

Abstract
Cytochrome c (Cytc)1is a cellular life and death decision molecule that regulates cellular energy supply and apoptosis through tissue specific post-translational modifications. Cytc is an electron carrier in the mitochondrial electron transport chain (ETC) and thus central for aerobic energy production. Under conditions of cellular stress, Cytc release from the mitochondria is a committing step for apoptosis, leading to apoptosome formation, caspase activation, and cell death. Recently, Cytc was shown to be a target of cellular signaling pathways that regulate the functions of Cytc by tissue-specific phosphorylations. So far five phosphorylations sites of Cytc have been mapped and functionally characterized, Tyr97, Tyr48, Thr28, Ser47, and Thr58. All five phosphorylations partially inhibit respiration, which we propose results in optimal intermediate mitochondrial membrane potentials and low ROS production under normal conditions. Four of the phosphorylations result in inhibition of the apoptotic functions of Cytc, suggesting a cytoprotective role for phosphorylated Cytc. Interestingly, these phosphorylations are lost during stress conditions such as ischemia. This results in maximal ETC flux during reperfusion, mitochondrial membrane potential hyperpolarization, excessive ROS generation, and apoptosis. We here present a new model proposing that the electron transfer from Cytc to cytochrome c oxidase is the rate-limiting step of the ETC, which is regulated via post-translational modifications of Cytc. This regulation may be dysfunctional in disease conditions such as ischemia-reperfusion injury and neurodegenerative disorders through increased ROS, or cancer where post-translational modifications on Cytc may provide a mechanism to evade apoptosis.
Keywords: Cytochrome c, cell signaling, phosphorylation, respiration, apoptosis, reactive oxygen species
1. Introduction
The mitochondrial electron transport chain (ETC) consumes oxygen as part of the oxidative phosphorylation (OxPhos) process to produce the majority of cellular ATP, the energy molecule of life. An average human produces 65 kg of ATP per day under resting conditions, and energy production can increase several-fold during exercise (Rich, 2003). Because of its essential role, OxPhos should be tightly regulated for optimal functioning of an organism. The ETC is comprised of four enzyme complexes and two electron carriers: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), the small non-protein electron carrier ubiquinone, bc1 complex (complex III), the electron carrier cytochrome c (Cytc), and cytochrome c oxidase (COX; complex IV) (Fig. 1). Electrons enter the ETC from NADH via complex I. Complex II links the ETC with the citric acid cycle, directly feeding electrons from FADH2 into the ubiquinone/ubiquinol pool. Except for complex II, complexes I, III, and IV pump protons across the inner mitochondrial membrane to generate the proton motive force (Δp), which consists of the pH gradient (ΔpH) and the mitochondrial membrane potential (ΔΨm), the latter of which is the primary component of Δp in mammals. The proton gradient is utilized by ATP synthase (complex V) to produce ATP. Dysregulation of the ETC can result in many pathological conditions due to lack of energy or mitochondrial membrane potential hyperpolarization, resulting in the generation of harmful reactive oxygen species (ROS) and the release of Cytc, which triggers apoptosis. Once Cytc is released into the cytosol from the mitochondria, it interacts with the protein apoptosis protease activating factor-1 (Apaf-1), which results in the formation of the apoptosome, leading to downstream caspase activation and cell death (Fig. 1). It should also be noted that Cytc, a small 104 amino acid globular protein in mammals with a covalently attached heme group, has many other functions. These functions either promote cell survival or cell death, including ROS scavenging, redox-coupled protein import via Erv1-Mia40 pathway, ROS formation via p66Shc, and cardiolipin peroxidase activity (Hüttemann et al., 2011). Cardiolipin is a mitochondria-specific phospholipid that binds to Cytc at multiple sites (Fig. 2A). This interaction allows Cytc to catalyze cardiolipin peroxidation, which also facilitates Cytc release from the mitochondria to the cytosol. Cytc thus serves as a hub for cellular life and death decisions. Given this crucial role, tight regulation of Cytc is needed to control its pro-life function in the ETC and its pro-death function in apoptosis. We here discuss the regulation of Cytc, focusing on phosphorylation and its effects on respiration and apoptosis. Based on recent data we propose as a new model that Cytc phosphorylation controls overall flux in the ETC and apoptosis, which determines the balance between ATP, ROS, and apoptosis.
Fig. 1. Proposed pivotal role of Cytc in respiration and apoptosis.
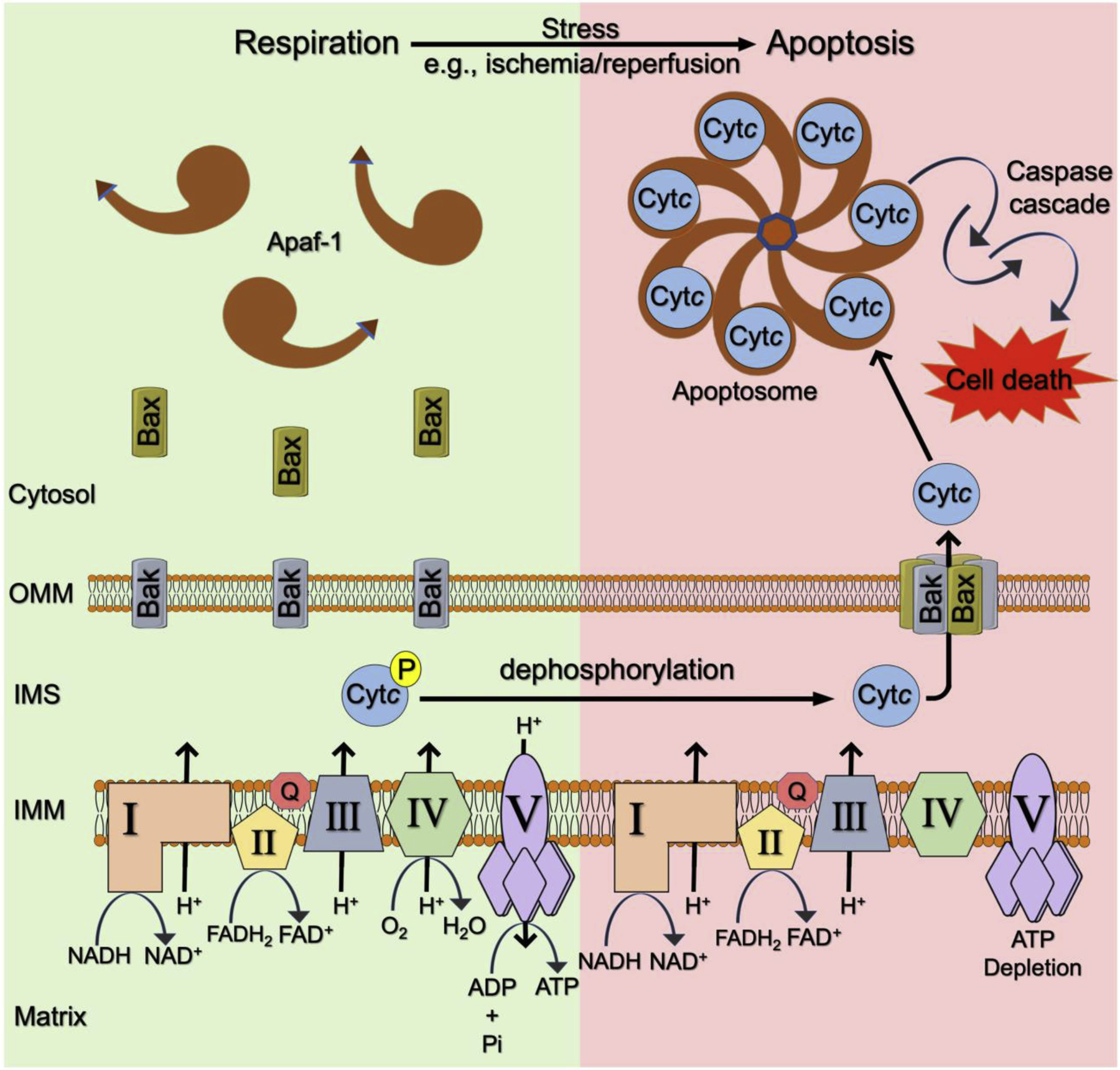
Cytc acts as an electron carrier between complex III (bc1 complex) and complex IV (cytochrome c oxidase) in the ETC resulting in efficient ATP production at complex V (ATP synthase). Under basal conditions, Cytc is phosphorylated in a tissue-specific manner. Phosphorylated Cytc is less likely to get released from the mitochondria and activate apoptosis in the cytosol. Under conditions of cellular stress, such as ischemia-reperfusion injury, dephosphorylated Cytc is eventually released from the mitochondria via channels including Bax/Bak pores. In the cytosol dephosphorylated Cytc interacts with Apaf-1 forming the apoptosome, which then activates capase-9 and the downstream caspase cascade, executing cell death. OMM; outer mitochondrial membrane, IMS; intermembrane space, IMM; inner mitochondrial membrane.
Fig. 2. Proposed Cytc binding sites of cardiolipin, ATP, bc1 complex, COX, and Apaf-1.
