

Abstract
Background and objective: Renzapride (ATL-1251), a novel benzamide, is currently under clinical development for the treatment of irritable bowel syndrome (IBS). Previous in vitro and in vivo experimental studies have characterized renzapride as a full serotonin 5-HT4 receptor agonist on the gut and a 5-HT3 receptor antagonist. Clinical studies have confirmed the therapeutic efficacy, tolerability and safety of renzapride in patients with constipation-predominant IBS. This study set out to characterize the pharmacological profile of renzapride and its potential metabolic products at both 5-HT and other monoamine receptors in the gut.
Methods: The affinity of renzapride, its (+) and (−) enantiomers, and its primary metabolite, renzapride N-oxide and its enantiomers, for serotonin receptors was assessed by means of in vitro radioligand binding inhibition studies. After membranes prepared from animal tissue or membranes of cell lines transfected with cloned human receptors had been incubated with radiolabelled ligand with high affinity for a specific receptor, renzapride was added to competitively inhibit this binding. Levels of bound radioligand were measured by filtration and counting of the bound radioactivity. In instances where >50% inhibition of radioligand binding had occurred, the inhibition constant (Ki) was calculated. Metabolism of renzapride by liver microsomes was assessed by incubating 10 μmol/L renzapride with human liver microsome samples for 60 minutes at 37°C. After the reaction was stopped, the samples were centrifuged and the supernatant analysed for metabolites by high-pressure liquid chromatography (HPLC). The potential inhibitory effects of renzapride on cytochrome P450 (CYP) enzymes were assessed by incubating renzapride at various concentrations over a 1–500 μmol/L concentration range with microsomes genetically engineered to express a single CYP.
Results: Renzapride was selective for serotonergic receptors and, in particular, had high affinity for human 5-HT3 and guinea-pig 5-HT4 receptors (Ki 17 and 477 nm, respectively). Inhibitory properties at 5-HT2B receptors were also identified for renzapride, as well as some affinity for 5-HT2A and 5-HT2C receptors. Renzapride N-oxide and its enantiomers demonstrated much lower affinity for all 5-HT receptors compared with renzapride. Renzapride was metabolized by liver microsomes to a limited extent and there was no significant non-microsomal metabolism of renzapride. Renzapride did not inhibit the major CYP drug-metabolizing enzymes CYP2C9, CYP2D6, CYP1A2, CYP2A6, CYP2C19, CYP2E1 or CYP3A4 at concentrations consistent with use in a clinical setting.
Conclusions: These results confirm and extend earlier studies in animal and human receptors that show renzapride is a potent and generally full 5-HT4 receptor agonist and 5-HT3 receptor antagonist. The results reported in the present study indicate that the metabolites of renzapride are minor and are unlikely to contribute to its therapeutic profile or lead to interaction of renzapride with other drugs that inhibit the major drug-metabolizing enzymes in the liver at therapeutic doses. These data contribute to the understanding of the pharmacological actions and metabolic fate of renzapride in vivo.
Keywords: Irritable Bowel Syndrome, Human Liver Microsome, Tegaserod, Alosetron, Pool Human Liver Microsome
Introduction
Renzapride (ATL-1251, BRL 24924, (±)-endo-4-amino-5-chloro-2-methoxy-N-(1-azabicyclo [3.3.1]non-4-yl) benzamide) is a novel benzamide currently under clinical development for the treatment of irritable bowel syndrome (IBS), a condition in which the rationale for drugs that influence the actions of serotonin in the gastrointestinal tract is well established and understood.[1] In clinical studies, its use is associated with reduced overall gastrointestinal transit times, increased colonic motility and improved bowel function in patients with constipation-predominant IBS (IBS-C).[2,3] The safety and efficacy of renzapride in alleviating abdominal pain and discomfort as well as disordered motility in patients with IBS-C has also been demonstrated.[4]
Renzapride is one of several compounds that interact with serotonin receptors that have been or are being developed for the treatment of gastrointestinal disorders.[5] Others include tegaserod, a serotonin 5-HT4 receptor partial agonist indicated for IBS-C and chronic constipation,[6] and alosetron, a selective 5-HT3 receptor antagonist, indicated for diarrhoea-predominant IBS.[7] Agonism at 5-HT4 receptors located in the gastrointestinal tract is believed to mediate a prokinetic effect, accelerating gastrointestinal transit, whilst modifications in visceral hypersensitivity resulting in decreases in the perception of abdominal pain and/or discomfort, feelings of bloating etc., may be mediated via 5-HT3 receptor antagonism, peripherally and/or centrally.
In vitro functional studies on gastrointestinal smooth muscle preparations have characterized renzapride as a full or near full agonist at 5-HT4 receptors.[8–13] Furthermore, the inhibitory action of renzapride on the serotonin-evoked Bezold-Jarisch reflex in rats[12,14,15] indicates 5-HT3 receptor inhibitory properties. Renzapride is therefore a full agonist at 5-HT4 receptors and a 5-HT3 receptor antagonist; both pharmacological properties have significant actions on the gastrointestinal tract.
Renzapride has been shown to be a gastrointestinal prokinetic agent in the rat and dog,[12,16–21] and it increases gastric emptying in healthy human subjects.[22,23] In vitro radioligand binding studies using rodent tissues showed that renzapride has higher affinity for 5-HT3 and 5-HT4 receptors than for a range of other serotonergic and catecholaminergic receptors (see Briejer et al.[24] for review).
The affinities of renzapride for cloned human 5-HT3 and 5-HT4 receptors have been reported by a number of investigators.[25–33] Nine splice variants of the human 5-HT4 receptor (i.e. a-g, n and i), have been cloned and most are found in the gastrointestinal tract.[33,34] Renzapride has high affinity for splice variants of the 5-HT4 receptor present in the gut, and functional studies indicate that it is a full agonist at the majority of these, particularly in preparations with high receptor density.[27,28,32,35] The characterization of the pharmacological profile of renzapride at human receptors is therefore of key interest in understanding the mechanism of action of novel therapies for the treatment of gastrointestinal disorders.
Renzapride is a racemate of (+) and (−) enantiomers in equal proportions. The synthesis of the enantiomers and their preliminary pharmacological profiles in animal species has been described by King et al.[36] These studies indicated that the (+) and (−) enantiomers of renzapride possess similar pharmacological properties.
We report here on the affinities of renzapride and its enantiomers for a range of mainly human cloned 5-HT and catecholaminergic receptors and transporters. The functional activities of the enantiomers of renzapride at rodent 5-HT4 and 5-HT2B receptors and the affinities and in vitro pharmacological profiles of renzapride N-oxide, the major metabolite of renzapride in the rat, dog and man (unpublished observations), and its enantiomers are also investigated. We also report on the metabolism of renzapride by human liver microsomes and S9 fraction in vitro to determine the role of cytochrome P450 (CYP) and other liver enzymes on the biotransformation of this molecule. The likely propensity of renzapride to inhibit the major human microsomal drug-metabolizing CYP enzymes in vitro is also investigated to assess its potential for drug-drug interactions at the level of hepatic metabolism.
Materials and Methods
Racemic (±)-renzapride hydrochloride (batch 60850-02) was synthesized by Evotec Ltd, Abingdon, UK on behalf of Alizyme Therapeutics Ltd, Cambridge, UK. The (+) and (−) enantiomers of renzapride (batches Bx135-24 and Bx135-25, respectively) and racemic (±)-renzapride N-oxide (batches 178-015-1 and 178-018-8) were synthesized by Ultrafine Chemicals and Research Ltd, Manchester, UK on behalf of Alizyme Therapeutics Ltd. The single enantiomers (+) and (−) of renzapride N-oxide (batches MH 45582 and MH 45583, respectively), were synthesized by Beecham Research Laboratories, Betchworth, UK. Unless stated otherwise, in this report renzapride and renzapride N-oxide refer to their racemic forms. Drugs were administered in 0.4% dimethyl sulfoxide (DMSO) in aqueous mediums in the radioligand binding experiments and in 0.1% DMSO in aqueous mediums for the organ bath studies.
In Vitro Radioligand Binding Studies
Tissue and Cell Membrane Preparations for Radioligand Binding Assays
Renzapride, its (+) and (−) enantiomers, renzapride N-oxide and its (+) and (−) enantiomers were investigated for the inhibition of in vitro radio-ligand binding using membranes prepared from animal tissue or membranes of cell lines transfected with cloned human receptors. Standard procedures for the preparation of the animal membranes in modified Tris-HCl pH 7.4 buffer were used according to the publications shown in table I for each assay. For assays using cloned human receptors, cell lines as shown in table I were stably transfected with a plasmid encoding the human receptor using standard procedures based on the publications cited in table I for each assay.
Table I.
