

Abstract
The Apolipoprotein E (APOE) gene is a major genetic risk factor associated with Alzheimer’s disease (AD). APOE encodes for three main isoforms in humans (E2, E3, and E4). Homozygous E4 individuals have more than a 10-fold higher risk for developing late-onset AD, while E2 carriers are protected. A hallmark of AD is a reduction in cerebral glucose metabolism, alluding to a strong metabolic component in disease onset and progression. Interestingly, E4 individuals display a similar regional pattern of cerebral glucose hypometabolism decades prior to disease onset. Mapping the metabolic landscape may help elucidate the underlying biological mechanism of APOE-associated risk for AD. Efficient metabolic coupling of neurons and glia is necessary for proper neuronal function, and disruption in glial energy distribution has been proposed to contribute to neuronal cell death and AD pathology. One important function of astrocytes – canonically the primary source of apolipoprotein E in the brain – is to provide metabolic substrates (lactate, lipids, amino acids and neurotransmitters) to neurons. Here we investigate the effects of APOE on astrocyte glucose metabolism in vitro utilizing scintillation proximity assays, stable isotope tracer metabolomics, and gene expression analyses. Glucose uptake is impaired in E4 astrocytes relative to E2 or E3 with specific alterations in central carbon metabolism. Using [U-13C] glucose allowed analyses of astrocyte-specific deep metabolic networks affected by APOE and provided insight to the effects downstream of glucose uptake. Enrichment of 13C in early steps of glycolysis was lowest in E4 astrocytes (highest in E2), while synthesis of lactate from glucose was highest in E4 astrocytes (lowest in E2). We observed an increase in glucose flux through the pentose phosphate pathway (PPP), with downstream increases in gluconeogenesis, lipid, and de novo nucleotide biosynthesis in E4 astrocytes. There was also a marked increase in 13C enrichment in the TCA cycle of E4 astrocytes – whose substrates were also incorporated in biosynthesis pathways at a higher rate. Pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) are the two main enzymes controlling pyruvate entry to the TCA cycle. PC gene expression is increased in E4 astrocytes and the activity relative to PDH was also increased, compared to E2 or E3. Decreased enrichment in the TCA cycle of E2 and E3 astrocytes is suggestive of increased oxidation and non-glucose derived anaplerosis, which could be fueling mitochondrial ATP production. Conversely, E4 astrocytes appear to increase carbon flux into the TCA cycle to fuel cataplerosis. Together, these data demonstrate clear APOE isoform-specific effects on glucose utilization in astrocytes, including E4-associated increases in lactate synthesis, PPP flux, and de novo biosynthesis pathways.
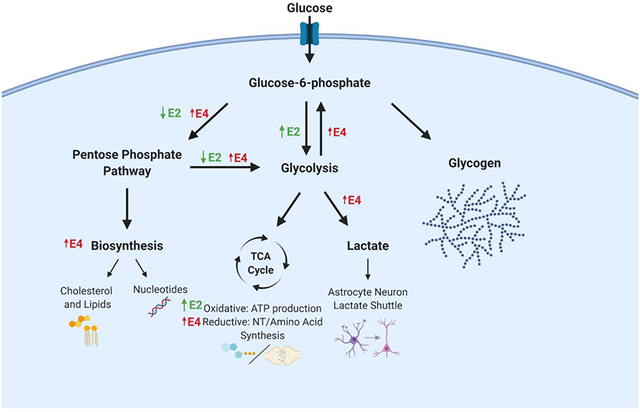
Graphical Abstract.
Glucose utilization in astrocytes expressing either human E2 (green arrows) or E4 (red arrows) relative to E3. E2 astrocytes display increased flux through glycolysis, a more oxidative TCA cycle, and decreased pentose phosphate pathway (PPP) flux. E4 astrocytes display increased glucose flux through PPP, in both re-entry into glycolysis (gluconeogenesis), increased biosynthesis, and increased lactate synthesis, with a less oxidative TCA cycle.
1. Introduction
There is a well-established association between late-onset Alzheimer’s disease (AD) and glucose metabolism. For example, AD patients display regional cerebral glucose hypometabolism as determined by 18-fluorodeoxyglucose positron emission tomography (FDG-PET), primarily in the posterior cingulate, parietal, and temporal cortices (Herholz et al., 2002; Minoshima et al., 1997). Clinical late-onset AD symptoms essentially never occur without glucose hypometabolism, and the extent of these metabolic changes are strongly correlated with the severity of clinical symptoms (Blass, 2002; Grady et al., 1986; Haxby et al., 1990). Additionally, postmortem metabolomics studies of AD brains often reveal alterations in glucose utilization and amino acid metabolism (Butterfield and Halliwell, 2019; Palmer, 1999; Xu et al., 2016).
