

Abstract
Purpose
We comprehensively evaluated the mutational spectrum of Leber congenital amaurosis (LCA) and investigated the molecular diagnostic rate and genotype–phenotype correlation in a Korean cohort.
Methods
This single-center retrospective case series included 50 Korean patients with LCA between June 2015 and March 2019. Molecular analysis was conducted using targeted panel-based next-generation sequencing, including deep intronic and regulatory variants or whole exome sequencing. The molecular diagnosis was made based on the inheritance pattern, zygosity, and pathogenicity.
Results
Among the 50 patients, 27 patients (54%) were male, and 11 (22%) showed systemic features. Genetic variants highly likely to be causative were identified in 78% (39/50) of cases and segregated into families. We detected two pathogenic or likely pathogenic variants in a gene linked to a recessive trait without segregation analysis in three cases (6.0%). GUCY2D (20%), NMNAT1 (18%), and CEP290 (16%) were the most frequently mutated genes in Korean LCA. Copy number variations were found in three patients, which accounted for 6% of LCA cases. A possible dual molecular diagnosis (Senior-Løken syndrome along with Leigh syndrome, and Joubert syndrome with transposition of the great arteries) was made in two patients (4%). Three of 50 patients were medically or surgically actionable: one patient for RPE65 gene therapy and two patients with WDR19 Senior-Løken syndrome for early preparation for kidney and liver transplantations.
Conclusions
This study demonstrated that approximately 4% of patients may have dual molecular diagnoses, and 6% were surgically or medically actionable in LCA. Therefore, accurate molecular diagnosis and careful interpretation of next-generation sequencing results can be of great help in patients with LCA.
Introduction
Leber congenital amaurosis (LCA) is a genetically heterogeneous retinal dystrophy with an incidence of approximately 2–3 per 100,000 births [1,2]. LCA is the most severe form of inherited retinal disorders and is accompanied by nystagmus and severe visual impairment within the first year of life. LCA accounts for approximately 5% of all inherited retinal disorders, and nearly 20% of children who attend special schools for blind individuals have LCA [3]. The mode of inheritance in LCA is typically autosomal recessive, although some form of LCA is known to be inherited as autosomal dominant [1,4]. Clinical diagnosis of LCA is straightforward based on the presence of nystagmus or wandering eye movement, oculodigital sign, sluggish or absent pupillary responses, and flat or severely diminished response on electroretinography (ERG) [5,6]. The development of next-generation sequencing (NGS) has enabled relatively simple characterization of the molecular features of LCA. To date, 25 genes have been shown to be associated with LCA (assessed July 2019, RetNet). Most of these genes are known to be important in retinal development or in the molecular pathways associated with phototransduction, retinoid cycle, molecular signal transduction, guanine synthesis, segment phagocytosis, photoreceptor morphogenesis, and intraphotoreceptor ciliary transport [7].
After the successful development of gene therapy for RPE65 (Gene ID 6121, OMIM 180069)-associated LCA, the molecular genetic diagnosis of LCA has received increased attention [8]. Although NGS can reveal causative mutations of LCA, the origin of 20% to 30% of cases remains unclear because of the genetic complexity of the disease, copy number variations, or variants in non-coding regions. Identifying genetic mutations is a key step in proper diagnosis with genetic counseling and contributes to the development of genetic therapeutic strategies. In East Asia, few studies have been conducted to explore the mutational spectrum of LCA. The most frequently mutated genes in patients from East Asia are CRB1 (Gene ID 23418, OMIM 604210), NMNAT1 (Gene ID 64802, OMIM 608700), GUCY2D (Gene ID 3000, OMIM 600179), and RPGRIP1 (Gene ID 57096, OMIM 605446) [9,10]. Three studies have evaluated the genetic profiles of Korean patients with LCA, but the total number of patients is too low to determine overall frequencies [11,12]. Therefore, this study was conducted to investigate the molecular profile of 50 consecutive patients with LCA and determine the genotype–phenotype correlation of LCA.
Methods
Patient recruitment
This retrospective consecutive case series recruited 50 unrelated Korean patients with LCA who underwent genetic testing between June 1, 2015, and March 31, 2019. Patients underwent detailed ophthalmic examinations, including optical coherence tomography and electroretinography, if applicable. Informed written consent was provided by the patients or their parents, or both, and peripheral blood samples were collected from all patients for genetic analysis. Whole blood was collected in the EDTA tube and transferred to the laboratory at room temperature to extract genomic DNA within 24 hours [13]. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, The Netherlands), followed by the manufacturer's instructions. The research protocol was approved by the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine (4–2019–0542). This study adhered to the tenets of the Declaration of Helsinki and ARVO statements of ethical principles for medical research involving human subjects.
