

Abstract
Pexidartinib (TURALIO™) is an orally administered small molecule tyrosine kinase inhibitor with selective activity against the colony-stimulating factor 1 (CSF1) receptor, KIT proto-oncogene receptor tyrosine kinase (KIT) and FMS-like tyrosine kinase 3 harboring an internal tandem duplication mutation (FLT3-ITD). In August 2019, the US FDA approved pexidartinib capsules for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery. This approval was based on positive results from the phase III ENLIVEN trial. Pexidartinib is being investigated in various malignancies as monotherapy or combination therapy. This article summarizes the milestones in the development of pexidartinib leading to its first approval for TGCT.
Pexidartinib (TURALIO™): Key points
| A small molecule tyrosine kinase inhibitor has been developed by Daiichi Sankyo for the treatment of TGCT |
| Received its first approval on 2 August 2019 in the USA |
| Approved for use in adult patients with symptomatic TGCT associated with severe morbidity or functional limitations and not amenable to surgery |
Introduction
Pexidartinib (TURALIO™) is a novel, orally available small molecule tyrosine kinase inhibitor (TKI) with potent and selective activity against the colony-stimulating factor 1 (CSF1) receptor [1–3]. CSF1, expressed in high levels in several types of solid tumors, facilitates the differentiation of monocytes into tumor-associated macrophages (TAMs) and the survival of TAMs within the tumor microenvironment [4–6]. TAMs in turn have immunosuppressive effects and promote tumor growth and metastases [4, 6]. Pexidartinib also inhibits KIT proto-oncogene receptor tyrosine kinase (KIT) and FMS-like tyrosine kinase 3 harboring an internal tandem duplication mutation (FLT3-ITD) [2, 3, 7, 8]. Pexidartinib has been developed by Daiichi Sankyo for the treatment of tenosynovial giant cell tumor (TGCT; also known as giant cell tumor of the tendon sheath or pigmented villonodular synovitis [9]). Most commonly arising in the synovium of joints or tendon sheaths of young adults, TGCT is a rare and locally aggressive neoplasm that overexpresses CSF1 [10, 11].
Pexidartinib 200 mg capsules were approved in the USA on 2 August 2019 for the treatment of adult patients with symptomatic TGCT associated with severe morbidity or functional limitations and not amenable to improvement with surgery [12]. Pexidartinib is the first systemic treatment to be approved by the FDA in this indication [12]. Approval was based on data from a pivotal phase III study (ENLIVEN), which constituted the first placebo-controlled trial of a systemic investigational treatment in TGCT [13]. The recommended dose of pexidartinib is 400 mg administered twice daily on an empty stomach (≥ 1 h before or ≥ 2 h after a meal or snack) [9].

Key milestones in the development of pexidartinib for the treatment of adults with symptomatic TGCT. MAA Marketing Authorization Application, NDA New Drug Application, ODAC Oncologic Drugs Advisory Committee
Capsules should be swallowed whole, rather than opened, broken or chewed. Treatment should continue until disease progression or unacceptable toxicity [9].
The US labelling of pexidartinib carries a boxed warning of hepatotoxicity [9]. Pexidartinib can induce serious and potentially fatal liver injury; liver tests should be conducted prior to initiating treatment with pexidartinib and then at specific intervals during treatment. If hepatotoxicity develops, pexidartinib should be withheld, administered at a reduced dose or permanently discontinued based on the severity of this. Due to the risk of hepatotoxicity, pexidartinib is only available in the USA to patients through a restricted Risk Evaluation and Mitigation Strategy (REMS) program [9].
Pexidartinib has been investigated in the phase I/II or II setting, either as monotherapy or in combination with other drugs, in the treatment of various cancers such as glioblastoma (in which TAMs and microglia contribute to glioma progression and can represent up to 30% of the tumor mass [14]), melanoma and metastatic breast cancer.
