

We performed the multi-omic characterization of gastric cancer to detect key molecular features for neoadjuvant chemotherapy.
Abstract
Neoadjuvant chemotherapy is a common treatment for patients with gastric cancer. Although its benefits have been demonstrated, neoadjuvant chemotherapy is underutilized in gastric cancer management, because of the lack of biomarkers for patient selection and a limited understanding of resistance mechanisms. Here, we performed whole-genome, whole-exome, and RNA sequencing on 84 clinical samples (including matched pre- and posttreatment tumors) from 35 patients whose responses to neoadjuvant chemotherapy were rigorously defined. We observed increased microsatellite instability and mutation burden in nonresponse tumors. Through comparisons of response versus nonresponse tumors and pre- versus posttreatment samples, we found that C10orf71 mutations were associated with treatment resistance, which was supported by drug response data and potentially through inhibition of cell cycle, and that MYC amplification correlated with treatment sensitivity, whereas MDM2 amplification showed the opposite pattern. Neoadjuvant chemotherapy also reshapes tumor-immune signaling and microenvironment. Our study provides a critical basis for developing precision neoadjuvant regimens.
INTRODUCTION
Gastric cancer (GC) is one of the most frequent cancer types and the second common cause of cancer-related deaths (8.2%) in the world (1). China has the largest number of patients with GC, and according to the National Central Cancer Registry of China, >679,000 new GC cases are diagnosed, and about 498,000 GC-related deaths occur per year (2, 3). Most of these patients are at advanced stages with a 5-year survival rate of <30% (4). Although surgical therapy is the primary treatment for GC, multimodality strategies are being used to improve patient survival. Neoadjuvant (or perioperative) chemotherapy is administered as an approach of “downstaging and downsizing” a locally advanced tumor before attempting a curative resection. Furthermore, for patients with GC at a high risk of developing distant metastases, neoadjuvant chemotherapy helps reduce the risk by eliminating potential cancer cells and informing sensitive therapeutic regimens for postoperative adjuvant chemotherapy (5). Compared to surgery alone, the benefits of this approach have been demonstrated by multiple clinical trials (6–8). Currently, neoadjuvant chemotherapy is routinely used for the management of patients with resectable advanced-stage GC.
However, a considerable proportion of patients with GC do not respond well to neoadjuvant chemotherapy. So far, only some clinical features such as age and preoperative weight loss can be used for patient selection, but with very limited power. Moreover, it remains unclear how neoadjuvant chemotherapy rewires cancer signaling and affects the tumor microenvironment. A thorough understanding of these questions is crucial in developing an optimized multimodality treatment plan. Therefore, there is an urgent need to characterize molecular markers that predict treatment response and decipher how GC evolves under the pressure of neoadjuvant chemotherapy. Focusing on a cohort of patients with GC whose responses to neoadjuvant chemotherapy were rigorously defined, we used a multi-omics, multiple-sampling strategy to address these questions in a systematic way.
RESULTS
Overall study design
The study comprised two major aims. The first aim was to identify the key molecular features associated with response to neoadjuvant chemotherapy. For this purpose, we performed the multi-omics sequencing of biopsy tumor samples from 35 patients with GC before neoadjuvant chemotherapy (table S1 shows detailed patient and sample information; Fig. 1A). For these cases, we performed whole-exome sequencing (WES) of the tumor samples, together with matched germline DNA samples, which was mainly used to identify somatic base substitutions and small indels. For 32 of the 35 cases (3 cases were excluded because of insufficient DNA amount), we also performed whole-genome sequencing, together with matched germline samples, which were used to identify somatic copy number alterations (SCNAs). In addition, we performed RNA sequencing (RNA-seq) for all tumor samples to characterize the mRNA expression profiles of protein-coding genes. After the biopsy, the patients received 5-fluorouracil + oxaliplatin–based neoadjuvant chemotherapy for 2 to 4 cycles. Then, on the basis of a rigorous evaluation of radiological and pathological evidence by one radiologist and three independent pathologists, the cases were classified into two groups: response (n = 17) and nonresponse (n = 18). Figure 1B shows representative radiological and pathological images. In the response group, the necrosis rates for all the cases were higher than 65%, whereas in the nonresponse group, the rates were all lower than 15% (Fig. 1C). The Mandard tumor regression grade (9) for cases in the response group was less than or equal to two, whereas it was four or five for cases in the nonresponse group (Fig. 1D). In general, patients in the response group were younger than those in the nonresponse group (Wilcoxon rank sum test, nresponse versus nnonresponse = 17:18, P = 0.047; fig. S1A), and the patients in the nonresponse group tended to have advanced stages (Wilcoxon rank sum test, nresponse versus nnonresponse = 17:18, P = 0.023; fig. S1B).
Fig. 1. Study overview.
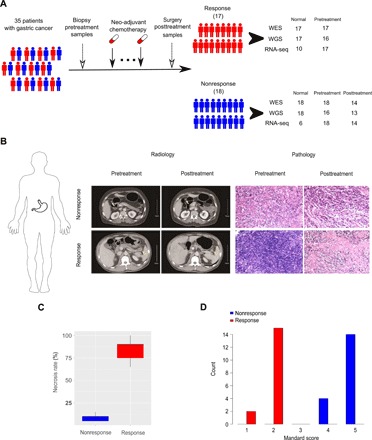
(A) Sample collection and multi-omics data generation. Our study included 35 patients with GC who received neoadjuvant chemotherapy before surgery. We collected pretreatment biopsy samples and posttreatment surgically resected tumor samples. On the basis of rigorously evaluated radiological and pathological evidence, patients were classified into a response group (n = 17) and a nonresponse group (n = 18). We obtained multi-omics data on the pretreatment samples and the nonresponse, posttreatment samples through whole-exome sequencing (WES), whole-genome sequencing (WGS), and RNA sequencing (RNA-seq). (B) The representative radiological and pathological images from responsive and nonresponsive patients. The yellow arrows indicate the lesion sites. (C) The necrosis rate distribution in the response and nonresponse groups. (D) Mandard tumor regression grading scores of the response and nonresponse groups.
