

Abstract
We report the development of a Pd(II)/(±)-MeO-SOX/2,5-dimethylbenzoquinone system that enables unprecedented access to anti-1,3 amino alcohol motifs in good yields (33 substrates, avg. 66% isolated yield, >20:1 dr) and high selectivities (avg. 10:1 dr). Switching ligands to (±)-CF3-SOX using a less bulky quinone oxidant, the kinetic syn-1,3 amino alcohol motif can be accessed in comparable yields and selectivities. Advantages of the stereodivergent nature of this reaction are seen in the synthesis of anti- and syn-1,3-amino alcohol vitamin D3 analogue intermediates in half the steps and higher overall yield to previous routes. Additionally, all eight possible stereoisomers of a chiral diamino alcohol core are generated from two amino acids. Mechanistic studies reveal that the anti-isomer is furnished through concurrent Pd(II)(SOX) catalyzed C—H amination and Pd(0)(SOX)-catalyzed isomerization cycles.
Graphical Abstract
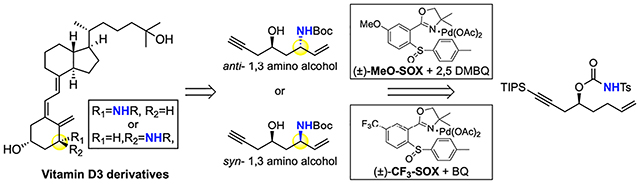
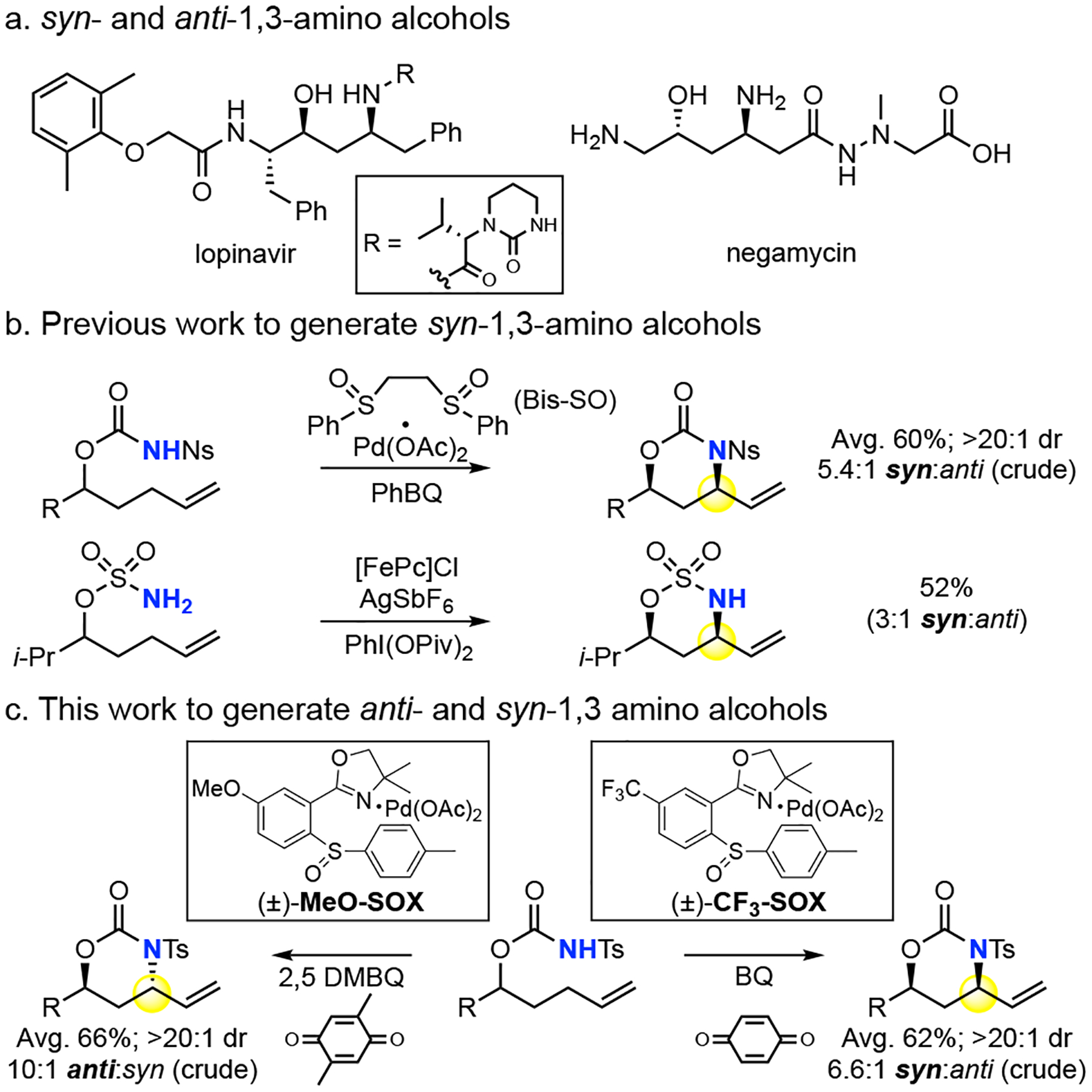
1, 3 amino alcohols are prominent structures in a di-verse range of biologically active natural products and pharmaceuticals. Direct C—H aminations, including metallonitrene systems1 and Pd(II)/bis-sulfoxide catalyzed allylic C—H amination2, are effective for synthe-sizing syn-1,3 amino alcohols with reduced oxidation-state manipulations and synthetic overhead. However, these methods have not been shown to furnish anti-1,3 amino alcohols. Classic methods for anti-1,3 amino alcohol synthesis involve C—C bond coupling reactions of pre-oxidized fragments to generate β-hydroxy imines3 or β-amino ketones4 followed by diastereoselective hydride reductions. Advances in transition metal catalyzed C–N cyclization of N-tosyl carbamates into allylic oxygenates5 and allenes6 still necessitate the use of pre-oxidized compounds.
Existing Pd(II)/bis-sulfoxide catalyzed intramolecular allylic C—H amination of N-nosyl carbamate substrates affords syn-1,3 amino alcohol precursors in preparative yields (avg. 60%) and diastereoselectivities (avg. 5.4:1 dr).2a These reactions proceed with a reversibly coordi-nating bis-sulfoxide ligand that relies on quinone oxidant to promote functionalization of a neutral π-allyl Pd.7 We questioned if the Pd(II)/SOX catalysis8,9, where functionalization proceeds via a cationic π-allylPd(SOX) intermediate9 gives an alternative stereooutcome. Herein we demonstrate that anti-1,3 amino alcohol motifs can be accessed for the first time via C—H amination using Pd(II)/(±)-MeO-SOX catalysis with a 2,5-dimethylbenzoquinone oxidant (2,5-DMBQ). Using the same substrate and switching the catalyst to Pd(II)/(±)-CF3-SOX and benzoquinone oxidant (BQ) affords the syn-1,3 amino alcohol motif.