Scientific Summary
Pharmacodynamics
Pexidartinib potently and selectively inhibits the CSF1R [biochemical half maximal inhibitory concentration (IC50) = 0.02 µmol/L [2]] and KIT (biochemical IC50 = 0.01 µmol/L [2]) receptor tyrosine kinases [1–3]. Only five other kinases were significantly inhibited by pexidartinib at 0.03 µmol/L and 1.0 µmol/L in a comprehensive screen of 226 different kinases; the biochemical IC50 values of these kinases were ≥ 8-fold higher than those of CSF1 or KIT [2]. Pexidartinib has been shown to inhibit FLT3 signaling more effectively in cells harboring FLT3-ITD than in cells only containing wild-type FLT3 (e.g. biochemical IC50 = 0.018 µmol/L vs. 1.8 µmol/L) [8].
Prospectively designed to engage the juxtamembrane region of the CSF1 receptor, pexidartinib was significantly (based on 95% CIs) more potent than PLX647 or imatinib when assayed against CSF1 receptor-dependent cells [3]. The three drugs were comparable in the extent to which they inhibited KIT-dependent cellular processes [3].
In immunohistochemical analyses of xenograft samples from pexidartinib- and control-treated mice, pexidartinib induced significant (p < 0.0001 vs. control) macrophage depletion in tumors, as indicated by ionized calcium-binding adapter molecule 1 (Iba-1) signal (a marker for macrophages) [15]. While another animal study found no effect of pexidartinib on intratumoral macrophage infiltration in vivo, gene expression profiling of TAMs indicated a shift in TAM polarization in pexidartinib-treated tumors compared with vehicle-treated tumors; this shift in polarization may contribute to inhibitory effects of pexidartinib on tumor growth [1]. Pexidartinib treatment significantly (p < 0.05) suppressed tumor growth relative to vehicle treatment in mouse models of various tumors [1, 14–16] and was shown to significantly (p = 0.026) increase median survival time in tumor-bearing mice (11.3 vs. 8.0 weeks with vehicle treatment) [1].
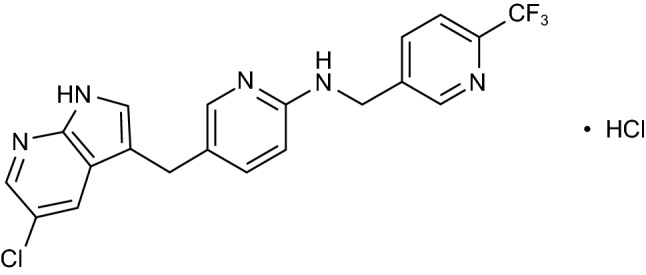
Chemical structure of pexidartinib
At double the mean maximum exposure of the approved dose (400 mg twice daily), pexidartinib is not associated with clinically relevant QTc interval prolongation [9].
Pharmacokinetics
Following administration of single oral doses of pexidartinib in healthy participants, pexidartinib exposure [peak plasma concentration (Cmax) and total area under the concentration-time curve (AUC0–∞)] increased in a generally dose-proportional manner over the dose range of 200–2400 mg [9, 17]. Median time to Cmax (Tmax) was 2.5 h. When administered with a high fat meal, the Cmax and AUC0–∞ of pexidartinib increased by 100% and Tmax was delayed by 2.5 h relative to fasting conditions [9, 17]. In patients receiving pexidartinib 400 mg twice daily, steady state was reached in ≈ 7 days and the median accumulation ratio was 3.6 [9]. Pexidartinib is highly bound to plasma protein (> 99%) and had a mean apparent volume of distribution of 187 L [9].
Pexidartinib is highly metabolized, primarily by CYP3A4 and UGT1A4 [9, 18]. After oral administration of a single radiolabeled dose of pexidartinib 400 mg, 65% of the radioactivity was recovered in the feces (44% as unchanged drug) and 27% was recovered in the urine as metabolites (≈ 10% as N-glucuronide) [9]. After a single oral dose of pexidartinib 400 mg, pexidartinib had a mean apparent clearance of 5.1 L/h and mean half-life of 26.6 h [9].