The second aim was to study the genomic evolution of gastric tumors under neoadjuvant chemotherapy. To achieve this, we obtained fresh tumor samples from 14 of the 18 patients in the nonresponse group through surgery after they received neoadjuvant chemotherapy, and we performed WES on these posttreatment samples (table S1 and Fig. 1A). For these cases, we also performed whole-genome sequencing (for 13 cases) and RNA-seq, respectively. In contrast, for the patients in the response group, since a good response to neoadjuvant chemotherapy resulted in lesions with low tumor cell content, no tumor tissues could be obtained. Together, this experimental design allowed us to identify multiple types of molecular aberrations (somatic base substitutions, SCNAs, and gene expression) involved in the tumor response to neoadjuvant chemotherapy from different perspectives (response versus nonresponse, pretreatment versus posttreatment).
Key mutational features associated with response to neoadjuvant chemotherapy
Using WES data (tumor: range, 165 to 285× and median, 210×; normal: range, 88 to 143× and median, 114×), we first investigated the mutational patterns of pretreatment samples in the two patient groups. We used a multiple caller–based MC3 approach to call substitutions and small indels, which has shown better performance than a single caller (10). For the 35 pretreatment tumor samples, we identified an average of 550 substitutions (range, 3 to 3444) and 121 small indels (range, 1 to 1236). These numbers are comparable to those observed in the GC samples in The Cancer Genome Atlas using the same mutation calling approach (fig. S2A and table S2) (10). We further validated the somatic base substitutions using RNA-seq data from the same samples, and for those substitution positions with sufficient RNA-seq coverage, >81% of them were validated, suggesting a high accuracy of our mutation calling.
We examined the composition of six possible base-pair substitutions and found that T > G substitutions exhibited the most distinct pattern between the response and nonresponse groups, especially when the substitution site was flanked by C and T (Fig. 2A and fig. S2B). Consistently, on decomposing the mutational spectrum into different mutational signatures, the Catalogue of Somatic Mutations in Cancer (COSMIC) signature 17 (T > G) (11), a signature previously observed in esophageal and stomach cancers and linked to a by-product of oxidative damage (12), showed a much higher contribution in the response group than that in the nonresponse group (Wilcoxon rank sum test, P = 0.049, nresponse versus nnonresponse = 17:18; Fig. 2B). We next examined the distributions of microsatellite instability (MSI) scores calculated by Microsatellite Analysis for Normal Tumor InStability (MANTIS) (13) in these tumors. We found that MSI score was significantly higher in the nonresponse group than that in the response group (Wilcoxon rank sum test, P = 0.022, nresponse versus nnonresponse = 17:18; Fig. 2C). Accordingly, we tested whether there was a higher mutation burden in the nonresponse group and confirmed this pattern (Student’s t test, nresponse versus nnonresponse = 17:18, P = 0.04; Figs. 2D and 3A). Preclinical data have shown that colorectal tumors with MSI-H status are resistant to 5-fluorouracil–based chemotherapy (14, 15). Clinically, only patients with MSI-negative colorectal cancer, and not those with MSI-H, benefit from adjuvant chemotherapy (16), rendering the MSI-H status a strong predictive factor for nonresponse to 5-fluorouracil–based chemotherapy (17). Our results provide the first evidence that MSI-H status can also serve as a predictive marker for nonresponse to neoadjuvant chemotherapy for patients with GC.
Fig. 2. Mutation signatures in pretreatment GC samples.
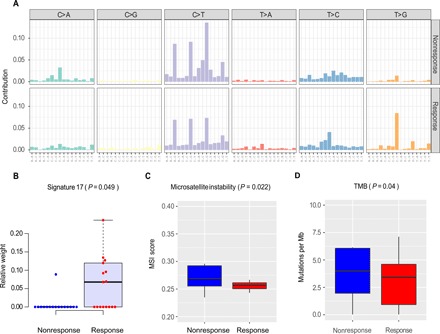
(A) Contributions of six possible substitution types at different nucleotide contexts. The relative weights of the COSMIC mutational signature 17 (B) and the microsatellite instability (MSI) scores (C) between the nonresponse and response groups. P value was based on Wilcoxon rank sum test. (D) Tumor mutation burden (TMB) distributions between the two groups. P value was based on one-tailed t test. (B to D) The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5× interquartile range of the lower and the upper quartiles, respectively.
Fig. 3. Significantly mutated genes in pretreatment GC samples.
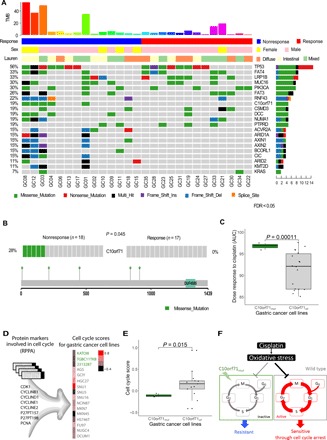
(A) Selected significantly mutated genes (SMGs) identified by MuSiC2 [false discovery rate (FDR) < 0.05] in pretreatment tumor samples. The bars on the top and on the right show the mutational rate observed for each patient and the composition of mutations in selected genes, respectively. Genes are ordered by their mutational frequencies, and different types of mutations are marked in different colors. (B) C10orf71 shows a significant mutation bias in the nonresponse samples (P < 0.045). The mutational sites are shown in the gene cartoon. (C) C10orf71 mutations are associated on the resistance to cisplatin in gastric cell lines; t test, P = 1.1 × 10−4. AUC, area under the curve. (D) Functional proteomic profiling of cell cycle based on eight protein markers in reverse-phase protein arrays. (E) C10orf71 mutations are associated on a lower cell cycle score in gastric cell lines; t test, P = 0.015. (F) A proposed mechanistic model in which C10orf71 mutations confer resistance to neoadjuvant chemotherapy through causing a less active cell cycle state.