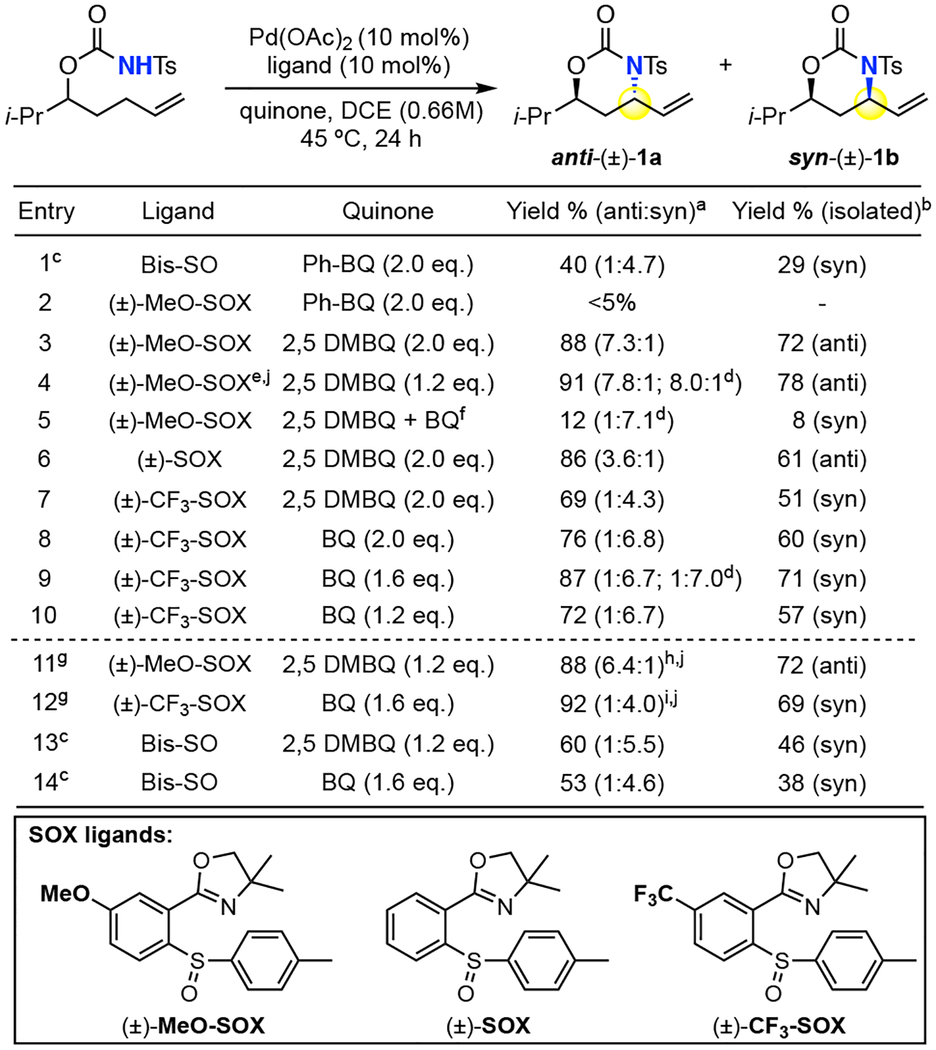
We initiated examining the reactivity of a N-tosyl carbamate substrate. Under Pd(II)/bis-sulfoxide catalysis2a (Table 1, entry 1), we observed low reactivity of this less acidic nitrogen nucleophile favoring syn-oxazinanone. Switching to (±)-MeO-SOX ligand, reported to promote intermolecular allylic C—H aminations with comparable nucleophiles,9 the reaction gave trace reactivity with phenylbenzoquinone (PhBQ) oxidant (entry 2). Analogous to previous reports,9 using highly substituted 2,5 DMBQ significantly increased the reaction productivity furnishing 88% yield of aminated product. The reaction also proceeded with good diastereoselectivity (7.3:1 dr) favoring the elusive thermodynamic5,6,10 anti-diastereomer (entry 3). Further exploration revealed that decreasing the equivalents of 2,5 DMBQ (2 → 1.2 equiv.) increased the yield and diastereoselectivity (entry 4). Low conversion under Pd(II)/MeO-SOX conditions with less hindered quinones appears to be due to an inhibitory effect on catalysis, perhaps by forming a η2-π/Pd complex that competes with essential substrate binding11: doping in 10 mol% BQ into otherwise standard anti-conditions results in significantly diminished yield of oxazinanone favoring the syn isomer (entry 5, vide infra). Continued evaluation of racemic SOX ligands revealed that simple (±)-SOX ligand gave less anti-diastereomer (4:1, entry 6), whereas one bearing an electron-withdrawing CF3 group ((±)-CF3 SOX) favored the kinetic syn-diastereomer (1:4, entry 7). Using BQ was beneficial to the yield and diastereoselectivity (76%, 1:7 dr, entry 8). Lowering the BQ loading (2 → 1.6 equiv.) increased the yield further to 87% (entry 9), whereas further decreases were not beneficial (entry 10). Brønsted acid additive (10 % diphenyl phosphinic acid)8a was used to promote reactivity for both the anti- and syn-conditions with substrates that showed sluggish reactivity (vide infra, Table 2, 16. Table 3, 29, 33–36, 43). For reactive substrates, acid additive may shorten reaction times (24h → 6h) albeit with diminishments in selectivity (entry 11, 12). Pd(II)/bis-sulfoxide catalysis under otherwise identical conditions showed improvements in yield but diastereoselectivity still favoring the syn-oxazinanone (entry 13, 14), underscoring the significance of the SOX ligand in the observed stereodivergence.
Table 1.
Reaction Development
|
Yield and dr determined by crude 1H NMR.
Isolated yield of pure diastereomer with >20:1 dr.
Commercial 1,2-Bis(phenylsulfinyl)ethane palladium(II) acetate catalyst (Pd(OAc)2/Bis-SO) used with additional 5% Bis-SO ligand.
Determined by HPLC.
Using enantiomeric pure (−)-(S)-MeO-SOX ligand, no significant kinetic resolution observed. See Scheme S9 in Supporting Information.
1.2 equiv 2.5 DMBQ and 0.1 equiv BQ was used.
10% Ph2P(O)OH was added; 6h.
No acid: 58% (2.0:1); isolated: 35% (anti).
No acid: 80% (1:6.8); isolated: 67% (syn).