Pexidartinib pharmacokinetics are not impacted to a clinically meaningful degree by age (18–84 years), sex, race (white or black) or mild hepatic impairment [total bilirubin ≤ upper limit of normal (ULN) with aspartate aminotransferase (AST) > ULN, or total bilirubin > 1 to 1.5 × ULN with any AST] [9]. Relative to that in patients with normal renal function [creatinine clearance (CLCR) ≥ 90 mL/min], pexidartinib exposure (AUC) was increased by ≈ 30% in patients with mild (CLCR 60–89 mL/min), moderate (CLCR 30–59 mL/min) and severe (CLCR 15–29 mL/min) renal impairment. Consequently, a dosage modification is recommended in patients with mild to severe renal impairment (pexidartinib 200 mg in the morning and 400 mg in the evening). Pexidartinib pharmacokinetics have not been sufficiently characterized in moderate hepatic impairment (total bilirubin > 1.5 to 3 × ULN and any AST) or investigated in severe hepatic impairment (total bilirubin > 3 to 10 × ULN and any AST); treatment with pexidartinib should not be initiated in patients with increased serum transaminases, total or direct bilirubin > ULN, or active liver or biliary disease [including elevated alkaline phosphatase (ALP)] [9].
In clinical studies, pexidartinib exposure was altered by coadministration with itraconazole, probenecid, rifampicin or esomeprazole [9]. Concomitant use of pexidartinib with strong CYP3A inhibitors (including grapefruit or grapefruit juice), strong UGT inhibitors, strong CYP3A inducers (including Saint John’s wort) or proton pump inhibitors should thus be avoided; if concomitant use with strong CYP3A or UGT inhibitors cannot be avoided, a reduced pexidartinib dosage (400 mg/day, administered as 200 mg twice daily) is recommended. Local prescribing information should be consulted for further information regarding potential drug interactions and dosage modifications [9].
Features and properties of pexidartinib
| Alternative names | Pexidartinib hydrochloride; Plexxikon 3397; PLX-3397; TURALIO |
| Class | 2 ring heterocyclic compounds; anti-dementias; anti-neoplastics; fluorine compounds; pyridines; pyrroles; small molecules |
| Mechanism of action | Fms-like tyrosine kinase 3 inhibitors; macrophage colony stimulating factor receptor antagonists; proto oncogene protein c-kit inhibitors |
| Route of administration | Oral |
| Pharmacodynamics | Inhibits colony-stimulating factor 1 (CSF1) receptor, KIT proto-oncogene receptor tyrosine kinase (KIT) and FMS-like tyrosine kinase 3 internal tandem duplication mutation (FLT3-ITD); depletes (or shifts polarization of) tumor-associated macrophages and inhibits tumor growth in mouse models |
| Pharmacokinetics | Generally dose-proportional pharmacokinetics; increased drug exposure if administered with food; drug exposure increased in patients with renal impairment; potential to interact with various other drugs if used concomitantly |
|
Adverse events (occurring in ≥ 15% of pexidartinib recipients) |
Hair color changes (depigmentation), fatigue, increased aspartate aminotransferase, increased alanine aminotransferase, dysgeusia, vomiting, periorbital edema, abdominal pain, decreased appetite, pruritus, hypertension and increased alkaline phosphatase |
| ATC codes | |
| WHO ATC code | LO1X-E (protein kinase inhibitors); LO4A-A (selective immunosuppressants); MO1 (anti-inflammatory and anti-rheumatic products); NO6D-X (other anti-dementia drugs) |
| EphMRA ATC code | L1H (protein kinase inhibitor anti-neoplastics); L4X (other immunosuppressants); M1 (anti-inflammatory and anti-rheumatic products); N6D (nootropics) |
| Chemical name | 5-[(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)methyl]-N-[[6-(trifluoromethyl)pyridin-3-yl]methyl]pyridin-2-amine monohydrochloride |
Therapeutic Trials
TGCT
Pexidartinib improved tumor response in adult patients with symptomatic, advanced TGCT for whom surgery was not recommended in the ENLIVEN trial [13]. ENLIVEN was a randomized, multinational phase III trial consisting of 24 weeks of double-blind treatment (part one) followed by an open-label extension (part two) [13]. In part one, patients with a worst pain or worst stiffness numeric rating scale (NRS) score of ≥ 4 and measureable disease as per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST; minimum tumor size 2 cm) were randomized to receive either oral pexidartinib (400 mg in the morning and 600 mg in the evening during the first 2 weeks, followed by 400 mg twice a day thereafter) or matched placebo. Efficacy was assessed in the modified intention-to-treat population (mITT; 61 pexidartinib recipients and 59 placebo recipients) and endpoints were analyzed hierarchically. In part two, patients who completed part one could receive open-label pexidartinib at their previous dose if they were randomized to pexidartinib in part one or to 800 mg/day (400 mg twice daily) if they had been randomized to placebo in part one. Treatment continued until disease progression or unacceptable toxicity [13]. At baseline, patients had a median age of 44 years and most patients (88%) had diffuse TGCT [9]; approximately half of the patients (53%) had ≥ 1 previous surgery for TGCT and 9% had previous TKI therapy [13]. The most common disease location was the knee (61% of patients) [13].