To identify individual mutated genes that potentially play a role in affecting the treatment response, we next identified significantly mutated genes (SMGs) using MuSiC2 (18) [P = 2.9 × 10−4, false discovery rate (FDR) = 0.05, table S3]. The top mutated genes included TP53, PI3KCA, RNF43, ARIDA1, and KRAS, as previously reported in other GC cohorts (Fig. 3A) (19). Among the SMGs identified in this and previous studies (19), C10orf71 was the only gene showing a distinct pattern between the response and nonresponse groups: It was mutated in five nonresponse samples but in none of the response samples (Fisher’s exact test, P = 0.04, FDR < 0.25), and no recurrent mutations were detected (Fig. 3B). To validate this observation, based on drug response data using a panel of 21 GC cells (20), we indeed observed that the cell lines harboring C10orf71 mutations were more resistant to cisplatin (the equivalent platinum drug included in the neoadjuvant chemotherapy regimen) than those wild-type cell lines (Student’s t test, nmut versus nWT = 3:18, P = 1.1 × 10−4; Fig. 3C and table S4). To gain more mechanistic insights, we analyzed functional proteomic data of gastric cell lines using reverse-phase protein arrays (RPPAs). We found that the cell lines with C10orf71 mutations showed a significantly lower cell cycle score (based on eight protein markers) than those wild-type cell lines (Student’s t test, nmut versus nWT = 3:18, P = 0.015; Fig. 3, D and E, and table S4). Since cell cycle arrest is one major action mechanism for platinum-based drugs (21), we proposed a model in which C10orf71 mutations confer resistance to neoadjuvant chemotherapy through causing a less active cell cycle state (Fig. 3F). Although further experiments are required to validate the proposed model, these results suggest that mutations in this gene are potential biomarkers for the resistance to neoadjuvant chemotherapy.
Key SCNAs associated with response to neoadjuvant chemotherapy
Using low-pass whole-genome sequencing data (tumor: range, 5 to 9× and median, 7×; normal: range, 5 to 9× and median, 7×), we next investigated the SCNAs that could distinguish the response group from the nonresponse group. We identified significantly amplified or deleted peaks for these two groups using GISTIC2.0 (FDR = 0.1) (22). While the deletion peak profiles of the response and nonresponse tumors were similar (fig. S3A), the response group contained two unique amplification peaks: one at 8q24.21, which contains a major driver gene, MYC, and another at 19q12, which harbors a cancer gene, CCNE1. Meanwhile, the nonresponse group contained one unique amplification peak at 12q15, which contains MDM2, a negative regulator of TP53 (Fig. 4A) (23). We observed similar peaks using parallel WES data (fig. S3B). This analysis suggests initial SCNA candidates for further analyses.
Fig. 4. Significant SCNAs and their downstream signaling effects in pretreatment GC samples.
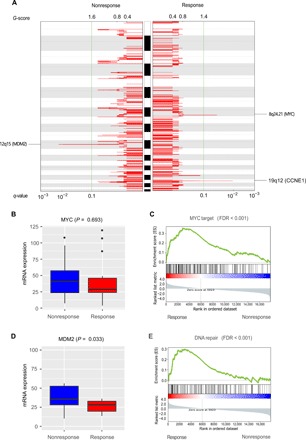
(A) Amplification signals for SCNAs plotted for response versus nonresponse groups. Two cancer genes, MYC and CCNE1, reside in the unique peaks at 8q24.21 and 19q21, respectively, in the response group, while MDM2, a negative regulator of TP53, resides in the unique peak at 12q15 in the nonresponse group. MYC (B) and MDM2 (D) mRNA expression levels in the nonresponse and response groups. P values were based on one-tailed Wilcoxon rank sum test. The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5× interquartile range of the lower and the upper quartiles, respectively. Enrichment of MYC target genes (C) and DNA repair pathway (E) in the up-regulated genes in the response group relative to the nonresponse group. FDR was based on gene set enrichment analysis.
We reasoned that if these amplification peaks played a role in affecting the response to neoadjuvant chemotherapy, then we would observe corresponding signals in their related downstream pathways. Therefore, using RNA-seq data, we next examined the mRNA expression profiles of these pretreatment samples. Although we did not observe a significantly differential expression of MYC between the two groups (Fig. 4B; t test, P = 0.6), the MYC target genes showed a significant enrichment of up-regulated genes in the response group relative to the nonresponse group (Fig. 4C; gene set enrichment analysis, nominal P = 0.0, FDR < 10−3), suggesting that MYC signaling was indeed activated in the response group. The association of MYC amplification with better response to similar chemotherapies has been reported in multiple diseases including breast cancer (24), small cell lung cancer (25), and colorectal cancer (26). Consistent with its amplification peak in the nonresponse group, MDM2 showed a significantly higher mRNA expression level in the group (Fig. 4D; Student’s t test, nresponse versus nnonresponse = 17:18, P = 0.033). Moreover, the DNA repair pathway showed a corresponding up-regulation in the response group relative to nonresponse, supporting a negative regulatory effect of MDM2 on DNA repair (Fig. 4E; gene set enrichment analysis, nominal P = 0.0, FDR < 10−3). The association of MDM2 amplification with worse clinical outcome to chemotherapy has been reported in breast cancer (27) and pancreatic cancer (28). We did not observe significant changes for CCNE1 gene itself or its related pathway. Collectively, these results suggest that some key SCNAs may contribute to the sensitivity (e.g., MYC amplification) or resistance (e.g., MDM2 amplification) to GC neoadjuvant chemotherapy.