Sequencing analysis
Molecular testing was performed with targeted next-generation sequencing (Department of Laboratory Medicine, Yonsei University College of Medicine) or whole exome sequencing at a core facility (DNA Link, Inc., Seoul, Korea). Sequencing was performed on an Illumina NextSeq 550 system (San Diego, CA) for targeted panel sequencing (n=39) and an Illumina NovaSeq 6000 system for whole exome sequencing (n=11). The targeted NGS panel included 113 genes associated with LCA, early onset retinal dystrophy, and infantile nystagmus, and 429 genes associated with inherited eye diseases (Appendix 1). The 429 gene targeted panel (version 2) also includes the deep intronic or regulatory variants associated with LCA (e.g., c.-70A>T and c.-69C>T in NMNAT1, and c.2991+1655A>G in the CEP290 (Gene ID 80184, OMIM 610142) gene, Appendix 2). Target enrichment was performed with custom-designed RNA oligonucleotide probes and a target enrichment kit (Celemics, Seoul, South Korea). Whole exome sequencing was performed with the xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc., Coralville, IA). Demultiplexed BAM files were aligned to the hg19 reference genome using BWA-aln [14]. Single-nucleotide variants and small insertions or deletions were called and crosschecked using the Genome Analysis ToolKit (GATK) version 3.8.0 with Haplotypecaller and VarScan version 2.4.0. Each variant suspected to be pathogenic, likely pathogenic, or a variant of uncertain significance was confirmed with visual inspection of the bam file using Integrative Genomics Viewer 2.3 software. Split-read-based detection of large structural variation was conducted using Pindel and Manta [15,16]. Read-depth-based detection of copy number variations (CNVs) was conducted using ExomeDepth version 1.1.10 [17], followed by visualization using a base-level read depth normalization algorithm designed by the authors. CopywriteR version 2.9.0 was used with a 1-Mb window option for off-target analysis and whole chromosomal CNV detection [18].
Variant filtering and classification
The variants with a minor allele frequency (MAF) >1% in the Genome Aggregation Database (gnomAD v2.1.1) were excluded from further investigation. The potential pathogenicity of each variant was determined according to the guidelines of the American College of Medical Genetics (ACMG), and three in silico prediction algorithms, including Sorting Intolerant From Tolerant (SIFT), PolyPhen2, and Combined Annotation Dependent Depletion, were used for pathogenicity prediction [19]. Molecular diagnosis was made based on the inheritance pattern, zygosity, and pathogenicity of the variant. Patients were divided into three groups: (1) probable molecular diagnosis: patients with pathogenic or likely pathogenic disease-associated variant(s) with segregation, (2) possible molecular diagnosis: patients with two heterozygous pathogenic or likely pathogenic mutations without segregation, or (3) unsolved: all other patients for whom no pathogenic or likely pathogenic disease-associated variants or patients harboring a single disease-associated variant in a gene linked with recessive traits.
Results
Patient demographics
The clinical and ophthalmic features of the 50 unrelated patients with LCA are listed in Appendix 3 and Appendix 4. Among the 50 patients, 27 patients (54%) were male, and 23 patients (46%) were female. The average age at genetic testing was 7.1±10.7 years (range, 0.3–39.8 years), and the median age was 1.7 years. All patients were single ethnicity (Korean), and no patients were of consanguineous parentage. All 50 patients had nystagmus or wandering eye movement within 6 months of age. Among them, 27 patients (54%) had wandering eye movement, 15 patients (30%) had pure horizontal jerk or pendular nystagmus, two patients (4%) had pure vertical type nystagmus, and six patients (12%) had multidirectional nystagmus. Pure vertical nystagmus was observed in two patients with mutations in RPGRIP1 (P32) and WDR19 (Gene ID 57728, OMIM 608151; P36).
Diagnostic rate of NGS
The overall diagnostic detection rate in this Korean LCA cohort was 84% (42/50) after targeted NGS or whole exome sequencing (Figure 1). Among the 42 patients, possible diagnosis was made in three patients (7.1%) due to unavailability of parental DNA. Eight patients were molecularly unsolved after NGS testing. A total of 82 putative pathogenic variants were found in 42 patients, and 22 variants (26.8%) were novel mutations (Appendix 5). Moreover, three patients (6%) were eligible for surgical or medical treatment. One patient (P31) with RPE65-associated LCA was a candidate for gene therapy, and two patients with Senior-Løken syndrome (P36 and P37) could undergo early preparation for kidney and liver transplantation.
Figure 1.
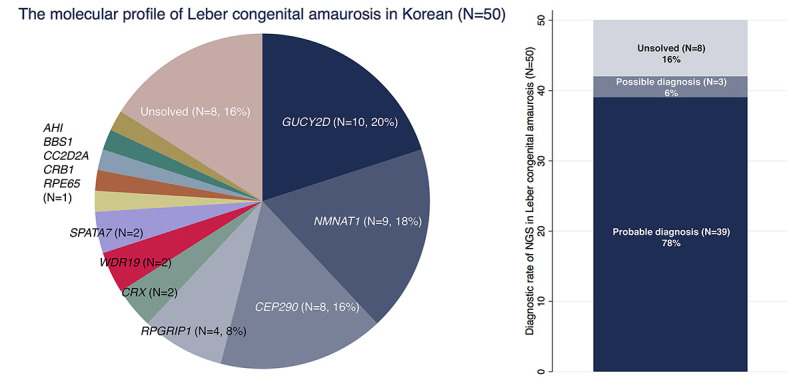
Molecular diagnosis of Leber congenital amaurosis in Korean patients. A: Distribution of mutated genes in Korean patients with Leber congenital amaurosis (LCA). B: Molecular diagnostic yield of next-generation sequencing in 50 patients with LCA.