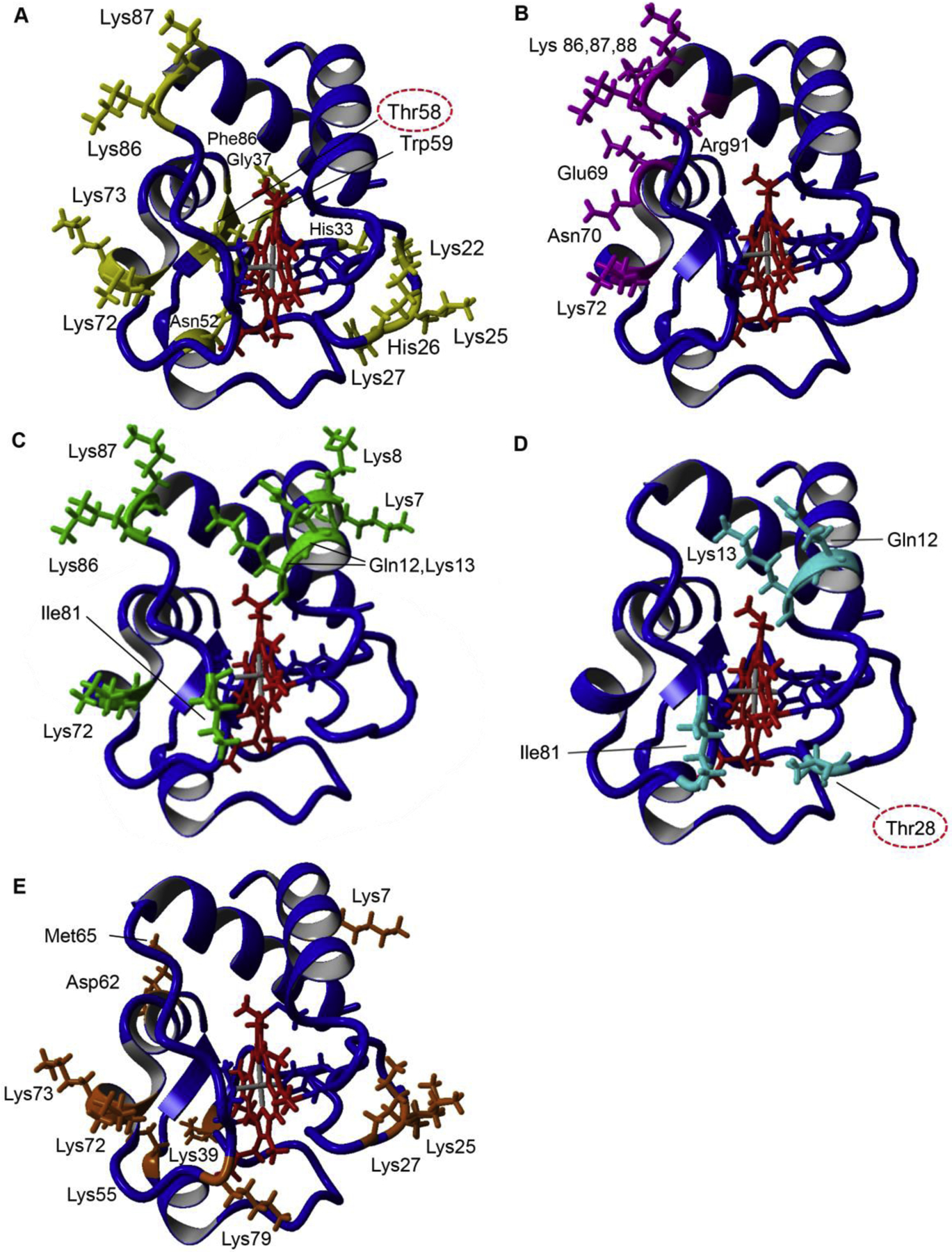
A: Cardiolipin binding sites of Cytc (yellow). Lys72, Lys73, Lys86, Lys87 (A-site) (Kagan et al., 2009), Lys22, Lys25, His26, Lys27, His33 (L-site) (Kagan et al., 2009), Asn52 (C-site) (Rytomaa and Kinnunen, 1994), Phe36, Gly37, Thr58, Trp59, and Lys60 (N-site) (O’Brien et al., 2015). B: ATP binding pocket of Cytc (magenta). Arg91 (Craig and Wallace, 1993; Tuominen et al., 2001), Glu69, Asn70, Lys72, Lys86, Lys87, Lys88, (McIntosh et al., 1996). C: COX binding sites of Cytc (green) were selected if defined as binding residues in at least two out of four publications, two docking simulations and two kinetic studies. Lys7, Lys8, Gln12, Lys13, Lys72, Ile81, Lys86, Lys87 (Roberts and Pique, 1999; Ferguson-Miller et al., 1978; Sato et al., 2016; Scharlau et al., 2019). D: bc1 complex core binding sites of Cytc (cyan) based on the yeast bc1-Cytc crystal structures and converted to mammalian Cytc residues. Gln12, Lys13, Thr28, Ile81(Lange and Hunte, 2002; Solmaz and Hunte, 2008). E: Apaf-1 binding sites of Cytc (orange), Lys7, Lys25, Lys39, Asp62, Met65 (Yu et al., 2001), Lys72 (Kluck et al., 2000), Lys27, Lys55, Lys73, Lys79 (Cheng et al., 2016). Amino acids that have been identified to be phosphorylated and are part of a binding site are circled.
2. Reversible phosphorylations of Cytc and COX
We modified protein purification protocols to preserve the in vivo phosphorylations of mitochondrial proteins, which made possible the discovery of novel phosphorylation sites on both COX and Cytc in mammals (Lee et al., 2006; Lee et al., 2009). About 18 phosphorylation sites have been mapped on COX with only a select few having been functionally studied (Helling et al., 2012a; Helling et al., 2012b). One such functionally characterized modification is Tyr304 phosphorylation that was mapped on catalytic subunit I of bovine liver COX, mediated by cAMP-dependent signaling (Lee et al., 2005) or inflammatory signaling via TNFα (Samavati et al., 2008), and resulted in strong enzyme inhibition. Despite decades of research on Cytc, its regulation through cell signaling-mediated phosphorylations was not uncovered until 2006 (Lee et al., 2006). Since then we and others have shown that these phosphorylations take place in a highly tissue-specific manner (Fig. 3). Tyr97 phosphorylation of Cytc was mapped for the first time in bovine heart tissue (Lee et al., 2006), followed by Tyr48, which was mapped in bovine liver (Yu et al., 2008), then Thr28 was mapped in bovine kidney (Mahapatra et al., 2017) and a second phosphorylation site, Thr58, was later also identified in rat kidney (Wan et al., 2019). Ser47 was mapped in both rat and porcine brain under basal conditions (Kalpage et al., 2019b) while Tyr97 was identified in rat and porcine brain post-insulin treatment (Sanderson et al., 2013a). Some of these phosphorylations were identified in high throughput mass spectrometry analyses in mammalian cells but not confirmed or functionally studied (Grimsrud et al., 2012; Hoffman et al., 2015; Lundby et al., 2013; Lundby et al., 2012; Parker et al., 2015; Sacco et al., 2016; Zhao et al., 2014; Zhao et al., 2011). These tissue-specific phosphorylations of Cytc fine-tune the multiple functions of Cytc to specific needs of various tissues and organs as discussed below in the sequence of their discovery and summarized in Table 1. As detailed below, all five reported Cytc phosphorylations lead to functional changes, including altered reaction kinetics with COX. A subset of them also result in a modified ability to trigger downstream caspase activation. Cytc is a small evolutionarily optimized protein, which may explain why modifications, even if they are not directly part of the interaction site based on crystallographic data or docking modeling simulations, may affect or interfere with optimal binding. For example, the interaction between Cytc and COX is mediated primarily by electrostatic interactions of positively charged lysine residues on Cytc and negatively charged residues on COX, in addition to hydrophobic interactions across the binding interface (Roberts and Pique, 1999; Schmidt et al., 2005). Depending on their specific location on Cytc, the introduction of negative charges upon phosphorylation, can affect and interfere with optimal binding of Cytc to COX, lowering the reaction rate. Other contributing factors may be a change of the Cytc redox potential and the attachment of the phosphate moiety, which may cause steric interference or structural changes on Cytc.
Fig. 3. Tissue-specific phosphorylation sites of Cytc (green) mapped in mammalian tissues under basal conditions.
