not reported. |
| Other bias | Unclear risk | Conference abstract. Very limited information. |
Liu 2006.
| Methods | Randomised controlled trial. | |
| Participants | 68 women randomised. Setting: Wuhan, China. Recruitment: June 2001‐July 2003. Inclusion criteria: women at 25 ‐ 37 weeks' gestation with severe gestational pruritus; serum total bile acids > 10 μmol/L and raised ALT or conjugated bilirubin. Exclusion criteria: other known causes of liver dysfunction. |
|
| Interventions |
UDCA (n = 34). 300 mg (18 mg/kg body weight) 3 times a day for 2 weeks. Placebo (n = 34). Combination of 10% glucose, vitamin C and inosine for 2 weeks. They were kept on a low‐fat diet and bed rest during the period of the study. |
|
| Outcomes |
Maternal: pruritus score, mode of birth, adverse effects, LFTs, total bile acids Pruritus score was self‐assessed every 3 days on a VAs: 0 = no pruritus; 1 = occasional; 2 = intermittent pruritus everyday with asymptomatic periods prevailing; 3 = intermittent pruritus everyday with preponderance of symptomatic periods; 4 = constant pruritus. However results were only reported as a number +‐ another number. Because it is not clear if these were means or medians, and if the +‐ was SD, SE or other measure of dispersion, these results are not analysable. Fetal/neonatal: antepartum testing prompting delivery; gestation at birth; passage of meconium; intrapartum fetal distress; Apgar scores at 1 and 5 minutes; birthweight, adverse events. |
|
| Notes | Fetal asphyxia was not defined. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "divided into treatment group and control group at random". No further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | It is unclear whether the clinicians/investigators and the participants were blinded to trial allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was no trial flow diagram. The trial was not registered. Follow‐up rates were not reported. |
| Selective reporting (reporting bias) | High risk | Stillbirths and neonatal deaths were not reported. Apgar scores, and adverse events were recorded but not reported. |
| Other bias | Low risk | No other additional bias noted. |
Luo 2008.
| Methods | Randomised controlled trial. | |
| Participants | 64 women randomised. Setting: Affiliated Hospital of Hanzhou Normal University, Hanzhou, China. Recruitment: June 2002‐July 2007. Inclusion criteria: neonatal jaundice and/or maternal? itching, rise in the levels of serum transaminase and CG. Exclusion criteria: any skin infection, prolonged liver disease, any other illnesses, high blood pressure, received other forms of treatment for ICP. |
|
| Interventions |
Transmetil + UDCA (n = 34). Transmetil (1 g + 5% Glucose 250 mL IV OD) + UDCA (250 mg Oral pill BD) for 10 days. UDCA (n = 30). UDCA 250 mg BD for 10 days. Patients took dexamethasone (10 mg OD) for 3 days before the treatment in both groups. |
|
| Outcomes |
Maternal: scale of itchiness (0‐4 Ribalta scale); levels of ALT, AST, total bile acids, amount of haemoglobin, CS rate. Fetal/neonatal: preterm birth, clearness of amniotic fluid (i.e. number of cases where the fluid was not clear), Apgar score, birthweight. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" ‐ no further details reported. |
| Allocation concealment (selection bias) | Unclear risk | "randomly assigned" ‐ no further details reported. |
| Blinding (performance bias and detection bias) All outcomes | High risk | The route of administration of interventions in the 2 groups were different and therefore blinding would not have been possible. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up or withdrawal in either group. |
| Selective reporting (reporting bias) | Unclear risk | Not all expected outcomes reported. |
| Other bias | Low risk | According to the translation, “Their traits and characteristics were not significantly different from each other". |
Nicastri 1998.
| Methods | Randomised controlled trial. | |
| Participants | 32 women randomised. Setting: Bari, Italy. Recruitment: March 1995 ‐ July 1996. Inclusion criteria: participants included women aged 19‐37 years, between 30‐37 weeks' gestation with history of pruritus after 28 weeks. |
|
| Interventions |
UDCA (n = 8). UDCA in 2 oral doses daily (600 mg/day) for 20 days. SAMe (n = 8). SAMe in the stable form of sulphate‐P‐toluenesulphonate diluted in 500 mL 5% dextrose and divided into 2 IV infusions (800 mg/day). UDCA+SAMe (n = 8). Combination of UDCA and SAMe in the doses specified above. Placebo (vitamin) (n = 8). LFTs and serum total bile acid levels were measured before and at the end of treatment. Pruritus was measured every 3 days up to 24 hours after delivery. Pruritus was scored as: 0 = absent pruritus; 1 = occasional pruritus; 2 = intermittent pruritus everyday with asymptomatic periods prevailing; 3 = intermittent pruritus everyday, with symptomatic periods prevailing; 4 = constant pruritus. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acids, side effects of the treatment. Fetal/neonatal: preterm birth; low birthweight; side effects of the treatment. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random permuted blocks. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not blinded. It is apparent from the study that blinding was not possible because the route of delivery of the interventions were different. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up reported. |
| Selective reporting (reporting bias) | High risk | Stillbirths and perinatal deaths, mean length of gestation, mode of birth and blood loss at birth were not reported. |
| Other bias | Low risk | No other additional bias noted. |
Palma 1997.
| Methods | Randomised controlled trial. | |
| Participants | 24 women randomised. Setting: Santiago, Chile. Recruitment: July 1993 ‐ June 1995. Inclusion criteria: severe gestational pruritus appearing at < 33 weeks' gestation and present daily for at least 2 weeks; fasting total bile salts > 12 μmol/L and ALT or AST > 40 IU/L. Exclusion criteria: chronic liver disorder; symptomatic cholelithiasis; metabolic diseases; dermatological or neuropsychiatric causes of pruritus; infections requiring antibiotics. |
|
| Interventions |
UDCA (n = 8). 1000 mg/day as 3 oral doses until birth. Placebo (starch) (n = 7). Orally, until birth. Participants were admitted in the hospital. Pruritus was assessed weekly by the same clinician using the following score: 0 = absence of pruritus; 1 = occasional pruritus; 2 = discontinuous pruritus everyday, prevailing asymptomatic lapses; 3 = discontinuous pruritus but prevailing asymptomatic lapses everyday; 4 = constant itching, day and night. Blood samples were collected for LFT and total bile salt levels. They had to be on a treatment for at least 3 weeks. |
|
| Outcomes |
Primary outcome: status of pruritus. Secondary outcomes: liver function and bile acid assays; mode of birth; PPH; fetal/neonatal deaths, fetal distress; gestation at birth; birthweight; adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised randomisation. Actual method of generation not reported, but judged likely to have been adequate |
| Allocation concealment (selection bias) | Low risk | UDCA and placebo capsules were provided by Dr Falk Pharma in coded boxes. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Identical UDCA and placebo capsules. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 9/24 women did not complete the trial and were excluded from analysis. 8 women did not complete 2 weeks of treatment (6 had spontaneous preterm vaginal births and another 2 women had CS due to signs of fetal distress). The ninth woman left hospital after 1 week of treatment. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient detail. |
| Other bias | Low risk | No other additional bias noted. |
PITCH 2012.