While many mysteries remain, much has been learned about cerebral metabolism over the past several decades, including a greater appreciation for the critical role astrocytes play in supporting neuronal function (Magistretti and Allaman, 2015). Central to this neuron-astrocyte metabolic coupling is the astrocytic utilization of glucose – the primary energy substrate for the brain – to both support their own metabolic needs as well as those of neurons (Magistretti and Allaman, 2015). Glucose metabolism in astrocytes begins with uptake of glucose from the cerebral vasculature. Upon entry, glucose is phosphorylated to glucose-6-phosphate (G6P) via hexokinase enzymes. G6P is then shunted to one of three main pathways: i) it is incorporated into glycolysis, ii) enters the pentose phosphate pathway (PPP), or iii) is stored as glycogen (Brown and Ransom, 2007). Glycolysis predominates as the main pathway for glucose metabolism in astrocytes, which generates ATP, NADH, and pyruvate or lactate (Hertz et al., 2007; Magistretti and Allaman, 2015). Lactate is thought to be shuttled to neurons as part of the astrocyte-neuron lactate shuttle. Pyruvate can be further metabolized in the TCA cycle to produce additional reducing equivalents that drive the electron transport chain for ATP production via oxidative phosphorylation, or for generation of amino acids utilized in biosynthesis and neurotransmitter synthesis (Pellerin et al., 1998; Simpson et al., 2007). PPP activity utilizes glucose to regenerate NADPH, an essential cofactor for both cellular redox management and in the biosynthesis of nucleotides and lipids. Finally, the role of glycogen metabolism in astrocytes has recently been broadened beyond a simple description as a glucose reserve during hypoglycemic states to include several essential functions in regards to memory formation and consolidation with implications in AD (Alberini et al., 2018; Bak et al., 2018; Brown and Ransom, 2007).
Importantly, disruption of any of the astrocyte-initiated glucose pathways described above can result in impairments in neuronal function (Fiebig et al., 2019). This fact may be reflected in epidemiological studies, as cerebral glucose hypometabolism precedes onset of late-onset AD in several high-risk groups (Reiman et al., 2001; Small et al., 2000). This includes carriers of the ε4 allele of apolipoprotein E (APOE) – the single strongest genetic risk factor for late-onset AD – in which “AD-like” regional patterns of glucose hypometabolism have been described as early as the second decade of life (Reiman et al., 2004). APOE exists in humans as three main isoforms in humans (E2, E3, and E4), with E4 – which is expressed by nearly 20% of the population – conferring up to a 15-fold increase in AD risk for homozygotes (Farrer et al., 1997; Raber et al., 2004). Conversely, E2 carriers (approximately 10% of the population) have a substantially lower risk of AD, and show slower mental decline and increased longevity (Chiang et al., 2010; Corder et al., 1994; Farrer et al., 1997; Garatachea et al., 2014; Wilson et al., 2002). While multiple cell types can synthesize APOE, astrocytes produce the majority of the CNS pool of the protein under normal conditions (Huang and Mahley, 2014).
While multiple studies show decreased FDG uptake in E4 carriers, the broader relationship between APOE and cerebral metabolism is complicated by findings from fMRI-based studies, as well its potential associations with amyloid pathology. In regards to the latter, some studies show a strong association between E4 and amyloid deposition on cerebral glucose uptake (Carbonell et al., 2016), others a weak association (Gonneaud et al., 2016), while yet others show that APOE’s association with cerebral glucose uptake is independent of amyloid (Jagust et al., 2012). Additionally, several fMRI-based analyses of E4 carriers show that E4 alters resting state connectivity and that E4 carriers have elevated baseline activity (Dickerson et al., 2005; Wishart et al., 2006), particularly in the hippocampus and areas of the default mode network (Liu et al., 2013). Mouse models of human APOE also reflect this complicated picture, with some studies showing reduced cerebral glucose uptake in E4 expressing mice (Johnson et al., 2017b; Lin et al., 2017), while others show regional hyperactivity and metabolic changes (Nuriel et al., 2017) and increased glucose uptake in old E4 mice, compared to those expressing E3 (Venzi et al., 2017). Intriguingly, some of these seemingly contradictory findings may be explained by viewing E4 carriage as a case of antagonistic pleiotropy, whereby potentially advantageous early life alterations in activity and connectivity result in detrimental metabolic changes in later life (Tuminello and Han, 2011).