Key somatic base substitution changes following neoadjuvant chemotherapy
Using the parallel WES data of 14 matched pre- and posttreatment sample pairs (average coverage, >200×), we examined the genomic evolution of GC under neoadjuvant chemotherapy. Overall, we did not detect significant changes in mutation burden or mutational signatures. For known cancer drivers, some tumors showed key mutational changes (mutation loss or gain), while others showed the same mutational profile before and after the treatment (Fig. 5A). To systematically identify the genes or pathways showing the most recurrent mutational changes, we used HotNet2 (29) to search for the protein interaction networks enriched for such signals. One top subnetwork identified consisted of IRS1, IRS2, PIK3CA, JAK1, and IL6ST (Fig. 5B). In particular, IRS1, which plays a key role in transmitting signals from the insulin and insulin-like growth factor-1 receptors to phosphatidylinositol 3-kinase/AKT pathway (30), consistently acquired new mutations in five posttreatment samples (Fisher’s exact test, P = 0.041), suggesting a recurrent resistance mechanism. Our analysis of response versus nonresponse pretreatment samples suggested that mutations in C10orf71 contribute to treatment resistance. If so, then we would expect increased allele mutation frequency or gain of new mutations in posttreatment samples, since tumor cells sensitive to the chemotherapy (i.e., those without such mutations) would likely be removed by selection during the treatment. Among the 14 sample pairs surveyed, four cases showed C10orf71 mutations in the pre- or posttreatment samples, two of which acquired new mutations following neoadjuvant chemotherapy. Across the eight mutation sites detected, we observed a significant increase in mutation allele frequency compared to the pretreatment samples (Fig. 5C; Student’s paired t test, n = 8 pairs, P < 0.05; even after adjusting for tumor purity, the same pattern still held true). Figure 5 (D and E) shows the schematic representation of the putative evolution of the two cases in which the acquired C10orf71 mutation increased its allele frequency gradually. Although the true evolutionary trajectory is hard to validate, this result suggests the critical role of C10orf71 mutations in affecting the tumor response.
Fig. 5. Mutational evolution following neoadjuvant chemotherapy.
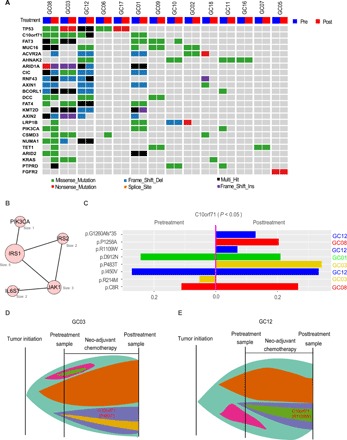
(A) Mutational profiles in cancer genes before and after neoadjuvant chemotherapy. (B) The top subnetwork enriched in mutational alterations following the treatment. The size of the circle indicates the number of samples with a mutation in the network. (C) The mutational allele frequencies in the coding region of C10orf71 before and after treatment in four patients. P value was based on paired t test. Mutations in different patients are shown in different colors. (D and E) A schematic representation of the putative evolution of the acquired C10orf71 mutations in the two patients.
Using exome-wide somatic base substitution data, we next tentatively inferred the evolution of the 14 GC cases using sciClone (31) and found a variety of clonal behaviors. Some cases showed limited changes in the major clonal composition, whereas the others showed a number of subclone loss and gain including driver mutations (fig. S4). Overall, we did not detect a significant difference in clone numbers (pretreatment, 3.7 versus posttreatment, 3.3., P = 0.43). Using whole-genome sequencing data of 13 matched pairs, we found very similar SCNA profiles between pretreatment and posttreatment samples (fig. S5).
Key gene expression and cell composition changes following neoadjuvant chemotherapy
Last, using parallel RNA-seq data of 14 pre- and posttreatment sample pairs (33 million reads per sample), we investigated the perturbed gene expression profiles following neoadjuvant therapy. We identified 869 differentially expressed genes (DESEQ2, n = 14 pairs, fold change, >2, P < 5 × 10−3, FDR < 0.05; Fig. 6A). These genes were significantly enriched in several down-regulated pathways, including inflammatory response, allograft rejection, KRAS signaling, and interleukin-6 (IL-6)/Janus kinase/signal transducer and activator of transcription 3 signaling (Fig. 6B). Our analysis of response versus nonresponse pretreatment samples suggested that MYC amplification and subsequent MYC signaling activation make tumor cells sensitive to the treatment. If so, then we would expect that in tumor samples with MYC amplification, the MYC signaling would be “deactivated” by purifying selection during evolution. We found that for seven MYC-amplified tumors, nearly half of their MYC target genes (47.2%, 93 of 197) were significantly down-regulated in nonresponse posttreatment tumors (Student’s paired t test, n = 7 pairs, P < 0.05), and compared to other genes, this proportion was much higher (Fisher’s exact test, P < 10−3). In contrast, only 3% of the MYC target genes were significantly up-regulated in these samples (Fig. 6C). These results further support a link between MYC amplification and sensitivity to neoadjuvant chemotherapy. We also examined the expression of several therapeutic targets currently being used in GC treatment and found that HER2, VEGFR1, and VEGFR2 were significantly down-regulated (paired Wilcoxon rank sum test, n = 14 pairs, P < 0.05, FDR < 0.1; Fig. 6D and fig. S6A).
Fig. 6. Changes in gene expression and tumor-infiltrating immune cell following neoadjuvant chemotherapy.
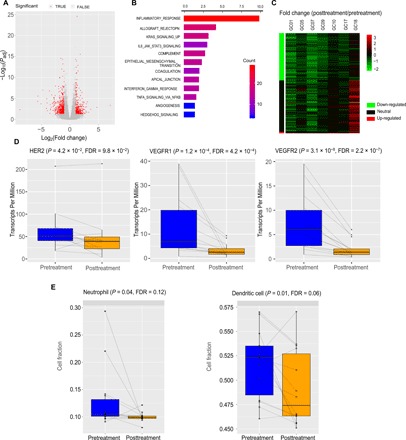
(A) Volcano plot showing differentially expressed genes between matched pre- and posttreatment samples. Significant genes are shown in red (fold change > 2, FDR < 0.05). (B) Pathways that are significantly down-regulated following neoadjuvant chemotherapy. Significantly differentially expressed genes were identified at fold change >2, FDR < 0.05. The bar color indicates the number of differentially expressed genes in the pathway. (C) A heatmap showing mRNA expression fold changes (posttreatment/pretreatment) of MYC target genes driven by the treatment in the MYC-amplified tumors. Genes with a significant differential expression (paired t test, P < 0.05) are marked in green (down-regulated) and red (up-regulated). (D) Differential expression of GC therapeutic targets in the pre- and posttreatment samples. P values were calculated on the basis of paired Wilcoxon rank sum test. (E) The fractions of neutrophil and dendritic cells in the pre- and posttreatment samples. P values were calculated on the basis of paired t test. (D and E) The middle line in the box is the median, the bottom and top of the box are the first and third quartiles, and the whiskers extend to 1.5× interquartile range of the lower and the upper quartiles, respectively.