| Methods | Multicentre, double‐blinded, randomised, controlled, factorial design trial. | |
| Participants | 125 women (111 for UDCA vs placebo comparison plus an additional 14 women in early vs expectant delivery not in UDCA vs placebo comparison) with ICP (pruritus and raised maternal serum bile acids) or pruritus and raised alanine transaminase (> 100 IU/L) recruited after 24 weeks' gestation. Setting: 9 maternity units in UK. |
|
| Interventions |
Comparison A: 1. UDCA n = 56 (60 babies). Starting dose 500 mg BD increased in increments of 500 mg per day every 3‐14 days if there was no biochemical or clinical improvement until a maximum of 2 g per day. 2. placebo n = 55 (64 babies). Placebo capsules increased according to the same regimen. Comparison B: 1. Early term delivery (induction or delivery commenced between 37 + 0 and 37 + 6 (n = 30). 2. Expectant management (spontaneous labour awaited until 40 weeks or CS as indicated, usually after 39 weeks' gestation); n = 33. |
|
| Outcomes |
Primary outcomes: UDCA vs placebo comparison: maternal itching (average of the worst itch in previous 24 hours – 100 mm VAS ‐ between randomisation and delivery). Timing of delivery comparison: CS. Secondary outcomes: Average itch in last 24 hours (VAS); total bile acids, ALT, APT, mode of onset of labour, mode of birth, indication for delivery, blood loss at birth; gestational age at birth, “baby outcome”, birthweight, presence of meconium‐stained amniotic fluid, arterial cord pH, venous cord pH, Apgar score at 5 minutes, congenital anomalies, admission to neonatal unit (and duration), need for ventilation (and duration), convulsions, jaundice, adherence; maternal adverse events. |
|
| Notes | 48 of the 62 women in the delivery vs expectant management arm were also part of the part of the UDCA vs placebo arm. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated. |
| Allocation concealment (selection bias) | Low risk | Central allocation using a web‐based database. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | For the UDCA vs placebo comparison “investigator, pharmacists and participant were blind to group allocation”. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up in either the drug or delivery comparisons; however 4 women in the UDCA group and 3 in the placebo group discontinued the intervention (3 wanted open‐label UDCA and 1 chose to discontinue after an adverse event in the UDCA group; 2 and 1 respectively in the placebo group). For the delivery vs expectant management comparison, none in the delivery group discontinued the intervention and 20 in the expectant management group discontinued (non‐exclusive ‐ 7 fetal/maternal compromise 10 maternal request for delivery, 14 obstetrician decision for delivery). Post randomisation exclusion. None for UDCA vs placebo comparison. None for early vs expectant delivery comparison. |
| Selective reporting (reporting bias) | Low risk | Trial registered. Most expected outcomes reported. |
| Other bias | Unclear risk | The timed delivery comparison was not blinded to obstetrician, patient or outcome assessor. |
Ribalta 1991.
| Methods | Randomised controlled trial. | |
| Participants | 20 women randomised. Setting: Santiago, Chile. Inclusion criteria: women with ICP, age 21‐38 years with pruritus appearing before week 32 of gestation. Participants had elevated levels of liver function markers. Exclusion criteria: liver and dermatological diseases, acute cholecystitis, urinary tract infection, diabetes, other chronic diseases. |
|
| Interventions |
SAMe (n = 9). 800 mg/day IV administered daily over 3 hours for 20 days. Placebo (n = 9). Mannitol IV administered daily over 3 hours for 20 days. Participants were admitted to the obstetrics ward before 34 weeks' gestation and were kept as in‐patient until 3‐5 days post delivery. They were given a low‐fat diet. No other medications were prescribed to improve pruritus. The severity of pruritus was assessed before treatment and subsequently every 5 days using the following score: 0 = absence of pruritus; 1 = occasional pruritus; 2 = discontinuous pruritus every day, with prevailing relapses at night; 3 = permanent pruritus during day and night. They were assessed by the same observer. Fasting blood samples were obtained immediately before treatment, every 5 days until delivery and then 1‐3 days, 1 month and 3 months after delivery. |
|
| Outcomes |
Maternal: status of pruritus; assays of liver function and bile acids; mode of birth; adverse reactions. Fetal/neonatal: gestation at birth; birthweight; Apgar score at 1 and 5 minutes. |
|
| Notes | No numerical data were reported, results were only presented as graphs making it difficult to extrapolate results. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Sequence established at random by the suppliers." |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation. A single lot of identical looking ampoules containing SAMe and mannitol were supplied by BioResearch S.p.A (Milano, Italy). The boxes were coded using the random sequence generated by the suppliers. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The participants and the investigators were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2/20 women did not complete the study; 1 from each group (1 CS for meconium‐stained amniotic fluid and 1 woman unable to tolerate iv infusions). |
| Selective reporting (reporting bias) | High risk | Most outcomes were only presented graphically. |
| Other bias | Low risk | No other additional bias noted. |
Riikonen 2000.
| Methods | Randomised controlled trial. | |
| Participants | 39 women randomised. Setting: Helsinki, Finland. Inclusion criteria: women with a singleton pregnancy referred due to an elevated serum bile acid concentration (> 5 mol/L) and/or presence of typical pruritus of ICP, with no concomitant chronic disease. The participants had to be on treatment for at least 10 days to be included in the analysis. Exclusion criteria: dermatological cause of pruritus; viral hepatitis (hepatitis B and C), primary liver and gallbladder diseases. 1 woman was entered into the study despite the absence of symptoms and biochemical abnormality. She had developed ICP in 3 previous pregnancies and later developed ICP. |
|
| Interventions |
Guar gum (n = 19). 5‐15 g day, orally; the dose was increased from 5 to 15 g/day at 3 day intervals, until birth. Placebo (wheat flour) (n = 20). Participants were seen in the outpatient clinic up to 37 weeks' gestation and were admitted to hospital at 37 weeks. Fetus was monitored by CTG at every clinic visit and daily at the ward. The intensity of pruritus was estimated by 1 investigator and participant simultaneously. The investigator used the following score: 0 = no pruritus; 1 = mild pruritus; 2 = moderate pruritus disturbing sleep but not requiring antihistamine medication; 3 = severe pruritus requiring continuous antihistamine medication. The participants used a 10 cm long VAS. Fasting blood samples were collected for the assessment of LFTs and total bile acids from 1‐3 days before birth. If pruritus was severe, women were given prometazine hydrochloride 10‐30 mg/day. |
|
| Outcomes |
Maternal: status of pruritus (assessed by both clinician and woman); assays of liver function and bile acids; CS for abnormal CTG; adverse effects. Fetal/neonatal: gestation at birth; birthweight. |
|
| Notes | This study had an additional non‐randomised control group of 20 women (additional to the 39 participating in the randomised trial) to provide a comparison group for the serum values. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "In‐house built computer programme validated according to company standard operating procedures." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment method not described. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The investigators and the participants were blinded to the drug used. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 9/39 (23%) women (5 guar gum, 4 placebo) were excluded from "the intervention analyses" to birth within 10 days of treatment. Not clear which analyses these were e.g. n's not given for Table II (liver function results). |
| Selective reporting (reporting bias) | High risk | Outcomes such as perinatal death, fetal distress and spontaneous birth < 37 weeks were not reported. |
| Other bias | Low risk | No other additional bias noted. |
Roncaglia 2004.