Clinical trials of pexidartinib
| Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor |
|---|---|---|---|---|---|---|
| Pexidartinib, placebo | Symptomatic TGCT | III | Active, not recruiting | Multinational | NCT02371369; ENLIVEN; PLX108-10; 2014-000148-14 | Daiichi Sankyo |
| Pexidartinib | Recurrent glioblastoma | II | Completed | USA | NCT01349036; PLX108-04 | Plexxikon |
| Pexidartinib | KIT-mutated advanced acral and mucosal melanoma | II | Active, not recruiting | UK | NCT02071940; PIANO; 11_DOG12_56 | The Christie NHS Foundation Trust |
| Pexidartinib | Advanced castration-resistant prostate cancer | II | Completed | USA | NCT01499043; PLX108-06 | Plexxikon |
| Pexidartinib, radiation therapy, temozolomide | Newly diagnosed glioblastoma | I/II | Active, not recruiting | USA | NCT01790503 | Plexxikon |
| Pexidartinib, pembrolizumab | Advanced melanoma and other solid tumours | I/II | Completed | USA | NCT02452424 | Plexxikon |
| Pexidartinib | Refractory leukemias and refractory solid tumors including NF1 PN | I/II | Recruiting | USA | NCT02390752 | National Cancer Institute |
| Pexidartinib, eribulin | Metastatic breast cancer | I/II | Active, not recruiting | USA | NCT01596751 | Hope Rugo |
| Pexidartinib, sirolimus | Sarcoma (phase I) and MPNST (phase II) | I/II | Active, not recruiting | USA | NCT02584647 | Gulam Manji |
| Pexidartinib, PLX9486, sunitinib | Advanced solid tumors | I/II | Active, not recruiting | USA | NCT02401815 | Plexxikon |
| Pexidartinib | Relapsed or refractory acute myeloid leukemia | I/II | Completed | USA | NCT01349049 | Plexxikon |
| Pexidartinib | Advanced solid tumors | I | Active, not recruiting | USA | NCT01004861 | Plexxikon |
| Pexidartinib, paclitaxel | Advanced solid tumors | I | Completed | USA | NCT01525602 | Plexxikon |
| Pexidartinib, durvalumab | Metastatic/advanced pancreatic or colorectal cancers | I | Active, not recruiting | France | NCT02777710 | Centre Leon Berard |
| Pexidartinib, binimetinib | Advanced gastrointestinal stromal tumor | I | Active, not recruiting | USA | NCT03158103 | Memorial Sloan Kettering Cancer Center |
| Pexidartinib | Advanced solid tumors | I | Active, not recruiting | Taiwan | NCT02734433 | Daiichi Sankyo |
NF1 PN neurofibromatosis type 1 related plexiform neurofibromas, NHS National Health Service, MPNST malignant peripheral nerve sheath tumor, TGCT tenosynovial giant cell tumor, UK United Kingdom
At week 25 of ENLIVEN, the proportion of patients achieving an overall response (complete response or partial response) based on RECIST and centrally evaluated MRI scans was significantly greater with pexidartinib than with placebo [39% vs. 0%; between-group difference (BGD) 39% (95% CI 27–52); p < 0.0001] (primary endpoint). In the pexidartinib group, 15% of patients had a complete response and 25% had a partial response. When overall response rate was based on tumor volume score (TVS; a TGCT-specific measure) and centrally evaluated MRI scans, the proportion of patients achieving an overall response at week 25 was also significantly greater with pexidartinib than with placebo [56% vs. 0%; BGD 56% (95% CI 42–68); p < 0.0001]; 5% of pexidartinib recipients had a complete response and 51% had a partial response. Responses to pexidartinib were maintained over longer-term, open-label treatment. As of data cutoff (31 January 2018; median follow-up 22 months), the best overall response by RECIST had increased to 53% in patients randomized to pexidartinib. Overall response by TVS was 64%. Median duration of response by RECIST or TVS was not reached, with few patients having progressed [13].