GUCY2D and NMNAT1 are frequently mutated genes in Korean patients
The most frequently observed variants were c.2649delT in GUCY2D, c.709C>T in NMNAT1, c.6012–12T>A in CEP290, and c.3565_3571del in RPGRIP1. Previous studies reported that CEP290 c.6012–12T>A is a common allele in Japanese and Korean patients with Joubert syndrome [11,20,21]. In gnomAD, the MAF of CEP290 c.6012–12T>A was 0.001080 in Korean patients and 0.00001785 overall. The MAF of RPGRIP1 c.3565_3571del was also high in the Korean population (MAF 0.001048 in Korean patients, 0.00001625 overall). Among the 50 patients, nine patients (18.0%) had mutations in NMNAT1. This rate is much higher than the rate in Western countries (4.9%) [22], and higher than in other East Asian such as Japanese (8.8%) and Chinese (2.3%) cohorts [9,23]. Except three cases, six patients with mutations in NMNAT1 showed the same compound heterozygous c.196C>T/c.709C>T mutations (P23, P24, P25, P26, P27, and P28). All nine patients with NMNAT1-associated LCA had the c.709C>T:p.(Arg237Cys) variant. This variant showed a high MAF (0.001048) in Korean gnomAD (gnomAD global MAF 0.00004951). Three unrelated patients with mutations in GUCY2D c.2649del were identified in the present cohort, but this variant was absent from gnomAD.
Possible dual molecular diagnosis in two patients
In this study, two patients with homozygous mutations in WDR19 c.3533G>A showed retinal dystrophy at early ages, accompanied by nephronophthisis and Caroli disease (Figure 2). One patient (P36) had no intellectual disability, while the other patient (P37) had severe mental retardation and mild hypotonia. In P37, brain magnetic resonance imaging (MRI) showed bilateral T2 hyperintensity of the corpus striatum predominantly in the putamen with diffuse volume loss of the brain, indicating metabolic mitochondrial disorders. Targeted NGS revealed compound heterozygous mutations in POLG (Gene ID 5428, OMIM 174763) c.1113G>T:p.(Lys371Asn)/c.2890C>T:p.(Arg964Cys) along with mutations in WDR19. POLG encodes the catalytic subunit of DNA polymerase gamma, which is essential for mitochondrial DNA replication and repair. Mutations in POLG have been linked to a diverse spectrum of clinical phenotypes, such as encephalomyopathies, resulting in autosomal recessive or dominant inheritance [24].
Figure 2.

Two patients with nephronophthisis and Caroli disease with homozygous mutations in WDR19 c.3533G>A. A–D: Fundus photograph, optical coherence tomography, renal ultrasonography, and abdominal computed tomography of a 7-year-old patient with Leber congenital amaurosis (LCA) with a homozygous mutation in WDR19 c.3533G>A (P36). Increased kidney echogenicity, multiple cystic formation of the kidney, and dilated intrahepatic bile duct were noted. She was neurologically normal. E–H: Fundus photograph, renal and abdominal ultrasonography, and brain magnetic resonance imaging (MRI) image of a 4-year-old patient with possible dual diagnosis of mutations in WDR19/POLG (P37). Brain MRI showed a bilateral T2 hyperintense signal in the putamen, which was not reported in Senior-Løken syndrome (black arrow). These findings suggest a possible dual diagnosis.
Patient 9 (P9) exhibited transposition of the great arteries in prenatal ultrasonography and underwent arterial switch operation at postnatal day 5. At the age of 4 months, she had no eye contact, nystagmus, and extinguished ERG. Brain MRI showed molar tooth sign. NGS analysis revealed compound heterozygous mutations in c.3847C>T/c.6271–1G>A CEP290, but transposition of the great arteries has not been reported in Joubert syndrome. Further investigation revealed a novel heterozygous mutation in TBX1 (Gene ID 6899, OMIM 602054) c.734A>G:p.(Tyr245Cys). TBX1 is associated with transposition of the great arteries, and haploinsufficiency of TBX1 causes malformation of the great vessel [25]. This variant is extremely rare (2/251482) in gnomAD, located in the DNA-binding domain, and predicted to be deleterious according to three different in silico prediction programs. However, the mother of the proband also had this heterozygous variant but was phenotypically normal. Therefore, this mutation could be related to incomplete penetrance or maternal mosaicism. The pathogenicity of this variant could not be determined.