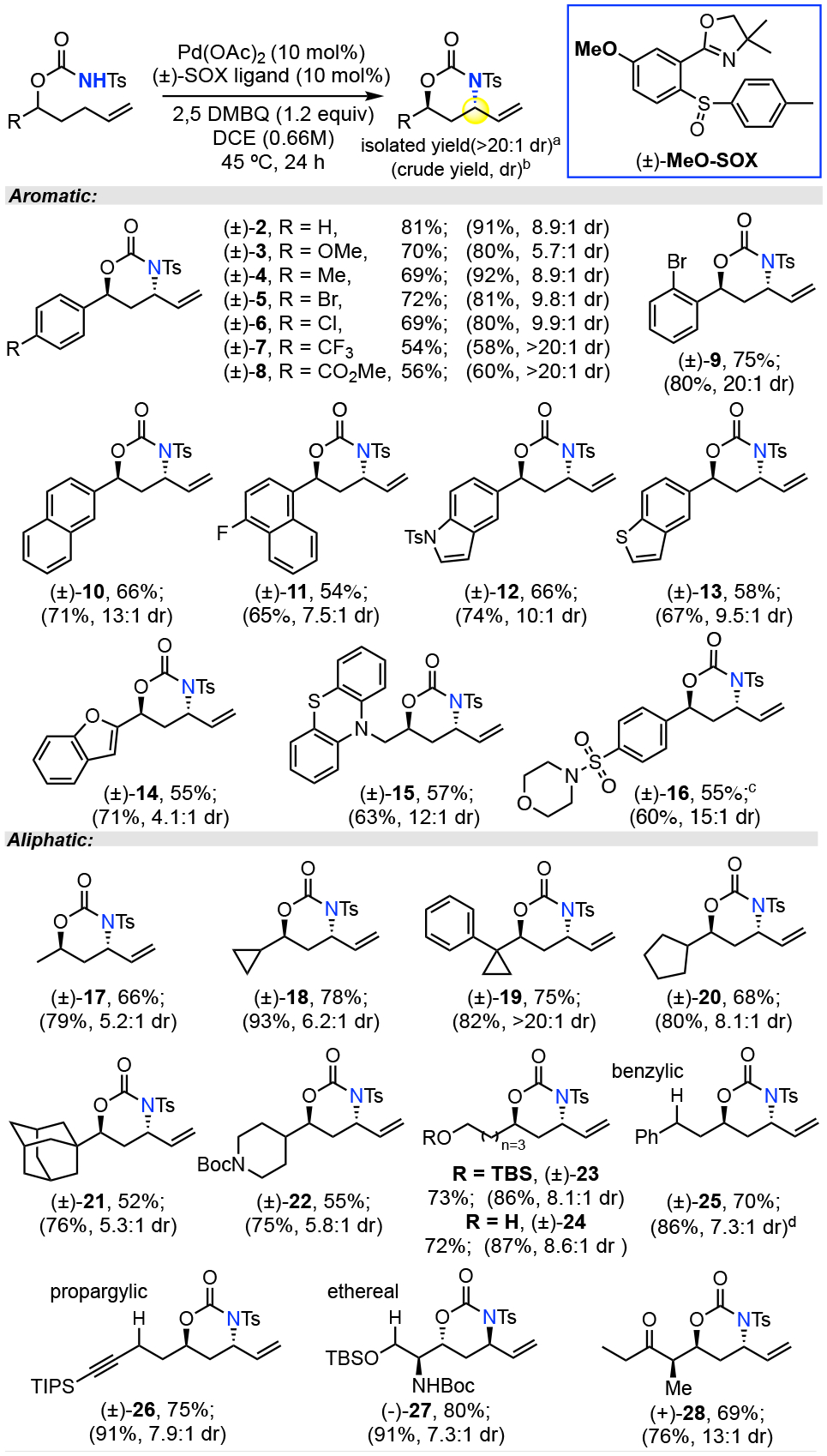
Table 2.
Scope for anti-1,3 Amino Alcohol Motifs
|
Isolated yield of anti diastereomer (>20:1 dr) over 3 runs.
Crude yield and dr determined by 1HNMR.
10% Ph2P(O)OH added to increase reactivity.
8.6:1 dr by HPLC
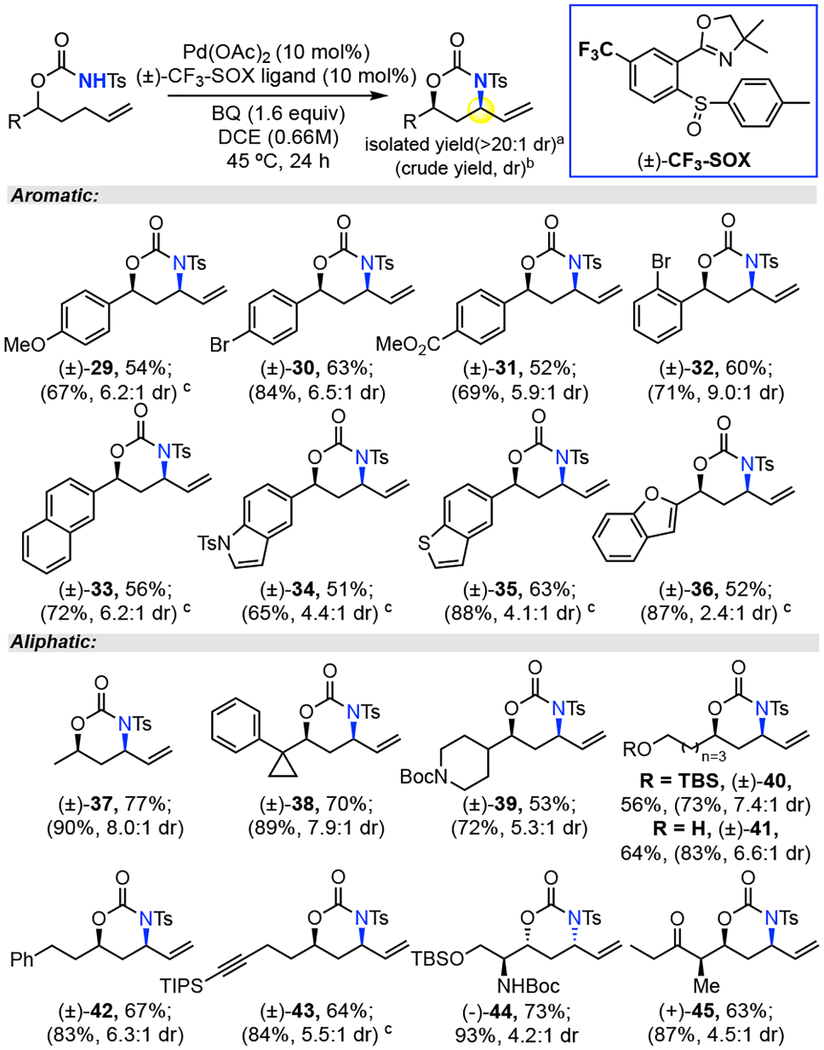
Table 3.
Scope for syn-1,3 Amino Alcohol Motifs
|
Isolated yield of syn diastereomer (>20:1 dr) over 3 runs.