To study the impact of neoadjuvant chemotherapy on the tumor microenvironment, we inferred the tumor-infiltrating immune cell abundance (including B cells, CD8 T cells, CD4 T cells, neutrophils, macrophages, and dendritic cells) from mRNA expression data using Tumor IMmune Estimation Resource (TIMER) (32). We found significantly decreased cell compositions for neutrophils and dendritic cells (Fig. 6E; Student’s paired t test, n = 14 pairs, P < 0.05, FDR < 0.12) and a marginal decrease for CD8 T cells (fig. S6B; Student’s paired t test, n = 14 pairs, P < 0.1, FDR < 0.2) posttreatment. Together, our RNA-seq analyses suggest that neoadjuvant chemotherapy can not only reshape immune signaling in the GC cells but can also modify their immune microenvironment.
DISCUSSION
Neoadjuvant chemotherapy is a widely applied treatment option for patients with GC, and several large clinical trials have demonstrated its benefits in improving prognosis and identifying responsive patients before therapy initiation, thus allowing better treatment planning. Here, we conducted a well-designed study with the aims of (i) identifying potential biomarkers that can select patients who would benefit from this treatment and (ii) elucidating the related resistance mechanisms. Our study has several key features. First, the clinical response to GC neoadjuvant chemotherapy was rigorously defined by three independent pathologists; only those samples with a consensus assessment were included, thereby reducing the effects of phenotypic noise on data mining. Second, we used a multi-omics characterization strategy by combining whole-exome, whole-genome, and RNA-seq, enabling us to detect different types of molecular aberrations. Furthermore, integrated analyses across different datasets helped us identify high-confidence signals. Third, for nonresponse tumors, we sequenced matched pre- and posttreatment samples, and their comparison is orthogonal to the comparison of response versus nonresponse tumors, further validating the patterns of interest. This integrated design and the multilayer analytic strategy greatly helped us overcome the potential limitation of sample size, commonly observed in similar studies.
Our study identified several molecular features associated with the tumor response to neoadjuvant chemotherapy, including C10orf71 and IRS1 mutations, and MYC and MDM2 amplifications. In particular, MSI status is currently being used to predict therapy response in colorectal cancer, and we observed a similar pattern, strongly supporting the potential of this feature as a clinical marker to manage therapy in patients with GC. Further efforts are warranted to validate the power of these potential marker candidates and build robust multifeature predictive models using independent GC patient cohorts. Our analysis also reveals the effects of neoadjuvant chemotherapy on tumor immune signaling and tumor immune microenvironment, which may have significant clinical implications in the use of immunotherapy. Together, our study represents a significant step toward optimized treatment strategies for individual patients with GC.
MATERIALS AND METHODS
Patient recruitment and sample cohort
Patients with GC (>T2N+M0, Union for International Cancer Control-American Joint Committee on Cancer, eighth edition) in this study were recruited at Peking University Cancer Hospital (Beijing, China). The study was conducted in accordance with the Declaration of Helsinki and approved by the Peking University Cancer Hospital Ethics Committee (Institutional Review Board approval number, 2019KT05). All patients provided written informed consent before treatment, sample collection, and analysis. We collected tumor and matched adjacent nontumor tissues through biopsy before the neoadjuvant treatment. For patients without available adjacent nontumor tissues, we collected blood samples instead. All patients then received the fluorouracil-based treatment regimen of capecitabine/S-1 + oxaliplatin {XELOX (Oxaliplatin+Capecitabine) (oxaliplatin, 130 mg/m2, intravenously, day 1; and capecitabine, 1000 mg/m2, orally, days 1 to 14) or SOX [Oxaliplatin+S-1(Tegafur/gimeracil/oteracil)] (oxaliplatin 130 mg/m2, intravenously, day 1; and S-1, 40–60 mg, twice a day, orally, days 1 to 14)} for 2 to 4 cycles and were evaluated for response to treatment before surgery. The pathological features were evaluated according to Mandard tumor regression grading (TRG) score, which was performed on all the patients on the basis of the standard criteria by three independent, blinded pathologists (Zhongwu Li, Yiqiang Liu, and Zhang Li). A response was identified if there was no evidence that tumors had upstaged and had a Mandard TRG score of 1 or 2; nonresponse was classified as tumors that were upstaged and had a Mandard TRG score of 4 or 5; and tumors with a Mandard TRG score of 3 were excluded (9). To reduce the classification noise, we only focused on patients with consensus assessments from the three pathologists and lastly included 35 patients (17 response versus 18 nonresponse) as the study cohort. We further collected tumor samples after surgery. For the patients with a response, no posttreatment tumor samples were collected.
Multi-omics data generation
For the 35 patients with GC, genomic DNA from tissue (or blood) samples was extracted using the AllPrep DNA Mini Kit (QIAGEN) and the QIAamp DNA blood mini kit (QIAGEN), respectively. For WES, 1 μg of DNA was sheared to short fragments [150 to 250 base pairs (bp)] using Bioruptor (Diagenode). The resulting DNA fragments were repaired. Adaptors were then ligated to both ends of the fragments. DNA fragments of the targeted size were selected. Afterward, polymerase chain reaction (PCR) was performed, and the resulting mixture was purified. Exome capture was performed using SureSelect Human All Exon V6 (Agilent) according to the manufacturer’s protocol. The hybridized mixtures were then amplified with PCR. Validated DNA libraries were then sequenced on Illumina NovaSeq 6000. For whole-genome sequencing, 1 μg of genomic DNA was sheared to 150 to 250 bp using Bioruptor (Diagenode). Then, DNA libraries were generated using the standard protocols of the TruSeq nano DNA kit (Illumina). The libraries were sequenced with paired-end runs using Illumina NovaSeq 6000 to a minimal depth of 6× base coverage. For RNA-seq, we extracted total RNA from fresh tissue using the AllPrep RNA mini kit (QIAGEN). For each sample, 3 μg of total RNA was used to generate the libraries through the TruSeq RNA v2 kit (Illumina). The libraries were sequenced on Illumina NovaSeq 6000 and 33 million 2 × 150–bp paired ends per sample on average.