One limiting factor in the pursuit of elucidating the role of APOE is that while PET imaging provides information on the regional uptake of glucose, it is not sufficient to distinguish downstream metabolic pathways (i.e. how that glucose is being utilized) nor does it have the resolution to determine cell specificity (i.e. by what cell types). Discerning these two key pieces of information in the context of APOE genotype may help elucidate the missing link between AD risk and glucose hypometabolism. Previously used methodologies to identify metabolic changes in AD and cognitively impaired brains are various metabolomics-based approaches (Orešič et al., 2011; Wilkins and Trushina, 2017). However, while traditional metabolomics approaches are able to provide a valuable snapshot of the metabolic pathways involved, they are unable to identify the precursor-product relationships that drive such perturbations. This type of precursor-product “tracing” of metabolites through various pathways is only possible with methods such as stable isotope resolved metabolomics (SIRM) (Fan et al., 2011).
In the current study, we employed SIRM to study astrocyte glucose metabolism by tracing universally labeled 13C glucose ([U-13C] glucose) in astrocytes expressing human E2, E3, or E4. This 13C tracer approach allowed us to identify multiple APOE isoform-dependent alterations in astrocyte central carbon metabolism, including alterations in glucose flux through glycolysis, PPP, and utilization of the TCA cycle. Specifically, glycolytic flux in E4 astrocytes indicates larger contributions to the PPP, increased lactate synthesis, altered patterns of TCA cycle activity and anaplerosis, and an increase in multiple biosynthetic pathways.
2. Materials and methods
2.1. Tissue culture and in vitro [U-13C] glucose metabolite labeling
Immortalized astrocytes derived from human APOE targeted-replacement mice homozygous for either E2, E3, or E4 (kind gift from Dr. David Holtzman) (Morikawa et al., 2005). For the tracer experiment, astrocytes seeded as triplicates into 10 cm tissue-culture dishes and maintained in maintenance media Advanced Dulbecco’s Modified Eagle Media (DMEM) supplemented with 10% FBS, 4% sodium pyruvate, and 1% Geneticin® (Thermo Fisher Scientific). At roughly 50% density, maintenance media was removed and cells were washed three times with sterile PBS. Tracer media (consisting of glucose free DMEM supplemented with 10mM [U-13C] glucose (Cambridge), 10% dialyzed FBS and 0.4% Geneticin®) was then added. Cells were incubated at 5% CO2 and 37 °C for 24 hours in order to reach isotopic steady state - meaning 13C enrichment into a given metabolite was stable relative to experimental error (Buescher et al., 2015). After 24 hours, media was removed and plates were washed on ice 3X with ice cold PBS. 1mL of ice cold CH3CN was added and cells were incubated at −20°C for 10 min to quench cellular metabolism and initiate cellular lysis. Plates were then scraped and cellular lysates collected in cryovials, snap frozen in liquid nitrogen, and then stored at −80°C until mass spectrometry analysis. Total cellular protein was measured using a BCA Protein Assay Kit (Thermo). Reported values were normalized to protein isolated from corresponding well.
3.2. Interpretation of 13C fractional labeling
To gain insight into the intracellular fate of glucose in astrocytes expressing human E2, E3, or E4, we analyzed the distribution of a [U-13C] glucose tracer into 117 distinct metabolites using an ion chromatography (IC) mass spectrometry (MS) approach (Higashi et al., 2014; Lane et al., 2008; Sun et al., 2017), further detailed below, to determine the intracellular fate of glucose in vitro (Figure 1A).
Figure 1. Experimental design and fractional labeling methodology.