Crude yield and dr determined by 1HNMR.
10% Ph2P(O)OH added to increase reactivity.
Evaluation of a broad range of substrates under Pd(II)/(±)-MeO-SOX catalysis afforded after isolation an average of 66% yield of > 20:1 anti-oxazinanone (Table 2). Arylated 1, 3-amino alcohol motifs, widely represented in bioactive compounds such as CERT antagonist HPA-1212, sedacryptine13 and nikkomycin Z14, afforded preparative yields and diastereoselectivities irrespective of electronic substitution (Table 2, 2-9). In general, electron neutral or rich aromatic substrates furnished anti-products with the highest yields (2-4) whereas highly electron deficient aromatics afforded products in the highest crude diastereoselectivities (7, 8). Aryl substrates bearing different substituted 1- or 2- naphthalene undergo allylic C—H amination with preparative yields and selectivities (10, 11). Medicinally important, oxida-tively labile heteroaromatic moieties including indole, benzothiophene, benzofuran, dibenzothiazine, and phenyl sulfonyl morpholine are all well tolerated in Pd(II)/(±)-MeO-SOX catalysis (12-16).
Aliphatic 1,3 amino alcohol motifs are common building blocks among bioactive small molecules including lopinavir15, ritonavir16 and negamycin17 (Figure 1a). A broad range of aliphatic substrates also afford anti-1,3-amino alcohol precursors in preparative yields and diastereoselectivities. Diastereoselectivity is not strongly impacted by the steric bulk of the alkyl substituent adja-cent to the carbamate. Alkyl substituents ranging in size from small methyl groups to larger cyclic alkanes (e.g. cyclopropanes, cyclopentane, adamantane) all underwent productive allylic C—H amination (17-21) with no correlation between steric bulk and diastereoselectivity. Aliphatic substrates derived from tertiary alcohols proceeded with good yields but poor diastereoselectivity under both the anti- and syn-conditions (see Supporting Information, Scheme S8). Common heterocycles such as Boc-protected piperidine are well-tolerated (22). Remote primary alcohols, both silyl protected and unpro-tected, give allylic C—H amination product with no observed alcohol oxidation (23, 24), showcasing chemoselectivity not common for Pd(II) oxidation systems.18 In contrast to metallo-nitrene based C—H aminations1, Pd(II)/sulfoxide-catalyzed amination shows high chemoselectivity for allylic C—H bonds over benzylic, propargylic and ethereal C—H bonds (25-27). The or-thogonality of this method to existing β-hydroxy imines3 or β-amino ketones4 reduction and rhodium-hydride catalyzed cyclization6 is highlighted by its tolerance of proximal alkyne and ketone functionalities (26, 28). Substrates with proximal stereocenters, even acidic ones, undergo allylic C—H amination with no detected epimerization (28).
Figure 1.
C—H aminations for 1,3 amino alcohol motifs
Several of the aromatic and aliphatic N-tosyl carbamate substrates were additionally evaluated under Pd(OAc)2/(±)-CF3-SOX/BQ catalysis. Gratifyingly, we observed by only altering the catalyst and oxidant, we were able to obtain the syn-oxazinanone products. Although the diastereoselectivities were not entirely turned over, all the substrates examined afforded after isolation an average of 62% yield of >20:1 syn-oxazinanone (Table 3). Significantly, although the previous Pd(II)/bis-sulfoxide catalyzed allylic C—H amination also affords syn-1,3 amino alcohol motifs in useful- albeit lower-yields and diastereoselectivities, installation of the more acidic N-nosyl carbamates requires synthesis of nosyl isocyanate (Table 1, entry 1).2 In contrast, N-tosyl carbamates, synthesized in one step from commercial tosyl isocyanate and homo-allylic alcohol, afford via Pd(II)/SOX catalysis either anti- or syn-1,3 amino alcohol motifs.