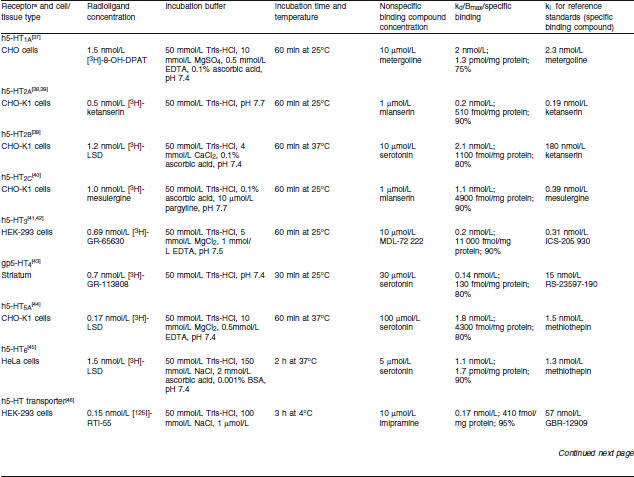
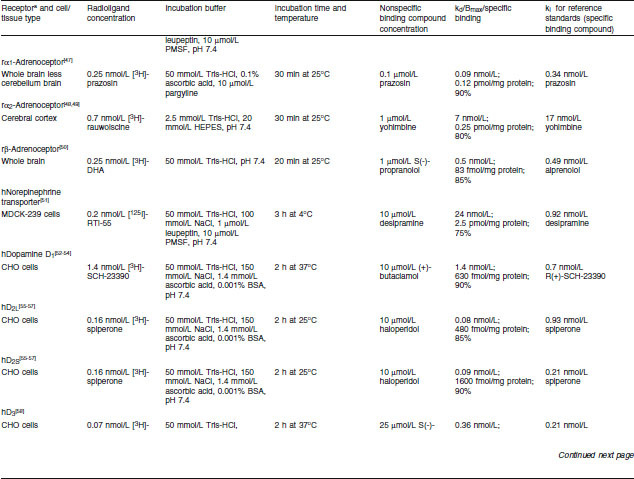
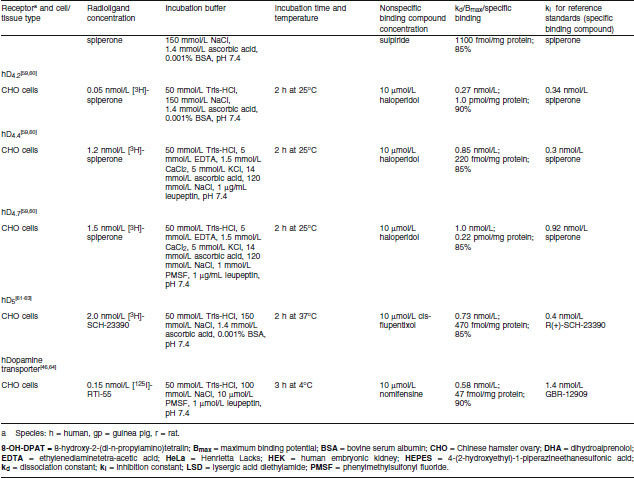
Inhibition of radioligand-specific binding to serotonin 5-HT receptors — assay methods
Binding Assay Conditions
Membrane fractions from the tissue homogenates, of which the protein concentrations had been determined by a standard procedure, were incubated with a radiolabelled ligand with high affinity for a particular receptor in 0.4% DMSO as shown in table I. Incubation buffers, pH and times to equilibrium for each assay are also shown in table I. Membranes were collected after the predetermined time by rapid filtration and the filter-bound radioactivity was counted by standard techniques.
Specific receptor binding was determined from nonspecific binding by the addition of excess unlabelled compound, shown in table I, which competes with the radioligand for binding to the receptor site. Specific binding was determined by subtraction of nonspecific binding from the total binding. Initial studies were conducted with one test and one replicate of the test compound (10 μmol/L), i.e. n = 2. If inhibition of specific binding was greater than 50%, inhibition constants were determined (see below) using five or six concentrations of all test compounds.
Data Analysis
IC50 (concentration that produces 50% inhibition) values, where presented, were determined by a non-linear, least-squares regression analysis using Data Analysis Toolbox™ (MDL Information Systems, San Leandro, CA, USA). Inhibition constants (ki), where presented, were calculated according to the equation of Cheng and Prusoff[65] using the observed IC50 of the tested compound, the concentration of radioligand employed in the assay and the historical values for the dissociation constant (kd) of the ligand (obtained experimentally at MDS Pharma Services, Taiwan Ltd, Taipei, Taiwan).
Organ Bath Experiments
Rat Stomach Strip
Strips of stomach fundus smooth muscle from Wistar rats (weight 275 ± 25 g) were prepared as described by Cohen and Fludzinski[66] and suspended in Krebs solution at 37°C at pH 7.4 in 5 mL organ baths.
Inhibition of α-Methyl-5-Serotonin-Induced Contractions
Isometric contractions of the tissue to 0.1 μmol/L α-methyl-5-serotonin were recorded. The effect of the 10-μmol/L test compound, administered in a 10-μL volume with a contact time of 5 minutes, on 0.1-μmol/L α-methyl-5-serotonin-induced contractions was determined. IC50 values were determined with three concentrations increasing by 0.5 log units.
Rat Isolated Oesophagus
Isolated oesophagus was prepared from Wistar rats (weight 275 ± 25 g), according to the method of Reeves et al.[11] and suspended in a 10-mL organ bath, perfused with Krebs solution containing indometacin 3 μmol/L and ketanserin 1 μmol/L at pH 7.4 and 37°C. Test compounds were administered in a volume of 10 μL.
Relaxation of Carbachol-Induced Contractions
Contraction of the oesophagus was induced by carbachol 3 μmol/L and recorded isometrically. The relaxant effect of serotonin 0.3 μmol/L was determined and the serotonin-like receptor agonist relaxant effect of test compounds determined at three concentrations increasing by 0.5 log units. Results were expressed as a percentage of the effect of serotonin 0.3 μmol/L. One test and a replicate were conducted at each drug concentration. Approximate EC50 (concentration that produces half the maximal effective response) values were determined graphically.
Human Liver Microsome Studies
Chemicals and Reagents
β-Nicotinamide adenine dinucleotide (β-NAD), β-NAD phosphate (β-NADP), β-NAD reduced form (β-NADH), glucose-6-phosphate, glucose-6-phosphate dehydrogenase (EC 1.1.1.49, type VII from Baker’s yeast), uridine-5-diphosphoglucuronic acid (UDPGA, trisodium salt), glutathione reduced form (GSH, free acid) and adenosine-3-phosphate 5-phosphosulphate (PAPS, lithium salt) were supplied by Sigma Aldrich Chemical Company, Dorset, UK.
CYP selective substrates were obtained from the following sources:
[14C]-Phenacetin 5.7 mCi/mmol, Sigma Aldrich Chemical Company, St Louis, MI, USA; coumarin, Sigma Aldrich Chemical Company, Dorset, UK; [14C]-tolbutamide 61 mCi/mmol, [14C]-S-mephenytoin, 56 mCi/mmol, [14C]-chlorzoxazone 57 mCi/mmol, and [14C]-testosterone 56 mCi/mmol, were supplied by Amersham Pharmacia Biotech, UK Ltd, Little Chalfont, UK. Bufuralol was a gift from Roche Products Ltd, Hertfordshire, UK. E-4031 was purchased from Wako Chemicals USA, Inc., Richmond, VA, USA.
The incubation phosphate buffer for the microsome studies was prepared by dissolving known amounts of dipotassium hydrogen phosphate and potassium dihydrogen phosphate in Millipore Pure water to give a 100-mmol/L solution that was adjusted to pH 7.4 by the addition of potassium hydroxide (0.1 mol/L). The buffer was stored at 4°C when not in use. All microsomal incubations undertaken in this study used a β-NADPH (β-nicotinamide adenine dinucleotide phosphate reduced form) regenerating system as enzyme co-factor for CYP. The co-factor solution typically contained glucose-6-phosphate (7.8 mg), glucose-6-phosphate dehydrogenase (6 units) and β-NADP (1.7 mg) dissolved in 1 mL of sodium bicarbonate solution (2% [w/v]).
Human liver microsomes used in this study were obtained from Human Biologics International, Scottsdale, AZ, USA, as part of this company’s HepatoScreen™ Test Kit. This contained liver microsomes from 14 individual donors and pooled microsomes containing microsomes from the same and other donors. The microsomes were supplied at a concentration of 20 mg/mL and were stored at −80°C while not in use. For incubations, microsomes were thawed and held on ice. Expressed CYP enzymes were purchased from Cypex Ltd, Dundee, UK. These are microsomes genetically engineered to express a single CYP enzyme and are derived from heterologous bacterial expression (bactosomes, Escherichia coli).
Metabolism of Renzapride by Human Liver Microsomes in Vitro
Pooled Human Liver Microsome Studies
In order to determine the optimal incubation conditions for the metabolism of renzapride by human liver microsomes, two preliminary studies were carried out. In the first, the final incubation concentration of renzapride was 1, 10 or 100 μmol/L with 0.5 mg microsomal protein/mL, and an incubation time of up to 60 minutes. In the second study, renzapride 10 μmol/L was selected as the final incubation concentration for 60 minutes with 2 and 4 mg microsomal protein/mL.
Incubation mixtures contained renzapride, microsomal protein and phosphate buffer (390 or 340 μL, 100 mmol/L, pH 7.4) to give a pre-incubation volume of 450 μL. Following a pre-incubation period of 5 minutes, microsomal reactions were initiated by the addition of enzyme co-factor solution, 50 μL, to give a final incubation volume of 500 μL. All pre-incubation and incubation stages were performed at 37°C in an oscillating water bath with vials open to the atmosphere. Microsomal reactions were terminated by the addition of acetonitrile (100 μL). Samples were stored at −20°C until assayed. After thawing, samples were centrifuged at ambient temperature to pellet the precipitated microsomal protein. An aliquot of each sample supernatant (200 μL) was removed into a glass vial for analysis by high-performance liquid chromatography (HPLC). Results, expressed as the percentage of metabolite-1 (M-1, the putative renzapride N-oxide) formed from renzapride (no protein control incubations), are the means of two observations per group.
In order to permit quantification of renzapride from area ratios, a standard curve was constructed (figure 1), with a dynamic range (linear response) of 2–500 mg/mL and a lower limit of quantification of 5 mg/mL.
Fig. 1.
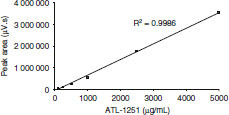
Renzapride standard curve determined by high-pressure liquid chromatography with UV detector (270 nm).
Since an authentic standard for renzapride N-oxide was not available at the time of these experiments and given that renzapride and M-1 represented the only drug-related material present in these incubation mixtures, the approximate concentrations of M-1 present were estimated by subtraction, i.e. the combined area ratios of the two analytes were assumed to equal 100%. This methodology clearly has limitations in terms of being able to accurately quantify M-1, although for the purposes of this study it was able to be used to provide a qualitative estimate of the amount of the putative N-oxide metabolite formation in these incubations.
Individual Donor Microsome Study
Renzapride 10 μmol/L was incubated with individual human liver microsomes from 14 donors (final incubation concentrations 4 mg microsomal protein/mL incubation mixture) for 0 and 60 minutes. The incubation conditions with respect to final volume, buffer, initiating co-factor solution, termination of reaction and assay of samples were as described for the preliminary studies above (see Pooled Human Liver Microsome Studies section).
The integrated peak area data were used to calculate an estimated rate of M-1 formation with liver microsomes from each individual donor (pmol/min/mg microsomal protein) using the 60-minute value for M-1 formation only. This was necessary because high time zero minutes (T = 0 min) M-1 formation was observed, presumably because of inefficient stopping of microsomal reactions. Results, expressed as the percentage of M-1 formed from renzapride (no protein control incubations), are single observations from each donor microsome preparation. By way of example:
Final [renzapride] in incubation mixture (i.e. 100% of the peak area ratio) = 10 μmol/L (5 × 10−9 mol); final [microsomal protein] = 4 mg/mL; final incubation volume = 0.5 mL; total incubation time = 60 min. Therefore, 1% of renzapride = 5 × 10−11 mol (50 pmol).
