

Supplemental Digital Content is Available in the Text.
Key Words: 2DR, dolutegravir, integrase strand transfer inhibitor, nucleoside reverse transcriptase inhibitor, treatment-naive
Background:
The 2-drug regimen dolutegravir + lamivudine was noninferior to dolutegravir + tenofovir disoproxil fumarate/emtricitabine in achieving HIV-1 RNA <50 copies/mL in treatment-naive adults in the 48-week primary analysis of the GEMINI trials. We present results from the prespecified 96-week secondary analyses.
Setting:
One hundred eighty-seven centers in 21 countries.
Methods:
GEMINI-1 and GEMINI-2 are identical, double-blind phase III studies. Participants with screening HIV-1 RNA ≤500,000 copies/mL were randomized 1:1 to once-daily dolutegravir + lamivudine or dolutegravir + tenofovir disoproxil fumarate/emtricitabine.
Results:
At week 96, dolutegravir + lamivudine (N = 716) was noninferior to dolutegravir + tenofovir disoproxil fumarate/emtricitabine (N = 717) in achieving HIV-1 RNA <50 copies/mL (Snapshot algorithm; −10% noninferiority margin) in the pooled analysis (proportion of responders, 86.0% vs 89.5%, respectively; adjusted treatment difference [95% CI], −3.4% [−6.7 to 0.0007]), GEMINI-1 (−4.9% [−9.8 to 0.03]), and GEMINI-2 (−1.8% [−6.4 to 2.7]). Proportions of participants in the HIV-1 RNA ≥50 copies/mL Snapshot category were largely unchanged from week 48 to 96. Eleven participants taking dolutegravir + lamivudine and 7 taking dolutegravir + tenofovir disoproxil fumarate/emtricitabine met confirmed virologic withdrawal criteria through week 96; none had treatment-emergent resistance mutations. Dolutegravir + lamivudine had a lower rate of drug-related adverse events than dolutegravir + tenofovir disoproxil fumarate/emtricitabine (19.6% vs 25.0%; relative risk ratio, 0.78; 95% CI: 0.64 to 0.95). Renal and bone biomarker changes favored dolutegravir + lamivudine.
Conclusions:
Consistent with 48-week data, dolutegravir + lamivudine demonstrated long-term, noninferior efficacy vs dolutegravir + tenofovir disoproxil fumarate/emtricitabine without increased risk of treatment-emergent resistance, supporting its use in treatment-naive HIV-1–infected individuals.
INTRODUCTION
Two-drug regimens (2DRs) can potentially reduce long-term cumulative drug exposure and decrease treatment-associated costs for HIV-1–infected individuals, who require lifelong therapy.1 The core antiretroviral agent in a 2DR must have high potency and a high barrier to resistance.1 As such, early studies investigating 2DRs as initial or maintenance therapy for HIV infection evaluated the pairing of the potent, well-tolerated nucleoside reverse transcriptase inhibitor (NRTI) lamivudine with pharmacologically boosted protease inhibitors (PIs), which have a high barrier to resistance.2–6 Although noninferior efficacy was shown against 3-drug regimens (3DRs), PIs are associated with adverse metabolic effects, long-term toxicities, and drug–drug interactions, limiting their appeal as components of lifelong therapy.7,8 Thus, a need remains for well-tolerated, potent 2DRs with a high barrier to resistance.
The integrase strand transfer inhibitor (INSTI) dolutegravir has a high barrier to resistance, making it a well-suited candidate for inclusion in a 2DR,9 particularly when paired with lamivudine,10 as previously observed.11,12 In primary week 48 analyses of the 2 phase III studies GEMINI-1 and GEMINI-2 in treatment-naive adults, dolutegravir + lamivudine was noninferior to dolutegravir + tenofovir disoproxil fumarate/emtricitabine in achieving HIV-1 RNA <50 copies/mL according to the US Food and Drug Administration (FDA) Snapshot algorithm.13 Importantly, resistance mutations associated with INSTIs or NRTIs did not emerge in the few participants who had virologic failure. These data led to the approval of the fixed-dose combination of dolutegravir/lamivudine as a once-daily, single-tablet 2DR by the FDA and the European Medicines Agency.14,15 In addition, the 2019 update to the US Department of Health and Human Services treatment guidelines for HIV-1 infection supports the use of dolutegravir + lamivudine as initial treatment in patients for whom abacavir, tenofovir disoproxil fumarate, or tenofovir alafenamide either cannot be used or are not optimal.16 European AIDS Clinical Society guidelines also indicate that when preferred regimens are not feasible or available,17 dolutegravir + lamivudine can be used. Both guidelines indicated the need for longer-term data to support the use of dolutegravir + lamivudine in a broader patient population. Here, we report longer-term results from the GEMINI-1 and GEMINI-2 planned secondary analyses at 96 weeks.
METHODS
Study Design
GEMINI-1 (NCT02831673) and GEMINI-2 (NCT02831764) are ongoing, identically designed, phase III, randomized, double-blind, noninferiority studies conducted at 187 centers in 21 countries. This report describes results through the final visit of the double-blind randomized phase at week 96. Protocols for GEMINI-1 and GEMINI-2 are available at https://www.viiv-clinicalstudyregister.com/study/204861#ps and https://www.viiv-clinicalstudyregister.com/study/205543#ps, respectively. Methods, including information regarding ethical compliance, have previously been described.13
Participants and Study Treatment
Eligible participants were aged ≥18 years with HIV-1 infection, ≤10 days of previous antiretroviral therapy (ART), and screening plasma HIV-1 RNA 1000 to 500,000 copies/mL. Women of reproductive potential were eligible if they were not pregnant or lactating and using highly effective contraception (defined by study protocol). Exclusion criteria included presence of pre-existing major viral resistance mutations to NRTIs, non-NRTIs, or PIs18 and active Centers for Disease Control and Prevention stage 3 HIV disease19 (except for cutaneous Kaposi sarcoma and CD4+ cell count <200 cells/mm3). Participants were assessed for eligibility during a screening period of ≤35 days. Eligible participants were randomized 1:1 to receive a once-daily 2DR of dolutegravir 50 mg plus lamivudine 300 mg or a once-daily 3DR of dolutegravir 50 mg plus tenofovir disoproxil fumarate 300 mg/emtricitabine 200 mg. Participants were stratified by screening HIV-1 RNA (≤100,000 or >100,000 copies/mL) and screening CD4+ cell count (≤200 or >200 cells/mm3) and treated in a double-blind randomized phase from day 1 to week 96 during which lamivudine and tenofovir disoproxil fumarate/emtricitabine tablets were provided overencapsulated to visually match each other, followed by an open-label randomized phase from week 96 to 148.