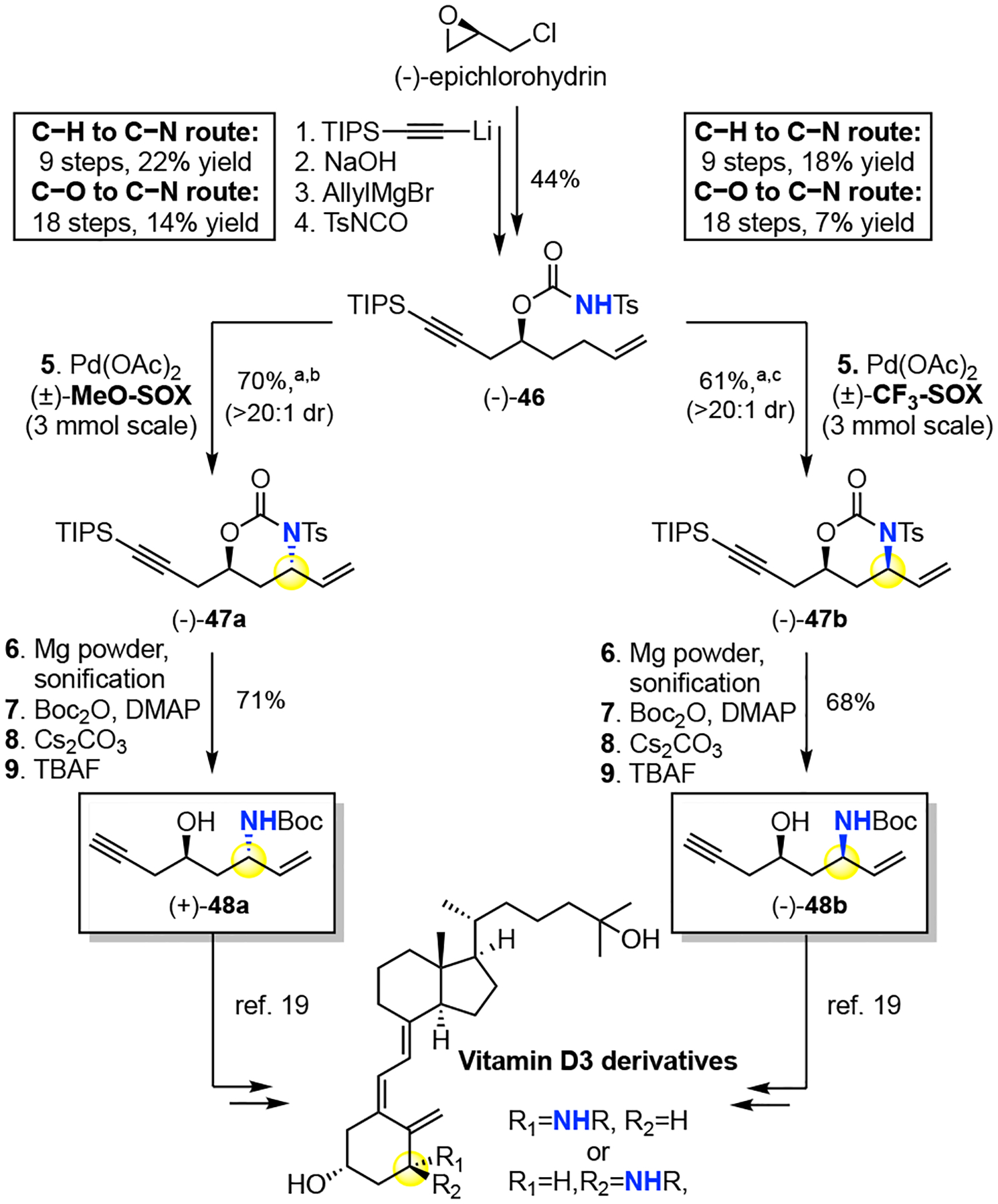
The impact of stereochemistry on small molecule function underscores the benefits of stereodivergency. For example, in the discovery phase of Vitamin D3 analogue synthesis19 for treating metabolic diseases, both syn- and anti-1,3 amino alcohol fragments towards modified A-rings were examined (Figure 2). The traditional route started from an oxygenated chiral precursor (L)-malic acid and proceeded via a lengthy functional group manipulations sequence. The allylic C—N bond was forged via a poorly diastereoselective intramolecular allylic carbamate rearrangement to furnish the syn- and anti-amino alcohols as a 2:1 mixture in ca. 18 steps ca. 14% yield for the anti-48a and 7 % for the syn-48b (Figure 2).19 In contrast, starting from commercial epichlorohydrin, the hydrocarbon core of chiral homoallylic carbamate 46 for the allylic C—H amination routes proceeds in 4 steps. Nitrogen is directly and stereoselectively installed into the hydrocarbon scaffold by using either Pd(II)/(±)-MeO-SOX or Pd(II)/(±)-CF3-SOX catalysis to afford preparative yields of the desired anti-47a or syn-47b products, respectively. Mild reductive N-desulfonylation6,20, Boc protection, base-mediated oxazinanone hydrolysis, followed by acetylene desilylation affords the desired anti- and syn-1,3 amino alcohol motifs (+)-48a, (−)-48b in 9 steps and 22% and 18% overall yields, respectively. Stereochemically pure anti- and syn-1,3 amino alcohols are furnished in higher overall yields and half the steps of the traditional route, making the C—H amination route favorable.19
Figure 2.
Streamlining synthesis.
aIsolated yield(>20:1 dr) over 2 runs. Crude yield and dr determined by 1H NMR: b78%,19:1 dr. c81%, 4.1:1 dr.
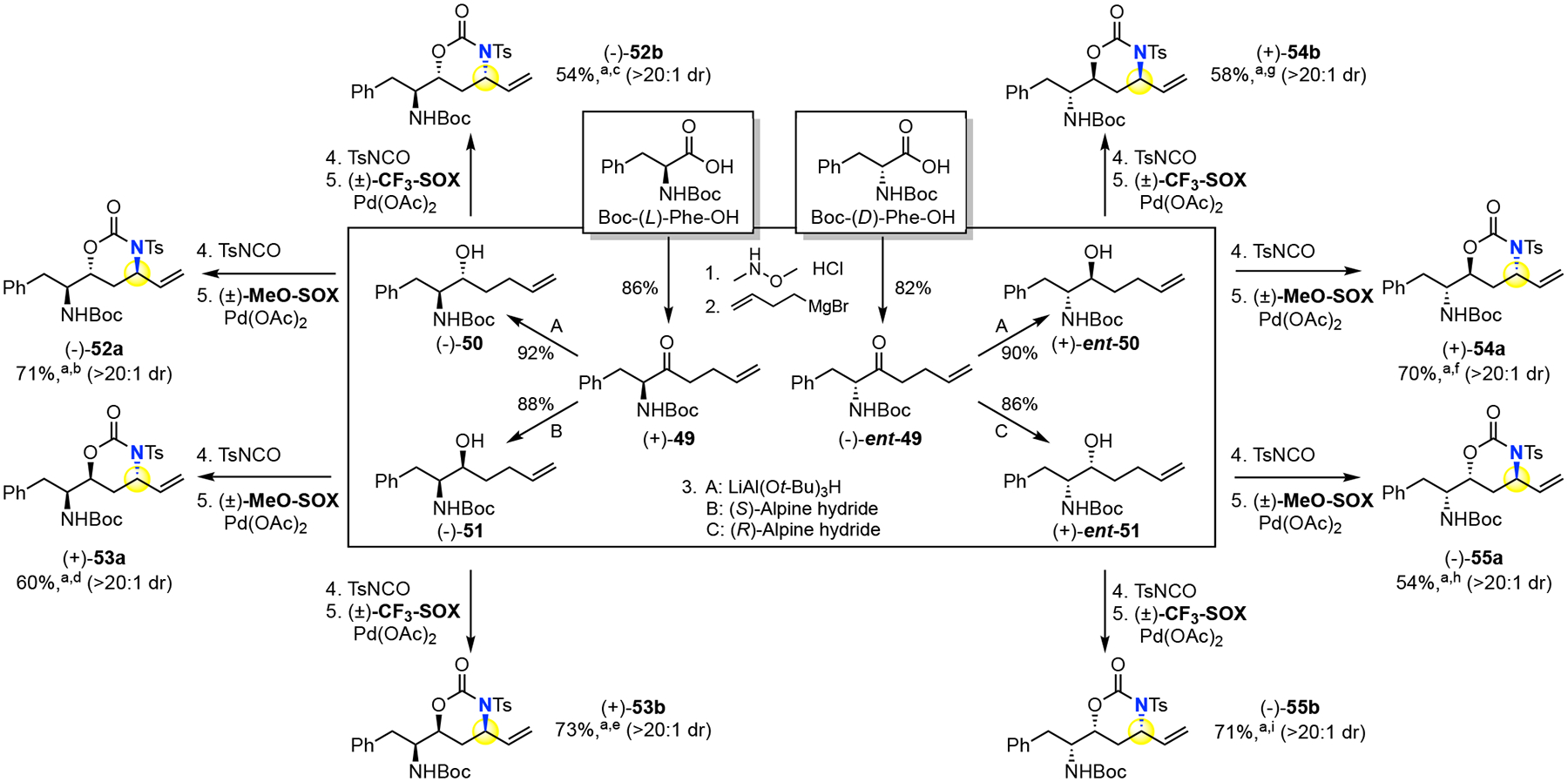
A distinctive feature of the stereodivergent allylic C—H amination is predictable and controllable diastereomeric outcome (anti or syn), even in the presence of proximal stereogenic centers. In chiral substrates, we have shown the absolute stereochemistry of the aminated site is controlled by the Pd(II)/SOX system in combination with the carbamate stereocenter (Table 2 and 3, entry 27, 28 and 44, 45). We envision that this feature will enable facile access to all possible stereoisomers of amino acid derived chiral diamino alcohol cores. These chiral subunits are found in hydroxyethylene dipeptide isostere pharmaceuticals (e.g. lopinavir15 and ritonavir16, Figure 1a). Alkylation of the Weinreb amide derivatives of Boc protected (L)- and (D)-phenylalanine furnished homoallylic amino ketones (+)-49 and (−)-ent-49 (Figure 3). Diastereoselective reduction of the ketones with LiAl(Ot-Bu)3H provided anti amino alcohols (−)-50 and (+)-ent-50 and Alpine hydride gave syn amino alcohols (−)-51 and (+)-ent-51.21 Installation of the N-tosyl carbamate was followed by stereodivergent allylic C—H amination using either Pd(II)/(±)-MeO-SOX/or Pd(II)/(±)CF3-SOX catalysis. In all cases examined, Pd(II)/(±)-MeO-SOX amination afforded the anti-oxazinanone and Pd(II)/(±)-CF3-SOX the syn-oxazinanone with preparative yields of the major diastereomer (54–73%, 52–55a, 52–55b). This streamlined route to all possible stereoisomers is competitive with previous routes that use pre-oxidized intermediates (α-amino-γ-lactone,22 enaminone,15 epoxides,23 etc.).
