

Abstract
A catalytic system has been developed for the direct alkylation of α-C–H bonds of aniline derivatives with strained C–C σ-bonds. This method operates through a photoredox mechanism in which oxidative formation of aminoalkyl radical intermediates enables addition to a bicyclobutane derivative, giving rise to α-cyclobutyl N-alkylaniline products. This mild system proceeds through a redox- and proton-neutral mechanism and is operational for a range of substituted arylamine derivatives.
Keywords: photoredox reaction, catalysis, C–C bond activation, anilines, alkylation, iridium catalysis
Graphical Abstract
The high stability of C–C single bonds typically renders them chemically inert; however, conformationally strained-ring systems display unique reactivity profiles. Early work by Wiberg and co-workers and by Gaoni demonstrated that a range of strained polycyclic hydrocarbons (some of which are shown in Figure 1) can be accessed synthetically,1–3 and that they can serve as effective alkylating agents.4,5 Indeed, because installation of cyclobutyl groups (cyclobutylation) can be accomplished through direct addition to bicyclo[1.1.0]butane (BCB) derivatives, this represents a potentially valuable method for the synthesis of cyclobutane-containing natural products or drug candidates.
Figure 1.

Strategy for cyclobutylation through addition to strained bicyclic alkanes. The strain energy for each molecule is derived by comparing δHf with a hypothetical strainless model, as discussed by Wiberg.19
In addition to anionic nucleophiles,6–8 organometallics,9–11 and amines,12,13 strained BCBs are effective coupling partners for radical species.7,14 Over the past few years, our laboratory has been interested in developing new methods that permit selective reactions through the use of photoredox catalysis. As part of this program, we recently developed a system for the direct addition of nonactivated amines to peptides through the formation of the corresponding α-amino radicals.15 This process is highly selective and it functions through a redox- and proton-neutral mechanism, such that stoichiometric additives are not required. We questioned whether these aminoalkyl radical intermediates16 could be intercepted by strain-activated BCB derivatives in a direct cyclobutylation reaction with amines. Although a number of impressive methods for cyclobutylation of radical intermediates have been described, most recently by the groups of Lin17 and Aggarwal,18 this approach (outlined in Figure 1) would be unique, in part, because it would potentially function on a range of nonactivated amine-containing substrates, without the need for programmed radical formation (i.e., halogen installation, etc.).
We considered the catalytic pathway shown in Figure 1, in which photoinduced electron transfer from the amine substrate to a photoredox catalyst would give rise to a radical cation. Deprotonation of the newly acidified C–H bond would afford a key α-amino radical species.20–23 Attack of this intermediate species on a strain-activated bicyclo[1.1.0]butane would afford a new C–C bond, and single-electron reduction and protonation would deliver the desired α-cyclobutylamine This system would operate in a redox- and proton-neutral manner, without the need for exogenous chemical additives.
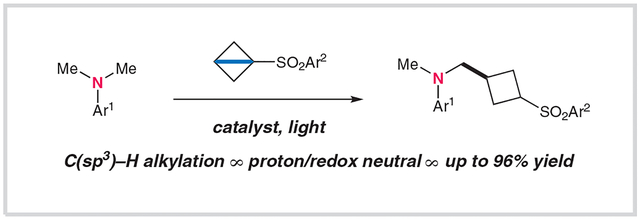
To evaluate the feasibility of this proposed transformation, we treated N,N-dimethylaniline with a small series of BCB reagents 1a–c to afford the corresponding cyclobutylated products 2a–c (Table 1). In the presence of 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)·PF6 [P1; dF = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine; dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] and blue light in N,N-dimethylacetamide (DMA), phenylsulfone 1a gave the desired product 2a in a promising yield (28%, as determined by NMR with an internal standard). Increasing the electron-withdrawing capacity of the sulfonyl activating group resulted in improved reactivity with aminoalkyl radicals. Specifically, the fluorinated phenylsulfone derivatives 1b and 1c gave the corresponding cyclobutane products 2b and 2c in yields of 64 and 69%, respectively. In this system, the aniline substrate was used as the excess reagent (five equivalents with respect to the BCB reagent) to suppress further activation and cyclobutylation of the desired products 2. Importantly, this process was not operational in the absence of a photoredox catalyst or light (Table 1, entries 1 and 2; 0% yield). Other catalysts could also be employed to accomplish this transformation, although less-oxidizing iridium catalysts (such as P2; entry 3: 35% yield) or organic dyes [including penta-9H-carbazol-9-ylbenzonitrile (5CzBn; P3) (entry 4: 64% yield)] were less efficient across an array of substrates. A variety of aprotic organic solvents could be utilized here with acceptable levels of reaction efficiency (entries 5–7; 46–51% yield).