| Methods | Randomised controlled trial. | |
| Participants | 46 women randomised. Setting: Monza, Italy. Recruitment: June 1996 ‐ December 2001. Inclusion criteria: women < 36 weeks' gestation complaining of gestational pruritus starting in the second or third trimester of pregnancy, persisting to birth and disappearing after, bile acids > 6 μmol/L or serum transaminases > 41 mg/dL. Exclusion criteria: other medical conditions known to be associated with pruritus. |
|
| Interventions |
SAMe (n = 22). 500 mg orally BD until birth. UDCA (n = 24). 300 mg orally BD until birth. No other medications apart from the study medications were used to improve pruritus and LFTs. Pruritus was scored using a semi‐quantitative scale of 1‐4. 1 = occasional pruritus; 2 = daily intermittent pruritus with preponderance of asymptomatic periods; 3 = daily intermittent pruritus with preponderance of symptomatic periods; 4 = persistent pruritus, day and night. LFTs and bile acid levels were evaluated every 7‐10 days and 1 and 3 months post delivery. Non‐stress tests and amniotic fluid volume assessment was done twice weekly. A biophysical profile was performed if the non‐stress test was non‐reactive. |
|
| Outcomes |
Primary outcome: reduction of serum bile acids concentration. Secondary outcomes: serum levels of transaminases and bilirubin; status of pruritus; blood loss; CS; gestation at delivery; rate of preterm delivery; meconium passage at birth; birthweight < 10th centile; Apgar score < 7 at 5 minutes; umbilical artery pH < 7.10; admission to the neonatal intensive care unit; adverse effects. |
|
| Notes | Labour was induced at 37 weeks' gestation or earlier in the presence of abnormal tests of fetal well being, obstetric complications or severe maternal symptoms unresponsive to therapy. There were 3 sets of twins (1 set in the UDCA group and 2 sets in the SAMe group); only 1 twin per set, chosen at random, was included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random number tables. |
| Allocation concealment (selection bias) | Unclear risk | "Assigned by computer‐generated random number tables." |
| Blinding (performance bias and detection bias) All outcomes | High risk | Reports that there was "no concealment of treatment allocation" which we interpret as not being blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses to follow‐up ‐ not reported but 1 pruritus score from each group is missing. |
| Selective reporting (reporting bias) | Unclear risk | Some expected outcomes (e.g. perinatal mortality) not reported. |
| Other bias | Low risk | No other additional bias noted. |
Shi 2002.
| Methods | Quasi‐randomised controlled trial. | |
| Participants | 58 women randomised. Recruitment: 1999 ‐ 2000. Inclusion criteria: women with ICP, not on any relevant treatment. Exclusion criteria: women with PIH, fatty liver and hepatitis. |
|
| Interventions |
DXLP (n = 29). 9 g thrice daily orally for 7 days. Yiganling (n = 29). 4 tablets 3 times a day for 7 days. |
|
| Outcomes |
Maternal: status of pruritus, jaundice; serum CGA, TB, ALT, AST, ALP, LDH, lipids profile; mode of birth. Fetal/neonatal: neonatal mortality; preterm birth at < 37 weeks; meconium‐stained liquor, birthweight. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Alternation according to hospital admission. |
| Allocation concealment (selection bias) | High risk | Alternation according to hospital admission. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were no losses to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes were reported for 25 (86%) participants for ALT and AST, 27 (93%) for ALP and for 21 (72%) women for bilirubin levels out of 29 participants receiving Danxiaoling and for 16 of 29 (55%) participants for bilirubin in the Yiganling group. The reasons for exclusion were unclear. |
| Other bias | Low risk | No other additional bias noted. |
Zhang 2012.
| Methods | Multicentre randomised controlled trial. | |
| Participants | 138 women recruited, among them 18 cases eliminated, data available for 120 women. Setting: 5 centres in Sichuan and Chongqing,China. Recruitment: July 2009 to March 2011. Inclusion criteria: ICP patients at 28 to 35 weeks of singleton pregnancy. Exclusion criteria: not reported. |
|
| Interventions |
Women randomised into 3 groups: UDCA (n = 41). 250 mg po Qid of UDCA. SAMe (n = 38). 1000 mg IV Qid of SAMe. SAMe+UDCA (n = 41). UDCA & SAMe (dosage not specified). |
|
| Outcomes |
Maternal: pruritus scores; total bile acid; ALT; AST; TB; delivery mode; adverse drug reactions. Fetal/neonatal: gestational ages; Apgar scores at 1 and 5 minutes; perinatal death. |
|
| Notes | Conference abstract. Very limited information.138 women recruited, among them 18 cases eliminated, data available for 120 women. Numerical results not reported for most of the outcomes ‐ just quotes whether there were differences between groups and P values. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Data limited – reported as abstract. |
| Allocation concealment (selection bias) | Unclear risk | Data limited – reported as abstract. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Data limited – reported as abstract. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 18 cases were eliminated ‐ but not sure at which stage they were eliminated, i.e. before or after randomisation. |
| Selective reporting (reporting bias) | Unclear risk | Data limited – reported as abstract. |
| Other bias | Unclear risk | Data limited – reported as abstract. |
ALP: alkaline phosphatase ALT: alanine transferase APTT: activated partial thromboplastin time AST: aspartate transaminase BD: twice daily CG: cholyglycine cm: centimetre CMV: cytomegalovirus CS: caesarean section CTG: cardiotocography DXLP: Danxiaoling Pill EBV: Epstein Barr virus g: gram HSV: herpes simplex virus ICP: intrahepatic cholestasis of pregnancy IM: intramuscular IV: intravenous kg: kilogram LDH: lactate dehydrogenase LFT: liver function test mg: milligram mL: millilitre μmol/L: micromoles per litre OD: once daily PCO2: carbon dioxide partial pressure pH: potential hydrogen PIH: pregnancy‐induced hypertension PO: per oral PO2: oxygen partial pressure PPH: postpartum haemorrhage QID: four times daily SAMe: S‐adenosylmethionine SD: standard deviation SEM: standard error of the mean TB: total bilirubin TDS: ter die sumendum (three times daily) U/L: units per litre UDCA: ursodeoxycholic acid VAS: visual analogue scale vs: versus YCHD: Yinchenghao decoction
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Elias 2001 | This study no longer appears on controlled‐trials.com (search date 14 February 2013). We've coded it as an excluded study rather than deleting it, since it was cited in the original version of this review, and to keep a record of why it was removed. |
| Shi 2006 | This is a clinical and experimental study looking at the effect of WLP in treating intrahepatic cholestasis of pregnancy. In the clinical aspect of the study, women in the control group received a combination of 5% glucose (250 mL), dexamethasone (5 mg), vitamin C2, compound injection of red sage root, potassium magnesium aspartate (0.3 g) and Barbital (0.06 g). Women in the test group received WLP in addition to the above components. This made the study very complex as it contained components that may individually affect the outcomes in intrahepatic cholestasis in pregnancy. The experimental part of the study was conducted on rat models. |
g: gram mg: milligram mL: millilitre WLP: Wuling Pill
Characteristics of studies awaiting assessment [ordered by study ID]
Wang 2003.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Untraceable. |
Characteristics of ongoing studies [ordered by study ID]
Mazzella 2010.