Figure 3.
Stereodivergent synthesis of diamino alcohol motifs.
aIsolated yield of major diastereomer (>20:1 dr) over two steps. Crude yield and dr determined by 1H NMR: b89%, 9.1: dr. c68%, 6.5:1 dr. d85%, 3.3:1 dr. e86%, 13:1 dr. f84%, 9.3:1. g72%, 7.4:1 dr. h79% 3.5:1 dr. i82%, 14:1 dr.
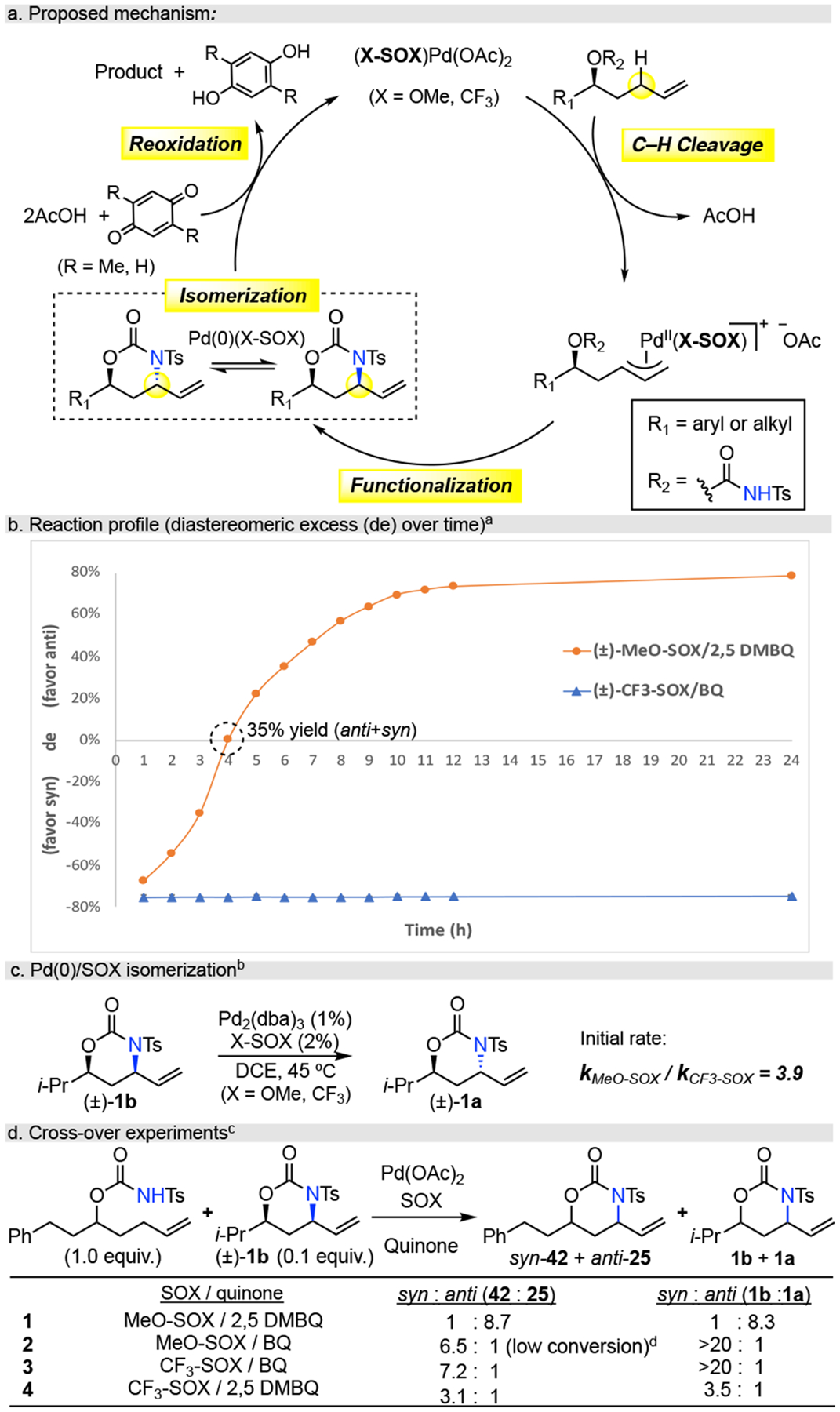
We first examined if the preference to form the more stable anti-heterocycle5,6,10 originates from C—H amination or in an off-cycle Pd reaction. Pd(II)/SOX-catalyzed allylic C—H functionalization proceeds via heterolytic allylic C—H cleavage to afford a cationic π-allylPd(SOX) intermediate9 that is subject to rapid π-σ-π isomerization8a (Figure 4a). The later indicates that C—N bond formation is the stereo-determining step in C—H amination. Evaluation of the diastereometric excess of Pd(OAc)2/(±)-MeO-SOX/2,5-DMBQ reaction over time shows that it initially generates the syn-isomer with the anti-isomer becoming favored after four hours when the reaction is at ca. 35% total yield (Figure 4b, See Supporting Information for full reaction profile). In contrast, the Pd(OAc)2/(±)-CF3-SOX/BQ reaction profile shows no change in syn-stereoselectivity (Figure 4b). This data suggests that under both catalytic systems, the syn-oxazinanone is the preferred kinetic product of C—H amination and that a Pd(0)-isomerization to the thermody-namically favored anti-oxazinanone5,6,10,24 occurs for the (±)-MeO-SOX but not for the (±)-CF3-SOX system.
Figure 4.