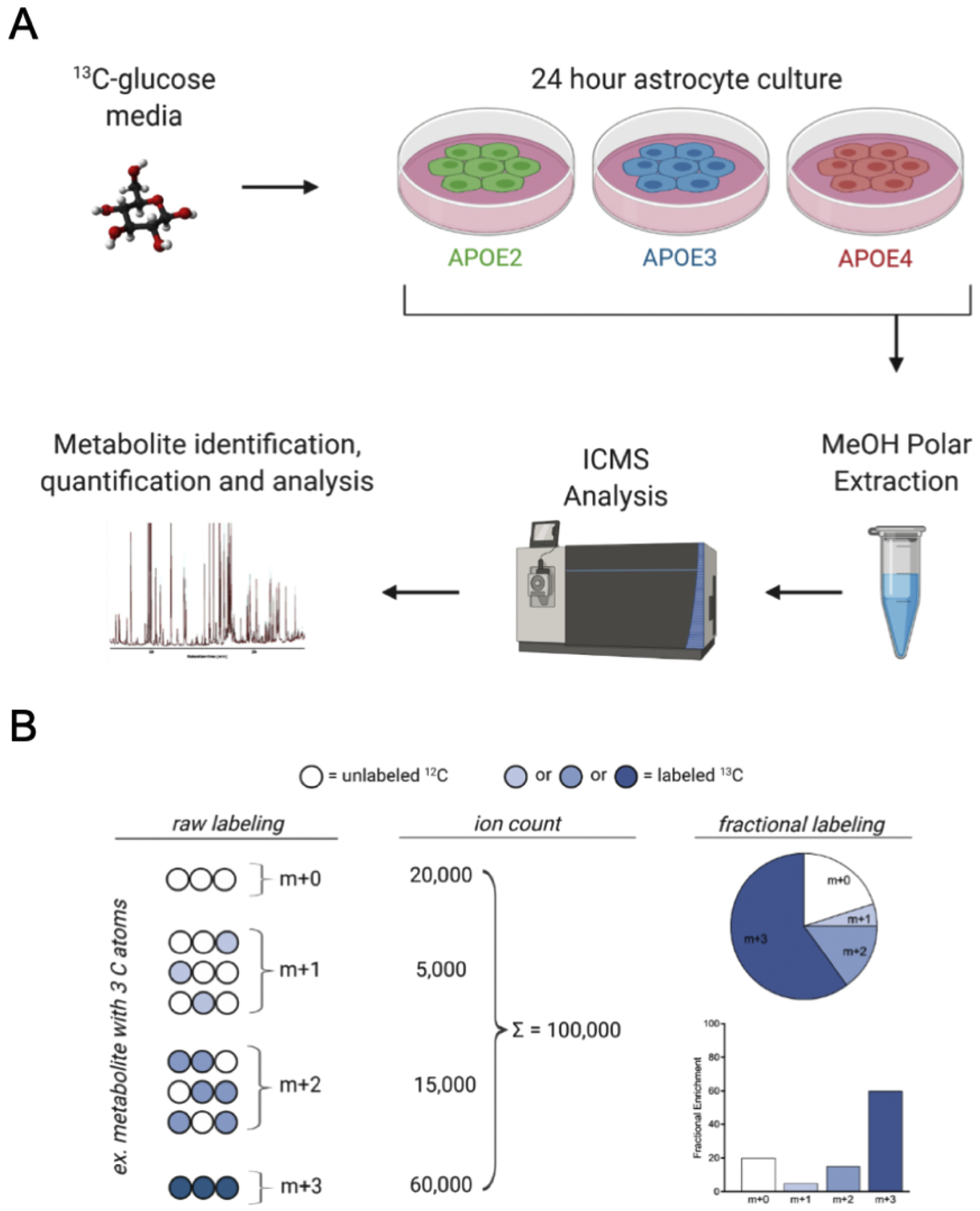
A) Stable Isotope Resolved Metabolomics Experimental design. Immortalized astrocytes expressing human E2, E3 or E4 were cultured for 24 hours in glucose-free media supplemented with 10 mM [U-13C] glucose tracer. Polar metabolites were extracted from the cells and 13C distribution among >100 metabolites was determined via ICMS (please see Methods for details). B) Example description of fractional labeling. Fractional enrichment of a metabolite is the abundance of an individual isotopologue (number of 13C atoms present) divided by the sum of all isotopologues for that metabolite. The distribution of labeling can indicate flux through a particular pathway or highlight contributions from specific enzymes based on the labeling pattern observed. Figure adapted from Buescher et al., 2015
Glucose tracing data is represented as fractional enrichment, which reflects the percent of each isotopologue contribution to the metabolite’s total pool. Isotopologues are defined as compounds that only differ in their isotopic distribution. M+0 represents a metabolite containing zero 13C atoms, while m+1 contains one 13C atom and so on and so forth. This is graphically represented in Figure 1B. These fractional labeling patterns provide information on glucose flux through specific pathways as well as estimations of enzyme activity (Buescher et al., 2015). For example, the ratio of citrate and pyruvate fractions can serve as surrogate enzyme activity measures for PDH and PC (PDH: citrate m+2/pyruvate m+3; PC: citrate m+3/pyruvate m+3) (Courtney et al., 2018). Following natural abundance correction (see reference below), we can conclude that the presence of a 13C atom in a given isotopologue is derived from glucose, as the only source of 13C was from exogenously administered [U-13C] glucose in the cell culture media.