After 60 minutes’ incubation M-1 formation = 15.55% (estimated from relative peak area ratio). Therefore, the amount of M-1 present after 60 minutes’ incubation = 15.55 × 50 pmol = 777.5 pmol.
To adjust for microsomal protein content: 0.5 mL of a 4 mg/mL suspension = 2mg protein present in each incubation. Therefore, 777.5 pmol M-1 are formed with 2 mg protein = 388.75 pmol M-1 formed per mg protein.
To adjust for rate of reaction over time: 388.75 pmol M-1 are formed after 60 minutes’ incubation per mg protein = 6.48 pmol M-1 formed per min per mg protein (6.5 pmol/min/mg).
Metabolism of Renzapride by Genetically Expressed Human Cytochrome P450 (CYP) Enzymes in Vitro
Renzapride (1–500 μmol/L) was incubated with microsomes genetically engineered to express a single CYP (1 mg microsomal protein/mL incubation mixture) for 0 and 120 minutes. Incubation mixtures contained renzapride (10 μL of 0.5 mmol/L solution), microsomal protein suspension (CYP1A2 83 μL, CYP2C9 81 μL, CYP2C19 71 μL, CYP2D6 28 μL and CYP3A4 35 μL) and phosphate buffer (100 mmol/L, pH 7.4) to a volume of 450 μL. Following a pre-incubation period of 5 minutes, microsomal reactions were initiated by the addition of enzyme co-factor solution (50 μL) to give a final incubation volume of 500 μL. All pre-incubation, incubation, termination and sample storage conditions were as described previously (see Pooled Human Liver Microsome Studies section).
Thawed reaction mixture samples were diluted 2 : 1 with ammonium acetate buffer (10 mmol/L, pH 4) and centrifuged for 5 minutes at ambient temperature to pellet the precipitated microsomal protein. Aliquots (200 μL) of the supernatant were removed into glass vials for analysis by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) for parent renzapride, the N-oxide derivative of renzapride and monohydroxylated renzapride. Results, expressed as the percentage of M-1 formed from renzapride (no protein control incubations), are single observations of each CYP enzyme.
Effect of Proadifen, a Non-Selective Inhibitor of CYP Enzymes, on the Metabolism of Renzapride by Human Liver Microsomes in Vitro
Renzapride 10 μmol/L was incubated with pooled human liver microsomes (4 mg microsomal protein/mL incubation mixture) in the presence and absence of the nonspecific CYP enzyme inhibitor proadifen 1 mmol/L for 60 minutes. Incubation mixtures contained renzapride, microsomal protein, inhibitor and phosphate buffer (100 mmol/L, pH 7.4) to give a pre-incubation volume of 450–500 μL. Following a pre-incubation period of 10 minutes, microsomal reactions were initiated by the addition of enzyme co-factor solution or renzapride. Control incubations contained methanol (5 μL) in place of specific inhibitor or super-pure water in place of renzapride or sodium bicarbonate (2% [w/v]) solution in place of enzyme co-factors. Positive control incubations were [14C]-testosterone 125 μmol/L.
All pre-incubation and incubation stages were as described previously (see Pooled Human Liver Microsome Studies section). Microsomal reactions were terminated after the desired time period by acetonitrile 500 μL or methanol 200 μL. Samples were stored at −20°C. Thawed samples were centrifuged for 10 minutes at ambient temperature to pellet the precipitated microsomal protein. An aliquot of each supernatant (200 μL) was removed into a glass vial for analysis by HPLC. Results, expressed as the percentage of M-1 formed from renzapride (no protein control incubations), are the means of two observations.
Metabolism of (+)- and (−)-Renzapride by Human Liver S9 Fractions in Vitro
Renzapride
Renzapride 10 μmol/L was incubated with human liver S9 fraction (1 mg protein/mL) incubation mixture for 0 and 60 minutes. One series of incubation mixtures contained S9 protein (10.5 μL), phosphate buffer (869.5 μL, 100 mmol/L, pH 7.4) and enzyme co-factor solution, supplemented with UDPGA, β-NAD, β-NADH, GSH and PAPS (100 μL) to give a pre-incubation volume of 980 μL.
A second series of incubation mixtures without supplemental co-factor solution contained S9 protein (10.5 μL) and phosphate buffer (969.5 μL, 100 mmol/L, pH 7.4) only, to give a pre-incubation volume of 980 μL. Control incubations contained phosphate buffer (100 mmol/L, pH 7.4) in place of S9 fraction. Following a pre-incubation period of 5 minutes, S9 reactions were initiated by the addition of renzapride (20 μL of 0.5 mmol/L solution), to give a final incubation volume of 1 mL. All pre-incubation, incubation and termination stages were performed as described previously (see Pooled Human Liver Microsome Studies section). Samples were stored at −80°C. For the zero (0) time-point incubations, renzapride and acetonitrile were added simultaneously in an attempt to rapidly reduce the enzymic activity of the S9 preparations, thereby ensuring an accurate time zero (minutes) sample. The samples were thawed on ice and diluted 2 : 1 with ammonium acetate buffer (10 mmol/L, pH 4) and centrifuged for 5 minutes at ambient temperature to pellet the precipitated S9 protein. Aliquots (200 μL) of the sample supernatant were removed into glass vials for analysis by LC-MS/MS. Results, expressed as the percentage of M-1 formed from renzapride (no protein control incubations), are single observations per group.
Enantiomers of Renzapride
The (−) and (+) enantiomers of renzapride, 10 μmol/L, were incubated with human liver S9 fraction (1 mg protein/mL incubation mixture) for 0 and 60 minutes. Incubation mixtures contained S9 protein (5.2 μL), phosphate buffer (924.8 μL, 100 mmol/L, pH 7.4) and were supplemented with an enzyme co-factor solution (50 μL,) as described previously (see Pooled Human Liver Microsome Studies section), to give a pre-incubation volume of 980 μL. Control incubations contained phosphate buffer (100 mmol/L, pH 7.4) in place of S9 fraction. Following a pre-incubation period of 5 minutes, S9 reactions were initiated by the addition of either (−)- or (+)-renzapride (20 μL of a 0.5-mmol/L solution), to give a final incubation volume of 1 mL. All pre-incubation and incubation stages were as described previously (see Pooled Human Liver Microsome Studies section). S9 reactions were terminated at time zero (0) minutes by the simultaneous addition of either (−)- or (+)-renzapride and acetonitrile (50 μL) to the incubation mixture and at time 60 minutes by the addition of acetonitrile (50 μL). Samples were stored at −80°C. The samples were thawed on ice and centrifuged for 5 minutes at ambient temperature to pellet the precipitated S9 protein. Aliquots (200 μL) of the sample supernatant were removed into glass vials for analysis by HPLC. Results, expressed as the percentage of M-1 formed from renzapride (no protein control incubations), are single observations per group.
Effect of Renzapride on the Metabolism of Specific Substrates for Individual CYP Enzymes by Human Liver Microsomes in Vitro
Renzapride was incubated with pooled human liver microsomes for 20–30 minutes in the presence of CYP selective substrates for the following CYP enzymes: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4.[66] Table II shows the substrates used and specific assay conditions for each CYP enzyme. Incubation mixtures contained renzapride, microsomal protein, enzyme co-factor solution (if required) and phosphate buffer. Following a pre-incubation period of 5 minutes, microsomal reactions were initiated as stated in table II to give a final incubation volume of 497.5–500 μL. Control incubations were as stated in table II. Positive control incubations contained a specific CYP inhibitor in place of renzapride.
Table II.
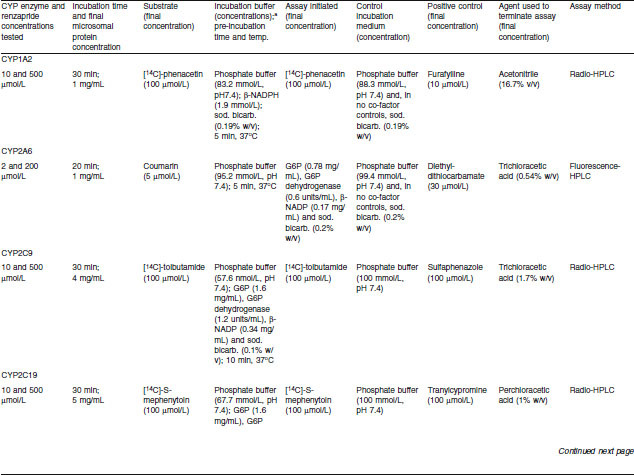
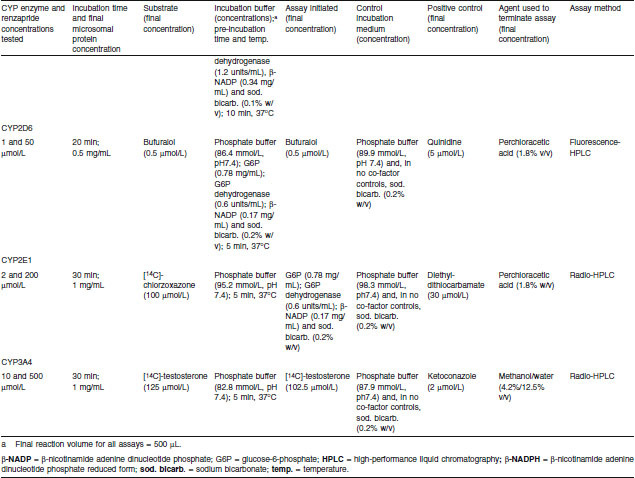
Effects of renzapride on metabolism of selective cytochrome P450 (CYP) substrates by human microsomes — assay conditions
All pre-incubation and incubation stages were performed as described previously (see Pooled Human Liver Microsome Studies section). Microsomal reactions were terminated after the desired time period and samples were stored at −20°C. Thawed samples were centrifuged at ambient temperature to pellet the precipitated microsomal protein. An aliquot of each sample supernatant (200 μL) was removed into a glass vial for analysis by radio- or fluorescence-HPLC. Results are expressed as the percentage inhibition of CYP-related activity by renzapride or positive control (means of two observations).