Mechanistic studies
ade = ((anti−syn)/(anti+syn)×100)% bNo isomerization observed without SOX ligand. cdr determined by HPLC. dBQ inhibits Pd(II)/MeO-SOX catalysis, also see Table 1, entry 5.
We hypothesized that the Pd(0)-catalyzed isomerizatioin may be more effectively promoted by the electron rich (±)-MeO-SOX than the electron deficient (±)-CF3-SOX ligand. To test the ligand effect on Pd(0)-isomerization, pure syn-oxazinanone (±)-1b was exposed to Pd2(dba)3 in the presence of both (±)-MeO-SOX and/(±)-CF3-SOX. Evaluation of the initial rates of anti-oxazinanone (±)-1a formation showed that Pd2(dba)3/(±)-MeO-SOX is ca. four times faster than Pd2(dba)3/(±)-CF3-SOX in promoting isomerization from the syn- to the anti-oxazinanone (Figure 4c). Whereas no isomerization of syn-oxazinanone (±)-1b occurs without ligand, phosphine ligands have been reported to promote this isomerization with Pd(0) under anaerobic condition.5,24
We additionally hypothesized that the Pd(0)-catalyzed isomerization may be more effective with the sterically bulky 2,5-DMBQ than with BQ oxidant. The Pd(OAc)2/(±)-MeO-SOX/2,5-DMBQ reaction was evaluated in the presence of 10% syn-oxazinanone (±)-1b (Figure 4d). Remarkably, syn-1b at low concentrations is competitive with suprastoichiometric 2,5 DMBQ in re-acting with Pd(0) as evidenced by its isomerization to favor anti-1a in comparable diastereomeric ratio to that observed under standard catalytic conditions (Figure 4d, entry 1, Table 1, entry 4). This suggests that the Pd(0) species is relatively long-lived under these conditions due to sluggish reactivity with the sterically hindered quinone. Supporting this hypothesis, when BQ oxidant is used, the reaction favors formation of syn-42 and syn-1b does not undergo isomerization (entry 2). In Pd(OAc)2/(±)-CF3-SOX catalysis, the use of 2,5 DMBQ erodes the syn:anti ratio under the reaction conditions in both product formation and in isomerization of syn-oxazinanone (±)-1b, but does not turn it over to favor the anti-product (entry 3,4). Collectively, the pairing of an electron rich SOX ligand with a sterically hindered quinone oxidant promotes Pd(0) opening of the syn-oxazinanone to enabled the first anti-selective allylic C—H amination. Alternatively, the Pd(0)-isomerization path-way can be attenuated to preserve the kinetic syn-oxazinanone product by using an electron deficient SOX ligand with an unhindered quinone oxidant.
We report a general method for stereodivergent synthesis of both anti- and syn-1,3 amino alcohol motifs via Pd(II)/SOX catalysis. The diastereoselectivity is tunable via the combination of SOX ligand and quinone oxidant. Mechanistic studies indicate that both Pd(II)/(±)-MeO-SOX and Pd(II)/(±)-CF3-SOX catalysis promotes C—H amination to the kinetic syn-oxazinanone whereas Pd(0)/(±)-MeO-SOX using sterically hindered 2,5 DMBQ oxidant can promote isomerization of the carbamate heterocycle to the thermodynamic anti-isomer. The SOX ligand’s capacity for supporting concurrent Pd(II) and Pd(0) processes is notable and will be the topic of further study.
Supplementary Material
ACKNOWLEDGEMENTS:
Financial support NIGMS MIRA (R35 GM122525). We thank R. Quevedo for repeating our experimental procedure in Table 2, 25.
Footnotes
The authors declare the following competing financial interest(s): The University of Illinois has filed a patent application on SOX ligands for allylic C–H functionalizations.
REFERENCES
- (1).(a) Liang C; Collet F; Robert-Peillard F; Muller P; Dodd RH; Dauban P Toward a synthetically useful stereoselective C-H amination of hydrocarbons. J. Am. Chem. Soc 2008, 130, 343. [DOI] [PubMed] [Google Scholar]; (b) Zalatan DN; Du Bois J A chiral rhodium carboxamidate catalyst for enantioselective C-H amination. J. Am. Chem. Soc 2008, 130, 9220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Harvey ME; Musaev DG; Du Bois J A diruthenium catalyst for selective, intramolecular allylic C-H amination: reaction development and mechanistic insight gained through experiment and theory. J. Am. Chem. Soc 2011, 133, 17207. [DOI] [PubMed] [Google Scholar]; (d) Paradine SM; White MC Iron-catalysed intramolecular allylic C-H amination. J. Am. Chem. Soc 2012, 134, 2016. [DOI] [PubMed] [Google Scholar]; (e) Paradine SM; Griffin JR Zhao J, Petronico AL Miller SM; White MC A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp3)–H amination. Nat. Chem 2015, 7, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Collet F; Lescot C; Dauban P Catalytic C-H amination: the stereoselectivity issue. Chem. Soc. Rev 2011, 40, 1926. [DOI] [PubMed] [Google Scholar]; (g) Roizen JL; Harvey ME; Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res 2012, 45, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Rice GT; White MC Allylic C-H amination for preparation of syn-1,3-amino alcohol motifs. J. Am. Chem. Soc 2009, 131, 11707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qi X; Rice GT; Lall MS; Plummer MS; White MC Diversification of a β-lactam pharmacophore via allylic C-H amination: accelerating effect of Lewis acid co-catalyst. Tetrahedron. 2010, 66, 4816. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nahra F; Liron F; Prestat G; Mealli C; Messaoudi A; Poli G Striking AcOH acceleration in direct intramolecular allylic amination reactions. Chem. Eur. J 2009, 15, 11078. [DOI] [PubMed] [Google Scholar]
- (3).Kochi T; Tang TP; Ellman JA Development and application of a new general method for the asymmetric synthesis of syn- and anti-1,3-amino alcohols. J. Am. Chem. Soc 2003, 125, 11276. [DOI] [PubMed] [Google Scholar]
- (4).(a) Keck GE; Truong AP Directed reduction of β-amino ketones to syn or anti 1,3-amino alcohol derivatives. Org, Lett 2002, 4, 3131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davis FA; Gaspari PM; Nolt BM; Xu P Asymmetric synthesis of acyclic 1,3-amino alcohols by reduction of N-sulfinyl β-amino ketones. Formal synthesis of (−)-Pinidinol and (+)-Epipinidinol. J. Org. Chem 2008, 73, 9619. [DOI] [PubMed] [Google Scholar]
- (5).Broustal G; Ariza X; Campagne J-M; Garcia J; Georges Y; Marinetti A; Robiette R A stereoselective approach to 1,3-amino alcohols protected as cyclic carbamates: kinetic vs. thermodynamic control. Eur. J. Org. Chem 2007, 4293. [Google Scholar]
- (6).Spreider PA; Haydl AM; Heinrich M; Breit B Rhodium-catalyzed diastereoselective cyclization of allenyl-sulfonylcarba-mates: A stereodivergent approach to 1,3-aminoalcohol derivatives. Angew. Chem. Int. Ed 2016, 55, 15569. [DOI] [PubMed] [Google Scholar]
- (7).(a) Chen MS; Prabagaran N; Labenz NA; White MC Serial ligand catalysis: a highly selective allylic C-H oxidation. J. Am. Chem. Soc 2005, 127, 6970. [DOI] [PubMed] [Google Scholar]; (b) Fraunhoffer KJ; White MC syn-1,2-Amino alcohols via diastereoselective allylic C-H amination. J. Am. Chem. Soc 2007, 129, 7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Ammann SE; Liu W; White MC Enantioselective allylic C-H oxidation of terminal olefins to isochromans by palladium(II)/chiral sulfoxide catalysis. Angew. Chem., Int. Ed 2016, 55, 9571. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu W; Ali SZ; Ammann SE; White MC Asymmetric allylic C-H alkylation via palladium(II)/cis-ArSOX catalysis. J. Am. Chem. Soc 2018, 140, 10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ma R; White MC C-H to C-N cross-coupling of sulfonamides with olefins. J. Am. Chem. Soc 2018, 140, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).DFT studies examining the anti/syn equilibrium in isopropyl and phenyl substituted N-tosyl oxazinanones anti-1a/syn-1b and anti-2/syn-2 consistently show a higher stability of the anti. See references 5 and 6.