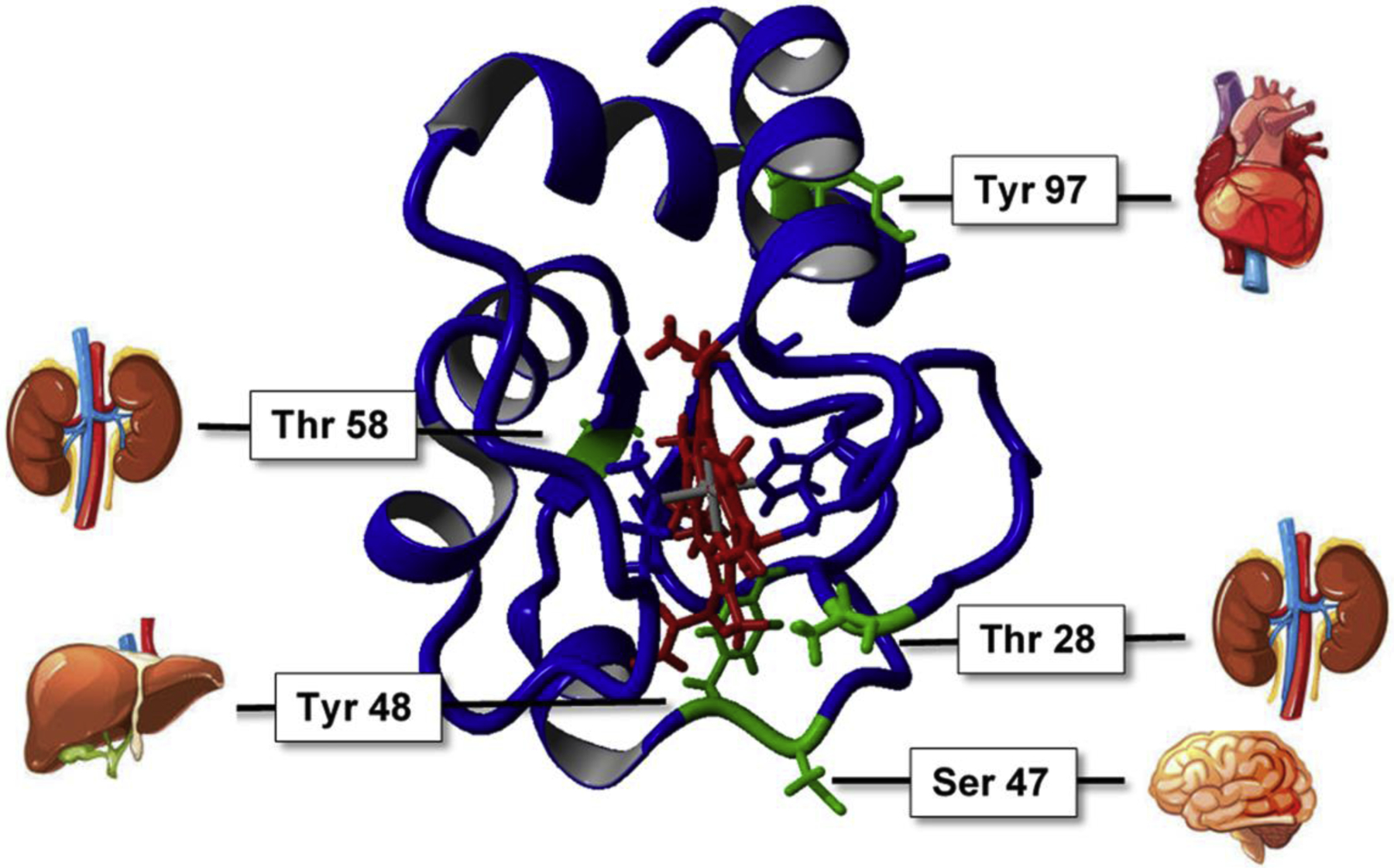
Sequence of their discovery: Tyr97 (heart), Tyr48 (liver), Thr28 (kidney), Ser47 (brain), Thr58 (kidney). The heme group is highlighted in red.
Table 1.
Summary of functionally characterized phosphorylation sites of Cytc
| Phosphorylation site | Low-throughput identification | Experimental models | Functional and structural characterization |
|---|---|---|---|
| Threonine 28 | Electrospray ionization-mass spectrometry (bovine kidney) (Mahapatra et al., 2017). | In vivo phosphorylated protein purified from bovine kidney, phosphomimetic T28E recombinant protein, mouse lung fibroblast cells expressing T28E Cytc (Mahapatra et al., 2017); phosphomimetic T28D recombinant protein (Guerra-Castellano et al., 2016). | Lower Cytc-COX activity, lower redox potential, higher rate of reduction. Lower respiration, membrane potential and ROS production in intact cells. Phosphorylated by AMPK (Mahapatra et al., 2017). Lower binding affinity with Cytc1 in bc1 complex (Guerra-Castellano et al., 2016). |
| Serine 47 | Electrospray ionization-mass spectrometry (porcine brain tissue) (Kalpage et al., 2019b). | In vivo phosphorylated protein purified from porcine brain, phosphomimetic S47E recombinant protein (Kalpage et al., 2019b); phosphomimetic S47D recombinant protein (Guerra-Castellano et al., 2016). | Lower Cytc-COX activity, lower caspase-3 activity, lower cardiolipin peroxidase activity, reduced heme degradation (Kalpage et al., 2019b). |
| Tyrosine 48 | Electrospray ionization-mass spectrometry (bovine liver) (Yu et al., 2008). | In vivo phosphorylated protein purified from bovine liver (Yu et al., 2008), Phosphomimetic Y48E recombinant protein (Garcia-Heredia et al., 2011; Pecina et al., 2010), Y48pCMF phosphomimetic Cytc (Moreno-Beltran et al., 2017). | Lower Cytc-COX activity (Pecina et al., 2010; Yu et al., 2008), abolished caspase-3 activity, reduced cardiolipin peroxidase activity, reduced cardiolipin binding, lower redox potential (Pecina et al., 2010). Lower caspase-9 activity (Moreno-Beltran et al., 2017). |
| Threonine 58 (replaced with isoleucine in humans) | Electrospray ionization-mass spectrometry (rat kidney) (Wan et al., 2019). | Phosphomimetic T58E recombinant protein, mouse lung fibroblasts expressing T58E Cytc (Wan et al., 2019). | Lower Cytc-COX activity, lower caspase-3 activity, lower redox potential, lower rate of oxidation, lower cardiolipin peroxidase activity, higher rate of reduction, lower respiration, membrane potential and ROS production in intact cells. Decreased cell death when treated with H2O2 and staurosporine (Wan et al., 2019). |
| Tyrosine 97 | Electrospray ionization-mass spectrometry (bovine heart, insulin-treated porcine brain). | In vivo phosphorylated protein purified from bovine heart (Lee et al., 2006), phosphomimetic Y97E recombinant Cytc (Garcia-Heredia et al., 2011); Y97pCMF phosphomimetic Cytc (Guerra-Castellano et al., 2018); insulin-treated rat model of global brain ischemia (Sanderson et al., 2013a). | Lower Cytc-COX activity, shift of the characteristic 695 nm peak to 687 nm (change in heme environment) (Lee et al., 2006), less thermally stable (Garcia-Heredia et al., 2011), lower caspase-3 activity (Guerra-Castellano et al., 2018), lower Cytc release from mitochondria, decreased neuronal cell death (Sanderson et al., 2013a). |
2.1. Tyrosine 97 (Tyr97) phosphorylation of Cytc
Lee et al. (Lee et al., 2006) reported for the first time that Cytc is post-translationally modified by phosphorylation. Cytc purified from bovine heart was phosphorylated on Tyr97, which shifted the characteristic 695 nm heme-iron-Met80 absorption band, a marker of Cytc integrity (Dickerson and Timkovich, 1975), to 687 nm. This spectral change in Tyr97-phosphorylated Cytc suggests that this phosphorylation affects the heme environment of Cytc. In the reaction with COX, Tyr97-phosphorylated and unphosphorylated Cytc displayed sigmoidal and hyperbolic kinetics, with Km values for Cytc in the reaction with COX of 5.5 μM and 2.5 μM, respectively (Lee et al., 2006). In another study, no change in redox potential was observed with phosphomimetic Tyr97Glu Cytc, but there was a significant decrease in protein stability of Tyr97Glu Cytc compared to the WT as determined by melting temperature analysis (Garcia-Heredia et al., 2011). This is likely due to the lack of the aromatic ring in Tyr97Glu Cytc, as opposed to the introduction of the negative charge since the control phenylalanine mutant did not show decreased stability (Garcia-Heredia et al., 2011). Tyr97 phosphorylation was later characterized after the replacement of tyrosine with a more realistic phosphomimetic, p-carboxymethyl-l-phenylalanine (pCMF), incorporated using the evolved tRNA synthetase method (Guerra-Castellano et al., 2015). However, Tyr97pCMF resulted in increased COX activity in contrast to the in vivo Tyr97 phosphorylated Cytc. The Tyr97pCMF phosphomimetic replacement resulted in lower caspase-3 activity while no change in cardiolipin binding or cardiolipin peroxidase activity was observed (Guerra-Castellano et al., 2018). The decrease in apoptotic activity could be due to the close proximity of this residue to Lys7, which is a key residue for apoptosome formation (Yu et al., 2001) (Fig. 2E). Tyr97 is conserved across most species ranging from mammals to plants and microorganisms. There are several fungi that have Leu, Gln, and Phe residues that cannot be phosphorylated in place of Tyr97 (Hampsey et al., 1988; Heller and Smith, 1966). This may be explained by the lack of tyrosine kinase signaling in lower species. The kinase mediating this reaction in mammals is yet to be identified. Sequence alignment of the Tyr97 phospho-epitope with Tyr304 phospho-epitope of COX subunit I revealed that 5 of 10 residues in the alignment are identical, suggesting the possibility that both proteins are targeted by the same kinase (Lee et al., 2006). In vivo, insulin treatment of post-ischemic porcine and rat brains resulted in Tyr97 phosphorylation of Cytc (Sanderson et al., 2013a). Furthermore, the induction of Tyr97 phosphorylation after insulin treatment rescued roughly 50% of the CA1 hippocampal neurons in an animal model of global brain ischemia/reperfusion injury, further supporting the anti-apoptotic role of Tyr97 phosphorylation of Cytc. The decrease in neuronal death was associated with a decrease in Cytc release 24 hours post-reperfusion (Sanderson et al., 2013a), suggesting that the phosphorylation state of Cytc may also play a role in Cytc release from the mitochondria to trigger apoptosis. Both cAMP signaling and insulin signaling promote anti-apoptotic mechanisms, suggesting a role of these pathways in the regulation of Cytc (Martin et al., 2005; Sanderson et al., 2009; Sanderson et al., 2008).