Table 1.
Optimal Conditions for α-Cyclobutylation of N,N-Dimethylaniline
 | ||
|---|---|---|
| Entry | Deviation from above conditions (with 1c) | Yield (%) of 2c |
| 1 | Reaction without catalyst | 0 |
| 2 | Reaction without blue light | 0 |
| 3 | 1 mol% Ir[(ppy)2(dtbbpy)]-PF6a (P2) as catalyst | 35 |
| 4 | 5 mol% 5CzBnb (P3) as catalyst | 64 |
| 5 | MeCN as solvent | 48 |
| 6 | DCE as solvent | 46 |
| 7 | DMSO as solvent | 51 |
ppy = 2-phenylpyridine.
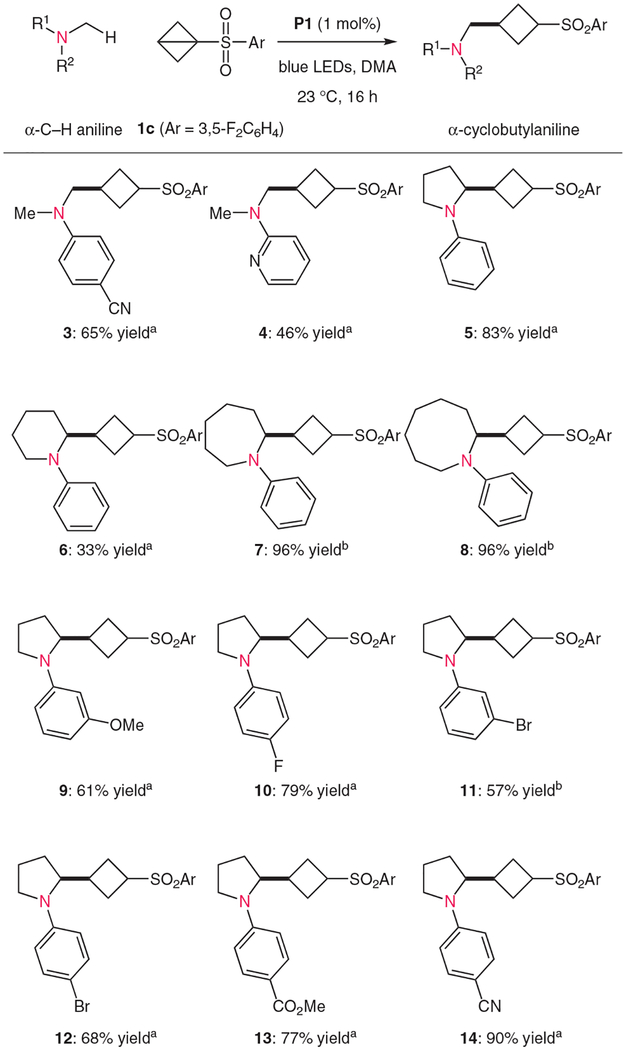
Having identified an effective protocol for the α-alkylation of anilines with BCB 1c, we evaluated the substrate scope of this process (again utilizing 1c as the limiting reagent with five equivalents of the aniline substrate).24 As shown in Table 2, electron-deficient dimethylaniline derivatives reacted under standard conditions to give the corresponding products 3 and 4 in yields of 65 and 46%, respectively. Cyclic aniline derivatives were also effectively transformed into the corresponding cyclobutylated products. For example, N-phenylpyrrolidine reacted well to afford 5 in 83% isolated yield. These conditions were less effective for the coupling of N-phenylpiperidine (which gave 6 in 33% yield, with 55% remaining starting material), presumably due to increased steric constraints. However, seven- and eight-membered saturated nitrogen heterocycles were excellent substrates, giving rise to the corresponding cyclobutanes 7 and 8 in near-quantitative yield (96% in each case).25 In accord with earlier reports,21 aniline derivatives with electron-donating groups in the ortho- or para-positions were not reactive in this system, where coupling attempts uniformly returned the starting materials. However, these conditions were effective for the transformation of electron-deficient aniline derivatives. Alkylation of a variety of N-arylpyrrolidines occurred smoothly; a methoxy substituent was well tolerated in the meta-position (9; 61% yield), as were fluoro and bromo substituents (10–12; 57–79% yield). As expected, the methoxycarbonyl- and nitrile-substituted phenylpyrrolidines were excellent substrates for this transformation, giving rise to products 13 and 14 in yields of 77 and 90%, respectively.