Assessments
Study visits were planned at baseline (day 1) and weeks 4, 8, 12, 16, 24, 36, 48, and every 12 weeks thereafter until week 144. Plasma for quantitative HIV-1 RNA analysis and storage was collected at all visits and quantitated using the Abbott RealTime HIV-1 assay (lower limit of quantitation, 40 copies/mL; Abbott Molecular, Des Plaines, IL). For participants with HIV-1 RNA ≥50 copies/mL at weeks 24, 48, or 96, a retest was conducted at weeks 28, 52, or 100, respectively. Participants met confirmed virologic withdrawal (CVW) criteria if a second and consecutive HIV-1 RNA value met any of the following definitions: decrease from baseline in HIV-1 RNA <1 log10 copies/mL, unless HIV-1 RNA <200 copies/mL, by week 12; confirmed plasma HIV-1 RNA ≥200 copies/mL at or after week 24; or HIV-1 RNA ≥200 copies/mL after confirmed consecutive HIV-1 RNA <200 copies/mL. These participants were discontinued from the study, and plasma samples from day 1 and the initial elevated, and therefore suspected, viral load were used for genotypic and phenotypic resistance tests (Monogram Biosciences, San Francisco, CA).
Adverse events (AEs), concomitant medications, and symptom-directed physical examinations were assessed at all study visits. AEs were coded using MedDRA, version 21.0. The maximum toxicity of AEs was graded using guidelines from the Division of AIDS, version 2.0.20 Testing for fasting lipids and glucose, urinalysis, and renal and bone biomarkers was conducted at baseline and weeks 24, 48, 96, and 144. Renal biomarkers included estimated glomerular filtration rate (based on Chronic Kidney Disease Epidemiology Collaboration equation [CKD-EPI] creatinine and CKD-EPI cystatin C), serum creatinine, urine protein/creatinine ratio, urine retinol-binding protein/creatinine ratio, and urine beta-2 microglobulin/creatinine ratio. Bone biomarkers included serum bone-specific alkaline phosphatase, serum osteocalcin, serum procollagen 1 N-terminal propeptide, and serum type 1 collagen C-telopeptide. The Columbia Suicide-Severity Rating Scale was used to monitor suicidal ideation and behavior starting from day 1.
Outcomes
The primary endpoint of each GEMINI study was the proportion of participants with plasma HIV-1 RNA <50 copies/mL at week 48 using the FDA Snapshot algorithm21 in the intention-to-treat–exposed (ITT-E) population. Endpoints for the week 96 secondary analysis included proportion of participants with HIV-1 RNA <50 copies/mL at week 96, change in CD4+ cell count from baseline, incidence of emergent mutations conferring genotypic and/or phenotypic resistance to dolutegravir + lamivudine or dolutegravir + tenofovir disoproxil fumarate/emtricitabine in participants meeting criteria for CVW, and proportion of participants with HIV-1 RNA <50 copies/mL in participant subgroups defined by demographic and baseline disease characteristics, including plasma viral load and CD4+ cell count. Safety endpoints included incidence and severity of AEs and proportion of participants who discontinued treatment because of AEs. Renal and bone biomarkers and lipids were monitored by assessing changes from baseline at week 96 (see Supplemental Digital Content 1, http://links.lww.com/QAI/B418 for complete list of secondary endpoints).
Statistical Analysis
All randomized participants who received ≥1 dose of study medication were included in the ITT-E population, which was used for the efficacy analyses. The safety population included all participants who received ≥1 dose of study medication and was analyzed according to actual treatment received. Week 96 secondary analyses of the individual studies as well as a pooled analysis were prespecified. The proportion of participants with HIV-1 RNA <50 copies/mL at week 96 was analyzed using a Cochran–Mantel–Haenszel test stratified by baseline plasma HIV-1 RNA (≤100,000 vs >100,000 copies/mL), baseline CD4+ cell count (≤200 vs >200 cells/mm3), and individual study (GEMINI-1 vs GEMINI-2). Baseline characteristics, response rates by study visit or participant subgroup (using Snapshot algorithm), and AEs were summarized using descriptive statistics. Treatment-related discontinuation equals failure (TRDF) was a preplanned analysis at week 96 that accounted for CVW, withdrawal due to lack of efficacy, withdrawal due to treatment-related AE, and participants who met protocol-defined stopping criteria. The proportion of participants without TRDF was estimated using the Kaplan–Meier nonparametric method. Change from baseline at week 96 in CD4+ cell count was analyzed using a mixed-effect repeated-measures model adjusting for study, treatment, visit, baseline plasma HIV-1 RNA, baseline CD4+ cell count, treatment-by-visit interaction, and baseline CD4+ cell count-by-visit interaction, with visit as the repeated factor. Changes in body weight and body mass index (BMI) from baseline to week 96 were summarized, and ad hoc summaries by sex are also presented. Change from baseline in lipids was analyzed using repeated-measures model adjusting for study, treatment, visit, baseline plasma HIV-1 RNA, baseline CD4+ cell count, age, baseline value, treatment-by-visit interaction, and baseline value-by-visit interaction, with visit as the repeated factor.