2.2. Tyrosine 48 (Tyr48) phosphorylation of Cytc
Tyr48 phosphorylation was primarily found on Cytc purified from cow liver (Yu et al., 2008). Tyr48 is conserved in eukaryotes and mammals (Zaidi et al., 2014). Functional studies using in vivo Tyr48-phosphorylated Cytc and a phosphomimetic mutant, Tyr48Glu Cytc, which carries a negative charge similar to the phosphate group, revealed reduced maximal turnover rates in the reaction with liver COX to about 50% (Pecina et al., 2010). Tyr48Glu replacement in Cytc completely abolished caspase-3 activity in a cell-free apoptosis detection system (Pecina et al., 2010), suggesting that this phosphorylation serves as a switch for apoptosis. Furthermore, Tyr48Glu Cytc also resulted in decreased pro-apoptotic cardiolipin peroxidase activity. These findings may be explained by the fact that the phosphomimetic Tyr48Glu reduced the binding of Cytc to cardiolipin by about 30% compared to unphosphorylated Cytc (Pecina et al., 2010). Another study that utilized Tyr48pCMF phosphomimetic Cytc further confirmed that this Cytc variant inhibits ETC flux and caspase-dependent apoptosis (Moreno-Beltran et al., 2017). Tyr48 is located at the lower median frontal area at the Ω loop (amino acid residues 40–57), which is close to the heme crevice, and susceptible to unfolding (De Rocco et al., 2014; Moreno-Beltran et al., 2017). Topography of Tyr48 and Tyr97 residues using NMR shows intensity changes when binding to cardiolipin compared to tyrosine residues Tyr67 and Tyr74, suggesting that changes in the former residues have a stronger influence on the molecular dynamics of the protein (Kapralov et al., 2011). Moreover, the midpoint redox potential of the phosphomimetic Tyr48Glu mutant was decreased by 45 mV compared to the WT control (Pecina et al., 2010), which is lower than that of cytochrome c1 in bc1 complex, suggesting that Tyr48 phosphorylation reduces the rate of electron transfer from cytochrome c1 to Cytc at the bc1 complex, which may also be beneficial for Cytc to function as a ROS scavenger (Pereverzev et al., 2003). Furthermore, a docking model of bc1 complex and Cytc proposed Tyr48 as a potential interactor with the bc1 complex (Kokhan et al., 2010), suggesting additional interference with ETC function upon phosphorylation of the site. Interestingly, human Tyr48Glu Cytc displayed an 80 mV reduction of the redox potential compared to WT Cytc (Garcia-Heredia et al., 2011) whereas the decrease was only 45 mV in rodent Cytc (Pecina et al., 2010). Tyrosine phosphorylation is mainly present in higher organisms and involved in mitochondrial and cancer signaling (Carpenter et al., 1978). Therefore, characterization of Tyr48 phosphorylation of Cytc may be an interesting future research direction to better understand the role of Cytc phosphorylation in cancer. It was reported that the Tyr48His mutation causes a mild human disease, thrombocytopenia, resulting in a lower level of blood platelets. The introduction of a positively charged histidine reduced the oxygen consumption rate and increased apoptotic activity (De Rocco et al., 2014). Furthermore, the Tyr48His mutation causes a structural transformation of Cytc from a hexacoordinated form to a pentacoordinated form which promotes cardiolipin peroxidase activity (Deacon et al., 2017). It remains unclear, however, why patients with the Tyr48His mutation present with a mild phenotype that is limited to platelets.
2.3. Threonine 28 (Thr28) phosphorylation of Cytc
A major portion of Cytc (~80%) purified from bovine kidney was found to be phosphorylated on residue Thr28 (Mahapatra et al., 2017). In vivo phosphorylated Cytc from the bovine kidney resulted in a 50% decrease in COX activity but there was no significant effect on caspase-3 activity compared to unphosphorylated bovine kidney Cytc. The Thr28Glu phosphomimetic mutant resulted in a 73% decrease in COX activity compared to recombinant unphosphorylated WT Cytc (Mahapatra et al., 2017). Surprisingly, in another study, Thr28Asp phosphomimetic increased COX activity, contrary to what we observed with in vivo phosphorylated Cytc and Thr28Glu Cytc. Across evolution, glutamate is permissible at this site of Cytc and found in several plant species, whereas aspartate is not (Zaidi et al., 2014). This suggests that glutamate is the more suitable phosphomimetic model to characterize Thr28 phosphorylation given its functional similarities to in vivo phosphorylated Cytc. Importantly, intact Cytc knockout cells expressing Thr28Glu Cytc also showed lower respiration rates compared to WT Cytc-expressing cells, mimicking the in vitro experiments with purified COX. Furthermore, cells expressing Thr28Glu Cytc had lower mitochondrial membrane potentials and ROS levels. This suggests that phosphorylation of Cytc at Thr28 leads to a partial inhibition of mitochondrial respiration resulting in an optimal intermediate mitochondrial membrane potential for efficient ATP generation with minimal ROS production (Kaim and Dimroth, 1999; Kalpage et al., 2019a). These findings suggested, for the first time, that modifications on Cytc can lead to an overall decrease in ETC flux in intact cells. The decrease in Cytc-COX activity and overall ETC flux with Thr28Glu Cytc may be in part attributed to changes in the midpoint redox potential as well as structural changes of Cytc. This residue is present in the COX binding domain of the protein, interacting with COX subunits I, II and VIIc based on a Cytc-COX docking model (Roberts and Pique, 1999). Furthermore, Thr28 is in close proximity to residue Asp50 of COX subunit I, which is one of the few residues of COX that structurally vary in the COX crystal structure based on its oxidized or reduced state (Sugitani and Stuchebrukhov, 2009). Asp50 is next to Asp51, a residue that was proposed to be involved in the proton pumping mechanism of COX as a proton ejection site (Tsukihara et al., 2003). Therefore, it is possible that Thr28 phosphorylation of Cytc inhibits the electron transfer-coupled proton pumping of COX. Thr28 of Cytc also interacts with Trp104 of COX subunit II, which is the site where electrons enter before reaching the binuclear CuA copper center of COX (Scharlau et al., 2019). Interference of Thr28 phosphorylation with these key residues of COX likely explains its inhibitory function on COX activity and overall ETC flux. Cytc Thr28 is part of the ‘negative classical gamma turn’ comprised of residues 27 through 29 and is important for the stability of Cytc (Sanishvili et al., 1995).
Thr28 phosphorylation of Cytc represents one of the few examples in the entire OxPhos system for which the corresponding kinase, i.e., AMP kinase (AMPK), has been identified (Mahapatra et al., 2017). AMPK is activated by the widely-used diabetes drug metformin (Zhou et al., 2001). Consistent with the Thr28 phosphorylation study, activation of AMPK with metformin leads to lower respiration in human renal proximal tubular epithelial cells (Takiyama et al., 2011). However, this decrease in OxPhos in the kidney is distinct compared to the generally accepted catabolic (ATP generation-promoting) role of AMPK in other organs (Carling, 2017).The pronounced regulation of kidney function by circadian rhythm oscillations might be a factor promoting anabolic pathways under some conditions (Firsov and Bonny, 2010; Zuber et al., 2009). AMPK has a high basal activity in the kidney (Bhargava and Schnellmann, 2017) likely due to the heavy reliance of this organ on OxPhos. Furthermore, AMPK plays a role in minimizing mitochondrial ROS (Rabinovitch et al., 2017), which was also observed with Cytc Thr28 phosphorylation (Mahapatra et al., 2017). Cytc purified from ischemic kidney was also fully dephosphorylated, similar to Cytc purified from ischemic brain (Kalpage et al., 2019b; Sanderson et al., 2013a), and both organs are known to be highly susceptible to ischemia/reperfusion injury (Kalogeris et al., 2016). Studies have shown that treatment with an AMPK activator, AICAR, prior to ischemia increases cell survival in a rat model of renal ischemia/reperfusion injury (Lempiainen et al., 2012). This supports our model of ischemia-reperfusion injury where we suggest that Cytc phosphorylation is required for the maintenance of optimal intermediate membrane potentials, which in turn would minimize ROS production upon reperfusion and thus limit tissue injury (Sanderson et al., 2013b).
2.4. Serine 47 (Ser47) phosphorylation of Cytc
Under basal conditions, Cytc in the mammalian brain was recently shown to be phosphorylated on Ser47 (Kalpage et al., 2019b). This phosphorylation is lost during ischemia, suggesting an important regulatory role of this modification in the brain. The Ser47 phosphorylation site was mapped in both porcine and rat brain tissue. Phos-tag gel electrophoresis showed that about 35% of the Cytc pool was phosphorylated. However, this is likely an underestimation as unavoidable ischemia during tissue harvesting may have started the dephosphorylation process. The role of the Ser47 phosphorylation was characterized using a glutamate phosphomimetic, Ser47Glu Cytc. Both Ser47 phosphorylated in vivo and phosphomimetic Cytc showed a 50% decrease in COX activity. Interestingly, Ser47 interacts with Lys58 of COX subunit VIIa based on docking simulations (Roberts and Pique, 1999). Subunit VIIa has a heart/skeletal muscle (heart-type) and liver/brain/kidney (liver-type) isoform pair. However, there was no difference in the relative change of activity of Ser47 phosphomimetic Cytc and the Km values when analyzed in the reaction with heart and brain COX (Kalpage et al., 2019b), suggesting that subunit VIIa isoforms do not modulate the effect of this Cytc phosphorylation.