Genotype–phenotype correlation in LCA
The fundus feature of NMNAT1-associated LCA was characterized by early onset round coloboma-like macular degeneration with pigmentary retinopathy (Appendix 6). Spectralis domain optical coherence tomography (Heidelberg Engineering, Heidelberg, Germany) revealed mild excavation of the macula in patients with mutations in NMNAT1. One patient (P22) had a compound heterozygous c.275G>A:p.(Trp92*)/c.709C>T:p.(Arg237Cys) variant in NMNAT1. This nonsense c.275G>A:p.(Trp92*) variant was novel, and colobomatous macular degenerations and oculodigital sign were more severe than in other patients with mutations in NMNAT1. Fundus photographs showed multiple bear-foot-like colobomatous macular degenerations. Another patient (P7) with the homozygous mutation in CEP290 c.6012–12T>A:p.(Arg2004Serfs*7) had severe ptosis at early infancy, severe psychomotor development delay, and extinguished ERG. The patient was treated with a frontalis sling with a silicone rod at the age of 10 months (Figure 3). A large case series of 99 patients with Joubert syndrome demonstrated that severe ptosis was observed usually in TMEM67 (Gene ID 91147, OMIM 609884), MKS1 (Gene ID 54903, OMIM 609883), TMEM216 (Gene ID 51259, OMIM 613277), CSPP1 (Gene ID 79848, OMIM 611654), RPGRIP1L (Gene ID 23322, OMIM 610937), and CELSR2 (Gene ID 1952, OMIM 604265) Joubert syndrome, not in CEP290 Joubert syndrome [26], and ptosis was not observed in any of the other patients with CEP290 in this cohort. This patient also had molar tooth sign on the brain MRI and nephronophthisis. This patient’s phenotype was consistent with Arima syndrome (OMIM:243910), which is considered a severe form of Joubert syndrome [27]. Arima syndrome has a specific homozygous variant (c.6012–12T>A) or compound heterozygous variants (c.1711+1G>A; c.6012–12T>A) in the CEP290 gene [27].
Figure 3.

Ptosis and severe neurologic feature of Arima syndrome caused by homozygous mutations in CEP290 c.6012–12T>A (P7). A: Severe ptosis was observed immediately after birth. B: Frontalis sling with silicone rod was used as treatment. C: Brain magnetic resonance imaging (MRI) revealed elongation of the superior cerebellar peduncle and cerebellar vermis hypoplasia.
Mutations in CRX caused LCA as either autosomal recessive or autosomal dominant traits
We identified two patients with novel mutations in CRX (Gene ID 1406, OMIM 602225). One patient (P11) had compound heterozygous c.101–1G>A/c.122G>A:p.(Arg41Gln) mutations, while the other (P12) had a novel heterozygous c.443del:p.(Gly148Alafs*39) mutation in CRX. The c.122G>A:p.(Arg41Gln) mutation is considered likely pathogenic in ClinVar, and previous studies identified that this variant is associated with autosomal dominant retinitis pigmentosa [28]. However, the parents of P11 had no sign of retinal dystrophy, and the daughter of P11 also had the c.122G>A:p.(Arg41Gln) variant in CRX, but she had no retinal dystrophy until the age of 12 years. The inheritance pattern in this family was consistent with an autosomal recessive trait. Therefore, mutations in CRX cause LCA or cone-rod dystrophy depending on the type of mutation, and the mutation can be inherited as either autosomal dominant or autosomal recessive (Figure 4).
Figure 4.

Mutations in CRX cause Leber congenital amaurosis (LCA) as either an autosomal recessive or autosomal dominant trait (P11). A: Fundus photograph showing macular dystrophy with peripheral retinal pigmentary changes. B: Optical coherence tomography showing diffuse loss of photoreceptor bands and thinning of the outer nuclear layer. C: The daughter of the proband (age 12 years) carrying the mutation in CRX c.122G>A had no evidence of retinal degeneration, suggesting that CRX can be inherited in an autosomal recessive manner. Although it has been reported that CRX c.122G>A variants cause cone-rod dystrophy in an autosomal dominant manner, the daughter of the proband showed no evidence of cone-rod dystrophy until the age of 12 years. D: Fundus photographs showing diffuse peripheral retinal pigmentary changes at early infancy (P12). E: Optical coherence tomography showing diffuse loss of photoreceptor bands and thinning of the outer nuclear layer. F: The heterozygous mutation in CRX c.443delG causes Leber congenital amaurosis (LCA), suggesting this novel mutation in CRX is inherited in an autosomal dominant manner.