RESULTS
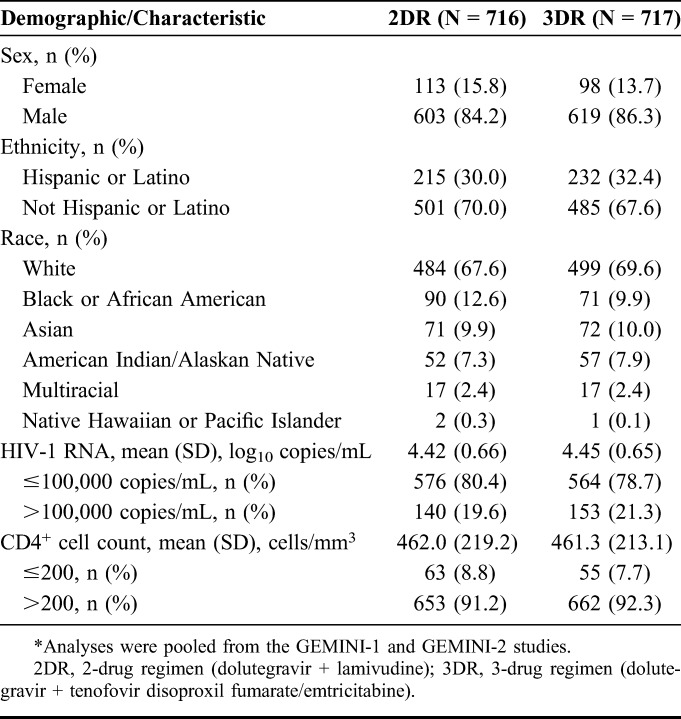
In GEMINI-1 and GEMINI-2, 1433 participants were randomized and received ≥1 dose of study medication (dolutegravir + lamivudine, N = 716; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, N = 717; see Figure, Supplemental Digital Content 2, http://links.lww.com/QAI/B418). As previously reported, baseline characteristics were well balanced between treatment groups.13 The majority of participants were male (85.3%; n = 1222) and white (68.6%; n = 983; Table 1). Overall, 20.4% (n = 293) of participants had baseline HIV-1 RNA >100,000 copies/mL, and 8.2% (n = 118) had CD4+ cell count ≤200 cells/mm.3
TABLE 1.
Demographics and Clinical Baseline Characteristics in the Pooled ITT-E Population From GEMINI-1 and GEMINI-2*
At the cutoff date for the 96-week analysis (April 4, 2019), 85.5% (n = 612) in the dolutegravir + lamivudine group and 88.8% (n = 637) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group remained on study. The most common reasons for study discontinuation were withdrawal of consent (2DR, 3.5% [n = 25]; 3DR, 2.9% [n = 21]), AE (2DR, 3.1% [n = 22]; 3DR, 2.2% [n = 16]), and lost to follow-up (2DR, 3.1% [n = 22]; 3DR, 2.0% [n = 14]; see Figure, Supplemental Digital Content 2, http://links.lww.com/QAI/B418).
Analysis of virologic outcomes by visit through week 96 shows a similar proportion of participants with HIV-1 RNA <50 copies/mL in either treatment group at each visit (Fig. 1A). At week 96, 86.0% (616/716) of participants in the dolutegravir + lamivudine group and 89.5% (642/717) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group achieved HIV-1 RNA <50 copies/mL (Snapshot algorithm; pooled analysis of the ITT-E population) for an adjusted treatment difference (95% CI) of −3.4% (−6.7 to 0.0007; Figs. 1B, C). Based on a prespecified −10% noninferiority margin, dolutegravir + lamivudine remained noninferior to dolutegravir + tenofovir disoproxil fumarate/emtricitabine at week 96 because the lower bound of the 95% CI for the adjusted treatment difference was greater than −10%. In GEMINI-1, HIV-1 RNA <50 copies/mL was achieved in 84.3% (300/356) of participants in the dolutegravir + lamivudine group and 89.4% (320/358) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group at week 96 (adjusted treatment difference [95% CI] was −4.9% [−9.8 to 0.03]); the corresponding numbers in GEMINI-2 were 87.8% (316/360) vs 89.7% (322/359; adjusted treatment difference [95% CI], −1.8% [−6.4 to 2.7]; see Table, Supplemental Digital Content 3, http://links.lww.com/QAI/B418). In the week 96 pooled analysis, proportions of participants in the HIV-1 RNA ≥50 copies/mL Snapshot category were 3.1% in the dolutegravir + lamivudine group and 2.0% in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group and were largely unchanged from week 48 to week 96 in both groups; 10.9% and 8.5%, respectively, had no virologic data (Fig. 1B).
FIGURE 1.
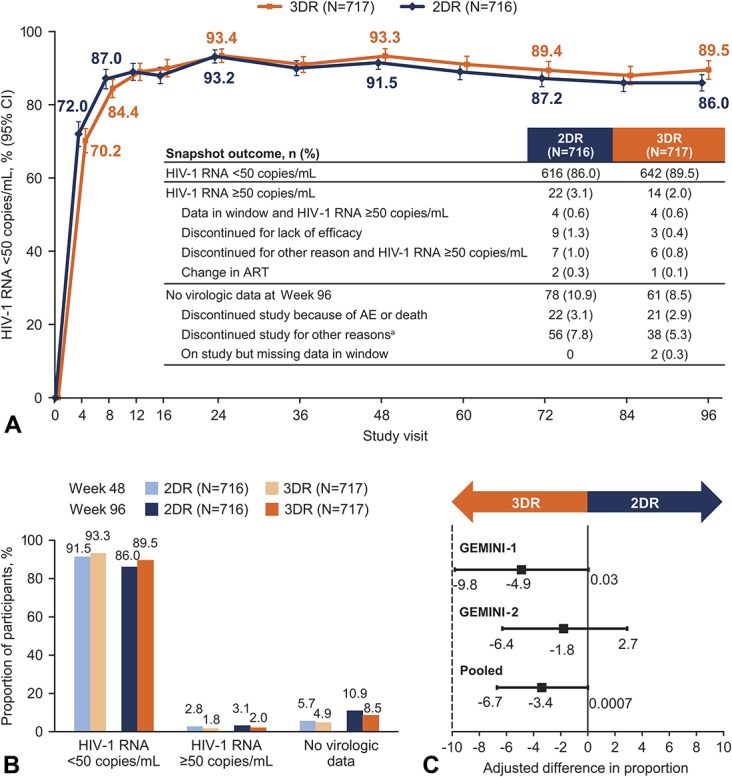
Snapshot analysis of the proportion of participants with plasma HIV-1 RNA <50 copies/mL (A) by visit and (B) at weeks 48 and 96 in the pooled ITT-E population. C, Adjusted treatment differences at week 96 of the Snapshot analysis in the ITT-E populations of the individual trials and the pooled analysis. aOther reasons for discontinuing study included (2DR, n [%] vs 3DR, n [%]) protocol deviation (10 [1.4%] vs 8 [1.1%]), lost to follow-up (18 [2.5%] vs 10 [1.4%]), physician decision (10 [1.4%] vs 4 [0.6%]), withdrew consent (18 [2.5%] vs 15 [2.1%]), and protocol-defined CVW (0 [0%] vs 1 [0.1%]). 2DR, 2-drug regimen (dolutegravir + lamivudine); 3DR, 3-drug regimen (dolutegravir + tenofovir disoproxil fumarate/emtricitabine).