In addition, Ser47-phosphorylated and phosphomimetic Cytc both resulted in a significant decrease (~70%) in caspase-3 activity. Furthermore, cardiolipin peroxidase activity was also lower in the presence of Ser47Glu phosphomimetic Cytc compared to unphosphorylated WT Cytc, further supporting the anti-apoptotic role of Ser47 phosphorylation. Ser47Glu phosphomimetic Cytc also demonstrated significantly lower degradation rates of the heme group in the presence of 3 mM H2O2, suggesting that when phosphorylated at this site Cytc has a better ability to withstand oxidative stress (Kalpage et al., 2019b). Another study analyzed the role of aspartate phosphomimetic Ser47Asp Cytc and similarly reported a decrease in both caspase-3 activity and cardiolipin peroxidase activity (Guerra-Castellano et al., 2016). Ser47Asp Cytc did not inhibit the reaction with COX as observed with Ser47Glu and in vivo Ser47 phosphorylated Cytc. The crystal structure of Ser47Glu Cytc (6N10.pdb) at 1.55 Å suggests that the Ser47-phosphorylated Cytc sidechain and the glutamate sidechain of Ser47Glu spatially arrange in a similar manner, suggesting that Ser47Glu is a better phosphomimetic model than Ser47Asp Cytc (Kalpage et al., 2019b). Overall, Ser47 phosphorylation of Cytc is a tissue protective modification that promotes cell survival under healthy conditions by lowering apoptotic activity. Furthermore, Ser47 phosphorylation of Cytc can prevent pathologically high respiration rates that would lead to hyperpolarization of the mitochondrial membrane potential and a burst of ROS causing cell death, as seen during ischemia/reperfusion injury (reviewed in (Sanderson et al., 2013b)). This suggests that a better understanding of the signaling pathway involved in Ser47 phosphorylation of Cytc may provide a pre- or post-conditional therapeutic target for brain ischemia/reperfusion injury.
2.5. Threonine 58 (Thr58) phosphorylation of Cytc
The Cytc Thr58 residue is conserved in the somatic Cytc isoform of some mammals but is replaced with an isoleucine residue in their testis isoform, which is also found in apes and humans who only have a single ubiquitous Cytc (Fig. 4). Thr58 phosphorylation was mapped in purified rat kidney Cytc by mass spectrometry. This phosphorylation was also found in a high-throughput phosphoproteomics study of rat kidney (Lundby et al., 2012). Unlike Thr28 phosphorylation, previously identified in five independent kidney preparations (Mahapatra et al., 2017), Thr58 phosphorylation was only found in some preparations. Considering that the kidney contains 26 different cell types (Poornejad et al., 2016), Thr58 phosphorylation may only occurs in a few cell types. Evolutionarily, threonine is the most conserved amino acid at residue 58 of Cytc (Zaidi et al., 2014). Isoleucine is the second most abundant and is present in mammals containing a testes-specific isoform and in humans, who only have a single functional Cytc gene that is ubiquitously expressed. In vitro and in vivo functional studies revealed that phosphomimetic Thr58Glu Cytc partially inhibits COX activity. Furthermore, the oxygen consumption rate in intact cells expressing Thr58Glu Cytc was decreased, suggesting that Cytc plays a rate-limiting role on overall ETC flux in this cell model. In addition, Thr58Glu replacement reduced the mitochondrial membrane potential and ROS production in intact cells, supporting our model in which Cytc phosphorylation limits membrane potential hyperpolarization, ROS production, and apoptosis. However, during stress conditions such as ischemia, this protective modification is lost, which may be the underlying mechanism explaining reperfusion injury, when oxygen reenters the tissue, and dephosphorylated Cytc drives membrane potential hyperpolarization, bursts of ROS, release of Cytc, and cell death (Sanderson et al., 2013b). In support of this model, the Thr58Glu phosphomimetic substitution also resulted in reduced caspase-3 activity in vitro, and intact cells stably expressing Thr58Glu Cytc showed protection from apoptosis when challenged with H2O2 and staurosporine. This data further supports an anti-apoptotic role of Thr58 phosphorylation of Cytc. Thr58 is located on the back side of the Cytc heme crevice (Fig. 3). However, electrostatic, steric, and conformational changes in Cytc may explain functional effects due to the small size of the Cytc molecule. Compared to WT, Thr58Glu Cytc was more resistant to heme degradation by H2O2. Thr58 was recently reported to be part of a cardiolipin binding site (O’Brien et al., 2015) (Fig. 2A). Cardiolipin peroxidase activity of Thr58Glu was inducible at high cardiolipin concentrations, suggesting that Cytc phosphorylation may stabilize the heme iron-Met80 bond and suppress cardiolipin peroxidation (Pecina et al., 2010), thus providing a safeguard mechanism with which Cytc release and apoptosis are properly regulated. Molecular dynamics simulations of Thr58Glu Cytc suggested that the structural stability of phosphomimetic Cytc is similar to that of Thr58 phosphorylated Cytc. With a lower oxidation rate and a higher reduction rate, phosphomimetic Thr58Glu Cytc reduces the electron transfer rate to COX in the ETC and serves as a better ROS scavenger (Wan et al., 2019).
Fig. 4. Alignment of ubiquitous Cytc from anthropoid primates (human, chimpanzee, macaque) and somatic and testes-specific Cytc isoforms of non-anthropoid mammals (bull, mouse, rat).
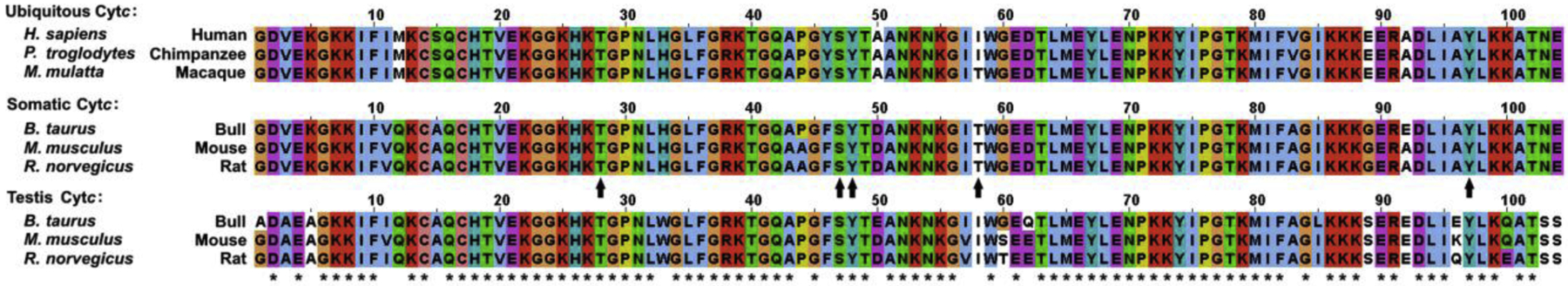
Stars indicate conserved residues. Arrows indicate mapped and functionally studied phosphorylation sites in mammals.
3. Is Cytc the rate-limiting step of the ETC?
The highly tissue-specific regulation of Cytc and the effects of specific Cytc phosphorylations on ETC and apoptotic function may have far-reaching implications. One question, which has been a subject of much debate, is which step within the ETC controls overall electron flux and is thus the bottleneck, or rate-limiting step, of the ETC. We here put forth a new concept and propose that the electron transfer from Cytc to COX at the terminal step of the ETC is the rate limiting step of this vital process in cellular bioenergetics. The terminal step of the ETC is highly regulated by allosteric ATP binding (Fig. 2B), tissue-specific isoforms, and post-translational modifications, which are found in both Cytc and COX. We propose that Cytc, which operates at the intersection of respiration and apoptosis, is an ideal candidate to regulate both processes, in particular through reversible phosphorylations. If this concept holds the test of time it would form the rationale for targeting Cytc to treat pathological conditions caused by mitochondrial dysfunction.
The COX-catalyzed reduction of oxygen to water is essentially irreversible and releases an excess of free energy that is twice as high as for complexes I and III (Hinkle et al., 1991). As discussed in detail below, several studies concluded that COX is the rate-limiting step of the ETC. However, the potential role of Cytc, which binds to and injects electrons into COX catalytic subunit II, has previously not been considered. This may be so in part, because traditional metabolic flux analyses use specific inhibitors of the individual complexes of the ETC such as rotenone, antimycin A, and cyanide for complexes I, III, and IV, respectively. These inhibitors are added at increasing amounts to determine the fraction of the individual complexes that are required to allow unimpeded flux. The lower the fraction needed to reduce overall flux in a given complex the more rate-limiting it is. However, no such inhibitor exists for Cytc so its potential rate-limiting role was never considered. For example, in most cases flux control coefficients of COX were determined by inhibitor titrations with cyanide or azide, which only assesses the contribution of oxygen reduction at the binuclear center of COX. In other words, the potential role of Cytc phosphorylation in overall ETC flux is hidden in the flux control coefficients of bc1 complex and COX. As discussed in the previous sections, experimental evidence suggests that modifications on Cytc can decisively affect ETC flux in intact cells in addition to regulating apoptosis.