High-Performance Liquid Chromatography (HPLC) and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry (LC-MS/MS) Assays
HPLC
Renzapride and authentic renzapride N-oxide, once available, were assayed using reverse-phase HPLC with on-line UV detection under the following conditions — column: Synergi, RP, 4 μm, 250 × 4.6 mm internal diameter (Phenomenex, Cheshire, UK); mobile phase: A = ammonium acetate (10 mmol/L, pH 4.0), B = 100% acetonitrile; gradient: linear gradient of 95% A for 5 min, then to 5% A over 10 min; UV detection: 270 nm; flow rate: 1 mL/min; temperature: 40°C. Chromatography data were collected using TurboChrom Client/Server V6.1.1 (Perkin Elmer Corp., Waltham, MA, USA).
Metabolite-1 was putatively identified as the N-oxide metabolite of renzapride formed in human liver microsomes in vitro by comparing the retention time of an authentic sample of renzapride N-oxide with the retention time for M-1 under the same chromatographic conditions.
LC-MS/MS
LC-MS/MS chromatography was performed using a Genesis C18, 4 μm, 50 × 2.1 mm analytical column (Jones Chromatography, Mid Glamorgan, UK). The mobile phase consisted of an ammonium acetate (10 mmol/L, pH 4.0)/acetonitrile mixture run isocratically at 0.2 mL/min. Samples were analysed using an API3000 LC-MS/MS (MDS Sciex, Toronto, Canada). The LC-MS/MS interface used was TurboIonspray operated in the positive ion detection mode. Instrumental conditions used for the analysis were:
Ion Spray voltage: 5000 V; desolvation temperature: 350°C; nebulizer gas flow: 8 L/min; desolvation gas flow: 5 L/min; collision gas: nitrogen; collision gas pressure: 4 (units); collision energy: 37 eV; declustering potential: 71 V; focusing potential: 350 V.
Samples were analysed using the following Multiple Reaction Monitoring (MRM) transitions:
channel 1: 324/184 amu (equivalent to parent renzapride); channel 2: 340/184 amu (equivalent to oxidation on the bicycloamino region of the renzapride molecule); channel 3: 340/200 amu (equivalent to oxidation on the aromatic region of the renzapride molecule).
Results
Radioligand Binding Studies
5-HT Receptors
The affinities of renzapride, its enantiomers, renzapride N-oxide for cloned human 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3 and guinea-pig 5-HT4 receptors are shown in table III. Renzapride and both its enantiomers had high affinity for human 5-HT3 receptors (Ki 17 nmol/L). Renzapride-N-oxide had markedly less affinity for this receptor (Ki 1980 nmol/L) and neither enantiomer showed higher affinity than the racemate. Renzapride and its enantiomers also had marked affinity for guinea-pig 5-HT4 receptors (ki 138–477 nmol/L). Again, the affinities of racemic renzapride-N-oxide and the enantiomers for guinea-pig 5-HT4 receptors were markedly less than that of the parent compound (for all compounds IC50 >10 μmol/L). Renzapride and its (+), (−) enantiomers had similar affinity for 5-HT2B receptors (Ki 667, 760, 481 and 324 nmol/L, respectively). Renzapride and its enantiomers showed similar modest micromolar affinity for human 5-HT2A and 5-HT2C receptors. Renzapride N-oxide had no significant affinity for these receptors (ki >10 μmol/L).
Table III.
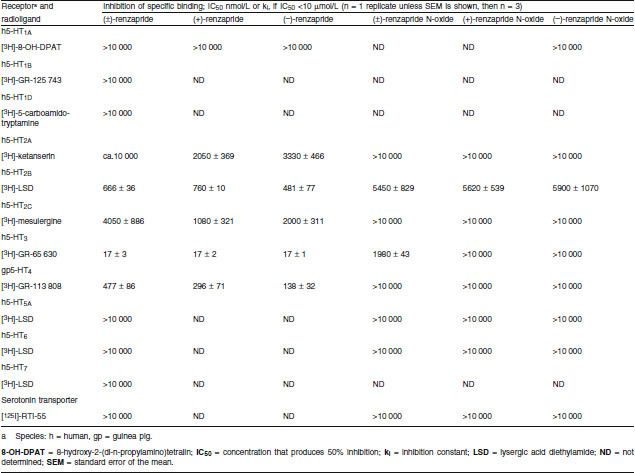
Renzapride, renzapride N-oxide, and their enantiomers: inhibition of radioligand binding to serotonin 5-HT receptors and serotonin transporter
Renzapride and renzapride N-oxide had no significant affinity for 5-HT1A, 5-HT1B, 5-HT1D, 5-HT5A, 5-HT6 or 5-HT7 receptors or the serotonin transporter (table III; for both compounds and each binding site ki >10 μmol/L).
Catecholaminergic Receptors
Neither renzapride nor renzapride N-oxide had any affinity for rat α1-, α2- and β-adrenoceptors or the human norepinephrine and dopamine transporters (ki >10 μmol/L). Renzapride and renzapride N-oxide had no significant affinity for human D4.2, D4.4 or D4.7 receptors or the human dopamine transporter (ki >10 μmol/L).
In Vitro Functional Studies
5-HT4 Receptor-Mediated Effects
Renzapride showed functional 5-HT-like agonism on rat isolated oesophagus (EC50 11 μmol/L). The (+) and (–) enantiomers also contracted the tissue (EC50 16 and 4.8 μmol/L, respectively).
5-HT2B Receptor-Mediated Effects
Inhibition of α-Methyl-5-Serotonin-Induced Contraction of Rat Stomach Fundus Strip
Renzapride inhibited α-methyl-5-serotonin-induced contractions of rat stomach fundus strip (IC50 5 μmol/L). The effects of the (+) and (−) enantiomers of renzapride and renzapride N-oxide were not investigated in this procedure.
Human Liver Microsome Studies
Metabolism of Renzapride by Human Liver Microsomes in Vitro
Preliminary incubation of renzapride with pooled human liver microsomes in a 0.5 mg/mL protein concentration indicated the presence by reverse-phase HPLC with online UV detection of a UV-absorbing (270 nm) component eluting just after renzapride at 15.3 minutes. This component, which was assigned M-1, had the same retention time as, and co-chromatographed with, an authentic renzapride N-oxide reference standard (figure 2).
Fig. 2.
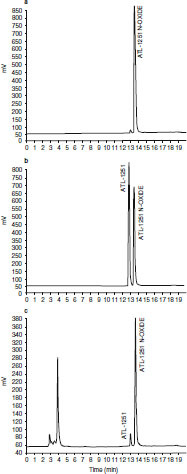
(a) High-pressure liquid chromatography with UV detector (HPLC-UV) chromatogram of (±)-renzapride N-oxide standard solution (1 mg/mL in acetonitrile). (b) HPLC-UV chromatogram of (±)-renzapride and (±)-renzapride N-oxide standard solutions (1 mg/mL in acetonitrile). (c) HPLC-UV chromatogram of sample from human liver microsome incubation (60 min) with (±)-renzapride and N-oxide reference standard (1 mg/mL in acetonitrile). ± signifies racemate of (+) and (−) enantiomers present in equal proportions.
As shown in table IV, the extent of M-1 formation after 60 minutes’ incubation was relatively low, representing approximately ≤3%, 21% and 34% of eluted material compared with no-protein controls at 0.5, 2 and 4 mg microsomal protein/mL incubation mixture, respectively. Furthermore, M-1 was produced only in the presence of enzyme co-factors and microsomal protein. The extent of formation of M-1 by individual human microsomes as measured by the percentage increase coincidentally eluting with the authentic N-oxide reference standard ranged from 2% to 16% (mean 7.5%) compared with no-protein controls after 60 minutes’ incubation. The mean rate of formation of renzapride N-oxide was 3.0 (range 0.7–6.6) pmol/min/mg protein.
Table IV.
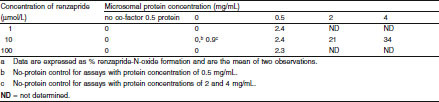
Metabolism of renzapride by pooled human liver microsomes; formation of renzapride N-oxide in vitro a
Metabolism of Renzapride by Expressed Human CYP Enzymes in Vitro
The incubation of renzapride with expressed human CYP1A2, CYP2C9, CYP2C19 and CYP2D6 enzymes resulted in the formation of no renzapride N-oxide (0%) by any of the enzymes, as detected by LC-MS/MS. There was evidence for the formation of trace amounts of oxidized metabolites corresponding to oxidation on the aromatic region of renzapride (data not shown). Incubation with expressed human CYP3A4 resulted in the formation of only a small amount of renzapride N-oxide (3%).
Effect of Proadifen on Metabolism of Renzapride by Human Liver Microsomes in Vitro
Proadifen 1 mmol/L had no effect on the formation of renzapride N-oxide by human liver microsomes. Renzapride N-oxide was formed to the same extent (8%) in the presence and absence of proadifen compared with no-co-factor controls.
Metabolism of Renzapride by Human S9 Hepatic Fraction in Vitro
The major metabolite formed in the incubation of renzapride (10 μmol/L) with human S9 fraction (27%) corresponded to oxidation on the bicycloamino region of the renzapride molecule (MRM ion transition 340/184) and was of similar chromatographic polarity to parent renzapride (based upon HPLC retention time). These data suggested that this S9-derived metabolite is renzapride N-oxide and is equivalent to M-1. There was evidence for the formation of oxidized metabolites corresponding to oxidation (P+16) on the aromatic region of renzapride (MRM transition 340/200). However, these species were present in only trace amounts. Again, the metabolism of renzapride by human liver S9 fraction required the presence of enzyme co-factors. Subsequently, the LC-MS/MS fragmentation pattern of an authentic sample of renzapride N-oxide confirmed the 340/184 ion transition as characteristic for this metabolite, supporting indications from the current study that M-1 is the N-oxide metabolite of renzapride (figure 3 and figure 4).
Fig. 3.