Most Snapshot failures that occurred after week 48 in both groups were discontinuations for nonvirologic or non–treatment-related reasons, including withdrawal of consent, lost to follow-up, protocol deviation, and physician decision (see Table, Supplemental Digital Content 3, http://links.lww.com/QAI/B418); this was most notable in the dolutegravir + lamivudine group in GEMINI-1. In the prespecified TRDF Kaplan–Meier analysis at week 96, 96.4% of participants in the dolutegravir + lamivudine group and 96.2% in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group did not discontinue for treatment-related reasons (unadjusted treatment difference [95% CI], 0.2% [−1.8 to 2.2]). In the ITT-E population, adjusted mean change from baseline to week 96 in CD4+ cell count was 269.0 cells/mm3 in the dolutegravir + lamivudine group and 259.2 cells/mm3 in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group.
Few participants met the prespecified criteria for CVW through week 96, with 11 participants (1.5%) in the dolutegravir + lamivudine group and 7 (1.0%) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group (see Table, Supplemental Digital Content 4, http://links.lww.com/QAI/B418). Viral loads ranged from 206 to 87,794 copies/mL at the visit where CVW criteria were met and from <50 to 3011 copies/mL at the follow-up withdrawal visits (in those with a separate withdrawal visit). Of these, 5 participants in the dolutegravir + lamivudine group and 2 in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group met CVW criteria between weeks 48 and 96 and 1 participant in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group met CVW criteria at week 12 but was not reported in the week 48 analysis because of a laboratory reporting error. The latter participant remained in the study and had HIV-1 RNA <50 copies/mL at week 96. No INSTI or NRTI resistance mutations emerged during treatment among any participants who met CVW criteria.
Proportion of participants with HIV-1 RNA <50 copies/mL in key subpopulations including race, sex, and age was generally comparable across treatment groups, consistent with the overall results (see Figure, Supplemental Digital Content 5, http://links.lww.com/QAI/B418). In participants with baseline HIV-1 RNA >100,000 copies/mL, 83.6% (117/140) and 86.3% (132/153) in the dolutegravir + lamivudine and dolutegravir + tenofovir disoproxil fumarate/emtricitabine groups, respectively, achieved HIV-1 RNA <50 copies/mL at week 96 (Fig. 2); the corresponding proportions were 86.6% (499/576) and 90.4% (510/564) for those with baseline HIV-1 RNA ≤100,000 copies/mL and 87.7% (573/653) and 89.7% (594/662) for those with baseline CD4+ cell count >200 cells/mm.3 Among participants with baseline CD4+ cell count ≤200 cells/mm,3 68.3% (43/63) in the dolutegravir + lamivudine group and 87.3% (48/55) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group achieved HIV-1 RNA <50 copies/mL at week 96. Of note, the higher rate of Snapshot virologic nonresponse in the baseline CD4+ cell count ≤200 cells/mm3 group on dolutegravir + lamivudine was primarily due to non–treatment-related reasons (see Table, Supplemental Digital Content 6, http://links.lww.com/QAI/B418). Consequently, there was little difference in the TRDF analysis at week 96 where rates of participants without treatment-related discontinuations with baseline CD4+ cell count ≤200 cells/mm3 were 92.6% in the dolutegravir + lamivudine group and 96.2% in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group. Furthermore, the number of participants meeting CVW criteria in this group was 3 and 2 in the 2DR and 3DR groups, respectively (see Table, Supplemental Digital Content 4, http://links.lww.com/QAI/B418). Although participants with screening HIV-1 RNA >500,000 copies/mL were excluded from the study, 2% of participants had HIV-1 RNA >500,000 copies/mL at the baseline visit (which was after the screening visit). Of these, 69.2% (9/13) in the dolutegravir + lamivudine group and 80.0% (12/15) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group achieved HIV-1 RNA <50 copies/mL at week 96; most Snapshot failures in both groups were due to discontinuations for nonvirologic or non–treatment-related reasons (see Table, Supplemental Digital Content 7, http://links.lww.com/QAI/B418).
FIGURE 2.
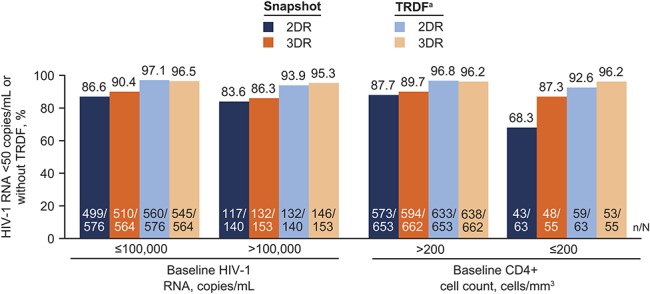
Snapshot and TRDF analyses of the proportion of participants with HIV-1 RNA <50 copies/mL or without TRDF at week 96 by baseline viral load and CD4+ cell count in the pooled ITT-E population from GEMINI-1 and GEMINI-2. 2DR, 2-drug regimen (dolutegravir + lamivudine); 3DR, 3-drug regimen (dolutegravir + tenofovir disoproxil fumarate/emtricitabine). aTRDF was a preplanned analysis at week 96. Percentages estimated from the TRDF Kaplan–Meier analysis.