CNVs in LCA
Using a read-depth algorithm, we effectively identified CNVs that met the ACMG standard guidelines in three individuals [29,30]. Plots of the normalized read-depth ratio of regions with CNVs in these patients are shown in Appendix 7. Two heterozygous deletions were identified in two individuals with mutations in NMNAT1. The homozygous c.709C>T mutation was initially suspected in P29, but detailed CNV analysis revealed an exon 4–5 deletion. Only one heterozygous c.709C>T variant was found using GATK best practice analysis in P30, and further investigation of the CNV identified a second mutation as a heterozygous deletion in exon 2 of NMNAT1 [31]. These intragenic deletions (an exon 4–5 deletion and an exon 2 deletion) involving NMNAT1 could be classified as likely pathogenic variants according to the PVS1 rule (0.9 points from 2E evidence) because they were predicted to disrupt reading frame and induce nonsense-mediated decay (NMD) [29,32]. Another form of CNV was found in a patient with a mutation in GUCY2D (P20). The fundus of the patient appeared normal, with no neurologic sign and extinguished ERG, which was consistent with GUCY2D LCA. NGS analysis revealed c.1790G>A:p.(Gly597Glu)/exon 4–5 duplication in GUCY2D. This GUCY2D exon 4–5 duplication occurred de novo (0.45 points from 4A evidence) and is absent from the database of genomic variants and gnomAD structural variants. The partial duplication was expected to cause reading frame disruption and NMD (0.45 points from 2I evidence); therefore, it could be classified as a likely pathogenic variant [29,32].
Discussion
In this study, we analyzed 50 consecutive patients to investigate the molecular spectrum of LCA. The diagnostic yield of NGS was 84%. We also found that GUCY2D, NMNAT1, and CEP290 were the most frequently mutated genes in the Korean population. Mutations in these genes occur in more than 50% of patients. Previous studies revealed that GUCY2D and CEP290 frequently contain genetic variants in Western countries [4], which is consistent with the present results. Interestingly, all patients with NMNAT1-associated LCA had the heterozygous c.709C>T variant. In gnomAD, the MAF of c.709C>T in NMNAT1 is relatively common in East Asian and Korean population [4]. The high MAF of NMNAT1 c.709C>T in Koreans may be related to the present results. Compared to populations in other countries, high MAFs of NMNAT1 c.709C>T, CEP290 c.6012–12T>A, and RPGRIP1 c.3565_3571del were found in the Korean population. The mutational spectrum of Korean patients with LCA appears to be similar to that in Japanese and Chinese populations, while NMNAT1 LCA is more prevalent in patients from Korea compared to those from other countries.
A previous study reported deep excavation of the macula with the absence of any distinct laminations in a 60-year-old patient with NMNAT1 LCA [33]. In the present study, mild excavation with preservation of the inner retinal layers was found in a 4-year-old with NMNAT1-associated LCA. The mechanism of retinal damage caused by mutations in NMNAT1 may occur preferentially in the outer retina, further progressing to the inner retinal layers. Moreover, a patient who carries the nonsense c.275G>A:p.(Trp92*) variant in NMNAT1 had multiple bear-foot-like macular degenerations rather than one round coloboma-like macular degeneration as observed in other patients with NMNAT1. Therefore, the more deleterious mutation in NMNAT1 causes a more severe phenotype. We observed that coloboma-like macular degeneration occurred at the age of 4 months, and thus has a narrow therapeutic window. Further studies are needed to determine the exact mechanism of macular coloboma-like formation in NMNAT1 LCA.
Previous studies showed that early preparation for kidney transplantation can reduce hemolysis-dependent periods in Senior-Løken syndrome [34,35]. RPE65-associated LCA can be treated by gene therapy. We identified three patients (6%) who could be managed or treated differently based on NGS testing. In terms of precision medicine, NGS can guide which genes are surgically or medically actionable. Additionally, a previous study reported that 4.9% of patients had diagnoses involving two or more disease loci [36]. The present study revealed two patients (4%) with pathogenic or likely pathogenic variant(s) in two different disease loci. Whole exome sequencing reveals large numbers of variants of unknown significance. Therefore, meticulous phenotyping and careful interpretation of genetic analysis results are essential for ensuring the correct diagnosis.
CRX encodes a cone-rod homeobox protein that plays a key role in photoreceptor development and survival [37,38]. Mutations in CRX cause LCA, cone-rod dystrophy, or macular dystrophy depending on the type of mutation [39-41]. All cases show a heterozygous state except four case reports of homozygous disease in LCA and severe retinopathy [41,42]. The mutations in CRX that arise in the homeodomain (residue 39–99) are usually missense mutations; heterozygous variants in the homeodomain cause predominantly cone-rod dystrophy, followed by LCA. The CRX c.122G>A mutation has been reported to cause late-onset autosomal dominant cone-rod dystrophy, but a recent study reported that middle-aged heterozygous carriers of this mutation showed a normal phenotype [43]. We also found that a family member carrying the heterozygous c.122G>A mutation had no retinal dystrophy until 12 years of age, and CRX causes LCA as an autosomal recessive trait. The pathogenic mechanism in most cases is likely dominant negative, with gain of function. However, recent literature reported that homozygous complete deletion of the CRX gene causes the LCA phenotype [41]. Considering the incomplete penetrance and complexity of the inheritance pattern of CRX, ophthalmologists should use caution during genetic counseling of patients with mutations in CRX.