| Trial name or title | Ursodeoxycholic Acid And Cholestasis Of Pregnancy (CERTO). |
| Methods | Randomised controlled trial. |
| Participants | 118. |
| Interventions |
Ursodeoxycholic acid 300 mg capsules 20 mg/kg body weight/day divided in 3 administrations per day from enrolment until delivery. Placebo 300 mg capsules 20 mg/kg body weight/day divided in 3 administrations per day from enrolment until delivery. |
| Outcomes |
Primary outcome Number of participants with preterm delivery (before week 37). Secondary outcomes Pruritus on the visual analogue scale, transaminases, bile acids, fetal movement count, number of pregnancies with cardiotocography suggestive of fetal stress; Apgar score, number of pregnancies with green stained amniotic fluid. |
| Starting date | Estimated study start date: November 2010. Estimated primary completion date: November 2013. This study is not yet open for participant recruitment (checked on 14.02.2013). |
| Contact information | Professor Giuseppe Mazzella, S.Orsola‐Malpighi Hospital/University of Bologna Email: giuseppe.mazzella@unibo.it |
| Notes | ClinicalTrials.gov identifier: NCT01226823. |
kg: kilogram mg: milligram
Differences between protocol and review
Glantz 2005 performed a subgroup analysis of changes in pruritus and laboratory parameters in women with bile acid levels greater than or equal to 40 µmol/L at inclusion. We have included this in the update. We have also included data relating to meconium‐stained liquor and caesarean section.
Contributions of authors
Michael Stokes wrote the initial version of the background, with help from Philippa Middleton. Michael Stokes, Vinita Gurung and Philippa Middleton independently identified and scanned the reports of identified studies and assessed risk of bias in the included trials. Vinita Gurung, Philippa Middleton and Stephen Milan wrote the review. Bill Hague supplied content expertise and reviewed review drafts. Jim Thornton checked the accuracy of data entry, reviewed the draft review and provided editorial support.
Sources of support
Internal sources
Discipline of Obstetrics and Gynaecology, The University of Adelaide, Australia.
Department of Health and Ageing, Australia.
Department of Obstetrics and Gynaecology, University of Nottingham, UK.
External sources
-
National Institute for Health Research, UK.
NIHR Programme of centrally‐managed pregnancy and childbirth systematic reviews of priority to the NHS and users of the NHS: 10/4001/02
Australian Department of Health and Ageing, Australia.
National Health and Medical Research Council, Australia.
Declarations of interest
Jim Thornton and Vinita Gurung are the authors of PITCH 2012. Assessment, data extraction and data entry for this trial was conducted by Philippa Middleton and Stephen Milan.
Edited (no change to conclusions)
References
References to studies included in this review
Binder 2006 {published data only}
- Binder T, Salaj P, Zima T, Vitek L. Randomized prospective comparative study of ursodeoxycholic acid and S‐adenosyl‐L‐methionine in the treatment of intrahepatic cholestasis of pregnancy. Journal of Perinatal Medicine 2006;34:383‐91. [DOI] [PubMed] [Google Scholar]
- Binder T, Salaj P, Zina T, Vitek L. [Ursodeoxycholic acid, S‐adenosyl‐L‐methionine and their combinations in the treatment of gestational intrahepatic cholestasis (ICP)]. Ceska Gynekologie 2006;71(2):92‐8. [PubMed] [Google Scholar]
Diaferia 1996 {published data only}
- Diaferia A, Nicastri PL, Tartagni M, Loizzi P, Iacovizzi C, Leo A. Ursodeoxycholic acid therapy in pregnant women with cholestasis. International Journal of Gynecology and Obstetrics 1996;52(2):133‐40. [DOI] [PubMed] [Google Scholar]
Fang 2009 {published data only}
- Fang J, Gou WL, Li QL. [Effect of integrative Chinese and Western medicine in treating pregnant women with intrahepatic cholestasis]. [Chinese]. [Zhongguo Zhong Xi Yi Jie He Za Zhi Zhongguo Zhongxiyi Jiehe Zazhi/Chinese] Journal of Integrated Traditional and Western Medicine [Zhongguo Zhong Xi Yi Jie He Xue Hui, Zhongguo Zhong Yi Yan Jiu Yuan Zhu Ban] 2009;29(10):869‐71. [PubMed] [Google Scholar]
Floreani 1996 {published data only}
- Floreani A, Paternoster D, Melis A, Grella PV. S‐adenosylmethionine versus ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: preliminary results of a controlled trial. European Journal of Obstetrics & Gynecology 1996;67:109‐3. [DOI] [PubMed] [Google Scholar]
- Floreani A, Paternoster D, Melis A, Zappala F, Chiaramonte M, Grella PV. A controlled trial with S‐adenosylmethionine versus ursodeoxycholic acid in intrahepatic cholestasis of pregnancy [EASL abstract]. Journal of Hepatology 1996;25 Suppl 1:S136. [Google Scholar]
Frezza 1984 {published data only}
- Frezza M, Pozzato G, Chiesa L, Stramentinoli G, Padova C. Reversal of intrahepatic cholestasis of pregnancy in women after high dose S‐adenosyl‐L‐methionine. Hepatology 1984;4(2):274‐8. [DOI] [PubMed] [Google Scholar]
Frezza 1990 {published data only}
- Frezza M, Cammareri G, Padova C, Italian Study Group for ICP. Beneficial effects of S‐adenosylmethionine in pregnant women with cholestasis: results of a multicenter controlled trial (abstract). Journal of Hepatology 1987;5(Suppl 1):S27. [Google Scholar]
- Frezza M, Centini G, Cammareri G, Grazie C, Padova C. S‐adenosylmethionine for the treatment of intrahepatic cholestasis of pregnancy. Results of a controlled clinical trial. Hepato‐Gastroenterology 1990;37(Suppl 2):122‐5. [PubMed] [Google Scholar]
Glantz 2005 {published data only}
- Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis of pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology 2005;42:1399‐405. [DOI] [PubMed] [Google Scholar]
- Glantz A, Reilly SJ, Benthin L, Lammert F, Mattsson LA, Marschall HU. Intrahepatic cholestasis of pregnancy: amelioration of pruritus by UDCA is associated with decreased progesterone disulphates in urine. Hepatology 2008;47(2):376‐9. [DOI] [PubMed] [Google Scholar]
Huang 2004 {published data only}
- Huang JY, Liu H. Analysis on therapeutic effect of Western and Chinese drug in treating intrahepatic cholestasis pregnancy. Chinese Journal of Integrated Traditional & Western Medicine 2004;24(4):309‐11. [PubMed] [Google Scholar]
Kaaja 1994 {published data only}
- Kaaja RJ, Kontula KK, Raiha A, Laatikainen T. Treatment of cholestasis of pregnancy with peroral activated charcoal. A preliminary study. Scandinavian Journal of Gastroenterology 1994;29(2):178‐81. [DOI] [PubMed] [Google Scholar]
Kondrackiene 2005 {published data only}
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology 2005;129:894‐901. [DOI] [PubMed] [Google Scholar]
- Kondrackiene J, Kupcinskas L, Sumskiene J. Efficacy of moderate doses of ursodeoxycholic acid in comparison to cholestyramine in the treatment of intrahepatic cholestasis of pregnancy [abstract]. Journal of Hepatology 2001;34(1):186. [Google Scholar]