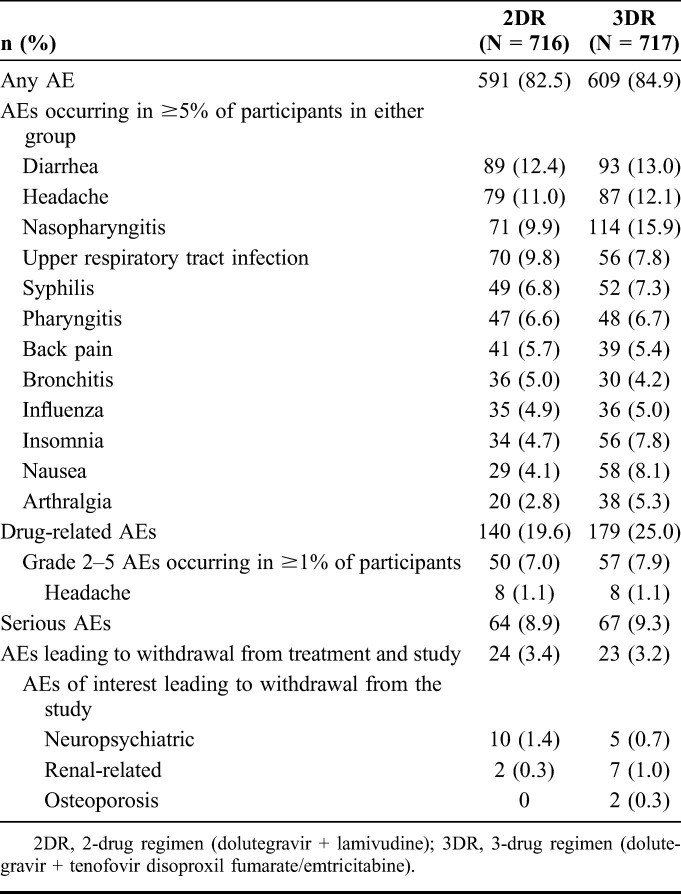
Through week 96, overall AE profiles were similar between treatment groups (relative risk [95% CI], 0.97 [0.93 to 1.02]). Consistent with week 48 results,13 the most common AEs in the pooled safety population were diarrhea, headache, nasopharyngitis, and upper respiratory tract infection (Table 2). Participants in the dolutegravir + lamivudine group had a lower rate of drug-related AEs compared with the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group (19.6% [140/716] vs 25.0% [179/717], respectively, relative risk [95% CI], 0.78 [0.64 to 0.95]); these differences were driven primarily by larger numbers of participants reporting drug-related grade 1 events, notably nausea, in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group. Rates of SAEs were similar between groups: dolutegravir + lamivudine, 8.9% (n = 64); dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 9.3% (n = 67). Five participants in the dolutegravir + lamivudine group (suicidal ideation, n = 2; psychotic disorder, n = 1; substance-induced psychotic disorder, n = 1; and hepatotoxicity, n = 1) and 4 in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group (suicidal ideation, n = 1; suicide attempt, n = 1; cholelithiasis, n = 1; and rhabdomyolysis, n = 1) experienced drug-related SAEs. Three fatal AEs occurred in the dolutegravir + lamivudine group, which were considered unrelated to study treatment (acute myocardial infarction, n = 1; Burkitt lymphoma, n = 1; and coronary artery disease, n = 1). AEs leading to withdrawal were reported in 3.4% of participants in the dolutegravir + lamivudine group (n = 24) and 3.2% in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group (n = 23; Table 2). Increased weight was reported as an AE in 1.8% (n = 13) of participants in the dolutegravir + lamivudine group and 1.4% (n = 10) in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group. Overall mean (SD) change in weight from baseline was 3.1 (5.7) kg in the dolutegravir + lamivudine group and 2.1 (7.4) kg in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group, and mean change in BMI was 1.04 and 0.67 kg/m2, respectively. Few participants with normal BMI at baseline became obese at week 96 (dolutegravir + lamivudine, 3; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 4; see Figure, Supplemental Digital Content 8, http://links.lww.com/QAI/B418). Mean change in weight from baseline was comparable between groups for female (dolutegravir + lamivudine, 1.50 kg; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 1.56 kg) and male participants (dolutegravir + lamivudine, 3.43 kg; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 2.18 kg). Similarly, mean change in BMI from baseline to week 96 was also comparable between groups for female (dolutegravir + lamivudine, 0.62 kg/m2; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 0.60 kg/m2) and male participants (dolutegravir + lamivudine, 1.11 kg/m2; dolutegravir + tenofovir disoproxil fumarate/emtricitabine, 0.69 kg/m2).
TABLE 2.
Summary of AEs in the Pooled Safety Population From GEMINI-1 and GEMINI-2
Overall, 6 pregnancies were reported on study, 2 since the week 48 analysis. Two pregnancies resulted in live births of healthy infants (1 at 39 weeks of gestation and 1 unknown gestation duration), 2 resulted in spontaneous abortions at 7 weeks and 4–5 weeks of gestation, and 2 resulted in elective abortions, both at 6 weeks of gestation. No apparent congenital abnormalities were reported.