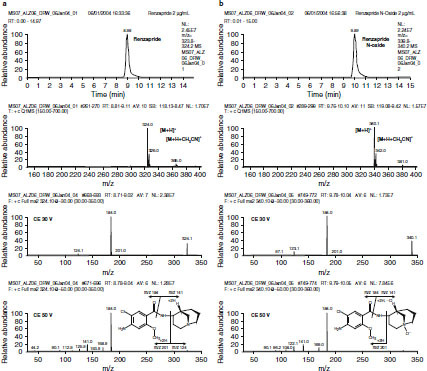
(a) Extracted ion chromatogram for m/z 324, positive ion scan and product ion mass spectra at collision energies (CE) of 30 V and 50 V for a standard of renzapride. (b) Extracted ion chromatogram for m/z 340, positive ion scan and product ion mass spectra at CE of 30 V and 50 V for a standard of renzapride N-oxide. M = parent renzapride molecule; MRM = Multiple Reaction Monitoring.
Fig. 4.
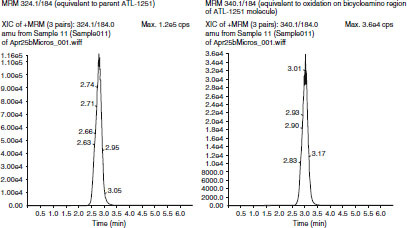
Liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) ion chromatogram of supplemented S9 fraction — 60-min incubation. MRM = Multiple Reaction Monitoring.
When the enantiomers of renzapride (10 μmol/L) were incubated with supplemented human liver S9, again a single metabolite, chromatographically equivalent to M-1 (renzapride N-oxide), was detected. There were no significant differences in the amount of N-oxide formation from the (−) and (+) enantiomers of renzapride, which were 21% and 16%, respectively, of the combined drug-related peak area ratios.
Effect of Renzapride on Metabolism of Selective Substrates of CYP Enzymes by Human Liver Microsomes in Vitro
The effect of renzapride on the metabolism of specific substrates of CYP enzymes by human liver microsomes is shown in table V. Renzapride inhibited the metabolism of specific substrates to the following extent: CYP1A2 11.7% at 500 μmol/L, CYP2A6 6.9% at 200 μmol/L, CYP2C9 58.1% at 500 μmol/L, CYP2C19 37.9% at 500 μmol/L, CYP2D6 54.4% at 50 μmol/L, CYP2E1 34% at 2 μmol/L and CYP3A4 16.4% at 500 μmol/L.
Table V.

Effect of renzapride on metabolism of cytochrome P450 (CYP)-selective substrates in human liver microsomes in vitro
Discussion
The studies reported above confirm and extend the results of earlier studies demonstrating that renzapride has high affinity for guinea-pig 5-HT4 and cloned human 5-HT3 receptors as well as some affinity for cloned human 5-HT2A and 5-HT2B receptors. The high affinity of renzapride for guinea-pig 5-HT4 receptors reported in the present study confirms the data reported for pig,[67] guinea-pig and rat tissues.[43] It has already been established that renzapride has high affinity for various splice variants of the human 5-HT4 receptor, i.e. 5-HT4(a), 5-HT4(b),[25,27,32] 5-HT4(c),[27] 5-HT4 (d)[27,30] and 5-HT4(e) receptors.[29]
Functional in vitro studies in gastrointestinal smooth muscle preparations show that renzapride acts as a full agonist for 5-HT4-mediated responses in guinea-pig ileum[12] and distal colon longitudinal muscle.[13] In this respect it is similar to cisapride but differs from tegaserod, which is a partial 5-HT4 receptor agonist in electrically stimulated isolated guinea-pig ileum.[8]
Using cloned human receptors, Blondel et al.[27] reported that renzapride was a partial agonist at 5-HT4(a) receptors and a full agonist at 5-HT4(b), 5-HT4(c) and 5-HT4(d) receptors, while Bach et al.[25] reported that renzapride was a full agonist at both 5-HT4(a) and 5-HT4(b) receptors at high receptor density, an observation confirmed recently by Pindon et al.[32] Mialet et al.[30] confirmed that renzapride was a full agonist at 5-HT4(d) receptors but was a partial agonist at 5-HT4(e) receptors. Renzapride has also been reported to be a full agonist at 5-HT4(i) receptors under experimental conditions of high receptor density, e.g. in the myenteric plexus of the gastrointestinal tract.[35] 5-HT4(a), 5-HT4(b), 5-HT4(c), 5-HT4(d), 5-HT4(hb) (a variant of the 5-HT4(b) isoform) and 5-HT4i, but not 5-HT4e, are expressed in human gut.[27,29,68] The specific role of each of the splice variants of the 5-HT4 receptor at the various levels of the gut and the relevance of binding to these receptor isoforms to the therapeutic actions of renzapride is unknown.
5-HT4 receptor agonism is a key property of the benzamide class of gastrointestinal prokinetic drugs, and is probably responsible for the stimulation of upper and lower gut motility observed with metoclopramide and cisapride in in vivo preclinical studies.[24] Similar properties have been described for renzapride in the rat[12,15,18,20] and dog.[16,17,19,21,69]
In in vitro studies of human gastric tissue, renzapride potentiates the effect of electrical field stimulation in isolated stomach strips[70] and serotonin-induced stimulation of acetylcholine release in isolated proximal stomach has been shown to be mediated by the 5-HT4 receptor.[71]
Renzapride, like serotonin, inhibited spontaneous activity of isolated colonic circular muscle in vitro in preparations of the human lower gut, an action shown to be mediated by the 5-HT4 receptor.[72] Serotonin-induced facilitation of colonic propulsion in man, via effects on circular and longitudinal muscle, is also believed to be mediated by this receptor subtype.[73,74] It has been suggested that 5-HT4 receptor agonists facilitate colonic propulsion via a coordinated inhibition of circumferential resistance and enhancement of longitudinal muscle contractility.[73] 5-HT4 receptor stimulation also leads to ion and fluid secretion into the lumen of human jejunum and ileum,[75–77] indicating that the actions of renzapride on the gut are unlikely to be restricted to effects on motility. In healthy subjects and IBS-C patients, renzapride stimulates gastric emptying, reduces small intestine transit time and increases colonic motility.[2,3,22,23] These actions are consistent with stimulation of 5-HT4 receptors in the smooth muscle of the upper and lower gut.
The high affinity of renzapride for human 5-HT3 receptors shown in the current study has been reported previously,[31] and these data confirm the earlier reported high affinity of the compound for 5-HT3 receptor binding sites using mouse neuroblastoma and porcine caudate nucleus tissue.[67,78] Pharmacological studies have shown that renzapride is a potent inhibitor of 5-HT3 receptors in vivo.[14,15]
Studies with 5-HT3 receptor antagonists indicate that blockade of the 5-HT3 receptor is associated with slowed colonic transit.[1,79] However, renzapride increases colonic motility in rodents, dogs, healthy human subjects and patients with constipation-predominant IBS. The present data indicate therefore that, in man, the agonist effect of renzapride on 5-HT4 receptors appears to predominate over 5-HT3-receptor inhibition with respect to gut motility. However, given the known role of 5-HT3 receptors in gastrointestinal sensation, including pain on intraluminal distension,[1] it would be predicted that renzapride may improve visceral sensitivity in IBS by an action at this receptor. Importantly, this is likely to be mediated through 5-HT3 and/or 5-HT4 receptors in the gastrointestinal tract, as opposed to via centrally located 5-HT3 receptors (as, at least in part, in the case of alosetron),[80] since very little of a renzapride dose appears to cross the blood-brain barrier or become distributed within brain tissue (unpublished observations). This may explain the apparent disconnect between the in vitro pharmacological profile and the pharmacodynamic effects in vivo of renzapride.
Clinical studies with renzapride in patients with IBS-C[1–3] and in patients with mixed-symptom IBS (unpublished observations) have confirmed that like the selective 5-HT4 receptor partial agonist tegaserod, renzapride increases bowel movement frequency, improves stool consistency and increases patient-reported relief of their overall IBS symptoms, especially in women.[3] However, unlike the selective 5-HT3-receptor antagonists (e.g. alosetron),[81] renzapride does not appear to be associated with constipatory adverse effects or with a significantly increased risk of colon ischaemia in this patient population, although much larger clinical studies and post-marketing surveillance are likely to be required to confirm whether or not this is actually the case.
The present study indicates that renzapride has only weak affinity for human 5-HT2A receptors. It has not been investigated whether renzapride is an agonist or antagonist at this site. The 5-HT2A receptor may play a role in mediating contractions of the canine colon.[82] Furthermore, 5-HT2A and 5-HT4 receptors have also been implicated in the stimulation of secretion in human colon.[76] It remains to be demonstrated, however, if the modest affinity of renzapride for the 5-HT2A receptor has any relevance to its clinical activity.
Renzapride has a modest affinity for human 5-HT2B receptors in vitro and further study showed it to be a functional 5-HT2B antagonist on rat fundus strip. Tegaserod has been shown to be a potent 5-HT2B receptor antagonist[83] and it has been postulated that 5-HT2B receptor antagonism may result in inhibition of serotonin-mediated gastrointestinal motility and visceral hyperactivity, especially in the human colon.[84] It remains to be clarified what contribution the 5-HT2B receptor antagonist properties of renzapride may make to the effects of the compound on colonic motility in man, particularly given the prokinetic role of the 5-HT4 receptor in this tissue.[72–74]
Renzapride has only moderate affinity for human 5-HT2C receptors. It has been previously reported to have no significant affinity for 5-HT2C receptors in porcine choroid plexus.[79] The relatively weak affinity of renzapride for 5-HT2C receptors indicates that activity at these receptors is unlikely to play a role in its action on the gastrointestinal tract or other tissues. Renzapride has no significant affinity for human 5-HT1A, 5-HT1B, 5-HT1D, 5-HT5A, 5-HT6 or 5-HT7 receptors. It also has no significant affinity for the serotonin transporter, confirming negative data in studies on the uptake of [3H]-serotonin into rat synaptosomes.[85]
The results reported in the current study indicate that renzapride is devoid of significant affinity for cloned rat α1-, α2- and β-adrenoceptors, and cloned human D1, D2L, D2S, D3, D4.2, D4.4, D4.7, D5 receptors, norepinephrine transporters and dopamine transporters. The data on D1 and D2 receptors confirm earlier reports using non-human species.[24]
The present study shows that the enantiomers of renzapride possess similar affinity for cloned human 5-HT3, 5-HT2A, 5-HT2B, and 5-HT2C receptors with no more than a 2-fold difference in potency at any of the receptor sites. The (–) enantiomer shows slightly more than a 2-fold greater affinity for guinea-pig 5-HT4 receptors than the (+) enantiomer and 3-fold greater potency in the functional study on isolated rat oesophagus. These data indicate that the (−) enantiomer of renzapride may be slightly more potent than the (+) enantiomer at 5-HT4 receptors. It has not been investigated whether the two enantiomers show the same full agonism at 5-HT4 receptors in tissues in which this has been demonstrated for the racemate. The in vitro and in vivo studies reported by King et al.[36] (see table VI) showed that the two enantiomers have very similar potency in pharmacological models in which agonism at 5-HT4 receptors is implicated: specifically, potentiation of the effects of in vitro electrical field stimulation of guinea-pig ileum and increase in intraluminal pressure in the Heidenhain pouch dog. The two enantiomers are also equipotent at increasing intragastric pressure in the fasted conscious rat (unpublished observations). It is concluded that whilst there may be some small differences in potency in in vitro models, the two enantiomers of renzapride show very similar potency in in vivo studies.