2.2. Polar metabolite analysis by Ion-Chromatography Mass Spectroscopy
Samples were prepared and run by the Research Center for Stable Isotope-Resolved Metabolomics (RCSIRM) at the University of Kentucky. Briefly, samples were thawed on ice and chloroform was added in a 2:1.5:1 CH3CN to water to chloroform ratio and shaken vigorously. The samples were then centrifuged at 3,500 × g for 20 min to produce an upper polar layer of metabolites. Polar metabolites were transferred, lyophilized and reconstituted in nanopure water. A Dionex ICS-5000+ ion chromatograph interfaced with a Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) operating at a resolution setting of 500,000 (FWHM at m/z 200) was used to identify 13C isotopologues. The chromatography was performed using a Dionex IonPac AG11-HC-4 μm RFIC&HPIC (2 × 50 mm) guard column upstream of a Dionex IonPac AS11-HC-4 μm RFIC&HPIC (2 × 250 mm) column. Chromatography and mass spectrometric settings with an acquisition m/z range of 80 to 700 were the same as described previously (Fan et al., 2016). Metabolites and their isotopologues were identified by chromatographic retention times and their m/z values compared with those of the standards. Spectra were curated for correct identification of isotopologues and subsequently integrated and exported via the TraceFinder 3.3 (Thermo, Waltham, MA, USA) software package. Peak areas were corrected for natural abundance as previously described (Moseley, 2010). Fractional enrichment was calculated to quantify 13C incorporation into downstream pathways.
2.3. Scintillation Proximity Assay
Scintillation Proximity Assay (SPA) measures uptake of radioactively labeled substrates as previously described(Wensaas et al., 2007). Immortalized astrocytes were seeded on Perkin Elmer Cytostar-T 96-well plates at a concentration of 50,000 cells/well in maintenance media. Approximately 24 hours after seeding the maintenance media was aspirated and cells were washed with sterile PBS. Cells were then starved for four hours in starvation media glucose-free DMEM, 10% FBS, 1mM sodium pyruvate, 0.4% Geneticin (Gibco) to promote cell cycle synchrony and maximal glucose kinetics. Following the starvation period, starvation media was replaced with starvation media containing deoxy-d-glucose, 2-[1,2-3H] (5.6 μCi/mL) and each well was measured in a MicroBeta scintillation counter (Perkin-Elmer) for 10 seconds every 15 minutes for two hours. Scintillation counts were normalized to cellular protein as quantified by a commercially available bicinchoninic acid (BCA) assay kit.
2.4. Microarray gene expression
RNA was isolated from in vitro astrocytes using Trizol (Thermofisher) according to manufacturer’s instructions. Briefly, E2, E3, and E4 immortal astrocytes under identical culture conditions in maintenance media were washed and treated with Trizol for 5 minutes to accomplish nucleoprotein complex dissociation. Chloroform was then added and samples were centrifgued at 12,000×g for 15 minutes to separate RNA from the lower organic phase. RNA was precipitated with addition of isopropanol, washed with 75% ethanol and dried briefly. RNA pellets were resuspended in 50uL of RNAse free water. Total RNA was reverse transcribed to cDNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems) on a SimpliAmp Thermocycler (Applied Biosystems). cDNA was quantified by NanoDrop and loaded at 100ng/well into a custom Taqman Gene Array plate with 2X Taqman Fast Advanced Mastermix and sealed with MicroAmp™ Optical Adhesive Film. Taqman qRT-PCR was performed over 40 cycles on a QuantStudio 3 Flex Real-Time PCR System under the following cycle conditions: enzyme activation at 95°C for 20 seconds, denaturation at 95°C for 1 second, and annealing and extension at 60°C for 20 seconds. Ct values were normalized to housekeeping gene 18S for each plate to obtain ΔCt values. Gene expression differences were found by comparing to 2-ΔCt values and expressed as percentage of the mean of E3 samples.
2.5. Statistics
All data are expressed as mean values +/− standard error. Comparisons between two groups were analyzed by two-tailed t-test. Multiple groups and/or multiple time points were analyzed using ANOVAs (Prism, Graphpad), or repeated measures ANOVA (time × groups). Statistical significance was determined using an error probability level of p < 0.05 corrected using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 1%.
3. Results
3.1. E4 astrocytes exhibit decreased glucose uptake while E2 have increased uptake
To begin to define the role of APOE in regulating glucose metabolism in astrocytes, we first measured the rate of glucose uptake in real time using a scintillation proximity assay (SPA). We observed a clear stepwise APOE effect on the rate of 3H-2DG uptake, with E4 astrocytes taking up significantly less, and E2 substantially more glucose, when compared to E3 astrocytes (Figure 2).