At week 96, changes in renal biomarkers significantly favored dolutegravir + lamivudine (Figs. 3A, B). Bone turnover biomarkers also favored dolutegravir + lamivudine, with significant increases observed with dolutegravir + tenofovir disoproxil fumarate/emtricitabine vs dolutegravir + lamivudine for all biomarkers (Fig. 3C). There were also fewer renal function–related AEs (2DR, n = 2; 3DR, n = 7). There were no osteoporosis AEs with the 2DR and 2 osteoporosis AEs with the 3DR that led to treatment discontinuation (Table 2). Changes in lipid parameters at week 96 were consistent with week 48 results.13 Total cholesterol, low-density lipoprotein cholesterol, and triglycerides increased in the dolutegravir + lamivudine group and decreased in the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group (see Figure, Supplemental Digital Content 9, http://links.lww.com/QAI/B418). Importantly, in both groups, increases in high-density lipoprotein (HDL) cholesterol with resultant decreases in total cholesterol/HDL cholesterol ratio were observed. Significant differences between treatment groups in adjusted mean change from baseline were observed for all lipid parameters. Forty participants (5.6%) in the 2DR group and 16 (2.2%) in the 3DR group initiated lipid-modifying agents after baseline.
FIGURE 3.
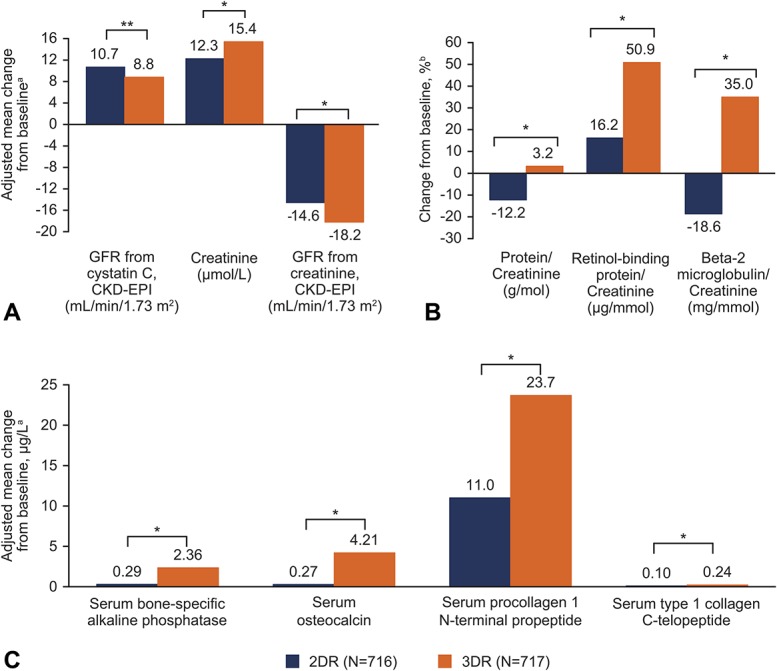
Profiles of renal and bone biomarkers. Adjusted mean change from baseline in (A) serum or plasma renal biomarkers and (B) ratios of urine renal biomarkers at week 96. C, Adjusted mean change from baseline in bone turnover biomarkers at week 96. CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration equation; 2DR, 2-drug regimen (dolutegravir + lamivudine); 3DR, 3-drug regimen (dolutegravir + tenofovir disoproxil fumarate/emtricitabine); GFR, glomerular filtration rate. aThe 96-week analysis used mixed-effect model repeat measurement. Mean change from baseline adjusted for study, treatment, visit, baseline viral load, baseline CD4+ cell count, age, sex, race, baseline biomarker value, treatment-by-visit interaction, and baseline biomarker value-by-visit interaction. For renal biomarkers, mean change from baseline was also adjusted for presence of diabetes and presence of hypertension. For bone biomarkers, mean change was also adjusted for BMI, smoking status, and current vitamin D use. No assumptions were made about the correlations between participant readings of biomarkers (the correlation matrix for within-participant errors was unstructured). bEstimated from geometric means ratio for baseline and week 96. *P < 0.001. **P < 0.005.
At week 96, changes from baseline in EQ-5D-5L utility score, visual analog scale, and health state utility score were similar between groups (see Table, Supplemental Digital Content 10, http://links.lww.com/QAI/B418).
DISCUSSION
The week 96 analysis of the GEMINI studies demonstrates the long-term virologic efficacy of the 2DR dolutegravir + lamivudine, as evidenced by its continued noninferiority vs dolutegravir + tenofovir disoproxil fumarate/emtricitabine, as initial therapy for HIV-1–infected individuals. High proportions of participants had HIV-1 RNA <50 copies/mL at week 96 in both the dolutegravir + lamivudine and dolutegravir + tenofovir disoproxil fumarate/emtricitabine groups, and response rates were similar between groups regardless of baseline viral load (including those with baseline HIV-1 RNA >500,000 copies/mL). Importantly, the proportion of participants with HIV-1 RNA ≥50 copies/mL at week 96 remained low and similar between treatment groups and from week 48 to 96. Most Snapshot failures that occurred after week 48 in both groups were discontinuations for nonvirologic or non–treatment-related reasons.
Consistent with the 48-week results, a lower rate of participants with baseline CD4+ cell count ≤200 cells/mm3 had HIV-1 RNA <50 copies/mL at week 96 in the dolutegravir + lamivudine group. Interpretation of this finding is limited by the relatively small number of participants in this subgroup (n = 118; 8% of the pooled ITT-E population). Nevertheless, most of the reasons for the lower response rate in this 2DR subset were not related to lack of efficacy or adverse drug reactions (see Table, Supplemental Digital Content 6, http://links.lww.com/QAI/B418). This is reflected in the results of the TRDF analysis, which show a high and similar proportion of participants without treatment-related discontinuations between treatment groups in the baseline CD4+ cell count ≤200 cells/mm3 subgroup.
One important question that has arisen during the development of dolutegravir-based 2DRs is the durability of the high barrier to resistance compared with dolutegravir-based 3DRs. This week 96 analysis has addressed this question and demonstrated that dolutegravir + lamivudine sustains a high barrier to resistance, with low numbers of participants experiencing CVW and zero emergence of resistance to INSTIs or NRTIs in either group. The lack of resistance development with the 2DR of dolutegravir + lamivudine over a prolonged treatment period is a very important finding because preservation of future treatment options for people with HIV, who can conceivably be on therapy for decades, is critical.