Leino 1998 {published data only}
- Leino R, Ekblad U, Irjala K, Erkkola R. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy. Prenatal and Neonatal Medicine 1998;3 Suppl 1:67. [Google Scholar]
Liu 2006 {published data only}
- Liu Y, Qiao F, Liu H, Liu D. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy. Journal of Huazhong University of Science and Technology. Medical Sciences 2006;26(3):350‐2. [DOI] [PubMed] [Google Scholar]
Luo 2008 {published data only}
- Luo DH. Clinical observation on the effect on of Transmetil combined with ursodeoxycholic acid for intrahepatic cholestasis of pregnancy. Modern Journal of Integrated Traditional Chinese and Western Medicine [Xian Dai Zhong Xi Yi Jie He Za Zhi] 2008;17(7):1011‐2. [Google Scholar]
Nicastri 1998 {published data only}
- Nicastri PL, Diaferia A, Tartagni M, Loizzi P, Fanelli M. A randomised placebo‐controlled trial of ursodeoxycholic acid and S‐adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. British Journal of Obstetrics and Gynaecology 1998;105:1205‐7. [DOI] [PubMed] [Google Scholar]
Palma 1997 {published data only}
- Meng LJ, Reyes H, Axelson M, Palma J, Hernandez I, Ribalta J, et al. Progesterone metabolites and bile acids in serum of patients with intrahepatic cholestasis of pregnancy: effect of ursodeoxycholic acid therapy. Hepatology 1997;26(6):1573‐9. [DOI] [PubMed] [Google Scholar]
- Palma J, Reyes H, Ribalta J, Hernandez I, Sandoval L, Alumna R, et al. Ursodeoxycholic acid in the treatment of cholestasis of pregnancy: a randomized, double‐blind study controlled with placebo. Journal of Hepatology 1997;27(6):1022‐8. [DOI] [PubMed] [Google Scholar]
- Palma J, Reyes H, Ribalta J, Hernandez I, Sandoval L, Silva O, et al. Ursodeoxycholic acid in cholestasis of pregnancy: final report of a randomized, double‐blind, placebo controlled study. Hepatology 1996;24(4 Pt 2):373A. [DOI] [PubMed] [Google Scholar]
PITCH 2012 {published data only}
- Chappell LC, Gurung V, Chambers J, Seed PT, Williamson C, Thornton JG. PITCH: Two randomised controlled trials in obstetric cholestasis: Ursodeoxycholic acid versus placebo and early delivery versus expectant management. Archives of Disease in Childhood: Fetal and Neonatal Edition 2011;96:Fa110‐Fa111. [Google Scholar]
- Chappell LC, Gurung V, Seed PT, Chambers J, Williamson C, Thornton JG. Ursodeoxycholic acid versus placebo, and early term delivery versus expectant management, in women with intrahepatic cholestasis of pregnancy: semifactorial randomised clinical trial. BMJ 2012;344:e3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung V, Chappell L, Seed P, Chambers J, Williamson C, Thornton J. Pitch: Ursodeoxycholic acid versus placebo, and early delivery versus expectant management in the management of intrahepatic cholestasis of pregnancy: Two randomised controlled trials. International Journal of Gynecology and Obstetrics 2012;119(Suppl 3):S363‐4. [Google Scholar]
- Gurung V, Williamson C, Chappell L, Chambers J, Briley A, Broughton Pipkin F, et al. Pilot study for a trial of ursodeoxycholic acid and/or early delivery for obstetric cholestasis. BMC Pregnancy and Childbirth 2009;9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson‐Piercy C. Obstetric cholestasis. BJOG: an international journal of obstetrics and gynaecology 2008;115(s1):20. [DOI] [PubMed] [Google Scholar]
- Thornton G, Gurung V, Chappel C, Williamson C, Chambers J. Ursodeoxycholic acid versus placebo, and early delivery versus expectant management, in women with obstetric cholestasis: Two semi‐factorial randomised clinical trials. Journal of Maternal‐Fetal and Neonatal Medicine 2012;25(S2):100‐1. [Google Scholar]
- Williamson C, Chappell LC, Gurung V, Chambers J, Seed PT, Thornton JG. Pitch: Two randomised controlled trials in intrahepatic cholestasis of pregnancy: Ursodeoxycholic acid vs. placebo and early delivery vs. expectant management. Hepatology 2011;54(4 Suppl):925A. [Google Scholar]
Ribalta 1991 {published data only}
- Ribalta J, Reyes H, Gonzalez MC, Iglesias J, Arrese M, Poniachik J, et al. S‐adenosyl‐L‐methionine in the treatment of patients with intrahepatic cholestasis of pregnancy: a randomized, double‐blind, placebo‐controlled study with negative results. Hepatology 1991;13(6):1084‐9. [PubMed] [Google Scholar]
Riikonen 2000 {published data only}
- Gylling H, Riikonen S, Nikkila K, Savonius H, Miettinen TA. Oral guar gum treatment of intrahepatic cholestasis and pruritus in pregnant women: effects on serum cholesterol and other non‐cholesterol sterols. European Journal of Clinical Investigation 1998;28(5):359‐63. [DOI] [PubMed] [Google Scholar]
- Riikonen S, Savonius H, Gylling H, Nikkila K, Tuomi AM, Miettinen TA. Oral guar gum, a gel‐forming dietary fiber relieves pruritus in intrahepatic cholestasis of pregnancy. Acta Obstetricia et Gynecologica Scandinavia 2000;79(4):260‐4. [PubMed] [Google Scholar]
Roncaglia 2004 {published data only}
- Roncaglia N, Arreghini A, Locatelli A, Bellini P, Andreotti C, Ghidini A. Obstetric cholestasis: outcome with active management. European Journal of Obstetrics and Gynaecology and Reproductive Biology 2002;100(2):167‐70. [DOI] [PubMed] [Google Scholar]
- Roncaglia N, Locatelli A, Arreghini A, Assi F, Cameroni I, Pezzullo JC, et al. A randomised controlled trial of ursodeoxycholic acid and S‐adenosyl‐L‐methionine in the treatment of gestational cholestasis. BJOG: an International Journal of Obstetrics and Gynaecology 2004;111:17‐21. [DOI] [PubMed] [Google Scholar]
- Roncaglia N, Locatelli A, Bellini P, Arreghini A, Andreotti C, Ghidini A. A randomised controlled trial of ursodeoxycholic acid and S‐adenosyl‐l‐methionine in the treatment of gestational cholestasis [abstract]. American Journal of Obstetrics and Gynecology 2000;182(1 pt 2):S167. [DOI] [PubMed] [Google Scholar]
Shi 2002 {published data only}
- Shi DY, Chen H, Xiao M. Observation on the effect of danxiaoling in supplementary treatment of intrahepatic cholestasis in pregnancy. Zhongguo Zhong Xi Yi Jie He Za Zhi 2002;22(2):116‐8. [PubMed] [Google Scholar]
Zhang 2012 {published data only}
- Zhang L, Qi HB, Li Z, Fu XD, Chen L. Multicenter randomized prospective controlled study of ursodeoxycholic acid and s‐adenosyl‐l‐methionine in the treatment of intrahepatic cholestasis of pregnancy. International Journal of Gynecology and Obstetrics 2012;119(Suppl 3):S526. [Google Scholar]
References to studies excluded from this review
Elias 2001 {published data only}
- Elias E. A double blind, randomized placebo controlled trial (DBRCT) or ursodeoxycholic acid in obstetric cholestasis (OC). National Research Register (www.controlled‐trials.com) (accessed 21 November 2001).