As with biochemical pathways in general, changes in the enzymatic activity of the rate limiting step can result in overall change of flux through the entire pathway and multiple studies have shown this also to be true for the ETC. Since metabolic flux analyses were not conducted for Cytc, we briefly introduce the topic by discussing what is known about COX, which – importantly – includes the electron transfer reaction from Cytc to COX. Several studies concluded that changes in COX activity can profoundly affect ETC flux. The rate-limiting step is often determined by the flux control coefficient, which is the change in relative flux/change in relative enzyme capacity (Gnaiger et al., 1998). Measurement of COX activity or capacity in intact cells through cyanide titrations revealed in a variety of human cell types that only a small excess COX capacity was present above what is required to carry out endogenous respiration (Villani and Attardi, 1997, 2000). A small excess COX capacity suggests a tight regulation of the ETC by COX. This conclusion was further supported by a cell line carrying mitochondria with a COX subunit 1 nonsense mutation. Cells carrying 35% of mutant mitochondrial DNA (mtDNA) resulted in 55% COX activity and 75% endogenous respiration whereas cells carrying 65% of mutant mtDNA resulted in a further decrease of COX activity (15%) and endogenous respiration (10%), suggesting that overall ETC flux is controlled by COX (Bruno et al., 1999). Respiration of human muscle fibers measured in the presence of glutamate and malate as substrates resulted in a 2.3-fold excess COX capacity. In the presence of both complex I and complex II substrates glutamate/malate and succinate, which further increases ETC flux, the excess COX capacity was decreased to 1.5-fold. Similarly, a lower excess COX capacity was present in skeletal muscle fibers of two ophthalmoplegia patients who carried deletions in 11% and 49% of their mtDNA. Both patients’ muscle fibers showed an increase in the flux control coefficient as predicted by lower excess COX capacity, further supporting a rate-limiting function of COX (Kunz et al., 2000). Metabolic flux control analyses in intact cells showed that COX has a lower excess capacity than that reported in isolated mitochondria (Piccoli et al., 2006). Metabolic flux control analysis of saponin-treated muscle fibers showed that COX activity is affected by oxygen availability to the tissue. Saponin-treated fibers are a model for mitochondrial function of muscle as it allows the extramitochondrial medium to be precisely controlled. Consequently, saponin-treated muscle fibers led to higher flux control coefficients indicating the importance of oxygen availability, the substrate of COX (Wiedemann and Kunz, 1998). Using normoxic (190 μmolar) media, a 50% reduction in COX activity did not affect overall respiration rates. However, under lower oxygen concentrations COX activity became more rate-limiting, likely explaining the pathological phenotypes observed in muscle COX deficiencies associated with mitochondrial diseases, neurodegenerative diseases, and the aging phenotype (Korzeniewski, 1997; Wiedemann and Kunz, 1998).
A mouse cell line carrying the COX subunit I Val421Ala mutation resulted in a 43–65% decrease in COX activity and a lower overall respiration rate in intact or permeabilized cells compared to the controls (Acin-Perez et al., 2003). These data suggest that excess COX capacity is only between 25–40% (1.2–1.4 fold) in this cell culture model, suggesting that this step can be rate-limiting for ETC flux. In order to address the role of tissue specificity on OxPhos regulation by COX, the flux control coefficient and excess COX capacity was determined in human saponin-permeabilized muscle fibers and digitonin-treated hippocampal homogenates. These tissue preparations mimic in vivo mitochondrial function. Skeletal muscle homogenates resulted in a higher flux control coefficient of 0.24 ± 0.07 compared to brain homogenates (0.12 ± 0.05). Furthermore, the muscle homogenates had a lower COX excess capacity of 1.9±0.2 compared to a higher COX excess capacity of 3.9±0.6 in the brain homogenates (Kudin et al., 2002). These results suggest that human muscle COX is more rate-limiting in OxPhos compared to human brain COX. Consistent with the above report, the COX respiratory threshold value, which is defined as the %-inhibition value of COX activity that has an effect on the overall rate of endogenous respiration, was measured in mitochondria isolated from 5 different rat tissue types. Both muscle and heart mitochondria had a lower COX respiratory threshold compared to liver, kidney and brain mitochondria (Rossignol et al., 1999). These differences in metabolic control may be attributed to the presence of 3 different tissue-specific isoforms in COX subunits VIa, VIIa, and VIII in heart and muscle COX (“heart-type COX”) compared to liver, kidney, and brain COX (“liver-type COX”). Several studies showed that liver-type COX has a significantly higher basal activity compared to heart-type COX, which provides a compensatory mechanism for tissues with less mitochondrial capacity and may explain the lower metabolic control of the brain isozyme compared to the heart isozyme of COX (Kalpage et al., 2019b; Sinkler et al., 2017; Vijayasarathy et al., 1998).
The role of the electrical component (ΔΨm) and the chemical component (ΔpH) of the mitochondrial electrochemical gradient on COX activity and overall respiration was investigated in intact human hepatoma HepG2 cells. The flux control coefficient of COX in the presence of nigericin, a potassium/proton antiporter, when ΔΨm is maximal, and in the presence of both nigericin and valinomycin (a potassium ionophore) when the electrochemical gradient is abolished, was comparable to the basal flux control coefficient of 0.73 (Dalmonte et al., 2009). In contrast, upon addition of valinomycin, which removes ΔΨm, a lower COX control coefficient of 0.30 was found, suggesting that it is primarily the ΔΨm component of the electrochemical gradient that is responsible for the tight regulation of COX in the overall process of the ETC (Dalmonte et al., 2009). Furthermore, COX activity is limited by the Cytc/COX molar ratio which has been reported to be 1.08 in hepatocytes (Jones et al., 1979) and 1.62 in rat cardiac myocytes (Kennedy and Jones, 1986). Finally, Jurkat cells treated with an anti-Fas antibody and digitonin led to Cytc release and induced apoptosis following a significant decrease in cellular respiration pertaining to the role of Cytc in the rate-limiting step of the ETC (Hajek et al., 2001). Our data, using Thr28Glu and Thr58Glu Cytc in intact cells, support the idea that a small modification on Cytc affects overall ETC activity. Future work should establish the role of Cytc on metabolic flux of the ETC under conditions that are more physiological and consider and maintain post-translational modifications of the protein found in tissues in vivo but that are lost when traditional mitochondrial purification protocols are used. In addition, Cytc-COX activity assays should omit tetramethyl-p-phenylenediamine (TMPD), which is often used as an electron transfer catalyst, and which renders the reaction unphysiological because TMPD can reduce Cytc when it is bound to COX (Cooper, 1990).
4. Conclusions
It appears to be crucial for cells to maintain a tightly regulated activity range of the last step of the ETC, catalyzed by Cytc and COX, to efficiently produce ATP and minimize ROS production under physiological conditions. This concept is supported by the fact that mammalian Cytc and COX are the only components of the OxPhos machinery that demonstrate all three regulatory mechanisms of metabolic enzymes: tissue-specific isoforms, allosteric regulation via the ATP/ADP ratio, and regulation by post-translational modifications (Fig. 3). The tissue-specificity of the mapped phosphorylation sites to date may seem unexpected. All phosphorylations lead to a reduction of the activity of the reaction catalyzed by Cytc and COX, which supports our concept that intermediate ETC flux and thus intermediate ΔΨm levels are optimal for efficient energy production while limiting ROS generation. However, their effect on the apoptotic pathway are distinct. As an example, Tyr48 phosphorylation, found in the liver, abolishes caspase 3 activation, which may be a safeguard mechanism of a detoxification organ that is constantly exposed to potentially harmful substances. Another example is Ser47 phosphorylation of Cytc, which also profoundly reduces the ability of Cytc to trigger apoptosis and is present in the brain, an organ with limited capacity for regeneration. Other mechanisms underlying tissue-specific phosphorylation of Cytc may be that certain kinases are only expressed in some tissues or their basal activity is high in a given organ, as we have found for AMPK-mediated Thr28 phosphorylation of Cytc in the kidney.
Cytc and COX have undergone significant adaptations during evolution of anthropoid primates to optimally meet species-specific energy demands. Of the 1500 amino acids in COX, about 300 were replaced in anthropoid primates compared to other mammals. Some of the charged residues in the Cytc binding site of COX were replaced by uncharged hydrophobic residues (Pierron et al., 2012; Schmidt et al., 2005). This adaption decreases the binding affinity between Cytc and COX (Fig. 2C), further supporting the rate-limiting function of this interaction. At the level of Cytc, the testes-specific isoform present in certain mammals became a pseudogene during primate evolution (Pierron et al., 2011) (Fig. 4). We propose that reversible phosphorylations of Cytc mediated by cell signaling pathways are primary regulatory mechanisms in higher organisms. This in turn determines ETC flux, ΔΨm, ATP production, and ROS generation, linking OxPhos to human disease through a lack of energy, ROS production, Cytc release, and activation of apoptosis. The recent discovery of novel regulatory phosphorylation sites on Cytc has advanced the understanding that Cytc plays a crucial role in regulating overall ETC flux and thus mitochondrial respiration, ΔΨm, ATP, and ROS.
Acknowledgements
This work was supported by the U.S. National Institutes of Health grants R01 GM116807, R01 NS091242, and STTR R42NS105238, the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award Nos. W81XWH-16-1-0175 and W81XWH-16-1-0516, the American Heart Association Award No. 20PRE35210130, and the Wayne State University Graduate Research Assistant Award. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agencies including the Department of Defense or the National Institutes of Health.