Table 2.
Scope of Anilines. a<2:1 dr, b2.3:1 dr.
|
In most cases, the products were isolated as roughly equal (<2:1) mixtures of cis- and trans-cyclobutane stereoisomers. Slightly higher selectivity was observed in a few cases (7, 8, and 11: ~2.3:1 ratio); although chromatographic separation of these diastereomers was often difficult, we were able to purify the major isomer of 7. Crystallization of this isomer permitted its analysis by X-ray crystallography, which demonstrated that the major isomer in this case was the cis-cyclobutane, consistent with protonation occurring from the less sterically hindered face of the cyclobutane ring. The synthetic utility of arylsulfonylcyclobutanes has been extensively demonstrated by the Baran group.13 In line with these reports, reductive desulfonylation of product 5 occurred smoothly in the presence of excess magnesium in methanol at room temperature to give 71% yield of the cyclobutyl product 15, as shown in Scheme 1.
Scheme 1.

Reductive desulfonation of arylsulfonylcyclobutane 5
In summary, we have developed a mild system for the direct C–H cyclobutylation of aniline derivates. This process operates through formation of an α-amino radical, followed by direct addition to a strained BCB derivative. The dis-closed protocol readily functionalizes a range of N-aryl amines and heterocyclic compounds of various ring sizes. In accord with previous reports, electron-deficient aniline derivates were shown to be efficient coupling partners. Because the resulting sulfonyl cyclobutane products have been shown to capable of being manipulated in a number of ways, we anticipate this process will be an attractive method for the generation of a range of α-cyclobutyl amine derivatives.
Supplementary Material
Funding Information
Financial support for this work was provided the National Institutes of Health (GM129495).
Footnotes
This paper is dedicated to Jürgen Klopp (manager of Liverpool F.C.) for his inspirational leadership.
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0039-1690197.
References and Notes
- (1).Wiberg KB; Ciula RP J. Am. Chem. Soc 1959, 81, 5261. [Google Scholar]
- (2).Gaoni Y Tetrahedron Lett. 1981, 22, 4339. [Google Scholar]
- (3).Wiberg KB; Walker FH J. Am. Chem. Soc 1982, 104, 5239. [Google Scholar]
- (4).Gaoni Y; Tomazic A J. Org. Chem 1985, 50, 2948. [Google Scholar]
- (5).Wiberg KB Chem. Rev 1989, 89, 975. [Google Scholar]
- (6).Blanchard EP Jr.; Cairncross A J. Am. Chem. Soc 1966, 88, 487. [Google Scholar]
- (7).Hall HK Jr.; Blanchard EP Jr.; Cherkofsky SC; Sieja JB; Sheppard WA J. Am. Chem. Soc 1971, 93, 110. [Google Scholar]
- (8).Gaoni Y Tetrahedron 1989, 45, 2819. [Google Scholar]
- (9).Gaoni Y Tetrahedron Lett. 1982, 23, 5215. [Google Scholar]
- (10).Panish R; Chintala SR; Boruta DT; Fang Y; Taylor MT; Fox JM J. Am. Chem. Soc 2013, 135, 9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fawcett A; Biberger T; Aggarwal VK Nat. Chem 2019, 11, 117. [DOI] [PubMed] [Google Scholar]
- (12).Gianatassio R; Lopchuk JM; Wang J; Pan C-M; Malins LR; Prieto L; Brandt TA; Collins MR; Gallego GM; Sach NW; Spangler JE; Zhu H; Zhu J; Baran PS Science 2016, 351, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lopchuk JM; Fjelbye K; Kawamata Y; Malins LR; Pan C-M; Gianatassio R; Wang J; Prieto L; Bradow J; Brandt TA; Collins MR; Elleraas J; Ewanicki J; Farrell W; Fadeyi OO; Gallego GM; Mousseau JJ; Oliver R; Sach NW; Smith JK; Spangler JE; Zhu H; Zhu J; Baran PS J. Am. Chem. Soc 2017, 139, 3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wiberg KB; Lampman GM; Ciula RP; Connor DS; Schertler P; Lavanish J Tetrahedron 1965, 21, 2749. [Google Scholar]