Figure 2. APOE effects astrocyte glucose uptake.
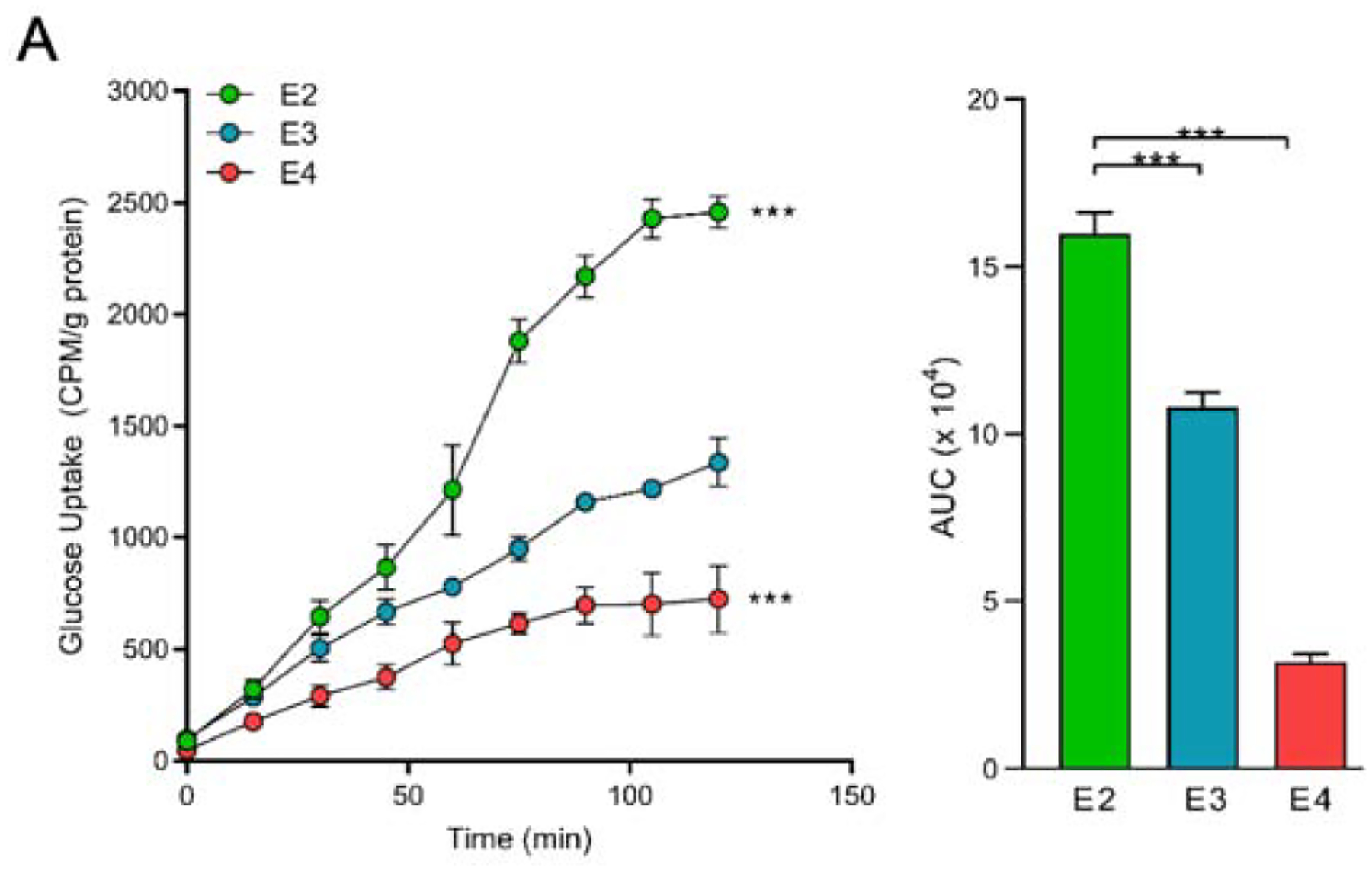
A) E2 astrocytes take up more glucose, while E4 astrocytes take up less glucose, relative to E3. Immortalized astrocytes expressing human APOE were treated with 3H-2-deoxyglucose (5.6uCi/mL) in vitro and uptake was measured by a scintillation proximity assay (SPA) over 120 minutes. Total area under the curve (AUC) was determined. Values represent mean +/− SEM (n=3). Data analyzed by two-way ANOVA of repeated measures (SPA) or t-test (AUC). *** p < 0.001.