Previous studies reported that the diagnostic yield of targeted NGS study for LCA is typically 50% to 80% [4,44], which is consistent with the present results. We detected CNVs in three patients using customized ExomeDepth software, which accounted for 7.1% of solved cases. Among the 50 patients, the cases of eight patients remained unsolved. Whole genome sequencing confirmed a molecular diagnosis of inherited retinal disease in 11 of 33 individuals who had not obtained a molecular diagnosis through targeted NGS testing [45]. Additionally, exome reanalysis with periodic assimilation identified pathogenic variants in 30% to 40% of exome-negative cases [46,47]. Therefore, further genomic analysis, such as whole genome sequencing or exome reanalysis, is required to detect large structural variants, variants in new discovered genes, Alu insertion, or non-coding variants.
This study had several limitations. First, it was a single-center, retrospective study consisting of 50 unrelated patients. A larger sample is needed to estimate the overall genetic profile of LCA in Korean patients. Second, targeted panel sequencing or whole exome sequencing may miss variants in deep intronic regions, non-coding regions (e.g., non-coding exon 1 in GUCY2D and non-coding exon 1 in NMNAT1), and low complex repeated sequence regions. Although the targeted panel included known deep intronic and regulatory variants (c.-70A>T and c.-69C>T in NMNAT1, and c.2991+1655A>G in CEP290), these variants were not detected in the present study cohort. It is known that the CEP290 c.2991+1655A>G variant is a founder mutation in non-Finnish Europeans, but not in other populations. Therefore, an ethnicity-specific targeted panel is needed to improve the molecular diagnostic rate. Third, we could not determine the transconfiguration of variants in three patients because DNA from family members was not available or the parents of the proband refused to undergo segregation analysis. Therefore, we classified these patients as possible diagnosis.
In conclusion, mutations in GUCY2D, NMNAT1, and CEP290 appeared to be the major genetic causes of LCA in Korean patients. The overall molecular pickup rate of LCA was 84%. We also found that 4% of patients had multiple molecular diagnoses in two different disease loci, and 6% of patients were surgically or medically actionable. In Korea, NMNAT1-associated LCA appears to be more prevalent than in other countries because of the high MAF of NMNAT1 c.709C>T in the Korean population. Because early retinal degeneration of the macula occurs in NMNAT1-associated LCA, visual prognosis was worst among all LCA cases. Despite the success of RPE65 gene therapy trials, animal or human studies toward clinical trials are limited. Further studies are needed to define the natural history and primary outcome in NMNAT1-associated LCA.
Acknowledgments
Contributors: DS, SS, HTL and JH conceived, wrote and provided critical revision of the manuscript. SS, S-TL, JRC, HTL, JL, SHB, S-HH provided critical revision of the manuscript. Figures were produced by DS and JH. Funding: Supported by a fund (#2018-ER6902–00) from the Research of Korea Centers for Disease Control and Prevention. Dr. Hyun Taek Lim (htlim@amc.seoul.kr) and Dr. Jinu Han (jinuhan@yuhs.ac) are co-corresponding authors for this study.
APPENDIX 1. Target genes associated with inherited eye diseases.
To access the data, click or select the words “Appendix 1.”
APPENDIX 2. Non-coding deep intronic or regulatory variants covered by the panel.
To access the data, click or select the words “Appendix 2.”
APPENDIX 3. The clinical features of 42 patients who receive probable or possible diagnosis.
To access the data, click or select the words “Appendix 3.”
APPENDIX 4. The clinical features of 8 patients with unsolved cases.
To access the data, click or select the words “Appendix 4.”
Appendix 5. Table 1. Putative pathogenic variants identified in 42 patients with Leber congenital amaurosis.
To access the data, click or select the words “Appendix 5.”
APPENDIX 6. Typical macular colobomatous degeneration of NMNAT1 patient and distinct “multiple colobomatous” retinal degeneration in a patient with novel NMNAT1 variant.
To access the data, click or select the words “Appendix 6.”
APPENDIX 7. Detailed analysis of copy number variations (CNVs) in Korean patients with Leber congenital amaurosis.
To access the data, click or select the words “Appendix 7.”