Shi 2006 {published data only}
- Shi DY, Chen H, Tian XY. Clinical and experimental study on wuling pill in treating gestation period intrahepatic cholestasis. Chinese Journal of Integrated Traditional and Western Medicine 2006;26(2):114‐8. [PubMed] [Google Scholar]
References to studies awaiting assessment
Wang 2003 {published data only}
- Wang DZ, Chen J. Study of ursodeoxycholic acid in 64 patients with intrahepatic cholestasis of pregnancy. Journal of Practical Medical Techniques 2003;10(9):991‐2. [Google Scholar]
References to ongoing studies
Mazzella 2010 {published data only}
- Mazzella G, Azzaroli F. Ursodeoxycholic acid and cholestasis of pregnancy. ClinicalTrials.gov accessed 6 June 2011.
Additional references
Alsulyman 1996
- Alsulyman OM, Ouzounian JG, Ames‐Castro M, Goodwin TM. Intrahepatic cholestasis of pregnancy: perinatal outcome associated with expectant management. American Journal of Obstetrics and Gynecology 1996;175:957‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Azzaroli 2007
- Azzaroli F, Mennone A, Feletti V, Simoni P, Baglivo E, Montagnani M, et al. Clinical trial: modulation of human placental multidrug resistance proteins in cholestasis of pregnancy by ursodeoxycholic acid. Alimentary Pharmacology and Therapeutics 2007;26(8):1139‐46. [DOI] [PubMed] [Google Scholar]
Bacq 1997
- Bacq Y, Sapey T, Brechot MC, Pierre F, Fignon A, Dubois F. Intrahepatic cholestasis of pregnancy: a French prospective study. Hepatology 1997;26:358‐64. [DOI] [PubMed] [Google Scholar]
Berg 1986
- Berg B, Helm G, Petersohn L, Tryding N. Cholestasis of pregnancy. Clinical and laboratory studies. Acta Obstetricia et Gynecologica Scandinavica 1986;65(2):107‐13. [DOI] [PubMed] [Google Scholar]
Bernhard 1994
- Bernhard, JD. General principles, overview and miscellaneous treatments of itching. Itch Mechanisms and Management of Pruritus. New York: McGraw‐Hill, 1994:367‐81. [Google Scholar]
Beuers 2001
- Beuers U, Bilzer M, Chittattu A, Kullak‐Ublick GA, Keppler D, Paumgartner G, et al. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C‐dependent mechanisms in cholestatic rat liver. Hepatology 2001;33(5):1206‐16. [DOI] [PubMed] [Google Scholar]
Beuers 2006
- Beuers U, Pusl T. Intrahepatic cholestasis of pregnancy ‐ a heterogenous group of pregnancy‐related disorders?. Hepatology 2006;43(4):647‐9. [DOI] [PubMed] [Google Scholar]
Boelsterli 1983
- Boelsterli UA, Rakhit G, Balazs T. Modulation by S‐adenosyl‐L‐methionine of hepatic Na+,K+‐ATPase, membrane fluidity, and bile flow in rats with ethinyl estradiol‐induced cholestasis. Hepatology 1983;3(1):12‐7. [DOI] [PubMed] [Google Scholar]
Briggs 2001
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in Pregnancy and Lactation. 6th Edition. Philadelphia: Lippincott Williams & Wilkins, 2001. [Google Scholar]
Bromma 1995
- Bromma B, Scharein E, Darsow U, Ring J. Effects of menthol and cold on histamine‐induced itch and skin reactions in man. Neuroscience Letters 1995;187(3):157‐60. [DOI] [PubMed] [Google Scholar]
Cai 2006
- Cai H, Song YH, Xia WJ, Jin MW. Aqueous extract of Yin‐Chen‐Hao decoction, a traditional Chinese prescription, exerts protective effects on concanavalin A‐induced hepatitis in mice through inhibition of NF‐k B. Journal of Pharmacy and Pharmacology 2006;58(5):677‐84. [DOI] [PubMed] [Google Scholar]
Cantoni 1952
- Cantoni GL. The nature of the active methyl donor formed enzymatically from L‐methionine and adenosinetriphosphate. Journal of the American Chemical Society 1952;74(11):2942‐3. [Google Scholar]
Cunningham 1968
- Cunningham MD, Kelley LR, Peters ER. Phenobarbitone in cholestasis. Lancet 1968;291(7551):1089. [DOI] [PubMed] [Google Scholar]
Davies 1995
- Davies MH, Silva RC, Jones SR, Weaver JB, Elias E. Fetal mortality associated with cholestasis of pregnancy and the potential benefit of therapy with ursodeoxycholic acid. Gut 1995;37:580‐4. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
de Swiet 2002
- Swiet M. Medical Disorders in Obstetric Practice. Oxford: Blackwell Publishing, 2002. [Google Scholar]
Diac 2006
- Diac M, Kenyon A, Nelson‐Piercy C, Girling J, Cheng F, Tribe RM, et al. Dexamethasone in the treatment of obstetric cholestasis: a case series. Journal of Obstetrics and Gynaecology 2006;26(2):110‐4. [DOI] [PubMed] [Google Scholar]
Eloranta 2001
- Eloranta ML, Heinonen S, Mononen T, Saarikoski S. Risk of obstetric cholestasis in sisters of index patients. Clinical Genetics 2001;60:42‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fisk 1988
- Fisk NM, Storey GN. Fetal outcome in obstetric cholestasis. British Journal of Obstetrics and Gynaecology 1988;95(11):1137‐43. [DOI] [PubMed] [Google Scholar]
Gaudet 2000
- Gaudet R, Merviel P, Berkane N, Schouppe S, Cocheton JJ, Uzan S. Fetal impact of cholestasis of pregnancy: experience at Tenon Hospital and literature review. Fetal Diagnosis Therapy 2000;15(4):191‐7. [DOI] [PubMed] [Google Scholar]
Germain 2002
- Germain A, Carvajal JA, Glasinovic JC, Kato KS, Williamson C. Intrahepatic cholestasis of pregnancy: an intriguing pregnancy‐specific disorder. Journal of the Society for Gynaecologic Investigation 2002;9:10‐4. [DOI] [PubMed] [Google Scholar]
Gonzalez 1989
- Gonzalez MC, Reyes H, Arrese M, Figueroa D, Lorca B, Andresen M, et al. Intrahepatic cholestasis of pregnancy in twin pregnancies. Journal of Hepatology 1989;9:84‐90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Heikkinen 1981
- Heikkinen J, Maentausta O, Ylostalo P, Janne O. Changes in serum bile acid concentrations during normal pregnancy, in patients with intrahepatic cholestasis of pregnancy and in pregnant women with itching. British Journal of Obstetrics and Gynaecology 1981;88(3):240‐5. [PubMed] [Google Scholar]
Heinonen 1999