Mutational data analysis
WES read pairs were trimmed and only read pairs with <3% N bases and >50% high-quality bases were kept. The resulting high-quality reads were aligned to the human reference genome (Homo_sapiens_assembly19) using Burrows-Wheeler Aligner 0.7.17 (33). To improve the alignment accuracy, we used the Genome Analysis Toolkit (version 3.8.1) (34) to process Binary Alignment Map (BAM) files through the steps including marking duplicates, local realignment around high-confidence insertion and deletions, and base quality recalibration. On the basis of ~7000 high-frequency single-nucleotide polymorphism sites, we confirmed matched pre-, posttreatment, and normal samples from the same patients. We used the variant calling pipeline developed by The Cancer Genome Atlas MC3 project to identify high-confidence somatic base substitutions and indels (10). Briefly, this pipeline uses six callers to call substitution mutations and three callers to identify indels with detailed annotation information. We only kept substitution mutations and indels supported by at least two callers for further analyses. We further validated WES somatic base substitutions based on RNA-seq data from the same samples. TopHat2 was used to generate the alignment (35). For each substitution position, we counted the coverage and the numbers of mutated reads based on RNA-seq BAM files from the same sample. For the positions with sufficient RNA-seq coverage (≥10×), we considered those with at least two reads supporting the base substitutions of interest validated. Mutational signatures in pretreatment samples were determined using the R package deconstructSigs v1.8.0 based on COSMIC signatures as mutation signature matrix (36). The MSI status for each tumor was evaluated using MANTIS (13), and 2539 loci obtained from the mSINGS package (37) were used for this analysis. We used MuSiC2 (18) to identify SMGs (FDR < 0.05) in all pretreatment samples. Maftools (38) was used to explore the SMGs that showed a significant mutational bias between nonresponse and response samples based on Fisher’s exact test. To detect the gene group enriched with base substitution changes following the treatment, we used HotNet2 (29) to identify significantly mutated subnetworks based on heat scores of each protein. We only kept genes with altered events; an altered event was defined as different nonsilent variants of the same patient before and after treatment. Heat scores were limited to major drivers or genes with at least two altered events. HINT + HI2012, MultiNet, and iRefIndex interaction network were used, and consensus subnetworks were visualized. Fisher’s exact test was used to determine the statistical significance.
Drug response assays and analysis
The cisplatin response data (area under the dose response curve) of the 21 GC cell lines with Cancer Cell Line Encyclopedia (CCLE) mutational profiling data were obtained from Genomics of Drug Sensitivity in Cancer (20). Among the cell lines, 3 contained nonsilent single-nucleotide variations in C10orf71 and 18 were wild type. Student t test was used to assess the difference between the cell lines with C10orf71 mutation and those with the wild type.
Reverse-phase protein array profiling data analysis
RPPA data were generated by the RPPA core facility at MD Anderson Cancer Center as previously described (39). RPPA slides were first quantified using ArrayPro (Meda Cybernetics) to generate signal intensities, then processed by SuperCurve to estimate the relative protein expression level, and were normalized by median polish. Protein markers were classified into 11 signaling pathways, and the pathway scores were calculated with the direction of protein members considered (40). Student t test was used to assess the difference between the cell lines with C10orf71 mutation and those with the wild type, and FDR was used for multiple testing correction.
Analysis of SCNAs
We used whole-genome sequencing data to infer copy number values in pre- and posttreatment samples. WES read pairs were trimmed following the criterion used for WES data. BWA 0.7.17 was used to align reads to the human reference genome (Homo_sapiens_assambly19). SCNAs were then estimated on the basis of matched normal-tumor pairs through Control-FREEC (version 11.5), with a window size of 5 kb (41). On the basis of the corresponding segmentation values, we used GISTIC2.0 (22) to identify regions with a statistically significant frequency of copy number alterations; the amplification and deletion thresholds were set to 0.1. Using the same tools, we performed a parallel analysis on the WES data as an independent dataset to validate the SCNA peaks identified through whole-genome sequencing.
Clonal structure analysis of pretreatment and posttreatment samples
To explore the clonal evolution driven by neoadjuvant chemotherapy, we inferred the dynamic changes of clonal structures using sciClone (31). The copy number values based on WES data inferred through Control-FREEC were used to exclude SNVs from copy-number alteration regions for the clonal analysis. We further validated the inferred subclonal evolution using another tool, PyClone (42).
Gene-expression analysis
We used an alignment-free tool, Kallisto (43), to quantify the gene expression based on RNA-seq data. We converted the output of Kallisto to gene-level counts and Transcripts Per Million (TPMs) using tximport (44). To identify differentially expressed genes between the response and nonresponse groups, we performed a t test for each gene and ranked them on the basis of the t statistic. We used gene set enrichment analysis based on the prerank to identify enriched hallmark pathways (45). To identify genes differentially expressed between the tumor samples of different treatment stages of nonresponders, we fitted a multifactor model with patient ID as the blocking factor and then conducted Wald test for the treatment stage effect using DESeq2 (46). Genes with FDR < 0.05 and fold change >2.0 or < 0.5 were selected as differentially expressed genes. We performed overrepresentation analysis (47) of MSigDB hallmark gene sets implemented by clusterProfiler (48) for up-regulated and down-regulated genes in the posttreatment samples separately. We further used Student’s t tests to compare the differential gene expression of MYC target genes before and after the treatment in MYC-amplified nonresponders. We inferred tumor-infiltrating immune cell abundance from mRNA expression data using TIMER (32). We performed paired Student’s t tests to identify differences in cell composition between the matched pre- and posttreatment samples.