References
- 1.Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. 2004;49:379–98. doi: 10.1016/j.survophthal.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Stone EM. Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Pham C, Sheth SJ, Keeffe JE, Carden SM. New trends in childhood vision impairment in a developed country. J AAPOS. 2017;21:496–8. doi: 10.1016/j.jaapos.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Chacon-Camacho OF, Zenteno JC. Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases. 2015;3:112–24. doi: 10.12998/wjcc.v3.i2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Laey JJ. Leber’s congenital amaurosis. Bull Soc Belge Ophtalmol. 1991;241:41–50. [PubMed] [Google Scholar]
- 6.Franceschetti A, Dieterle P. Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia. Confin Neurol. 1954;14:184–6. [PubMed] [Google Scholar]
- 7.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, Feathers KL, Luong VA, Yzer S, Balaggan K, Viswanathan A, de Ravel TJ, Casteels I, Holder GE, Tyler N, Fitzke FW, Weleber RG, Nardini M, Moore AT, Thompson DA, Petersen-Jones SM, Michaelides M, van den Born LI, Stockman A, Smith AJ, Rubin G, Ali RR. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med. 2015;372:1887–97. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Wang X, Zou X, Xu S, Li H, Soens ZT, Wang K, Li Y, Dong F, Chen R, Sui R. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis Cohort. Invest Ophthalmol Vis Sci. 2015;56:3642–55. doi: 10.1167/iovs.14-15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rim JH, Lee ST, Gee HY, Lee BJ, Choi JR, Park HW, Han SH, Han J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017;135:1376–85. doi: 10.1001/jamaophthalmol.2017.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Rim JH, Hwang IS, Kim J, Shin S, Lee ST, Choi JR. Diagnostic application of clinical exome sequencing in Leber congenital amaurosis. Mol Vis. 2017;23:649–59. [PMC free article] [PubMed] [Google Scholar]
- 12.Seong MW, Kim SY, Yu YS, Hwang JM, Kim JY, Park SS. Molecular characterization of Leber congenital amaurosis in Koreans. Mol Vis. 2008;14:1429–36. [PMC free article] [PubMed] [Google Scholar]
- 13.Rainen L, Arbique J, Asthana D, Earley M, Geiszler R, Krieg-Schneider F, Mannhalter C, Ogino S, Parish G, Ballas C. Clinical Laboratory Standards Institute Document MM13-A: Collection, transport, preparation, and storage of specimens for molecular methods approved guideline. Wayne, PA: Clinical and Laboratory Standards Institute 2005. [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, Saunders CT. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 17.Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO, Thrasher AJ, Kumararatne D, Doffinger R, Nejentsev S. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, Nevedomskaya E, Xu G, de Ruiter J, Lolkema MP, Ylstra B, Jonkers J, Rottenberg S, Wessels LF, Adams DJ, Peeper DS, Krijgsman O, Copywrite R. DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. doi: 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–d94. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsurusaki Y, Kobayashi Y, Hisano M, Ito S, Doi H, Nakashima M, Saitsu H, Matsumoto N, Miyake N. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013;58:113–5. doi: 10.1038/jhg.2012.117. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, Ieda D, Saitoh S, Hiyane M, Suzumura H, Yagyu K, Shiraishi H, Nakajima M, Fueki N, Habata Y, Ueda Y, Komatsu Y, Yan K, Shimoda K, Shitara Y, Mizuno S, Ichinomiya K, Sameshima K, Tsuyusaki Y, Kurosawa K, Sakai Y, Haginoya K, Kobayashi Y, Yoshizawa C, Hisano M, Nakashima M, Saitsu H, Takeda S, Matsumoto N. Molecular genetic analysis of 30 families with Joubert syndrome. Clin Genet. 2016;90:526–35. doi: 10.1111/cge.12836. [DOI] [PubMed] [Google Scholar]
- 22.Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, Staniszewska M, Shukla R, Palavalli L, Mohand-Said S, Waseem NH, Jalali S, Perin JC, Place E, Ostrovsky J, Xiao R, Bhattacharya SS, Consugar M, Webster AR, Sahel JA, Moore AT, Berson EL, Liu Q, Gai X, Pierce EA. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–5. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosono K, Nishina S, Yokoi T, Katagiri S, Saitsu H, Kurata K, Miyamichi D, Hikoya A, Mizobuchi K, Nakano T, Minoshima S, Fukami M, Kondo H, Sato M, Hayashi T, Azuma N, Hotta Y. Molecular Diagnosis of 34 Japanese Families with Leber Congenital Amaurosis Using Targeted Next Generation Sequencing. Sci Rep. 2018;8:8279. doi: 10.1038/s41598-018-26524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Da Pozzo P, Cardaioli E, Rubegni A, Gallus GN, Malandrini A, Rufa A, Battisti C, Carluccio MA, Rocchi R, Giannini F, Bianchi A, Mancuso M, Siciliano G, Dotti MT, Federico A. Novel POLG mutations and variable clinical phenotypes in 13 Italian patients. Neurol Sci. 2017;38:563–70. doi: 10.1007/s10072-016-2734-3. [DOI] [PubMed] [Google Scholar]