- Heinonen S, Kirkinen P. Pregnancy outcome with intrahepatic cholestasis. Obstetrics and Gynecology 1999;94(2):189‐93. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Insel 2010
- Insel P, Ross D, McMahon K, Bernstein M. Nutrition. Fourth. London, United Kingdom: Jones and Barlett, 2010:154. [Google Scholar]
Jiang 1986
- Jiang ZH, Qiu ZD, Liu WW, Liu YH, Wang QN, Miao HZ, et al. Intrahepatic cholestasis of pregnancy and its complications. Analysis of 100 cases in Chongqing area. Chinese Medical Journal 1986;99(12):957‐60. [MEDLINE: ] [PubMed] [Google Scholar]
Johnston 1979
- Johnston WG, Baskett TF. Obstetric cholestasis; a 14 year review. Journal of Obstetrics and Gynecology 1979;133(3):299‐301. [DOI] [PubMed] [Google Scholar]
Jones 1990
- Jones EA, Bergasa NU. The pruritus of cholestasis: from bile acids to opiate antagonists. Hepatology 1990;11:884‐7. [DOI] [PubMed] [Google Scholar]
Kauppila 1979
- Kauppila A, Tuimala R, Ylikorkala O, Reinila M, Ylostalo P. Placental steroid synthesis from DHEAS during dexamethasone therapy. Obstetrics and Gynecology 1979;54:39‐41. [DOI] [PubMed] [Google Scholar]
Kenyon 2001
- Kenyon AP, Piercy CN, Girling J, Williamson C, Tribe RM, Shennan AH. Pruritus may precede abnormal liver function tests in pregnant women with obstetric cholestasis: a longitudinal analysis. BJOG: an international journal of obstetrics and gynaecology 2001;108:1190‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kenyon 2002
- Kenyon AP, Piercy CN, Girling J, Williamson C, Tribe RM, Shennan AP. Obstetric cholestasis, outcome with active management: a series of 70 cases. BJOG: an international journal of obstetrics and gynaecology 2002;109:282‐8. [DOI] [PubMed] [Google Scholar]
Kenyon 2005
- Kenyon AP, Girling JC. Obstetric cholestasis. Progress in Obstetrics and Gynaecology 2005;16:37‐56. [Google Scholar]
Kirkinen 1984
- Kirkenen P, Ylostalo P, Heikkinen J, Maentausta O. Gallbladder function and maternal acids in intrahepatic cholestasis of pregnancy. European Journal of Obstetrics & Gynecology and Reproductive Biology 1984;18:29‐34. [DOI] [PubMed] [Google Scholar]
Klaasen 1970
- Klaasen CD. Effects of phenobarbital on the plasma disappearance and biliary excretion of drugs in rats. Journal of Pharmacology and Experimental Therapeutics 1970;175:289‐300. [PubMed] [Google Scholar]
Krasopoulos 1980
- Krasopoulos JC, Bari VA, Needle MA. The adsorption of bile salts on activated carbon. Lipids 1980;15(5):365‐70. [DOI] [PubMed] [Google Scholar]
Laatikainen 1975
- Laatikainen T, Ikonen E. Fetal prognosis in obstetric hepatosis. Annales Chirurgiae et Gynaecologiae Fenniae 1975;64:155‐64. [MEDLINE: ] [PubMed] [Google Scholar]
Lammert 2000
- Lammert F, Marshall HU, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management. Hepatology 2000;33:1012‐21. [DOI] [PubMed] [Google Scholar]
Lee 2007
- Lee TY, Changa HH, Chen JH, Hsueh ML, Kuo JJ. Herb medicine Yin‐Chen‐Hao‐Tang ameliorates hepatic fibrosis in bile duct ligation rats. Journal of Ethnopharmacology 2007;109(2):318‐24. [DOI] [PubMed] [Google Scholar]
Lee 2008
- Lee RH, Kwok KM, Ingles S, Wilson ML, Mullin P, Incerpi M, et al. Pregnancy outcomes during an era of aggressive management for intrahepatic cholestasis of pregnancy. American Journal of Perinatology 2008;25:341‐5. [DOI] [PubMed] [Google Scholar]
Lee 2009
- Lee TY, Chang HH, Kuo JJ, Shen JJ. Changes of hepatic proteome in bile duct ligated rats with hepatic fibrosis following treatment with Yin‐Chen‐Hao‐Tang. International Journal of Molecular Medicine 2009;23:477‐84. [DOI] [PubMed] [Google Scholar]
Leevy 1997
- Leevy CB, Koneru B, Klein KM. Recurrent familial prolonged intrahepatic cholestasis of pregnancy associated with chronic liver disease. Gastroenterology 1997;113:966‐72. [DOI] [PubMed] [Google Scholar]
Locatelli 1999
- Locatelli A, Roncaglia N, Arreghini A, Bellini P, Vergani P, Ghidini A. Hepatitis C virus infection is associated with a higher incidence of cholestasis of pregnancy. British Journal of Obstetrics and Gynaecology 1999;106(5):498‐500. [DOI] [PubMed] [Google Scholar]
Meng 1997
- Meng LJ, Reyes H, Palma J, Hernandez I, Ribalta J, Sjovall J. Effects of ursodeoxycholic acid on conjugated bile acids and progesterone metabolites in serum and urine of patients with intrahepatic cholestasis of pregnancy. Journal of hepatology 1997;27(6):1029‐40. [PUBMED: 9453429] [DOI] [PubMed] [Google Scholar]
Milkiewicz 2002
- Milkewicz P, Elias E, Williamson C, Weaver J. Obstetric cholestasis. BMJ 2002;324:123‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mitsuyoshi 1999
- Mitsuyoshi H, Nakashima T, Sumida Y, Yoh T, Nakajima Y, Ishikawa H, et al. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochemical and Biophysical Research Communications 1999;263:537‐42. [DOI] [PubMed] [Google Scholar]
Morgan 1993
- Morgan LM, Tredger JA, Shavila Y, Travis JS, Wright J. The effect of non‐starch polysaccharide supplementation on circulating bile acids, hormone and metabolite levels following a fat meal in human subjects. British Journal of Nutrition 1993;70:491‐501. [DOI] [PubMed] [Google Scholar]
Paternoster 2002
- Paternoster DM, Fabris F, Palù G, Santarossa C, Bracciante R, Snijders D, et al. Intra‐hepatic cholestasis of pregnancy in hepatitis C virus infection. Acta Obstetricia et Gynecologica Scandinavica 2002;81(2):99‐103. [PubMed] [Google Scholar]
Qui 1983
- Qui ZD, Wang QN, et al. Intrahepatic cholestasis of pregnancy: Clinical analysis and follow up of 22 cases. Chinese Medical Journal (English) 1983;96:902‐6. [PubMed] [Google Scholar]
RCOG 2011
- Royal College of Obstetricians and Gynaecologists. Obstetric Cholestasis (Guideline No. 43). London: RCOG, 20. [Google Scholar]
Reid 1976
- Reid R, Ivey KJ, Rencoret RH, Storey B. Fetal complications of obstetric cholestasis. British Medical Journal 1976;1(6014):870‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Reyes 1976