Pexidartinib also improved patient symptoms and functional outcomes in ENLIVEN [13]. Relative to placebo, pexidartinib significantly improved mean change from baseline in range of motion at week 25 (p = 0.0043) and mean change from baseline in physical functioning at week 25 (as assessed by the Patient-Reported Outcomes Measurement Information System – Physical Function scale; p = 0.0019). In addition, the mean decrease from baseline in worst stiffness NRS score at week 25 was significantly greater with pexidartinib (p < 0.0001). With respect to pain, the proportion of pain responders (i.e. patients with a ≥ 30% improvement in mean pain score without a ≥ 30% increase in analgesic use) did not significantly differ between treatment groups [13].
Pexidartinib was effective in treating TGCT in patients who crossed over to open-label pexidartinib 400 mg twice daily after receiving placebo in ENLIVEN (n = 30) [13]. At week 25 of pexidartinib therapy, the overall response rate was 30% based on RECIST and 57% based on TVS. As of data cutoff, this had increased to 53% based on RECIST and 67% based on TVS (median response duration not reached). Improvements in functional outcomes and stiffness were consistent with those seen with pexidartinib during placebo-controlled treatment [13].
Pexidartinib was associated with an overall response rate of 52% (95% CI 32–73; all partial responses) according to RECIST in patients with advanced TGCT in the extension of the phase I dose-escalation trial (NCT01004861) (in patients with advanced, incurable solid tumors that included a cohort of patients with TGCT) [3]. The disease control rate (complete response, partial response or stable disease) was 83% and median progression-free survival for these patients was not reached at the time of data cutoff (median treatment duration 8 months). Patients in this extension (n = 23) received open-label oral pexidartinib at a dose of 1000 mg per day until disease progression or unacceptable toxicity [3].
In a pooled analysis of long-term data from ENLIVEN and the phase I dose-escalation extension trial, pexidartinib was associated with an overall response rate (complete response or partial response) of 54% as assessed by RECIST and 64% as assessed by TVS after a median treatment duration of 17 months; median duration of response was not reached [19]. Patients had received pexidartinib at an initial dose of 800 or 1000 mg per day in either study (n = 130); 61 patients were ongoing in the studies at data cutoff [19].
Glioblastoma
Pexidartinib did not demonstrate efficacy in the treatment of recurrent glioblastoma in an open-label, single-arm, multicenter phase II study (NCT01349036) [20]. Pexidartinib was associated with a 6-month progression-free survival rate of 8.8% (95% CI 3.5–21.6; no significant improvement vs. historical controls) [primary endpoint]. There were no complete or partial imaging responses. The 6-month overall survival rate was 88.3% and median overall survival was 9.4 months. Patients received oral pexidartinib at a dose of 1000 mg per day (n = 37 analyzed). At baseline, patients had a median age of 58.5 years and median Karnofsky Performance Status score of 90 [20].
Pexidartinib was associated with a median progression-free survival of 6.7 months (vs. 7.5 months in historical controls) [primary endpoint] in patients with newly diagnosed glioblastoma in the phase II portion of an open-label phase Ib/II trial (NCT01790503) [21]. Estimated median overall survival in pexidartinib recipients was 15.4 months (vs. 18.9 months in controls). Patients (n = 42 evaluable) received oral pexidartinib 400 mg twice daily five days per week during standard of care (SOC) therapy (radiation therapy and temozolomide) and seven days per week after SOC therapy [21].
Solid Tumors
Oral pexidartinib in combination with paclitaxel was of clinical benefit (i.e. produced a complete response, partial response or stable disease) in 50% of patients with advanced solid tumors (n = 38 evaluable) in a first-in-human phase Ib trial (NCT01525602); one patient (with peritoneal carcinoma) had a complete response and five patients (with breast, rectal, bladder or ovarian cancer) had partial responses [6]. In part one of the study (a dose-escalation component), pexidartinib 1600 mg per day was selected as the recommended phase II dose [6].