Safety results from the week 96 analysis of GEMINI-1 and GEMINI-2 were consistent with week 48 results.13 Overall, there were few treatment-related discontinuations and a lower rate of drug-related AEs with the 2DR vs the 3DR. No new safety signals were observed between weeks 48 and 96. Although mean weight increased in both groups from baseline, no participants discontinued the study because of weight-related AEs. Changes in lipid parameters at week 96 relative to baseline generally were in favor of the dolutegravir + tenofovir disoproxil fumarate/emtricitabine group, consistent with the known effect of tenofovir disoproxil fumarate on cholesterol.22 The total cholesterol/HDL ratio, which is often used to estimate long-term cardiovascular risk, decreased in both groups over 96 weeks, although the difference between groups significantly favored the tenofovir disoproxil fumarate–containing regimen. Notably, a greater proportion of participants initiated lipid-lowering agents in the 2DR group than in the 3DR group. The favorable effects on renal and bone biomarkers observed for dolutegravir + lamivudine vs dolutegravir + tenofovir disoproxil fumarate/emtricitabine in the week 48 analysis were maintained through week 96. Consistent with these trends, there were fewer renal-related and osteoporosis AEs with the 2DR that led to treatment discontinuation. These treatment group differences might be attributed to the known effects of tenofovir disoproxil fumarate on renal and bone health.23,24 Tenofovir alafenamide may have fewer renal and bone toxicities than tenofovir disoproxil fumarate, but due to consideration of standard of care and availability in participating countries at the time of study setup, the GEMINI trials used tenofovir disoproxil fumarate in the 3DR.25
Limitations of the GEMINI trials include the demographics of the study population, which was predominantly white (69%), male (85%), and aged <50 years at enrollment (90%), and may limit the generalizability of the results. However, virologic efficacy in key subpopulations including race, sex, and age was generally comparable across treatment groups. As described above, a low number of participants were enrolled with baseline CD4+ cell count ≤200 cells/mm,3 limiting the interpretability of the results in this population with advanced HIV disease. Notably, there were no restrictions based on baseline viral load or CD4+ cell count in the approvals from the FDA and the European Medicines Agency.14,15 Few participants had very high viral loads (HIV-1 RNA >500,000 copies/mL) at treatment initiation since participants with screening HIV-1 RNA >500,000 copies/mL were excluded from the study. Participants with evidence of hepatitis B virus infection (since lamivudine monotherapy is generally not considered an adequate therapy for hepatitis B) and those with any major drug-resistance mutations were also excluded.
GEMINI-1 and GEMINI-2 are the largest phase III trials of a 2DR in ART-naive individuals with HIV-1 infection, which resulted in high precision of the noninferiority analysis. Results through 96 weeks demonstrate long-term durability with continued absence of treatment-emergent resistance and sustained noninferiority of dolutegravir + lamivudine compared with a standard and potent 3DR of dolutegravir + tenofovir disoproxil fumarate/emtricitabine. In conclusion, the week 96 results of the GEMINI-1 and GEMINI-2 trials provide further support for use of this 2DR as a treatment option for ART-naive people with HIV-1 infection.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the study participants; their families and caregivers; investigators (see list of investigators in Supplemental Digital Content 11, http://links.lww.com/QAI/B418) and site staff who participated in the study; and the ViiV Healthcare, GlaxoSmithKline, Pharmaceutical Product Development, and Parexel study team members. Editorial assistance was provided under the direction of the authors by Jonathan Morgan, PhD, CMPP, and Jennifer Rossi, MA, ELS, MedThink SciCom, and was funded by ViiV Healthcare.
Footnotes
Supported by ViiV Healthcare.
Presented in part at the 10th IAS Conference on HIV Science; July 21–24, 2019; Mexico City, Mexico; Slides WEAB0404LB.
P.C. has served on advisory boards for GlaxoSmithKline (GSK), ViiV Healthcare, and Merck; served as an investigator for Abbott, Gilead, ViiV Healthcare, GSK, Merck, and Richmond; and has received honoraria for his speaking or chairing engagements from Abbott, GSK, Gilead, Merck, and ViiV Healthcare. J.S.M. has received lecture fees, sponsorship, and honoraria from Gilead, Stendhal, AbbVie, ViiV Healthcare, Janssen, and Merck Sharp & Dohme (MSD; all before 2019). J.R.A. has received advisory fees, speaker fees, and grant support from ViiV Healthcare, Janssen, Gilead, MSD, Alexa, and Teva. A.A. has served as a paid consultant to Gilead, Janssen, Merck, and ViiV Healthcare and received research funding from Gilead, Janssen, and ViiV Healthcare. A.E.C. has received advisory fees from GSK, ViiV Healthcare, and Gilead; conference sponsorship from Gilead and Janssen; and speaker travel fees from GSK. C.-C.H. has received honoraria for speaking at educational events or consulting from AbbVie, Bristol-Myers Squibb (BMS), Gilead, Janssen, and ViiV Healthcare and has received research funding from BMS, Janssen, Gilead, Merck, and ViiV Healthcare. J.K.R. has received grant/research support from Gilead; served as a consultant/advisor to Abbott, AbbVie, Bionor, Gilead, Hexal, Janssen, Merck, and ViiV Healthcare; and was a speaker at educational events for AbbVie, Gilead, Janssen, and Merck. P.-M.G. has received grants from BMS and Janssen and has received honoraria and consulting fees from Gilead, ViiV Healthcare, and AbbVie. J.S., C.Y.M., M.U., A.R.T., K.A.P., B.W., M.G., M.A., J.v.W., and K.Y.S. are employees of ViiV Healthcare and own stock in GSK. R.U. and D.B. are employees of GSK and own stock in GSK. The remaining author has no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Back D. 2-Drug regimens in HIV treatment: pharmacological considerations. Germs. 2017;7:113–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arribas JR, Girard PM, Landman R, et al. Dual treatment with lopinavir-ritonavir plus lamivudine versus triple treatment with lopinavir-ritonavir plus lamivudine or emtricitabine and a second nucleos(t)ide reverse transcriptase inhibitor for maintenance of HIV-1 viral suppression (OLE): a randomised, open-label, non-inferiority trial. Lancet Infect Dis. 2015;15:785–792. [DOI] [PubMed] [Google Scholar]