3.2. E4 astrocytes show increased non-oxidative PPP activity and lactate generation
Using a [U-13C] glucose tracer in vitro, we found substantial 13C incorporation into glycolysis in all APOE isoforms as demonstrated by high levels of 13C enrichment in all measured metabolites (>50% for all genotypes) (Figure 3). The first step in glucose metabolism after transport into an astrocyte is phosphorylation to G6P, where E2 astrocytes displayed the highest fractional enrichment in G6P at m+6 (Figure 3B). E4 astrocytes had the lowest enrichment in G6P at m+6, due to increased fractions at m+3 and m+5. Both fructose-6-phosphate (F6P) and fructose-1,6-bisphosphate (F1,6BP) labeling show a similar pattern as G6P, with E4 having the highest m+3 and m+5 fractions while E2 has the greatest m+6 fraction (Figure 3C). We did not observe significant differences between genotypes in the three-carbon glycolytic intermediates 1,3-bisphosphoglycerate, 2/3-phosphoglycerate, and phosphoenolpyruvate. Interestingly, glucose flux into the late stages of glycolysis (pyruvate and lactate) was significantly higher in E4 astrocytes. This is reflected in the increased fraction of fully labeled (m+3) pyruvate and lactate in E4, compared to E2 and E3 astrocytes (Figure 3E–F).
Figure 3. APOE alters glucose flux through glycolysis and the pentose phosphate pathway.
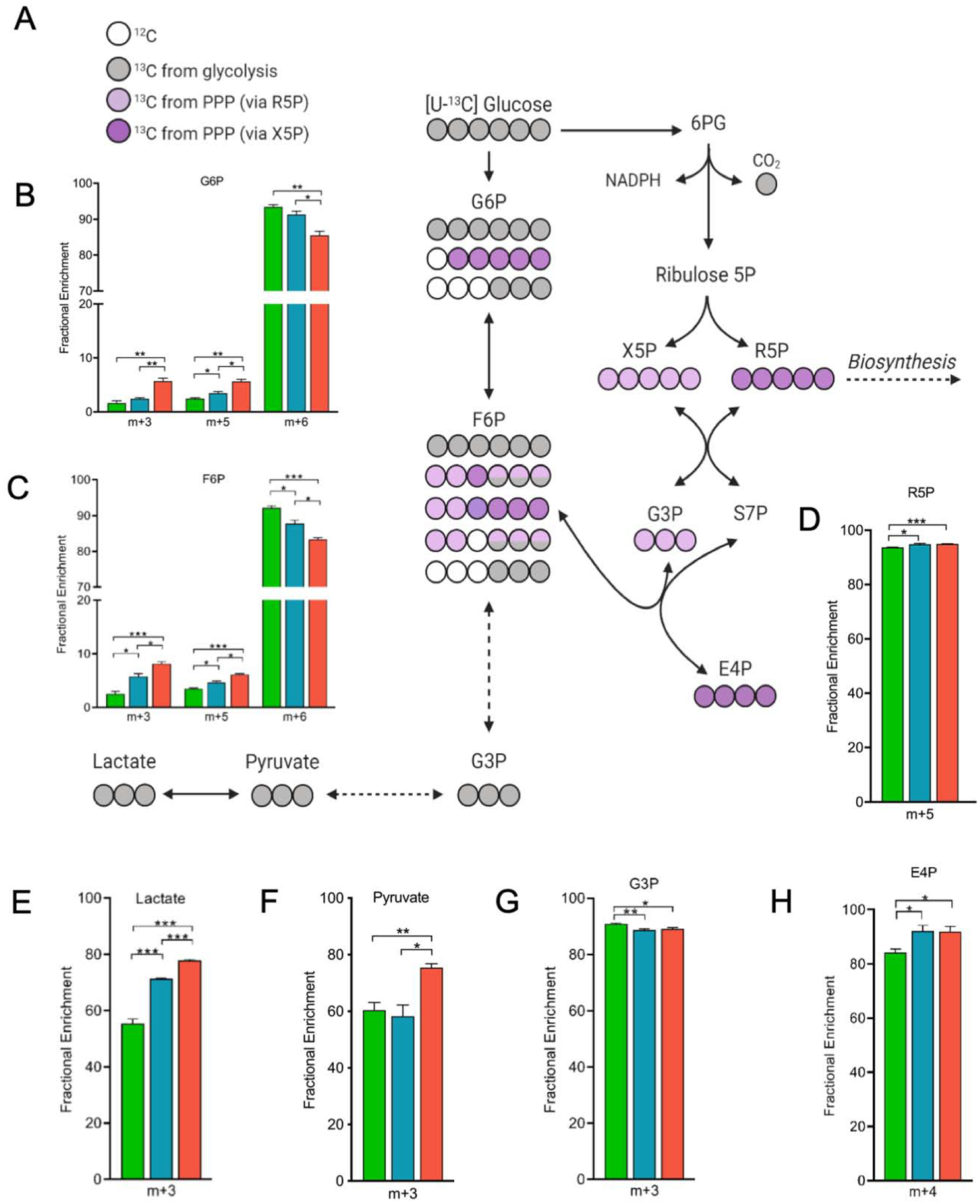
A) Tracing of glucose carbon through glycolysis, pentose phosphate pathway (PPP), and gluconeogenesis in astrocytes expressing E2, E3 or E4 grown in glucose tracer supplemented media for 24 hours. Carbons are color coded based on the pathway and/or enzyme the carbon was provided from (Grey circles: 13C; light purple: 13C from xyulose-5-phospahte (X5P); dark purple: 13C from ribose-5-phosphate (R5P); white circle: non-labeled 12C.)