Abbreviations:
- Cytc
cytochrome c
- COX
cytochrome c oxidase
- ΔΨm
mitochondrial membrane potential
- ETC
electron transport chain
- OxPhos
oxidative phosphorylation
- ROS
reactive oxygen species
- TMPD
tetramethyl-p-phenylenediamine
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acin-Perez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernandez-Silva P, Perez-Martos A, Montoya J, Lopez-Perez MJ, Sancho J, Enriquez JA, 2003. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet 12(3), 329–339. [DOI] [PubMed] [Google Scholar]
- Bhargava P, Schnellmann RG, 2017. Mitochondrial energetics in the kidney. Nat Rev Nephrol 13(10), 629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno C, Martinuzzi A, Tang Y, Andreu AL, Pallotti F, Bonilla E, Shanske S, Fu J, Sue CM, Angelini C, DiMauro S, Manfredi G, 1999. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am J Hum Genet 65(3), 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D, 2017. AMPK signalling in health and disease. Curr Opin Cell Biol 45, 31–37. [DOI] [PubMed] [Google Scholar]
- Carpenter G, King L Jr., Cohen S, 1978. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 276(5686), 409–410. [DOI] [PubMed] [Google Scholar]
- Cheng TC, Hong C, Akey IV, Yuan S, Akey CW, 2016. A near atomic structure of the active human apoptosome. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CE, 1990. The steady-state kinetics of cytochrome c oxidation by cytochrome oxidase. Biochim Biophys Acta 1017(3), 187–203. [DOI] [PubMed] [Google Scholar]
- Craig DB, Wallace CJ, 1993. ATP binding to cytochrome c diminishes electron flow in the mitochondrial respiratory pathway. Protein Sci 2(6), 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmonte ME, Forte E, Genova ML, Giuffre A, Sarti P, Lenaz G, 2009. Control of respiration by cytochrome c oxidase in intact cells: role of the membrane potential. J Biol Chem 284(47), 32331–32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rocco D, Cerqua C, Goffrini P, Russo G, Pastore A, Meloni F, Nicchia E, Moraes CT, Pecci A, Salviati L, Savoia A, 2014. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim Biophys Acta 1842(2), 269–274. [DOI] [PubMed] [Google Scholar]
- Deacon OM, Karsisiotis AI, Moreno-Chicano T, Hough MA, Macdonald C, Blumenschein TMA, Wilson MT, Moore GR, Worrall JAR, 2017. Heightened Dynamics of the Oxidized Y48H Variant of Human Cytochrome c Increases Its Peroxidatic Activity. Biochemistry 56(46), 6111–6124. [DOI] [PubMed] [Google Scholar]
- Dickerson R, Timkovich R, 1975. Cytochrome c, in: Boyer P (Ed.) The Enzymes. Academic Press, New York, pp. 397–472. [Google Scholar]
- Ferguson-Miller S, Brautigan DL, Margoliash E, 1978. Definition of cytochrome c binding domains by chemical modification. III. Kinetics of reaction of carboxydinitrophenyl cytochromes c with cytochrome c oxidase. J Biol Chem 253(1), 149–159. [PubMed] [Google Scholar]
- Firsov D, Bonny O, 2010. Circadian regulation of renal function. Kidney Int 78(7), 640–645. [DOI] [PubMed] [Google Scholar]
- Garcia-Heredia JM, Diaz-Quintana A, Salzano M, Orzaez M, Perez-Paya E, Teixeira M, De la Rosa MA, Diaz-Moreno I, 2011. Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti-apoptotic switch. J Biol Inorg Chem 16(8), 1155–1168. [DOI] [PubMed] [Google Scholar]
- Gnaiger E, Lassnig B, Kuznetsov A, Rieger G, Margreiter R, 1998. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol 201 (Pt 8), 1129–1139. [DOI] [PubMed] [Google Scholar]
- Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, Yandell BS, Attie AD, Coon JJ, Pagliarini DJ, 2012. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab 16(5), 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra-Castellano A, Diaz-Moreno I, Velazquez-Campoy A, De la Rosa MA, Diaz-Quintana A, 2016. Structural and functional characterization of phosphomimetic mutants of cytochrome c at threonine 28 and serine 47. Biochim Biophys Acta 1857(4), 387–395. [DOI] [PubMed] [Google Scholar]
- Guerra-Castellano A, Diaz-Quintana A, Moreno-Beltran B, Lopez-Prados J, Nieto PM, Meister W, Staffa J, Teixeira M, Hildebrandt P, De la Rosa MA, Diaz-Moreno I, 2015. Mimicking Tyrosine Phosphorylation in Human Cytochrome c by the Evolved tRNA Synthetase Technique. Chemistry (Weinheim an der Bergstrasse, Germany) 21(42), 15004–15012. [DOI] [PubMed] [Google Scholar]
- Guerra-Castellano A, Diaz-Quintana A, Perez-Mejias G, Elena-Real CA, Gonzalez-Arzola K, Garcia-Maurino SM, De la Rosa MA, Diaz-Moreno I, 2018. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc Natl Acad Sci U S A 115(31), 7955–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek P, Villani G, Attardi G, 2001. Rate-limiting step preceding cytochrome c release in cells primed for Fas-mediated apoptosis revealed by analysis of cellular mosaicism of respiratory changes. J Biol Chem 276(1), 606–615. [DOI] [PubMed] [Google Scholar]
- Hampsey DM, Das G, Sherman F, 1988. Yeast iso-1-cytochrome c: genetic analysis of structural requirements. FEBS Lett 231(2), 275–283. [DOI] [PubMed] [Google Scholar]
- Heller J, Smith EL, 1966. Neurospora crassa cytochrome c. II. Chymotryptic peptides, tryptic peptides, cyanogen bromide peptides, and the complete amino acid sequence. J Biol Chem 241(13), 3165–3180. [PubMed] [Google Scholar]
- Helling S, Hüttemann M, Kadenbach B, Ramzan R, Vogt S, Marcus K, 2012a. Discovering the phosphoproteome of the hydrophobic cytochrome c oxidase membrane protein complex. Methods Mol Biol 893, 345–358. [DOI] [PubMed] [Google Scholar]
- Helling S, Hüttemann M, Ramzan R, Kim SH, Lee I, Muller T, Langenfeld E, Meyer HE, Kadenbach B, Vogt S, Marcus K, 2012b. Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics 12(7), 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle PC, Kumar MA, Resetar A, Harris DL, 1991. Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry 30(14), 3576–3582. [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, Stockli J, Burchfield JG, Jensen TE, Jothi R, Kiens B, Wojtaszewski JF, Richter EA, James DE, 2015. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab 22(5), 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Pecina P, Rainbolt M, Sanderson TH, Kagan VE, Samavati L, Doan JW, Lee I, 2011. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 11(3), 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP, Orrenius S, Mason HS, 1979. Hemoprotein quantitation in isolated hepatocytes. Biochim Biophys Acta 576(1), 17–29. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G, 2009. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med 46(11), 1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaim G, Dimroth P, 1999. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J 18(15), 4118–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ, 2016. Ischemia/Reperfusion. Compr Physiol 7(1), 113–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalpage HA, Bazylianska V, Recanati MA, Fite A, Liu J, Wan J, Mantena N, Malek MH, Podgorski I, Heath EI, Vaishnav A, Edwards BF, Grossman LI, Sanderson TH, Lee I, Hüttemann M, 2019a. Tissue-specific regulation of cytochrome c by post-translational modifications: respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J 33(2), 1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalpage HA, Vaishnav A, Liu J, Varughese A, Wan J, Turner AA, Ji Q, Zurek MP, Kapralov AA, Kagan VE, Brunzelle JS, Recanati MA, Grossman LI, Sanderson TH, Lee I, Salomon AR, Edwards BFP, Hüttemann M, 2019b. Serine-47 phosphorylation of cytochrome c in the mammalian brain regulates cytochrome c oxidase and caspase-3 activity. FASEB J (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapralov AA, Yanamala N, Tyurina YY, Castro L, Samhan-Arias A, Vladimirov YA, Maeda A, Weitz AA, Peterson J, Mylnikov D, Demicheli V, Tortora V, Klein-Seetharaman J, Radi R, Kagan VE, 2011. Topography of tyrosine residues and their involvement in peroxidation of polyunsaturated cardiolipin in cytochrome c/cardiolipin peroxidase complexes. Biochim Biophys Acta 1808(9), 2147–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy FG, Jones DP, 1986. Oxygen dependence of mitochondrial function in isolated rat cardiac myocytes. Am J Physiol 250(3 Pt 1), C374–383. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Ellerby LM, Ellerby HM, Naiem S, Yaffe MP, Margoliash E, Bredesen D, Mauk AG, Sherman F, Newmeyer DD, 2000. Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J Biol Chem 275(21), 16127–16133. [DOI] [PubMed] [Google Scholar]
- Kokhan O, Wraight CA, Tajkhorshid E, 2010. The binding interface of cytochrome c and cytochrome c(1) in the bc(1) complex: rationalizing the role of key residues. Biophys J 99(8), 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski B, 1997. Thermodynamic regulation of cytochrome oxidase. Mol Cell Biochem 174(1–2), 137–141. [PubMed] [Google Scholar]
- Kudin A, Vielhaber S, Elger CE, Kunz WS, 2002. Differences in flux control and reserve capacity of cytochrome c oxidase (COX) in human skeletal muscle and brain suggest different metabolic effects of mild COX deficiencies. Mol Biol Rep 29(1–2), 89–92. [DOI] [PubMed] [Google Scholar]
- Kunz WS, Kudin A, Vielhaber S, Elger CE, Attardi G, Villani G, 2000. Flux control of cytochrome c oxidase in human skeletal muscle. J Biol Chem 275(36), 27741–27745. [DOI] [PubMed] [Google Scholar]
- Lange C, Hunte C, 2002. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc Natl Acad Sci U S A 99(5), 2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Hüttemann M, 2005. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem 280(7), 6094–6100. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Yu K, Doan JW, Grossman LI, Hüttemann M, 2006. New prospects for an old enzyme: mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry 45(30), 9121–9128. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Yu K, Samavati L, Pecina P, Pecinova A, Hüttemann M, 2009. Isolation of regulatory-competent, phosphorylated cytochrome c oxidase. Methods Enzymol 457, 193–210. [DOI] [PubMed] [Google Scholar]