- (15).Aycock RA; Pratt CJ; Jui NT ACS Catal. 2018, 8, 9115. [Google Scholar]
- (16).McNally A; Prier CK; MacMillan DWC Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wu X; Hao W; Ye K-Y; Jiang B; Pombar G; Song Z; Lin S J. Am. Chem. Soc 2018, 140, 14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Silvi M; Aggarwal VK J. Am. Chem. Soc 2019, 141, 9511. [DOI] [PubMed] [Google Scholar]
- (19).Wiberg KB Angew. Chem. Int. Ed. Engl 1986, 25, 312. [Google Scholar]
- (20).For early examples of photochemical addition of aminoalkyl radicals to conjugated olefins see:; (a) Cookson RC; Hudec J; Mirza NA Chem. Commun 1968, 180. [Google Scholar]; (b) Yoon U-C; Kim J-U; Hasegawa E; Mariano PS J. Am. Chem. Soc 1987, 109, 4421. [Google Scholar]
- (21).For seminal papers on amine α-C–H activation by using photoredox see:; (a) Kohls P; Jadhav D; Pandey G; Reiser O Org. Lett 2012, 14, 672. [DOI] [PubMed] [Google Scholar]; (b) Miyake Y; Nakajima K; Nishibayashi YJ Am. Chem. Soc 2012, 134, 3338. [DOI] [PubMed] [Google Scholar]; (c) Ruiz Espelt L; Wiensch EM; Yoon TP J. Org. Chem 2013, 78, 4107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Douglas JJ; Cole KP; Stephenson CRJ J. Org. Chem 2014, 79, 11631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Trowbridge A; Reich D; Gaunt MJ Nature 2018, 561, 522. [DOI] [PubMed] [Google Scholar]; (f) Flodén NJ; Trowbridge A; Willcox D; Walton SM; Kim Y; Gaunt MJ J. Am. Chem. Soc 2019, 141, 8426. [DOI] [PubMed] [Google Scholar]
- (22).For enantioselective examples see:; (a) Ruiz Espelt L; McPherson IS; Wiensch EM; Yoon TP J. Am. Chem. Soc 2015, 137, 2452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Murphy JJ; Bastida D; Paria S; Fagnoni M; Melchiorre P Nature 2016, 532, 218. [DOI] [PubMed] [Google Scholar]
- (23).For recent papers on amine α-C–H activation by using umpolung reactions of imines see:; (a) Wu Y; Hu L; Li Z; Deng L Nature 2015, 523, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu B; Deng L Angew. Chem. Int. Ed 2018, 57, 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).α-Cyclobutylanilines 2–14; General ProcedureA screw-top test tube equipped with a stirrer bar was charged with [Ir{dF(CF3)ppy}2(dtbbpy)]·PF6 (1 mol%), 1C (1 equiv), and, if solid, the appropriate aniline (5 equiv). The tube was sealed with a PTFE/silicon septum and connected to a Schlenck line. The atmosphere was exchanged by applying a vacuum and backfilling with N2 (this process was conducted a total of three times). Under a N2 atmosphere, the tube was charged by syringe with previously degassed solvent (DMA; 2 mL/mmol 1C) and, if liquid, the appropriate aniline. The resulting solution was stirred at 700 rpm and irradiated with blue LEDs (4 cm from the lamp) for 16 h. The reaction was then quenched with sat. aq NaHCO3 (10 mL), and the mixture was extracted with EtOAc (3 × 15 mL). The combined extracts were washed sequentially with 5% aq LiCl (3 × 15 mL) and brine (2 × 15 mL), dried (Na2SO4), and concentrated in vacuo. The residue was purified by chromatography (silica gel) to give the desired product. See the Supporting Information for full details, along with physical and spectroscopic data for the products.