In phase I studies, pexidartinib monotherapy (NCT01004861) [3], pexidartinib plus sirolimus (NCT02584647) [22], pexidartinib plus binimetinib (NCT03158103) [23], pexidartinib plus PLX9486 (NCT02401815) [24] and pexidartinib plus durvalumab (NCT02777710) [25] showed some antitumor activity in adult patients with solid tumors such as TGCT, unresectable malignant peripheral nerve sheath tumor (MPNST) and advanced gastrointestinal stromal tumor (GIST). Pexidartinib monotherapy was active against tumors in pediatric patients with neurofibromatosis type I related plexiform neurofibromas (NCT02390752; the recommended phase II dose was 800 mg/m2 per day) [26] and produced an objective tumor response in one patient with TGCT in a trial in Asian patients with advanced solid tumors (NCT02734433; n = 8 evaluable) [27].
Adverse Events
Oral pexidartinib monotherapy or combination therapy had a manageable tolerability profile in the clinical trials discussed in Sect. 2.3 [3, 6, 13, 20–22, 24, 27].
During part one of the pivotal ENLIVEN trial, treatment-emergent adverse events (TEAEs) of any grade occurred in 98% of pexidartinib recipients versus 93% of placebo recipients and TEAEs of grade 3 or 4 severity occurred in 44% versus 12% [13]. The most common TEAEs of any grade (occurring in ≥ 15% of pexidartinib recipients and more than with placebo) were hair color changes (67% of pexidartinib recipients vs. 3% of placebo recipients), fatigue (54% vs. 36%), increased AST (39% vs. 0%), increased alanine aminotransferase (ALT; 28% vs. 2%), dysgeusia (25% vs. 2%), vomiting (20% vs. 5%), periorbital edema (18% vs. 2%), abdominal pain (16% vs. 10%), decreased appetite (16% vs. 10%), pruritus (16% vs. 3%), hypertension (15% vs. 10%) and increased ALP (15% vs. 0%). The most common grade 3 or 4 TEAEs (occurring in ≥ 5% of pexidartinib recipients) were increased ALT (10% of pexidartinib recipients vs. 2% of placebo recipients), increased AST (10% vs. 0%), increased ALP (7% vs. 0%) and hypertension (5% vs. 0%). TEAEs led to permanent treatment discontinuation in 13% of pexidartinib recipients; in most cases, discontinuation was due to liver-related TEAEs. TEAEs required treatment interruption or dose reduction in 38% of pexidartinib recipients and 10% of placebo recipients; these TEAEs were most commonly hepatic (e.g. increased AST, increased ALT or cholestatic hepatotoxicity) [13].
Serious adverse events were reported in 13% of pexidartinib recipients (vs. 2% of placebo recipients) during part one of ENLIVEN [13]. Three pexidartinib recipients (5%) developed signs of serious liver injury (i.e. ALT or AST ≥ 3 × ULN with total bilirubin ≥ 2 × ULN with ALP > 2 × ULN); following treatment discontinuation, their ALT, AST and total bilirubin improved to < 2 × ULN in 1–7 months [9]. Patient enrolment in ENLIVEN was terminated early due to cases of serious hepatotoxicity, although the trial continued [28]. There was a death due to type A aortic dissection in a patient with a history of cardiovascular disease during part two of ENLIVEN; this was not considered to be treatment-related [13].
As pexidartinib can induce serious and potentially fatal liver injury, the US labelling of pexidartinib carries a boxed warning of hepatotoxicity (Sect. 1) [9]. Across all clinical trials involving patients with TGCT, there have been five cases of serious hepatic adverse reactions (mixed or cholestatic hepatotoxicity) assessed as probably drug-related (as of May 2019; n = 140 exposed to pexidartinib for a median treatment duration of 19 months); these included the three aforementioned cases during ENLIVEN part one, one additional patient during ENLIVEN part two and one patient from NCT03291288 (a pharmacokinetic study) [29]. All of these cases were reversible (four within 2 months, while one case of ductopenia resolved after 7 months) [29]. In non-TGCT populations, 2/768 pexidartinib recipients experienced irreversible cholestatic liver injury across clinical trials [9]. There was an exposure-response relationship between steady-state pexidartinib exposure and serum transaminase levels (ALT and AST) in trials; the risk of increased serum transaminases is higher at higher pexidartinib exposures. As administering pexidartinib with food increases drug exposure (Sect. 2.2), pexidartinib should be taken on an empty stomach (Sect. 1). Consult local prescribing information for further information, including recommended dosage modifications in patients who develop hepatotoxicity while receiving pexidartinib [9].