- (11).Pattillo CC; Strambeanu II; Calleja P; Vermeulen NA; Mizuno T; White MC Aerobic linear allylic C-H amination: overcoming benzoquinone inhibition. J. Am. Chem. Soc 2016, 138, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ueno M; Huang YY; Yamano A; Kobayashi S Revised stereochemistry of ceramide-trafficking inhibitor HPA-12 by X-ray crystallography analysis. Org. Lett 2013, 15, 2869. [DOI] [PubMed] [Google Scholar]
- (13).Hootele C; Colau B; Halin F Sedum alkaloids II: sedacryptine, a new minor base from sedum acre. Tetrahedron Lett. 1980, 21, 5061. [Google Scholar]
- (14).Gaughran JP; Lai MH; Kirsch DR; Silverman SJ Nikkomycin Z is a specific inhibitor of Saccharomyces cerevisiae chitin synthase isozyme Chs3 in vitro and in vivo. J Bacteriology. 1994, 176, 5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Stoner EJ; Cooper AJ; Dickman DA; Kolaczkowski L; Lallaman JE; Liu J-H; Oliver-Shaffer PA; Patel KM; Paterson JB; Plata DJ; Riley DA; Sham HL; Stengel PJ; Tien J-H Synthesis of HIV protease inhibitor ABT-378 (Lopinavir). Org. process Res. Dev 2000, 4, 264. [Google Scholar]
- (16).Kempf DJ; Sham HL; Marsh KC; Flentge CA; Betebenner D; Green BE; McDonald E; Vasavanonda S; Saldivar A; Wideburg NE; Kati WM; Ruiz L; Zhao C; Fino L; Patterson J; Molla A; Plattner JJ; Norbeck DW Discovery of ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem 1998, 41, 602. [DOI] [PubMed] [Google Scholar]
- (17).Kondo S; Shibahara S; Takahashi S; Maeda K; Umezawa H; Ohno M Negamycin, a novel hydrazide antibiotic. J. Am. Chem. Soc 1971, 93, 6305. [DOI] [PubMed] [Google Scholar]
- (18).(a) Jensen DR; Pugsley JS; Sigman MS palladium-catalyzed enantioselective oxidations of alcohols using molecular oxygen. J. Am. Chem. Soc 2001, 123, 7475. [DOI] [PubMed] [Google Scholar]; (b) Ferreira EM; Stoltz BM The Palladium-catalyzed oxidative kinetic resolution of secondary alcohols with molecular oxygen. J. Am. Chem. Soc 2001, 123, 7725. [DOI] [PubMed] [Google Scholar]; (c) Werner EW; Mei TS; Burckle AJ; Sigman MS Enantioselective Heck arylations of acyclic alkenyl alcohols using a redox-relay strategy. Science. 2012, 338, 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang D; Weinstein AB; White PB; Stahl SS Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem. Rev 2018, 118, 2636. [DOI] [PubMed] [Google Scholar]
- (19).Watanabe M,; Asano R; Nagasawa K; Uesugi M Vitamin D3 derivatives and pharmaceutical use thereof. WO2016103722A1 2016.
- (20).(a) Nyasse B; Grehn L; Ragnarsson U Mild, efficient cleavage of arenesulfonamides by magnesium reduction. Chem. Commun 1997, 1017. [Google Scholar]; Tolerance for reducible functionality see:; (b) Guo LD; Huang XZ; Luo SP; Cao WS; Ruan YP; Ye JL; Huang PQ Organocatalytic, asymmetric total synthesis of (−)-haliclonin A. Angew. Chem., Int. Ed 2016, 55, 4064. [DOI] [PubMed] [Google Scholar]
- (21).(a) Våbenø J; Brisander M; Lejon T; Luthman K Diastereoselective reduction of a chiral N-Boc-protected δ-amino-α, β-unsaturated γ-keto ester Phe-Gly dipeptidomimetic. J. Org. Chem 2002, 67, 9186. [DOI] [PubMed] [Google Scholar]; (b) Mikkelsen LM; Jensen CM; Høj B; Blakskjær P; Skrydstrup T Further studies in the acyl-type radical additions promoted by SmI2: mechanistic implications and stereoselective reduction of the keto-functionality. Tetrahedron. 2003, 59, 10541. [Google Scholar]
- (22).(a) Ghosh AK; McKee SP; Thompson WJ; Darke PL; Zugay JC Potent HIV-1 protease inhibitors: stereoselective synthesis of a dipeptide mimic. J. Org. Chem 1993, 58, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baker WR; Pratt JK Dipeptide isosteres. 2. Synthesis of hydroxyethylene dipeptide isostere diastereomers from a common &-lactone intermediate. Preparation of renin and HIV-1 protease inhibitor transition state mimics. Tetrahedron. 1993. 49 8739. [Google Scholar]
- (23).Benedetti F; Norbedo S Epoxyalcohol route to hydroxyethylene dipeptide isosteres: a new synthesis of the diaminoalcohol core of HIV-protease inhibitor ABT-538 (Ritonavir). Chem. Commun, 2001. 201. [Google Scholar]
- (24).Bando T; Harayama H; Fukazawa Y; Shiro M; Fugami K; Tanaka S; Tamaru Y Regio- and stereoselective synthesis of 1,3-hydroxyl amines via palladium-catalyzed carbonate-carbamate transformation with unique stereoselectivity: synthesis of 3-amino-4-penten-1-ols. J. Org. Chem 1994, 59, 1465. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.