Supplementary Material
Acknowledgments
We thank K. Mojumdar for editorial assistance. Funding: This study was supported by the Peking University Clinical Scientist Program; National Science Foundation Fund (31870805); The Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201701); “San Ming” Project of Shenzhen City (SZSM201612051); and the Suzhou New District, Jiangsu Province, China. Author contributions: H.L. and J.J. conceived and designed the research. Ziyu Li and X.G. contributed to the collection of specimens, design of the analysis, and discussion of clinical significance. X.P. and M.-J.C. led the data analysis. Zhe Li, Bin Wei, and H.L. contributed to the data analysis. Zhongwu Li, Y.L., and Li Zhang contributed to pathological evaluation. L.T. contributed to radiological evaluation. Other authors contributed to the collection of specimens and performed the experiments. Ziyu Li, X.G., X.P., H.L., and J.J. wrote the manuscript, with input from all authors. H.L. supervised the whole project. Competing interests: X.P., Zhe Li, Bin Wei, Baoye Wei, and Y.D. are full-time employees, and H.L. is a shareholder and scientific advisor of Precision Scientific Ltd. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The raw sequence data reported in this study have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers PRJCA001620, which are publicly accessible at http://bigd.big.ac.cn/gsa. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay4211/DC1
Fig. S1. The age and stage distributions of patients with GC in this study.
Fig. S2. Somatic base substitution summary.
Fig. S3. SCNA signal profiles identified by GISTIC2.0 in the nonresponse and response groups.
Fig. S4. Variant allele frequency in pre- and posttreatment samples based on WES data.
Fig. S5. SCNA signal profiles identified by GISTIC2.0 in pre- and posttreatment samples.
Fig. S6. Changes in gene expression and immune cell compositions before and after treatment.
Table S1. The summary of patient clinical information and sequencing data.
Table S2. Somatic mutations identified in this study.
Table S3. SMGs identified in this study.
Table S4. GC cell lines profiled for RPPAs.
REFERENCES AND NOTES
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A., Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Chen W., Zheng R., Baade P. D., Zhang S., Zeng H., Bray F., Jemal A., Yu X. Q., He J., Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Yang L., Zheng R., Wang N., Yuan Y., Liu S., Li H., Zhang S., Zeng H., Chen W., Incidence and mortality of stomach cancer in China, 2014. Chin. J. Cancer Res. 30, 291–298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkin D. M., Global cancer statistics in the year 2000. Lancet Oncol. 2, 533–543 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Newton A. D., Datta J., Loaiza-Bonilla A., Karakousis G. C., Roses R. E., Neoadjuvant therapy for gastric cancer: Current evidence and future directions. J. Gastrointest. Oncol. 6, 534–543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham D., Allum W. H., Stenning S. P., Thompson J. N., van de Velde C., Nicolson M., Scarffe J. H., Lofts F. J., Falk S. J., Iveson T. J., Smith D. B., Langley R. E., Verma M., Weeden S., Chua Y. J.; MAGIC Trial Participants , Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 355, 11–20 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Hartgrink H. H., van de Velde C. J. H., Putter H., Songun I., Tesselaar M. E., Kranenbarg E. K., de Vries J. E., Wils J. A., van der Bijl J., van Krieken J.; Cooperating Investigators of The Dutch Gastric Cancer Group , Neo-adjuvant chemotherapy for operable gastric cancer: Long term results of the Dutch randomised FAMTX trial. Eur. J. Surg. Oncol. 30, 643–649 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Ychou M., Boige V., Pignon J.-P., Conroy T., Bouché O., Lebreton G., Ducourtieux M., Bedenne L., Fabre J.-M., Saint-Aubert B., Genève J., Lasser P., Rougier P., Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: An FNCLCC and FFCD multicenter phase III trial. J. Clin. Oncol. 29, 1715–1721 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Mandard A. M., Dalibard F., Mandard J. C., Marnay J., Henry-Amar M., Petiot J. F., Roussel A., Jacob J. H., Segol P., Samama G., Ollivier J.-M., Bonvalot S., Gignoux M., Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma. Clinicopathologic correlations. Cancer 73, 2680–2686 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Ellrott K., Bailey M. H., Saksena G., Covington K. R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K. E., McLellan M., Sofia H. J., Hutter C., Getz G., Wheeler D., Din L.; MC3 Working Group; Cancer Genome Atlas Research Network , Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexandrov L. B., Jones P. H., Wedge D. C., Sale J. E., Campbell P. J., Nik-Zainal S., Stratton M. R., Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomkova M., Tomek J., Kriaucionis S., Schuster-Böckler B., Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biol. 19, 129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kautto E. A., Bonneville R., Miya J., Yu L., Krook M. A., Reeser J. W., Roychowdhury S., Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 8, 7452–7463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hewish M., Lord C. J., Martin S. A., Cunningham D., Ashworth A., Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat. Rev. Clin. Oncol. 7, 197–208 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Koopman M., Venderbosch S., van Tinteren H., Ligtenberg M. J., Nagtegaal I., Van Krieken J. H., Punt C. J., Predictive and prognostic markers for the outcome of chemotherapy in advanced colorectal cancer, a retrospective analysis of the phase III randomised CAIRO study. Eur. J. Cancer 45, 1999–2006 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Ribic C. M., Sargent D. J., Moore M. J., Thibodeau S. N., French A. J., Goldberg R. M., Hamilton S. R., Laurent-Puig P., Gryfe R., Shepherd L. E., Tu D., Redston M., Gallinger S., Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 349, 247–257 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Des Guetz G., Schischmanoff O., Nicolas P., Perret G.-Y., Morere J.