Ongoing Clinical Trials
In patients with TGCT, the ENLIVEN and NCT01004861 trials are approaching completion. A prospective observational study (NCT02948088) is following patients with diffuse TGCT, some of whom have received pexidartinib [30].
An open-label, single-arm phase II trial (PIANO; NCT02071940) of pexidartinib in advanced KIT-mutated acral and mucosal melanoma is currently ongoing in the UK. A number of phase I/II trials evaluating pexidartinib monotherapy (NCT02390752) or combination therapy (NCT01790503; NCT01596751; NCT02584647; NCT02401815) in various tumors and malignancies are currently ongoing in the USA. Several phase I trials in patients with advanced solid tumors (NCT01004861; NCT02777710; NCT03158103; NCT02734433) in the USA, France and Taiwan have not yet reached completion.
Current Status
Pexidartinib received its first approval on 2 August 2019 in the USA for the treatment of adult patients with symptomatic TGCT associated with severe morbidity or functional limitations and not amenable to improvement with surgery [9].
Compliance with Ethical Standards
Funding
The preparation of this review was not supported by any external funding.
Conflicts of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Yvette Lamb is a salaried employee of Adis International Ltd/Springer Nature, is responsible for the article content and declares no relevant conflicts of interest.
Footnotes
Enhanced material for this AdisInsight Report can be found at 10.6084/m9.figshare.9917990.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Change history
2/28/2020
The original article has been corrected.
Change history
2/28/2020
The original article has been corrected.
References
- 1.Ao JY, Zhu XD, Chai ZT, et al. Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular carcinoma. Mol Cancer Ther. 2017;16(8):1544–1554. doi: 10.1158/1535-7163.MCT-16-0866. [DOI] [PubMed] [Google Scholar]
- 2.DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428–437. doi: 10.1056/NEJMoa1411366. [DOI] [PubMed] [Google Scholar]
- 4.Shi G, Yang Q, Zhang Y, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27(1):244–260. doi: 10.1016/j.ymthe.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stafford JH, Hirai T, Deng L, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18(6):797–806. doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wesolowski R, Sharma N, Reebel L, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol. 2019;11:1–13. doi: 10.1177/1758835919854238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton E, Wong B, Zhang J. The novel inhibitor PLX3397 effectively inhibits FLT3-mutant AML. Blood. 2011;118:3632. doi: 10.1182/blood.V118.21.3632.3632. [DOI] [Google Scholar]
- 8.Smith CC, Zhang C, Lin KC, et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015;5(6):668–679. doi: 10.1158/2159-8290.CD-15-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daiichi Sankyo Inc. TURALIOTM (pexidartinib) capsules, for oral use: US prescribing information 2019. http://www.accessdata.fda.gov/. Accessed 30 Sep 2019.
- 10.Gelhorn HL, Tong S, McQuarrie K, et al. Patient-reported symptoms of tenosynovial giant cell tumors. Clin Ther. 2016;38(4):778–793. doi: 10.1016/j.clinthera.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giustini N, Bernthal NM, Bukata SV, et al. Tenosynovial giant cell tumor: case report of a patient effectively treated with pexidartinib (PLX3397) and review of the literature. Clin Sarcoma Res. 2018;8:14. doi: 10.1186/s13569-018-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Food and Drug Administration. FDA approves first therapy for rare joint tumor [media release]. 2 Aug 2019. http://www.fda.gov.