Table VI.
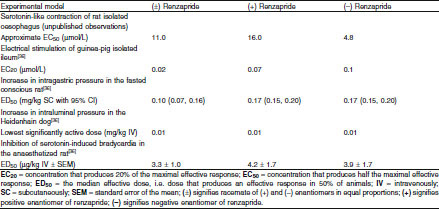
Comparative pharmacological profiles of renzapride and its enantiomers
The similar affinity of the two enantiomers for human 5-HT3 receptors confirms the finding of King et al.[36] that the enantiomers are equipotent at inhibiting the serotonin-induced Bezold-Jarisch reflex in the anaesthetized rat. These data are consistent with the observation that the two enantiomers of renzapride are equipotent as 5-HT3 receptor antagonists. The two enantiomers of renzapride, therefore, show a very similar spectrum of pharmacological activity at 5-HT receptors.
Renzapride N-oxide had only micromolar affinity for human 5-HT3 and 5-HT2B receptors and no significant affinity for guinea-pig 5-HT4 or human 5-HT1A, 5-HT1B, 5-HT1D, 5-HT5A, 5-HT6 or 5-HT7 receptors. No functional studies on the action of renzapride N-oxide at 5-HT3 and 5-HT2B receptors have been performed. Like renzapride, renzapride N-oxide also had no significant affinity for cloned rat α1-, α2- and β-adrenoceptors, and cloned human D1, D2L, D2S, D3, D4.2, D4.4, D4.7, D5 receptors and norepinephrine and dopamine transporters.
The present results on the radioligand binding profile of renzapride N-oxide therefore indicate that this principal metabolite of renzapride in man has a significantly weaker affinity for 5-HT receptors than renzapride, with no additional affinities for catecholaminergic receptors. The enantiomers of the metabolite show the same weak affinity for human 5-HT3 and 5-HT2B receptors.
The present studies on the metabolism of renzapride by human liver microsomes show that the compound is metabolized only to a very modest extent by this tissue and that this metabolism results in the formation of a predominant single metabolite (M-1). This metabolite has the same chromatographic and LC-MS/MS ion transition profile as authentic renzapride N-oxide. Since M-1 was observed only in incubations containing both microsomal protein and enzyme co-factors, it is highly probable that this was a metabolic product and not a result of chemical or thermal degradation. Incubation of renzapride with individual human donor microsomes showed again that the only metabolite identified was renzapride N-oxide, the extent of formation of which ranged from 2% to 16% under the conditions used.
Formation of the N-oxide of renzapride from the parent compound was not reduced by a non-selective inhibitor of CYP enzymes, indicating that these enzymes probably do not play a major role in the metabolism of renzapride. Incubation of renzapride with human S9 fraction, supplemented with additional co-factors, also showed evidence for the formation of renzapride N-oxide. Only traces of additional oxidative, and, therefore, probably phase I microsomal metabolites, were found in the S9 incubations, indicating that there is no significant non-microsomal metabolism of renzapride. Subsequent experiments have suggested that hepatic functional mixed oxidase enzymes (e.g. FMO-3) are likely to play the major role in the metabolism of renzapride in vivo (unpublished observations).
There was no difference in the rate of formation of the N-oxide by either the (−) or (+) enantiomers of renzapride by human S9 fraction in vitro, and it is therefore concluded that the metabolism of renzapride is not stereospecific. These data, when taken together with data demonstrating that the N-oxide metabolite comprises <10% of circulating drug-related material after a single, oral [14C]-labelled 4-mg dose in healthy female volunteers (unpublished observations), support the conclusion that renzapride N-oxide is unlikely to contribute to the clinical effects of renzapride in man.
In vitro studies with selective substrates of CYP indicate that renzapride does not significantly inhibit the human drug-metabolizing CYP enzymes CYP2C9, CYP1A2, CYP2A6, CYP2C19, CYP2E1 or CYP3A4 at therapeutically relevant concentrations. A definitive pharmacokinetic study in patients with IBS-C taking the proposed clinical dose of renzapride 4 mg once daily has confirmed the maximum circulating levels of renzapride to be 15.3 ng/mL (47.2 nmol/L) at time to maximum plasma concentration (1.4 hours).[2] In the current study, the inhibitory activity of renzapride measured against specific hepatic CYP enzymes in vitro are likely to be clinically non-relevant given that these concentrations of renzapride are far in excess (>10 000× for CYP1A2, CYP2C9, CYP2C19 and CYP3A4, >4000× for CYP2A6, >1000× for CYP2D6, and >40× for CYP2E1) of those likely to be achieved in patients in therapeutic use.
Conclusions
The present studies confirm and extend the results of earlier studies in human and non-human tissue that show that renzapride is selective for serotonin receptors. Its pharmacological profile in a range of human isolated tissues and preclinical species indicate that renzapride is a full 5-HT4 receptor agonist. It is also a 5-HT3 receptor antagonist, although in animals and in humans its prokinetic effects on the gastrointestinal tract, probably associated largely with peripheral 5-HT4 receptor agonism, appear to predominate. The peripheral 5-HT3 receptor antagonist properties of renzapride may contribute to beneficial effects on visceral hypersensitivity in patients with IBS.
As would be predicted from its pharmacological profile, clinical studies to date have shown that renzapride is safe and well tolerated, and in patients with IBS-C significantly increases relief from abdominal pain and discomfort and increases bowel movements.[2,4] Renzapride may also have therapeutic utility in IBS patients with mixed symptomatology (alternating constipation and diarrhoea) as a result of its 5-HT4 receptor agonist and 5-HT3 receptor antagonist properties.
The results of the in vitro studies of the enantiomers of renzapride reported above are consistent with earlier reports that, although there may be small differences in potency at 5-HT4 receptors between them, (+)- and (−)-renzapride enantiomers possess very similar pharmacological profiles in vivo, supporting continued development of this drug as the racemate.
In vitro studies with human liver microsomes indicate that renzapride is metabolized by this tissue only to a very limited extent, which reflects the in vivo situation in man, where the majority of the dose administered (>60%) is excreted in the urine, largely (ca. 80%) as unchanged, parent drug (unpublished observations). The major metabolic product of renzapride appears to be renzapride N-oxide, although this is a minor metabolite in vivo, accounting for <10% of circulating drug-related material. Additionally, in the current study, radioligand binding studies show that this metabolite is unlikely to contribute to the therapeutic actions of renzapride in man.
Although inhibitory activity of renzapride on five specific CYP enzymes was detected in the current study, this inhibition did not occur at therapeutically relevant concentrations. Further, a nonspecific inhibitor was shown to have no effect on the microsomal metabolism of renzapride. These results, combined with the fact that renzapride is rapidly cleared (half-life = 10.1 hours)[2] and is not highly bound to plasma proteins (i.e. 33.3% in pooled male and female human control plasma, 13.2% to human serum albumin and 5.1% to α-glycoprotein — unpublished observations), indicate that plasma concentrations of renzapride should not be influenced by drugs that inhibit the major CYP enzymes. Similarly, the lack of propensity for renzapride to inhibit a range of CYP enzymes in vitro at ‘clinical’ concentrations suggests that drug-drug interactions with therapeutic agents for which these enzymes are major metabolizing systems should not be an issue with renzapride in therapeutic use and no evidence of any such effects has been seen in clinical studies to date.
The preliminary studies reported herein contribute to our understanding of the pharmacological activities underlying the therapeutic mechanism of action of renzapride in patients with gastrointestinal disorders (e.g. IBS and chronic constipation), as well as the metabolic fate of renzapride in vivo and its potential for interfering with the metabolism of other drugs, all of which support its clinical utility.
Acknowledgements
The research work described in this article was funded entirely by Alizyme Therapeutics Ltd. The radioligand binding and organ bath experiments reported in this study were conducted on behalf of Alizyme in the laboratories of MDS Pharma Services, Taiwan Ltd, Taipei, Taiwan. The human liver microsome studies were conducted on behalf of Alizyme by BioDynamics Research Ltd, Northamptonshire, UK. The authors are both current employees and shareholders of Alizyme Therapeutics Ltd.