- Lempiainen J, Finckenberg P, Levijoki J, Mervaala E, 2012. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol 166(6), 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV, 2013. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci Signal 6(278), rs11. [DOI] [PubMed] [Google Scholar]
- Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV, 2012. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra G, Varughese A, Ji Q, Lee I, Liu J, Vaishnav A, Sinkler C, Kapralov AA, Moraes CT, Sanderson TH, Stemmler TL, Grossman LI, Kagan VE, Brunzelle JS, Salomon AR, Edwards BF, Hüttemann M, 2017. Phosphorylation of Cytochrome c Threonine 28 Regulates Electron Transport Chain Activity in Kidney: IMPLICATIONS FOR AMP KINASE. J Biol Chem 292(1), 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MC, Allan LA, Lickrish M, Sampson C, Morrice N, Clarke PR, 2005. Protein kinase A regulates caspase-9 activation by Apaf-1 downstream of cytochrome c. J Biol Chem 280(15), 15449–15455. [DOI] [PubMed] [Google Scholar]
- McIntosh DB, Parrish JC, Wallace CJ, 1996. Definition of a nucleotide binding site on cytochrome c by photoaffinity labeling. J Biol Chem 271(31), 18379–18386. [DOI] [PubMed] [Google Scholar]
- Moreno-Beltran B, Guerra-Castellano A, Diaz-Quintana A, Del Conte R, Garcia-Maurino SM, Diaz-Moreno S, Gonzalez-Arzola K, Santos-Ocana C, Velazquez-Campoy A, De la Rosa MA, Turano P, Diaz-Moreno I, 2017. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc Natl Acad Sci U S A 114(15), E3041–e3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien ES, Nucci NV, Fuglestad B, Tommos C, Wand AJ, 2015. Defining the Apoptotic Trigger: THE INTERACTION OF CYTOCHROME c AND CARDIOLIPIN. J Biol Chem 290(52), 30879–30887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BL, Yang G, Humphrey SJ, Chaudhuri R, Ma X, Peterman S, James DE, 2015. Targeted phosphoproteomics of insulin signaling using data-independent acquisition mass spectrometry. Sci Signal 8(380), rs6. [DOI] [PubMed] [Google Scholar]
- Pecina P, Borisenko GG, Belikova NA, Tyurina YY, Pecinova A, Lee I, Samhan-Arias AK, Przyklenk K, Kagan VE, Hüttemann M, 2010. Phosphomimetic substitution of cytochrome c tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 49(31), 6705–6714. [DOI] [PubMed] [Google Scholar]
- Pereverzev MO, Vygodina TV, Konstantinov AA, Skulachev VP, 2003. Cytochrome c, an ideal antioxidant. Biochem Soc Trans 31(Pt 6), 1312–1315. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Scrima R, Boffoli D, Capitanio N, 2006. Control by cytochrome c oxidase of the cellular oxidative phosphorylation system depends on the mitochondrial energy state. Biochem J 396(3), 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierron D, Opazo JC, Heiske M, Papper Z, Uddin M, Chand G, Wildman DE, Romero R, Goodman M, Grossman LI, 2011. Silencing, positive selection and parallel evolution: busy history of primate cytochromes C. PLoS One 6(10), e26269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierron D, Wildman DE, Hüttemann M, Letellier T, Grossman LI, 2012. Evolution of the couple cytochrome c and cytochrome c oxidase in primates. Adv Exp Med Biol 748, 185–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poornejad N, Schaumann LB, Buckmiller EM, Roeder BL, Cook AD, 2016. Current Cell-Based Strategies for Whole Kidney Regeneration. Tissue Eng Part B Rev 22(5), 358–370. [DOI] [PubMed] [Google Scholar]
- Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG, 2017. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep 21(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Rich P, 2003. Chemiosmotic coupling: The cost of living. Nature 421(6923), 583. [DOI] [PubMed] [Google Scholar]
- Roberts VA, Pique ME, 1999. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J Biol Chem 274(53), 38051–38060. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Malgat M, Mazat JP, Letellier T, 1999. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J Biol Chem 274(47), 33426–33432. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK, 1994. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem 269(3), 1770–1774. [PubMed] [Google Scholar]
- Sacco F, Humphrey SJ, Cox J, Mischnik M, Schulte A, Klabunde T, Schafer M, Mann M, 2016. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat Commun 7, 13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M, 2008. Tumor necrosis factor α inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem 283(30), 21134–21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Kumar R, Murariu-Dobrin AC, Page AB, Krause GS, Sullivan JM, 2009. Insulin activates the PI3K-Akt survival pathway in vulnerable neurons following global brain ischemia. Neurol Res 31(9), 947–958. [DOI] [PubMed] [Google Scholar]
- Sanderson TH, Kumar R, Sullivan JM, Krause GS, 2008. Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-XL binding. J Neurochem 106(3), 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Mahapatra G, Pecina P, Ji Q, Yu K, Sinkler C, Varughese A, Kumar R, Bukowski MJ, Tousignant RN, Salomon AR, Lee I, Hüttemann M, 2013a. Cytochrome c is tyrosine 97 phosphorylated by neuroprotective insulin treatment. PLoS One 8(11), e78627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M, 2013b. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47(1), 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanishvili R, Volz KW, Westbrook EM, Margoliash E, 1995. The low ionic strength crystal structure of horse cytochrome c at 2.1 Å resolution and comparison with its high ionic strength counterpart. Structure 3(7), 707–716. [DOI] [PubMed] [Google Scholar]
- Sato W, Hitaoka S, Inoue K, Imai M, Saio T, Uchida T, Shinzawa-Itoh K, Yoshikawa S, Yoshizawa K, Ishimori K, 2016. Energetic Mechanism of Cytochrome c-Cytochrome c Oxidase Electron Transfer Complex Formation under Turnover Conditions Revealed by Mutational Effects and Docking Simulation. J Biol Chem 291(29), 15320–15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharlau M, Geren L, Zhen EY, Ma L, Rajagukguk R, Ferguson-Miller S, Durham B, Millett F, 2019. Definition of the Interaction Domain and Electron Transfer Route between Cytochrome c and Cytochrome Oxidase. Biochemistry 58(40), 4125–4135. [DOI] [PubMed] [Google Scholar]
- Schmidt TR, Wildman DE, Uddin M, Opazo JC, Goodman M, Grossman LI, 2005. Rapid electrostatic evolution at the binding site for cytochrome c on cytochrome c oxidase in anthropoid primates. Proc Natl Acad Sci U S A 102(18), 6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkler CA, Kalpage H, Shay J, Lee I, Malek MH, Grossman LI, Hüttemann M, 2017. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxid Med Cell Longev 2017, 1534056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmaz SR, Hunte C, 2008. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J Biol Chem 283(25), 17542–17549. [DOI] [PubMed] [Google Scholar]
- Sugitani R, Stuchebrukhov AA, 2009. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim Biophys Acta 1787(9), 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiyama Y, Harumi T, Watanabe J, Fujita Y, Honjo J, Shimizu N, Makino Y, Haneda M, 2011. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1alpha expression and oxygen metabolism. Diabetes 60(3), 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S, 2003. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci U S A 100(26), 15304–15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuominen EK, Zhu K, Wallace CJ, Clark-Lewis I, Craig DB, Rytomaa M, Kinnunen PK, 2001. ATP induces a conformational change in lipid-bound cytochrome c. J Biol Chem 276(22), 19356–19362. [DOI] [PubMed] [Google Scholar]
- Vijayasarathy C, Biunno I, Lenka N, Yang M, Basu A, Hall IP, Avadhani NG, 1998. Variations in the subunit content and catalytic activity of the cytochrome c oxidase complex from different tissues and different cardiac compartments. Biochim Biophys Acta 1371(1), 71–82. [DOI] [PubMed] [Google Scholar]
- Villani G, Attardi G, 1997. In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc Natl Acad Sci U S A 94(4), 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani G, Attardi G, 2000. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic Biol Med 29(3–4), 202–210. [DOI] [PubMed] [Google Scholar]
- Wan J, Kalpage HA, Vaishnav A, Liu J, Lee I, Mahapatra G, Turner AA, Zurek MP, Ji Q, Moraes CT, Recanati MA, Grossman LI, Salomon AR, Edwards BFP, Hüttemann M, 2019. Regulation of Respiration and Apoptosis by Cytochrome c Threonine 58 Phosphorylation. Sci Rep 9(1), 15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann FR, Kunz WS, 1998. Oxygen dependence of flux control of cytochrome c oxidase -- implications for mitochondrial diseases. FEBS Lett 422(1), 33–35. [DOI] [PubMed] [Google Scholar]
- Yu H, Lee I, Salomon AR, Yu K, Hüttemann M, 2008. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim Biophys Acta 1777(7–8), 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Wang X, Purring-Koch C, Wei Y, McLendon GL, 2001. A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J Biol Chem 276(16), 13034–13038. [DOI] [PubMed] [Google Scholar]
- Zaidi S, Hassan MI, Islam A, Ahmad F, 2014. The role of key residues in structure, function, and stability of cytochrome-c. Cell Mol Life Sci 71(2), 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bak S, Pedersen AJ, Jensen ON, Hojlund K, 2014. Insulin increases phosphorylation of mitochondrial proteins in human skeletal muscle in vivo. J Proteome Res 13(5), 2359–2369. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leon IR, Bak S, Mogensen M, Wrzesinski K, Hojlund K, Jensen ON, 2011. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol Cell Proteomics 10(1), M110 000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE, 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108(8), 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, Bonny O, Firsov D, 2009. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A 106(38), 16523–16528. [DOI] [PMC free article] [PubMed] [Google Scholar]