- (25).2-{3-[(3,5-Difluorophenyl)sulfonyl]cyclobutyl}−1-phenylazepane (cis-7)By following general procedure, the reaction of 1C (116.6 mg, 0.5 mmol, 1 equiv), 1-phenylazepane (450 μl, 2.5 mmol, 5 equiv), and [Ir{dF(CF3)ppy}2(dtbbpy)]·PF6 (6.4 mg, 0.005 mmol, 0.01 equiv), followed by purification by flash column chromatography [silica gel, EtOAc–hexanes (0–20%)], gave a clear oil; yield: 128.8 mg (64%).1H NMR (600 MHz, CDCl3): δ = 7.38–7.32 (m, 2 H), 7.19 (dd, J = 8.5, 7.0 Hz, 2 H), 7.07–6.97 (m, 1 H), 6.72 (d, J = 8.3 Hz, 2 H), 6.61 (t, J = 7.2 Hz, 1 H), 3.86 (dt, J = 10.4, 6.9 Hz, 1 H), 3.63–3.52 (m, 2 H), 3.06 (dd, J = 15.6, 11.4 Hz, 1 H), 2.52–2.43 (m, 1 H), 2.41–2.30 (m, 2 H), 2.23 (dp, J = 11.9, 4.1 Hz, 1 H), 2.15 (dtd, J = 12.0, 7.9, 4.1 Hz, 1 H), 2.08 (dt, J = 14.4, 7.2 Hz, 1 H), 1.77–1.66 (m, 3 H), 1.60–1.52 (m, 1 H), 1.37–1.22 (m, 2 H), 1.22–1.13 (m, 1 H). 13C NMR (151 MHz, CDCl3): δ = 162.8 (dd, J = 255.8, 11.4 Hz), 148.5, 141.3 (t, J = 7.8 Hz), 129.4, 115.1, 112.1–111.7 (m), 110.8, 109.4 (t, J = 24.9 Hz), 58.6, 53.2, 43.2, 34.3, 31.9, 29.8, 26.9, 26.3, 25.9, 24.8. 19F (376 MHz, CDCl3): δ = −104.95 to −104.96 (m).X-ray-quality crystals of cis-7 were obtained by Et2O and hexane vapor diffusion. C22H25F2NO2S, Mr = 405.49, monoclinic, P21/n (No. 14), a = 9.56610(10), b = 21.4256(3), c = 19.3969(2) Å, β = 96.1240(10)°, V = 3952.89(8) Å3, T = 102(4) K, Z = 8, Z′ = 2, μ(MoKα) = 0.200; 135731 reflections measured, 20640 unique (Rint = 0.0684), which were used in all calculations. The final wR2 was 0.1495 (all data) and R1 was 0.0566 [I > 2σ(I)].Cambridge Crystallographic Data Centre (CCDC) contains the supplementary crystallographic data for compound cis-7. The data can be obtained free of charge from The CCDC via www.ccdc.cam.ac.uk/getstructures.2-{3-[(3,5-Difluorophenyl)sulfonyl]cyclobutyl}−1-phenylazepane (trans-7)By following general procedure, the reaction of 1C (116.6 mg, 0.5 mmol, 1 equiv), 1-phenylazepane (450 μl, 2.5 mmol, 5 equiv), and [Ir{dF(CF3)ppy}2(dtbbpy)]·PF6 (6.4 mg, 0.005 mmol, 0.01 equiv), followed by purification by flash column chromatography [silica gel, EtOAc–hexanes (0–20%)], gave a clear oil; yield: 64 mg (32%).1H NMR (600 MHz, CDCl3): δ = 7.40–7.35 (m, 2 H), 7.15 (dd, J = 8.9, 7.1 Hz, 2 H), 7.04 (tt, J = 8.4, 2.3 Hz, 1 H), 6.70 (d, J = 8.4 Hz, 2 H), 6.57 (t, J = 7.2 Hz, 1 H), 3.84 (td, J = 9.1, 6.4 Hz, 1 H), 3.69–3.58 (m, 2 H), 3.07 (ddd, J = 15.7, 11.5, 2.0 Hz, 1 H), 2.90–2.82 (m, 1 H), 2.66–2.58 (m, 1 H), 2.50–2.42 (m, 1 H), 2.24–2.13 (m, 2 H), 2.13–2.04 (m, 1 H), 1.74–1.65 (m, 3 H), 1.53–1.51 (m, 1 H), 1.37–1.22 (m, 2 H), 1.20–1.13 (m, 1 H).13C NMR (151 MHz, CDCl3): δ =162.9 (dd, J = 255.9, 11.4 Hz), 148.8, 141.3 (t, J = 8.0 Hz), 129.4, 115.1, 111.9 (dd, J = 21.7, 6.5 Hz), 110.9, 109.4 (t, J = 24.9 Hz), 58.6, 54.7, 43.0, 36.2, 32.2, 29.6, 26.0, 25.7, 25.7, 24.7.19F NMR (376 MHz, CDCl3): δ = −105.01 to −105.04 (m).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.