References
- 1.Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol. 2004;141:1285–93. doi: 10.1038/sj.bjp.0705762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Camilleri M, McKinzie S, Fox J, et al. Effect of renzapride on transit in constipation-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2004;2:895–904. doi: 10.1016/S1542-3565(04)00391-X. [DOI] [PubMed] [Google Scholar]
- 3.Tack J, Middleton SJ, Horne MC, et al. Pilot study of the efficacy of renzapride on gastrointestinal motility and symptoms in patients with constipation-predominant irritable bowel syndrome. Aliment Pharmacol Ther. 2006;23:1655–65. doi: 10.1111/j.1365-2036.2006.02940.x. [DOI] [PubMed] [Google Scholar]
- 4.George A, Meyers NL, Palmer RMJ. Efficacy and safety of renzapride in constipation-predominant IBS: a phase IIB study in the UK primary healthcare setting. Gut 2003; 52: SVI, A91
- 5.Sanger GJ, King FD. From metoclopramide to selective gut motility stimulants and 5-HT3 receptor antagonists. Drug Des Deliv. 1988;3:273–95. [PubMed] [Google Scholar]
- 6.Scott LJ, Perry CM. Tegaserod. Drugs. 1999;58:491–6. doi: 10.2165/00003495-199958030-00013. [DOI] [PubMed] [Google Scholar]
- 7.Balfour JA, Goa KL, Perry CM. Alosetron. Drugs. 2000;59:511–8. doi: 10.2165/00003495-200059030-00008. [DOI] [PubMed] [Google Scholar]
- 8.Buchheit KH, Buhl T. Prokinetic benzamides stimulate peristaltic activity in the isolated guinea pig ileum by activation of 5-HT4 receptors. Eur J Pharmacol. 1991;205:203–8. doi: 10.1016/0014-2999(91)90821-7. [DOI] [PubMed] [Google Scholar]
- 9.Dumuis A, Sebben M, Bockaert J. The gastrointestinal prokinetic benzamide derivatives are agonists at the non-classical 5-HT receptor (5-HT4) positively coupled to adenylate cyclase in neurons. Naunyn Schmiedebergs Arch Pharmacol. 1989;340:403–10. doi: 10.1007/BF00167041. [DOI] [PubMed] [Google Scholar]
- 10.Elswood CJ, Bunce KT, Humphrey PP. Identification of putative 5-HT4 receptors in guinea-pig ascending colon. Eur J Pharmacol. 1991;196:149–55. doi: 10.1016/0014-2999(91)90421-L. [DOI] [PubMed] [Google Scholar]
- 11.Reeves JJ, Bunce KT, Humphrey PPA. Investigation into the 5-hydroxytryptamine receptor mediating smooth muscle relaxation in the rat oesophagus. Br J Pharmacol. 1991;103:1067–72. doi: 10.1111/j.1476-5381.1991.tb12301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanger GJ. Increased gut cholinergic activity and antagonism of 5-hydroxytryptamine M-receptors by BRL 24924: potential clinical importance of BRL 24924. Br J Pharmacol. 1987;91:77–87. doi: 10.1111/j.1476-5381.1987.tb08985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wardle KA, Sanger GJ. The guinea-pig distal colon: a sensitive preparation for the investigation of 5-HT4 receptor-mediated contractions. Br J Pharmacol. 1993;110:1593–9. doi: 10.1111/j.1476-5381.1993.tb14006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida N, Omoya H, Kato S, et al. 5-HT3 receptor antagonist effects of DAT-582, (R) enantiomer of AS-5370. Eur J Pharmacol. 1992;216:435–40. doi: 10.1016/0014-2999(92)90442-7. [DOI] [PubMed] [Google Scholar]
- 15.Schiavone A, Volonte M, Micheletti R. The gastrointestinal motor effect of benzamide derivatives is unrelated to 5-HT3 receptor blockade. Eur J Pharmacol. 1990;187:323–9. doi: 10.1016/0014-2999(90)90359-E. [DOI] [PubMed] [Google Scholar]
- 16.Song CW, Lee KY, Kim CD, et al. Effect of cisapride and renzapride on gastrointestinal motility and plasma motilin concentration in dogs. J Pharmacol Exp Ther. 1997;281:312–6. [PubMed] [Google Scholar]
- 17.Nagakura Y, Kamato T, Nishida A, et al. Characterization of 5-hydroxytryptamine (5-HT) receptor subtypes influencing colonic motility in conscious dogs. Naunyn Schmiedebergs Arch Pharmacol. 1996;353:489–98. doi: 10.1007/BF00169167. [DOI] [PubMed] [Google Scholar]
- 18.Cooper SM, McClelland CM, McRichie B, et al. BRL 24924: a new and potent gastric motility stimulant [abstract] Br J Pharmacol. 1986;88:383P. [Google Scholar]
- 19.Bermudez J, Dunbar A, Sanger GJ, et al. Stimulation of canine gastric motility by BRL 24924, a new gastric prokinetic agent. J Gastrointest Motil. 1990;2:281–6. doi: 10.1111/j.1365-2982.1990.tb00036.x. [DOI] [Google Scholar]
- 20.Dunbar AW, McClelland CM, Sanger GJ. BRL 24924: a stimulant of gut motility which is also a potent antagonist of the Bezold-Jarisch reflex in anaesthetised rats. Br J Pharmacol. 1986;88:319P. [Google Scholar]
- 21.Gullikson GW, Loeffler RF, Virina MA. Relationship of serotonin-3 receptor antagonist activity to gastric emptying and motor-stimulating actions of prokinetic drugs in dogs. J Pharmacol Exp Ther. 1991;258:103–10. [PubMed] [Google Scholar]
- 22.Meyers NL, Palmer RMJ, Wray HA, et al. Effects of single oral doses of renzapride on gastrointestinal motility in fasted healthy subjects [abstract]. Gut 2002; 51: SIII, A137
- 23.Staniforth DH, Pennick M. Human pharmacology of renzapride: a new gastrokinetic benzamide without dopamine antagonist properties. Eur J Clin Pharmacol. 1990;38:161–4. doi: 10.1007/BF00265977. [DOI] [PubMed] [Google Scholar]
- 24.Briejer MR, Akkermans LM, Schuurkes JA. Gastrointestinal prokinetic benzamides: the pharmacology underlying stimulation of motility. Pharmacol Rev. 1995;47:631–51. [PubMed] [Google Scholar]
- 25.Bach T, Syversveen T, Kvingedal AM, et al. 5-HT4(a) and 5-HT4(b) receptors have nearly identical pharmacology and are both expressed in human atrium and ventricle. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:146–60. doi: 10.1007/s002100000299. [DOI] [PubMed] [Google Scholar]
- 26.Blondel O, Vandecasteele G, Gastineau M, et al. Molecular and functional characterization of a 5-HT4 receptor cloned from human atrium. FEBS Lett. 1997;412:465–74. doi: 10.1016/S0014-5793(97)00820-X. [DOI] [PubMed] [Google Scholar]
- 27.Blondel O, Gastineau M, Dahmoune Y, et al. Cloning, expression, and pharmacology of four human 5-hydroxytryptamine type 4 receptor isoforms produced by alternative splicing in the carboxyl terminus. J Neurochem. 1998;70:2252–61. doi: 10.1046/j.1471-4159.1998.70062252.x. [DOI] [PubMed] [Google Scholar]
- 28.Claeysen S, Faye P, Sebben M, et al. 5-HT4 receptors: cloning and expression of new splice variants. Ann NY Acad Sci. 1997;861:49–56. doi: 10.1111/j.1749-6632.1998.tb10172.x. [DOI] [PubMed] [Google Scholar]
- 29.Mialet J, Berque-Bestel I, Eftekhari P, et al. Isolation of the serotoninergic 5-HT4(e) receptor from human heart and comparative analysis of its pharmacological profile in C6-glial and CHO cell lines. Br J Pharmacol. 2000;129:771–81. doi: 10.1038/sj.bjp.0703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mialet J, Berque-Bestel I, Sicsic S, et al. Pharmacological characterization of the human 5-HT4(d) receptor splice variant stably expressed in Chinese hamster ovary cells. Br J Pharmacol. 2000;131:827–35. doi: 10.1038/sj.bjp.0703641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagakura Y, Akuzawa S, Miyata K, et al. Pharmacological properties of a novel gastrointestinal prokinetic benzamide selective for human 5-HT4 receptor versus human 5-HT3 receptor. Pharmacol Res. 1999;39:375–82. doi: 10.1006/phrs.1998.0454. [DOI] [PubMed] [Google Scholar]
- 32.Pindon A, van Hecke G, van Gompel P, et al. Differences in signal transduction of two 5-HT4 receptor splice variants: compound specificity and dual coupling with Galphas- and Galphai/o-proteins. Mol Pharmacol. 2002;61:85–96. doi: 10.1124/mol.61.1.85. [DOI] [PubMed] [Google Scholar]
- 33.Steward LJ, Boess FG, Steele JA, et al. Importance of phenylalanine 107 in agonist recognition by the 5-hydroxytryptamine (3A) receptor. Mol Pharmacol. 2000;57:1249–55. [PubMed] [Google Scholar]
- 34.Bockert J, Claeysen S, Compan V, et al. 5-HT4 receptors. Curr Drug Targets — CNS & Neurolog Dis. 2004;3:39–51. doi: 10.2174/1568007043482615. [DOI] [PubMed] [Google Scholar]
- 35.Brattelid T, Kvingedal AM, Krobert KA, et al. Cloning, pharmacological characterisation and tissue distribution of a novel 5-HT4 receptor splice variant, 5-HT4(i) Naunyn Schmiedebergs Arch Pharmacol. 2004;369:616–28. doi: 10.1007/s00210-004-0919-4. [DOI] [PubMed] [Google Scholar]
- 36.King FD, Hadley MS, Joiner KT, et al. Substituted benzamides with conformationally restricted side chains: 5. Azabicyclo[x.y.z] derivatives as 5-HT4 receptor agonists and gastric motility stimulants. J Med Chem. 1993;36:683–9. doi: 10.1021/jm00058a004. [DOI] [PubMed] [Google Scholar]
- 37.Martin GR, Humphrey PP. Receptors for 5-hydroxytryptamine: current perspectives on classification and nomenclature. Neuropharmacology. 1994;33:261–73. doi: 10.1016/0028-3908(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 38.Saucier C, Albert PR. Identification of an endogenous 5-hydroxytryptamine type 2A receptor in NIH-3T3 cells: agonist-induced down-regulation involves decreases in receptor RNA and number. J Neurochem. 1997;68:1998–2011. doi: 10.1046/j.1471-4159.1997.68051998.x. [DOI] [PubMed] [Google Scholar]
- 39.Bonhaus DW, Bach C, DeSouza A, et al. The pharmacology and distribution of human 5-hydroxytryptamine type 2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors. Br J Pharmacol. 1995;115:622–8. doi: 10.1111/j.1476-5381.1995.tb14977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf WA, Schutz LJ. The serotonin 5-HT2C receptor is a prominent serotonin receptor in basal ganglia: evidence from functional studies on serotonin-mediated phosphoinositide hydrolysis. J Neurochem. 1997;69:1449–58. doi: 10.1046/j.1471-4159.1997.69041449.x. [DOI] [PubMed] [Google Scholar]