- 25.Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K, Bosman E, Steffes G, Steel KP, Simrick S, Basson MA, Illingworth E, Scambler PJ. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. 2009;119:3301–10. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks BP, Zein WM, Thompson AH, Mokhtarzadeh M, Doherty DA, Parisi M, Glass IA, Malicdan MC, Vilboux T, Vemulapalli M, Mullikin JC, Gahl WA, Gunay-Aygun M. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology. 2018;125:1937–52. doi: 10.1016/j.ophtha.2018.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh M, Ide S, Iwasaki Y, Saito T, Narita K, Dai H, Yamakura S, Furue T, Kitayama H, Maeda K, Takahashi E, Matsui K, Goto YI, Takeda S, Arima M. Arima syndrome caused by CEP290 specific variant and accompanied with pathological cilium; clinical comparison with Joubert syndrome and its related diseases. Brain Dev. 2018;40:259–67. doi: 10.1016/j.braindev.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Martin-Merida I, Aguilera-Garcia D, Fernandez-San Jose P, Blanco-Kelly F, Zurita O, Almoguera B, Garcia-Sandoval B, Avila-Fernandez A, Arteche A, Minguez P, Carballo M, Corton M, Ayuso C. Toward the Mutational Landscape of Autosomal Dominant Retinitis Pigmentosa: A Comprehensive Analysis of 258 Spanish Families. Invest Ophthalmol Vis Sci. 2018;59:2345–54. doi: 10.1167/iovs.18-23854. [DOI] [PubMed] [Google Scholar]
- 29.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2019 doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–5. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 31.Lee ST, Han J. Missed Heterozygous Deletion in Study of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2019 doi: 10.1001/jamaophthalmol.2019.3755. [DOI] [PubMed] [Google Scholar]
- 32.Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–24. doi: 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han IC, Critser DB, Stone EM. Swept-Source OCT of a Macular Coloboma in NMNAT1-Leber Congenital Amaurosis. Ophthalmol Retina. 2018;2:1040. doi: 10.1016/j.oret.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012;75:88–97. doi: 10.1016/j.visres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coussa RG, Otto EA, Gee HY, Arthurs P, Ren H, Lopez I, Keser V, Fu Q, Faingold R, Khan A, Schwartzentruber J, Majewski J, Hildebrandt F, Koenekoop RK. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet. 2013;84:150–9. doi: 10.1111/cge.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, Xia F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE, Yang Y, Lupski JR. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017;376:21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivolta C, Berson EL, Dryja TP. Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum Mutat. 2001;18:488–98. doi: 10.1002/humu.1226. [DOI] [PubMed] [Google Scholar]
- 38.Chen S, Wang QL, Xu S, Liu I, Li LY, Wang Y, Zack DJ. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum Mol Genet. 2002;11:873–84. doi: 10.1093/hmg/11.8.873. [DOI] [PubMed] [Google Scholar]
- 39.Tran NM, Zhang A, Zhang X, Huecker JB, Hennig AK, Chen S. Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS Genet. 2014;10:e100411. doi: 10.1371/journal.pgen.1004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hull S, Arno G, Plagnol V, Chamney S, Russell-Eggitt I, Thompson D, Ramsden SC, Black GC, Robson A, Holder GE, Moore AT, Webster AR. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2014;55:6934–44. doi: 10.1167/iovs.14-14715. [DOI] [PubMed] [Google Scholar]
- 41.Ibrahim MT, Alarcon-Martinez T, Lopez I, Fajardo N, Chiang J, Koenekoop RK. A complete, homozygous CRX deletion causing nullizygosity is a new genetic mechanism for Leber congenital amaurosis. Sci Rep. 2018;8:5034. doi: 10.1038/s41598-018-22704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swaroop A, Wang QL, Wu W, Cook J, Coats C, Xu S, Chen S, Zack DJ, Sieving PA. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8:299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- 43.Chapi M, Sabbaghi H, Suri F, Alehabib E, Rahimi-Aliabadi S, Jamali F, Jamshidi J, Emamalizadeh B, Darvish H, Mirrahimi M, Ahmadieh H, Daftarian N. Incomplete penetrance of CRX gene for autosomal dominant form of cone-rod dystrophy. Ophthalmic Genet. 2019;8:266–8. doi: 10.1080/13816810.2019.1622023. [DOI] [PubMed] [Google Scholar]
- 44.Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, Braun TA, Mullins RF, Scheetz TE, Sheffield VC, Tucker BA. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology. 2017;124:1314–31. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, Lamb JA, Perveen R, Hall G, Newman WG, Bishop PN, Roberts SA, Leach R, Tearle R, Bayliss S, Ramsden SC, Nemeth AH, Black GC. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123:1143–50. doi: 10.1016/j.ophtha.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Need AC, Shashi V, Schoch K, Petrovski S, Goldstein DB. The importance of dynamic re-analysis in diagnostic whole exome sequencing. J Med Genet. 2017;54:155–6. doi: 10.1136/jmedgenet-2016-104306. [DOI] [PubMed] [Google Scholar]
- 47.Shashi V, Schoch K, Spillmann R, Cope H, Tan QK, Walley N, Pena L, McConkie-Rosell A, Jiang YH, Stong N, Need AC, Goldstein DB. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet Med. 2019;21:161–72. doi: 10.1038/s41436-018-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]