- 13.Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019;394(10197):478–487. doi: 10.1016/S0140-6736(19)30764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan D, Kowal J, Akkari L, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36(43):6049–6058. doi: 10.1038/onc.2017.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patwardhan PP, Surriga O, Beckman MJ, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res. 2014;20(12):3146–3158. doi: 10.1158/1078-0432.CCR-13-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mok S, Koya RC, Tsui C, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandell AG, Duchin KL, Desai M, et al. Effect of dose strength, food, and pH modifiers on the pharmacokinetics of the multi-kinase inhibitor pexidartinib [abstract no. PI-152] Clin Pharmacol Ther. 2018;103(Suppl S1):S54. [Google Scholar]
- 18.Daiichi Sankyo Inc. Briefing document for the May 14, 2019, US FDA oncologic drugs advisory committee meeting: Turalio (pexidartinib) capsules. 2019. http://www.fda.gov/. Accessed 30 Sep 2019.
- 19.Gelderblom H, Tap WD, Palmerini E, et al. Pexidartinib for advanced tenosynovial giant cell tumor (TGCT): long-term efficacy and safety from the phase 3 ENLIVEN and phase 1 PLX108-01 (TGCT cohort) studies [abstract no. 11042] J Clin Oncol. 2019;37(Suppl 15):11042. [Google Scholar]
- 20.Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18(4):557–564. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colman H, Raizer JJ, Walbert T, et al. Phase 1b/2 study of pexidartinib (PEX) in combination with radiation therapy (XRT) and temozolomide (TMZ) in newly diagnosed glioblastoma [abstract no. 2015] J Clin Oncol. 2018;36(Suppl 15):2015. doi: 10.1200/JCO.2018.36.15_suppl.2015. [DOI] [Google Scholar]
- 22.Manji GA, Van Tine BA, Lee SM, et al. Phase 1 combination therapy with pexidartinib (PEX) and sirolimus (S) to target tumor-associated macrophages in pigmented villonodular synovitis, malignant peripheral nerve sheath tumors, and other soft tissue sarcomas [abstract no. 11055] J Clin Oncol. 2019;37(Suppl 15):11055. [Google Scholar]
- 23.Rosenbaum E, Kelly C, D’Angelo SP, et al. A phase I study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist. 2019;24:1–8. doi: 10.1634/theoncologist.2019-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner AJ, Tap WD, Shields AF, et al. A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of PLX9486 alone and in combination (combo) with the KIT inhibitors pexidartinib (pexi) or sunitinib (su) in patients (Pts) with advanced solid tumors and gastrointestinal stromal tumor (GIST) [abstract no. 11509] J Clin Oncol. 2018;36(Suppl 15):11509. doi: 10.1200/JCO.2018.36.15_suppl.11509. [DOI] [Google Scholar]
- 25.Cassier PA, Garin G, Eberst L, et al. MEDIPLEX: a phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC) [abstract no. 2579] J Clin Oncol. 2019;37(Suppl 15):2579. doi: 10.1200/JCO.2019.37.15_suppl.2579. [DOI] [Google Scholar]
- 26.Hittson /l, Glod J, Amaya M, et al. Phase I study of pexidartinib (PLX3397) in children with refractory leukemias and solid tumors including neurofibromatosis type I (NF1) related plexiform neurofibromas (PN) [abstract no. 10546] J Clin Oncol. 2017;35(Suppl 15):10546. doi: 10.1200/JCO.2017.35.15_suppl.10546. [DOI] [Google Scholar]
- 27.Lee JH, Chen TW, Hsu CH, et al. A phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Invest New Drugs. 2019 doi: 10.1007/s10637-019-00745-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daiichi Sankyo Company. ENLIVEN phase 3 study of pexidartinib in tenosynovial giant cell tumor (TGCT) will continue to completion following enrollment discontinuation [media release]. 21 Oct 2016. http://www.daiichisankyo.com.
- 29.Bauer S, Lewis JH, Gelderblom H, et al. Pexidartinib for locally advanced tenosynovial giant cell tumor (TGCT): characterization of hepatic adverse reactions (ARs) [poster no. 1696P]. In: ESMO 2019.
- 30.Mastboom M, Palmerini E, Stacchiotti S, et al. First prospective observational study in diffuse-type tenosynovial giant cell tumors [abstract no. 11560] J Clin Oncol. 2018;36(Suppl 15):11560. doi: 10.1200/JCO.2018.36.15_suppl.11560. [DOI] [Google Scholar]