- 41.Miller K, Weisberg E, Fletcher PW, et al. Membrane-bound and solubilized brain 5HT3 receptors: improved radioligand binding assays using bovine area postrema or rat cortex and the radioligands [3H]-GR65630, [3H]-BRL43694, and [3H]-LY278584. Synapse. 1992;11:58–66. doi: 10.1002/syn.890110108. [DOI] [PubMed] [Google Scholar]
- 42.Boess FG, Steward LJ, Steele JA, et al. Analysis of the ligand binding site of the 5-HT3 receptor using site directed mutagenesis: importance of glutamate 106. Neuropharmacology. 1997;36:637–47. doi: 10.1016/S0028-3908(97)00044-0. [DOI] [PubMed] [Google Scholar]
- 43.Grossman CJ, Kilpatrick GJ, Bunce KT. Development of a radioligand binding assay for 5-HT4 receptors in guinea-pig and rat brain. Br J Pharmacol. 1993;109:618–24. doi: 10.1111/j.1476-5381.1993.tb13617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rees S, den Daas I, Foord S, et al. Cloning and characterisation of the human 5-HT5A serotonin receptor. FEBS Lett. 1994;355:242–6. doi: 10.1016/0014-5793(94)01209-1. [DOI] [PubMed] [Google Scholar]
- 45.Monsma FJ, Jr, Shen Y, Ward RP, et al. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol. 1993;43:320–7. [PubMed] [Google Scholar]
- 46.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–30. [PubMed] [Google Scholar]
- 47.Greengrass P, Bremner R. Binding characteristics of [3H]-prazosin to rat brain alpha-adrenergic receptors. Eur J Pharmacol. 1979;55:323–6. doi: 10.1016/0014-2999(79)90202-4. [DOI] [PubMed] [Google Scholar]
- 48.Boyajian CL, Leslie FM. Pharmacological evidence for alpha-2 adrenoceptor heterogeneity: differential binding properties of [3H]-rauwolscine and [3H]-idazoxan in rat brain. J Pharmacol Exp Ther. 1987;241:1092–8. [PubMed] [Google Scholar]
- 49.Broadhurst AM, Alexander BS, Wood MD. Heterogeneous [3H]-rauwolscine binding sites in rat cortex: two alpha 2-adrenoceptor subtypes or an additional non-adrenergic interaction? Life Sci. 1988;43:83–92. doi: 10.1016/0024-3205(88)90240-8. [DOI] [PubMed] [Google Scholar]
- 50.U’Prichard DC, Bylund DB, Snyder SH. (+/−)-[3H]-Epinephrine and (−)[3H]-dihydroalprenolol binding to beta 1- and beta 2-noradrenergic receptors in brain, heart, and lung membranes. J Biol Chem. 1978;253:5090–102. [PubMed] [Google Scholar]
- 51.Galli A, DeFelice LJ, Duke BJ, et al. Sodium-dependent norepinephrine-induced currents in norepinephrine-transporter-transfected HEK-293 cells blocked by cocaine and antidepressants. J Exp Biol. 1995;198:2197–212. doi: 10.1242/jeb.198.10.2197. [DOI] [PubMed] [Google Scholar]
- 52.Dearry A, Gingrich JA, Falardeau P, et al. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature. 1990;347:72–6. doi: 10.1038/347072a0. [DOI] [PubMed] [Google Scholar]
- 53.Sunahara RK, Niznik HB, Weiner DM, et al. Human dopamine D1 receptor encoded by an intronless gene on chromosome 5. Nature. 1990;347:80–3. doi: 10.1038/347080a0. [DOI] [PubMed] [Google Scholar]
- 54.Zhou QY, Grandy DK, Thambi L, et al. Cloning and expression of human and rat D1 dopamine receptors. Nature. 1990;347:76–80. doi: 10.1038/347076a0. [DOI] [PubMed] [Google Scholar]
- 55.Bunzow JR, Van Tol HH, Grandy DK, et al. Cloning and expression of a rat D2 dopamine receptor cDNA. Nature. 1988;336:783–7. doi: 10.1038/336783a0. [DOI] [PubMed] [Google Scholar]
- 56.Grandy DK, Marchionni MA, Makam H, et al. Cloning of the cDNA and gene for a human D2 dopamine receptor. Proc Natl Acad Sci U S A. 1989;86:9762–6. doi: 10.1073/pnas.86.24.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayes G, Biden TJ, Selbie LA, et al. Structural subtypes of the dopamine D2 receptor are functionally distinct: expression of the cloned D2A and D2B subtypes in a heterologous cell line. Mol Endocrinol. 1992;6:920–6. doi: 10.1210/mend.6.6.1323056. [DOI] [PubMed] [Google Scholar]
- 58.Sokoloff P, Giros B, Martres MP, et al. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–51. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 59.Van Tol HH, Wu CM, Guan HC, et al. Multiple dopamine D4 receptor variants in the human population. Nature. 1992;358:149–52. doi: 10.1038/358149a0. [DOI] [PubMed] [Google Scholar]
- 60.Van Tol HH, Bunzow JR, Guan HC, et al. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature. 1991;350:610–4. doi: 10.1038/350610a0. [DOI] [PubMed] [Google Scholar]
- 61.Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–9. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 62.Sunahara RK, Guan HC, O’Dowd BF, et al. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature. 1991;350:614–9. doi: 10.1038/350614a0. [DOI] [PubMed] [Google Scholar]
- 63.Weinshank RL, Adham N, Macchi M, et al. Molecular cloning and characterization of a high affinity dopamine receptor (D1 beta) and its pseudogene. J Biol Chem. 1991;266:22427–35. [PubMed] [Google Scholar]
- 64.Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–9. doi: 10.1016/0165-6147(93)90029-J. [DOI] [PubMed] [Google Scholar]
- 65.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 66.Cohen ML, Fludzinski LA. Contractile serotonergic receptor in rat stomach fundus. J Pharmacol Exp Ther. 1987;243:264–9. [PubMed] [Google Scholar]
- 67.Schiavi GB, Brunet S, Rizzi CA, et al. Identification of serotonin 5-HT4 recognition sites in the porcine caudate nucleus by radioligand binding. Neuropharmacology. 1994;33:543–9. doi: 10.1016/0028-3908(94)90085-X. [DOI] [PubMed] [Google Scholar]
- 68.Bender E, Pindon A, van Oers I, et al. Structure of the human serotonin 5-HT4 receptor gene and cloning of a novel 5-HT4 splice variant. J Neurochem. 2000;74:478–89. doi: 10.1046/j.1471-4159.2000.740478.x. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida N, Mizumoto A, Iwanaga Y, et al. Effects of 5-hydroxytryptamine type 3 receptor antagonists on gastrointestinal motor activity in conscious dogs. J Pharmacol Exp Ther. 1991;256:272–8. [PubMed] [Google Scholar]
- 70.Burke TA, Sanger GJ. Regionally selective cholinergic stimulation by BRL 24924 in the human isolated gut. Br J Clin Pharmacol. 1988;26:261–5. doi: 10.1111/j.1365-2125.1988.tb05275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leclere PG, Lefebvre RA. Presynaptic modulation of cholinergic neurotransmission in the human proximal stomach. Br J Pharmacol. 2002;135:135–42. doi: 10.1038/sj.bjp.0704471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tam FS, Hillier K, Bunce KT. Characterization of the 5-hydroxy-tryptamine receptor type involved in inhibition of spontaneous activity of human isolated colonic circular muscle. Br J Pharmacol. 1994;113:143–50. doi: 10.1111/j.1476-5381.1994.tb16186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prins NH, Akkermans LM, Lefebvre RA, et al. 5-HT(4) receptors on cholinergic nerves involved in contractility of canine and human large intestine longitudinal muscle. Br J Pharmacol. 2000;131:927–32. doi: 10.1038/sj.bjp.0703615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prins NH, Shankley NP, Welsh NJ, et al. An improved in vitro bioassay for the study of 5-HT4 receptors in the human isolated large intestinal circular muscle. Br J Pharmacol. 2000;129:1601–8. doi: 10.1038/sj.bjp.0703254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borman RA, Burleigh DE. Evidence for the involvement of a 5-HT4 receptor in the secretory response of human small intestine to 5-HT. Br J Pharmacol. 1993;110:927–8. doi: 10.1111/j.1476-5381.1993.tb13901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borman RA, Burleigh DE. Heterogeneity of 5-HT receptors mediating secretion in the human intestine. Ann NY Acad Sci. 1996;812:224–5. doi: 10.1111/j.1749-6632.1997.tb48183.x. [DOI] [PubMed] [Google Scholar]
- 77.Budhoo MR, Harris RP, Kellum JM. The role of the 5-HT4 receptor in Cl-secretion in human jejunal mucosa. Eur J Pharmacol. 1996;314:109–14. doi: 10.1016/S0014-2999(96)00474-8. [DOI] [PubMed] [Google Scholar]
- 78.van Wijngaarden I, Tulp MT, Soudijn W. The concept of selectivity in 5-HT receptor research. Eur J Pharmacol. 1990;188:301–12. doi: 10.1016/0922-4106(90)90190-9. [DOI] [PubMed] [Google Scholar]
- 79.Spiller RC. Effects of serotonin on intestinal secretion and motility. Curr Opin Gastroenterology. 2001;17:99–103. doi: 10.1097/00001574-200103000-00001. [DOI] [PubMed] [Google Scholar]
- 80.Berman SM, Chang L, Suyenobu B, et al. Condition-specific deactivation of brain regions by 5-HT3 receptor antagonist alosetron. Gastroenterology. 2002;123:969–77. doi: 10.1053/gast.2002.35990. [DOI] [PubMed] [Google Scholar]
- 81.Friedel D. Ischemic colitis during treatment with alosetron. Gastroenterology. 2001;120:557–60. doi: 10.1053/gast.2001.21177. [DOI] [PubMed] [Google Scholar]
- 82.Prins NH, Briejer MR, Schuurkes JA. Characterization of the contraction to 5-HT in the canine colon longitudinal muscle. Br J Pharmacol. 1997;120:714–20. doi: 10.1038/sj.bjp.0700954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beattie DT, Smith JA, Marquess D, et al. The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br J Pharmacol. 2004;143:549–60. doi: 10.1038/sj.bjp.0705929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borman RA, Tilford NS, Harmer DW, et al. 5-HT2B receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br J Pharmacol. 2002;135:1144–51. doi: 10.1038/sj.bjp.0704571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng CH, Costall B, Naylor RJ, et al. The effect of 5-HT receptor ligands on the uptake of [3H]5-hydroxytryptamine into rat cortical synaptosomes. Eur J Pharmacol. 1993;239:211–4. doi: 10.1016/0014-2999(93)90996-U. [DOI] [PubMed] [Google Scholar]