- 3.Cahn P, Andrade-Villanueva J, Arribas JR, et al. Dual therapy with lopinavir and ritonavir plus lamivudine versus triple therapy with lopinavir and ritonavir plus two nucleoside reverse transcriptase inhibitors in antiretroviral-therapy-naive adults with HIV-1 infection: 48 week results of the randomised, open label, non-inferiority GARDEL trial. Lancet Infect Dis. 2014;14:572–580. [DOI] [PubMed] [Google Scholar]
- 4.Di Giambenedetto S, Fabbiani M, Quiros Roldan E, et al. Treatment simplification to atazanavir/ritonavir + lamivudine versus maintenance of atazanavir/ritonavir + two NRTIs in virologically suppressed HIV-1-infected patients: 48 week results from a randomized trial (ATLAS-M). J Antimicrob Chemother. 2017;72:1163–1171. [DOI] [PubMed] [Google Scholar]
- 5.Raffi F, Babiker AG, Richert L, et al. Ritonavir-boosted darunavir combined with raltegravir or tenofovir-emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomised non-inferiority trial. Lancet. 2014;384:1942–1951. [DOI] [PubMed] [Google Scholar]
- 6.Riddler SA, Haubrich R, DiRienzo AG, et al. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med. 2008;358:2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl). 2015;7:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Molina JA, Pulido F, Di Giambenedetto S, et al. Individual patient data meta-analysis of randomized controlled trials of dual therapy with a boosted PI plus lamivudine for maintenance of virological suppression: GeSIDA study 9717. J Antimicrob Chemother. 2018;73:2927–2935. [DOI] [PubMed] [Google Scholar]
- 9.Brenner BG, Wainberg MA. Clinical benefit of dolutegravir in HIV-1 management related to the high genetic barrier to drug resistance. Virus Res. 2017;239:1–9. [DOI] [PubMed] [Google Scholar]
- 10.Quercia R, Perno CF, Koteff J, et al. Twenty-five years of lamivudine: current and future use for the treatment of HIV-1 infection. J Acquir Immune Defic Syndr. 2018;78:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cahn P, Rolón MJ, Figueroa MI, et al. Dolutegravir-lamivudine as initial therapy in HIV-1 infected, ARV-naive patients, 48-week results of the PADDLE (Pilot Antiretroviral Design with Dolutegravir LamivudinE) study. J Int AIDS Soc. 2017;20:21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taiwo BO, Zheng L, Stefanescu A, et al. ACTG A5353: a pilot study of dolutegravir plus lamivudine for initial treatment of human immunodeficiency virus-1 (HIV-1)-infected participants with HIV-1 RNA <500000 copies/mL. Clin Infect Dis. 2018;66:1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cahn P, Madero JS, Arribas JR, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet. 2019;393:143–155. [DOI] [PubMed] [Google Scholar]
- 14.Dovato [Package Insert]. Research Triangle Park, NC: ViiV Healthcare; 2019. [Google Scholar]
- 15.Dovato [Summary of Product Characteristics]. Uxbridge, United Kingdom: ViiV Healthcare; 2019. [Google Scholar]
- 16.AIDSinfo. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV: US Department of Health and Human Services; Available at: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv/0. Accessed July 10, 2019. [Google Scholar]
- 17.European AIDS Clinical Society. Guidelines Version 9.1. 2018. Available at: http://www.eacsociety.org/files/2018_guidelines-9.1-english.pdf. Accessed January 2, 2020. [Google Scholar]
- 18.Wensing AM, Calvez V, Günthard HF, et al. Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention. Revised Surveillance Case Definition for HIV infection—United States, 2014: US Department of Health and Human Services; Available at: https://www.cdc.gov/mmwr/pdf/rr/rr6303.pdf. Accessed August 5, 2019. [Google Scholar]
- 20.National Institute of Allergy and Infectious Diseases. Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.0. 2014. Available at: https://rsc.niaid.nih.gov/sites/default/files/daids-ae-grading-table-v2-nov2014.pdf. Accessed January 2, 2020. [Google Scholar]
- 21.US Food and Drug Administration. Human Immunodeficiency Virus-1 Infection: Developing Antiretroviral Drugs for Treatment. Guidance for Industry: US Department of Health and Human Services; 2015. Available at: https://www.fda.gov/media/86284/download. Accessed January 2, 2020. [Google Scholar]
- 22.Santos JR, Saumoy M, Curran A, et al. The lipid-lowering effect of tenofovir/emtricitabine: a randomized, crossover, double-blind, placebo-controlled trial. Clin Infect Dis. 2015;61:403–408. [DOI] [PubMed] [Google Scholar]
- 23.Cooper RD, Wiebe N, Smith N, et al. Systematic review and meta-analysis: renal safety of tenofovir disoproxil fumarate in HIV-infected patients. Clin Infect Dis. 2010;51:496–505. [DOI] [PubMed] [Google Scholar]
- 24.Mulligan K, Glidden DV, Anderson PL, et al. Effects of emtricitabine/tenofovir on bone mineral density in HIV-negative persons in a randomized, double-blind, placebo-controlled trial. Clin Infect Dis. 2015;61:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeJesus E, Haas B, Segal-Maurer S, et al. Superior efficacy and improved renal and bone safety after switching from a tenofovir disoproxil fumarate- to a tenofovir alafenamide-based regimen through 96 weeks of treatment. AIDS Res Hum Retroviruses. 2018;34:337–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.