- Reyes H, Ribalta J, Gonzalez‐Ceron M. Idiopathic cholestasis of pregnancy in a large kindred. Gut 1976;17:709‐13. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reyes 1992
- Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterology Clinics of North America 1992;21(4):905‐21. [PubMed] [Google Scholar]
Reyes 1997
- Reyes H. Review: Intrahepatic cholestasis. A puzzling disorder of pregnancy. Gastroenterology and Hepatology 1997;12:211‐6. [DOI] [PubMed] [Google Scholar]
Ribalta 1995
- Ribalta J, Reyes H, Hernandez I, Fuentes O, Baez M, Gonzalez M, et al. Can a selenium deficiency affect the pathogenesis of cholestasis in pregnancy?. Gastroenterology and Hepatology 1995;18:114‐20. [PubMed] [Google Scholar]
Rioseco 1994
- Rioseco AJ, Ivankovic MB, Alejandro M, Hamed F, Kato SR, Parer JT, et al. Intrahepatic cholestasis of pregnancy: a retrospective case‐control study of perinatal outcome. American Journal of Obstetrics and Gynecology 1994;170:890‐5. [DOI] [PubMed] [Google Scholar]
Robinson 1971
- Robinson S H, Yannoni C, Nagasawa S. Bilirubin excretion in rats with normal and impaired bilirubin conjugation: effect of Phenobarbital. Journal of Clinical Investigation 1971;50(12):2602‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rodrigues 1998
- Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. Journal of Clinical Investigation 1998;101:2790‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Roncaglia 2002
- Roncaglia N, Arreghini A, Locatelli A, Bellini P, Andreotti C, Ghidini A. Obstetric cholestasis: outcome with active management. European Journal of Obstetrics and Gynaecology and Reproductive Biology 2002;100(2):167‐70. [DOI] [PubMed] [Google Scholar]
Ropponen 2006
- Ropponen A, Sund R, Riikonen S, Ylikorkala, Aittomaki K. Intrahepatic cholestasis of pregnancy as an indicator of liver and biliary diseases: a population‐based study. Hepatology 2006;43:723‐8. [DOI] [PubMed] [Google Scholar]
Roszkowski 1968
- Roszkowski I, Pisarek‐Miedzinska D. Jaundice in pregnancy. II. Clinical course of pregnancy and delivery and condition of neonate. American Journal of Obstetrics and Gynecology 1968;101(4):500‐3. [PubMed] [Google Scholar]
Sackett 2000
- Sackett DL, Strauss SE, Richardson WS, Rosenberg W, Haynes RB. Therapy. Evidence‐based Medicine: How to Practice and Teach EBM. Second Edition. New York: Churchill Livingstone, 2000:105‐53. [Google Scholar]
Sadler 1995
- Sadler LC, Lane M, North R. Severe fetal intracranial haemorrhage during treatment with cholestyramine for intrahepatic cholestasis of pregnancy. BJOG: an international journal of obstetrics and gynaecology 1995;102(2):169‐70. [DOI] [PubMed] [Google Scholar]
Shaw 1982
- Shaw D, Frohlich J, Wittmann BA, Willms M. A prospective study of 18 patients with cholestasis of pregnancy. American Journal of Obstetrics and Gynecology 1982;142:621‐5. [DOI] [PubMed] [Google Scholar]
Simmer 1975
- Simmer HH, Frankland M, Greipel M. On the regulation of fetal and maternal 16‐a‐hydroxydehydroepiandrosterone and its sulphates by cortisol and ACTH in human pregnancy at term. American Journal of Obstetrics and Gynecology 1975;121:646‐52. [DOI] [PubMed] [Google Scholar]
Stiehl 1999
- Stiehl A, Benz C, Sauer P. Mechanism of hepatoprotective action of bile salts in liver disease. Gastroenterology Clinics of North America March 1999;28(1):195‐209. [DOI] [PubMed] [Google Scholar]
Stramentinoli 1981
- Stramentinoli G, Padova C, Gualano M, Rovagnati P, Galli‐Kienle M. Ethynylestradiol‐induced impairment of bile secretion in the rat: protective effects of S‐adenosyl‐L‐methionine and its implication in estrogen metabolism. Gastroenterology 1981;80(1):154‐8. [PubMed] [Google Scholar]
Vallejo 2006
- Vallejo M, Briz O, Serrano MA, Monte MJ, Marin JJ. Potential role of trans‐inhibition of the bile salt export pump by progesterone metabolites in the etiopathogenesis of intrahepatic cholestasis of pregnancy. Journal of Hepatology 2006;44:1150‐7. [DOI] [PubMed] [Google Scholar]
Walker 2002
- Walker IA, Nelson‐Piercy C, Williamson C. Role of bile acid measurement in pregnancy. Annals of Clinical Biochemistry 2002;39:105‐13. [DOI] [PubMed] [Google Scholar]
Williamson 2001
- Williamson C, Gorelik J, Eaton BM, Lab M, Korchev Y, Swiet M. The bile acid taurocholate impair rat cardiomyocyte function: a proposed mechanism for intra‐uterine fetal death in obstetric cholestasis. Clinical Science 2001;100:363‐9. [PubMed] [Google Scholar]
Williamson 2004
- Williamson C, Hems LM, Goulis DG, Walker I, Chambers J, Donaldson O, et al. Clinical outcome in a series of cases of obstetric cholestasis identified via a patient support group. BJOG: an international journal of obstetrics and gynaecology 2004;111:676‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wilson 1979
- Wilson BR, Haverkamp AD. Cholestatic jaundice of pregnancy: new perspectives. Obstetrics and Gynecology 1979;54(5):650‐2. [MEDLINE: ] [PubMed] [Google Scholar]
Ylostalo 1975
- Ylostalo P, Ylikorkala O. Hepatosis of pregnancy. A clinical study of 107 patients. Annales Chirurgiae et Gynaecologiae Fenniae 1975;64:128‐34. [MEDLINE: ] [PubMed] [Google Scholar]
Zecca 2006
- Zecca E, Luca D, Marras M, Caruso A, Tommaso B, Romagnoli C. Intrahepatic cholestasis of pregnancy and neonatal respiratory distress syndrome. Pediatrics 2006;117(5):1669‐72. [DOI] [PubMed] [Google Scholar]
Zecca 2008
- Zecca E, Luca D, Baroni S, Vento G, Tiberi E, Romagnoli C. Bile acid‐induced lung injury in newborn infants: a bronchoalveolar lavage fluid study. Pediatrics 2008;121:e146‐e149. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Burrows 2001
- Burrows RF, Clavisi O, Burrows E. Interventions for treating cholestasis in pregnancy. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD000493] [DOI] [PubMed] [Google Scholar]