-F., Uzzan B., Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur. J. Cancer 45, 1890–1896 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Dees N. D., Zhang Q., Kandoth C., Wendl M. C., Schierding W., Koboldt D. C., Mooney T. B., Callaway M. B., Dooling D., Mardis E. R., Wilson R. K., Ding L., MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 22, 1589–1598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network , Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W., Soares J., Greninger P., Edelman E. J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J. A., Thompson I. R., Ramaswamy S., Futreal P. A., Haber D. A., Stratton M. R., Benes C., McDermott U., Garnett M. J., Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dasari S., Tchounwou P. B., Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mermel C. H., Schumacher S. E., Hill B., Meyerson M. L., Beroukhim R., Getz G., GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chène P., Inhibiting the p53–MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 3, 102–109 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Pereira C. B. L., Leal M. F., Abdelhay E. S. F. W., Demachki S., Assumpção P. P., de Souza M. C., Moreira-Nunes C. A., da Silva Tanaka A. M., Smith M. C., Rodríguez Burbano R., MYC amplification as a predictive factor of complete pathologic response to docetaxel-based neoadjuvant chemotherapy for breast cancer. Clin. Breast Cancer 17, 188–194 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Mollaoglu G., Guthrie M. R., Böhm S., Brägelmann J., Can I., Ballieu P. M., Marx A., George J., Heinen C., Chalishazar M. D., Cheng H., Ireland A. S., Denning K. E., Mukhopadhyay A., Vahrenkamp J. M., Berrett K. C., Mosbruger T. L., Wang J., Kohan J. L., Salama M. E., Witt B. L., Peifer M., Thomas R. K., Gertz J., Johnson J. E., Gazdar A. F., Wechsler-Reya R. J., Sos M. L., Oliver T. G., MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to Aurora kinase inhibition. Cancer Cell 31, 270–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L., Xue M., Chung D. C., c-Myc is regulated by HIF-2α in chronic hypoxia and influences sensitivity to 5-FU in colon cancer. Oncotarget 7, 78910–78917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki A., Toi M., Yamamoto Y., Saji S., Muta M., Tominaga T., Role of MDM2 overexpression in doxorubicin resistance of breast carcinoma. Jpn. J. Cancer Res. 89, 221–227 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S.-H., Lee J.-C., Guo J.-C., Kuo S.-H., Tien Y.–. W., Kuo T.-C., Cheng A.-L., Yeh K.-H., Association of MDM2 expression with shorter progression-free survival and overall survival in patients with advanced pancreatic cancer treated with gemcitabine-based chemotherapy. PLOS ONE 12, e0180628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leiserson M. D., Vandin F., Wu H.-T., Dobson J. R., Eldridge J. V., Thomas J. L., Papoutsaki A., Kim Y., Niu B., McLellan M., Lawrence M. S., Gonzalez-Perez A., Tamborero D., Cheng Y., Ryslik G. A., Lopez-Bigas N., Getz G., Ding L., Raphael B. J., Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 47, 106–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dearth R. K., Cui X., Kim H.-J., Hadsell D. L., Lee A. V., Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle 6, 705–713 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Miller C. A., White B. S., Dees N. D., Griffith M., Welch J. S., Griffith O. L., Vij R., Tomasson M. H., Graubert T. A., Walter M. J., Ellis M. J., Schierding W., DiPersio J. F., Ley T. J., Mardis E. R., Wilson R. K., Ding L., SciClone: Inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLOS Comput. Biol. 10, e1003665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B., Severson E., Pignon J.-C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J. C., Rodig S., Signoretti S., Liu J. S., Liu X. S., Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L., TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenthal R., McGranahan N., Herrero J., Taylor B. S., Swanton C., deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salipante S. J., Scroggins S. M., Hampel H. L., Turner E. H., Pritchard C. C., Microsatellite instability detection by next generation sequencing. Clin. Chem. 60, 1192–1199 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Mayakonda A., Lin D.-C., Assenov Y., Plass C., Koeffler H. P., Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghandi M., Huang F. W., Jané-Valbuena J., Kryukov G. V., Lo C. C., McDonald E. R. III, Barretina J., Gelfand E. T., Bielski C. M., Li H., Hu K., Andreev-Drakhlin A. Y., Kim J., Hess J. M., Haas B. J., Aguet F., Weir B. A., Rothberg M. V., Paolella B. R., Lawrence M. S., Akbani R., Lu Y., Tiv H. L., Gokhale P. C., de Weck A., Mansour A. A., Oh C., Shih J., Hadi K., Rosen Y., Bistline J., Venkatesan K., Reddy A., Sonkin D., Liu M., Lehar J., Korn J. M., Porter D. A., Jones M. D., Golji J., Caponigro G., Taylor J. E., Dunning C. M., Creech A. L., Warren A. C., McFarland J. M., Zamanighomi M., Kauffmann A., Stransky N., Imielinski M., Maruvka Y. E., Cherniack A. D., Tsherniak A., Vazquez F., Jaffe J. D., Lane A. A., Weinstock D. M., Johannessen C. M., Morrissey M. P., Stegmeier F., Schlegel R., Hahn W. C., Getz G., Mills G. B., Boehm J. S., Golub T. R., Garraway L. A., Sellers W. R., Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J., Zhao W., Akbani R., Liu W., Ju Z., Ling S., Vellano C. P., Roebuck P., Yu Q., Eterovic A. K., Byers L. A., Davies M. A., Deng W., Gopal Y. N. V., Chen G., von Euw E. M., Slamon D., Conklin D., Heymach J. V., Gazdar A. F., Minna J. D., Myers J. N., Lu Y., Mills G. B., Liang H., Characterization of human cancer cell lines by reverse-phase protein arrays. Cancer Cell 31, 225–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O., Barillot E., Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth A., Khattra J., Yap D., Wan A., Laks E., Biele J., Ha G., Aparicio S., Bouchard-Côté A., Shah S. P., PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bray N. L., Pimentel H., Melsted P., Pachter L., Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Soneson C., Love M. I., Robinson M. D., Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyle E. I., Weng S., Gollub J., Jin H., Botstein D., Cherry J. M., Sherlock G., GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu G., Wang L.-G., Han Y., He Q.-Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay4211/DC1
Fig. S1. The age and stage distributions of patients with GC in this study.
Fig. S2. Somatic base substitution summary.
Fig. S3. SCNA signal profiles identified by GISTIC2.0 in the nonresponse and response groups.
Fig. S4. Variant allele frequency in pre- and posttreatment samples based on WES data.
Fig. S5. SCNA signal profiles identified by GISTIC2.0 in pre- and posttreatment samples.
Fig. S6. Changes in gene expression and immune cell compositions before and after treatment.
Table S1. The summary of patient clinical information and sequencing data.
Table S2. Somatic mutations identified in this study.
Table S3. SMGs identified in this study.
Table S4. GC cell lines profiled for RPPAs.