B-H) Fractional enrichments in the isotopologues of glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), R5P, lactate, pyruvate, glyceraldehyde-3-phosphate (G3P), and erythrose-4-phosphate (E4P). Not all possible labeled metabolites are shown. The x-axis denotes percentage of total isotopic distribution per isotopologue shown. Data analyzed by multiple t-tests. Values represent mean +/− SEM (n=3). * p < 0.05; **p < 0.01; *** p < 0.001.
Notably, we also observed a stepwise APOE effect on 13C enrichment in the non-oxidative stage of the PPP (E2<E3<E4), as evidence by fully labeled (m+5) ribose-5-phosphate (R5P) and fully labeled (m+4) erythrose-4-phosphate (E4P) (Figure 3D,H). The last oxidative reaction in the PPP is the production of ribulose 5-phosphate, which can reversibly isomerize to R5P or alternatively undergo a series of reactions that result in the production of other pentose phosphates as well as the glycolysis intermediates F6P and glyceraldehyde-3-phosphate (G3P). The m+5 labeling of F1,6BP, F6P, and G6P likely reflects re-entry of PPP products into glycolysis via the non-oxidative branch of the PPP (Fan et al., 2012), and shows a clear stepwise APOE effect on the rate of PPP contribution to glycolysis (E2<E3<E4). Similarly, the stepwise (E2<E3<E4) increase in m+3 labeling of the glycolysis intermediates F6P and G6P reflects gluconeogenesis (reverse glycolysis). Together, this suggests that compared to E3 astrocytes, E2 astrocytes have increased flux directly through glycolysis, while E4 astrocytes cycle more glucose through the PPP.
3.3. APOE alters TCA cycle activity
To further explore the effects of APOE on glucose metabolism in pathways downstream from glycolysis, we examined the fractional enrichment of TCA cycle intermediates and the anaplerotic sites involved therein. Pyruvate entry into the TCA cycle is facilitated by the enzymes pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC), which incorporate either two or three carbon atoms, respectively (Alves et al., 2015). Citrate m+2 fraction indicates contributions from PDH, which is highest in E2 and lowest in E4. Comparing PC activity to PDH activity provides an indication of TCA anaplerosis. E2 astrocytes had the lowest PC:PDH ratio of 0.75, followed by E3 at 0.83 and E4 at 0.89 (Figure 4A), suggesting that E4 astrocytes are undergoing more anaplerosis in order to replenish the TCA of lost substrates. A higher citrate m+5 in E4 astrocytes also suggests increased anaplerosis from contributions of both PC and PDH. When utilizing a fully labeled pyruvate, PC generates oxaloacetate m+3 and when combined with, via citrate synthase, an acetyl-CoA that is produced from PDH utilizing a fully labeled pyruvate ultimately produces citrate m+5 (Figure 4F). Interestingly, gene expression of PC was significantly higher in E4 astrocytes compared to E2 and E3 cells (Figure 4B). Additionally, the m+5 fraction of citrate is highest in E4 astrocytes (which have a PC:PDH ratio close to 1), further suggesting that PC is contributing more to TCA cycle flux in E4 astrocytes than E2 or E3 (Figure 4F).
Figure 4. Alterations in glucose utilization in the TCA cycle and associated enzymes with APOE genotype.
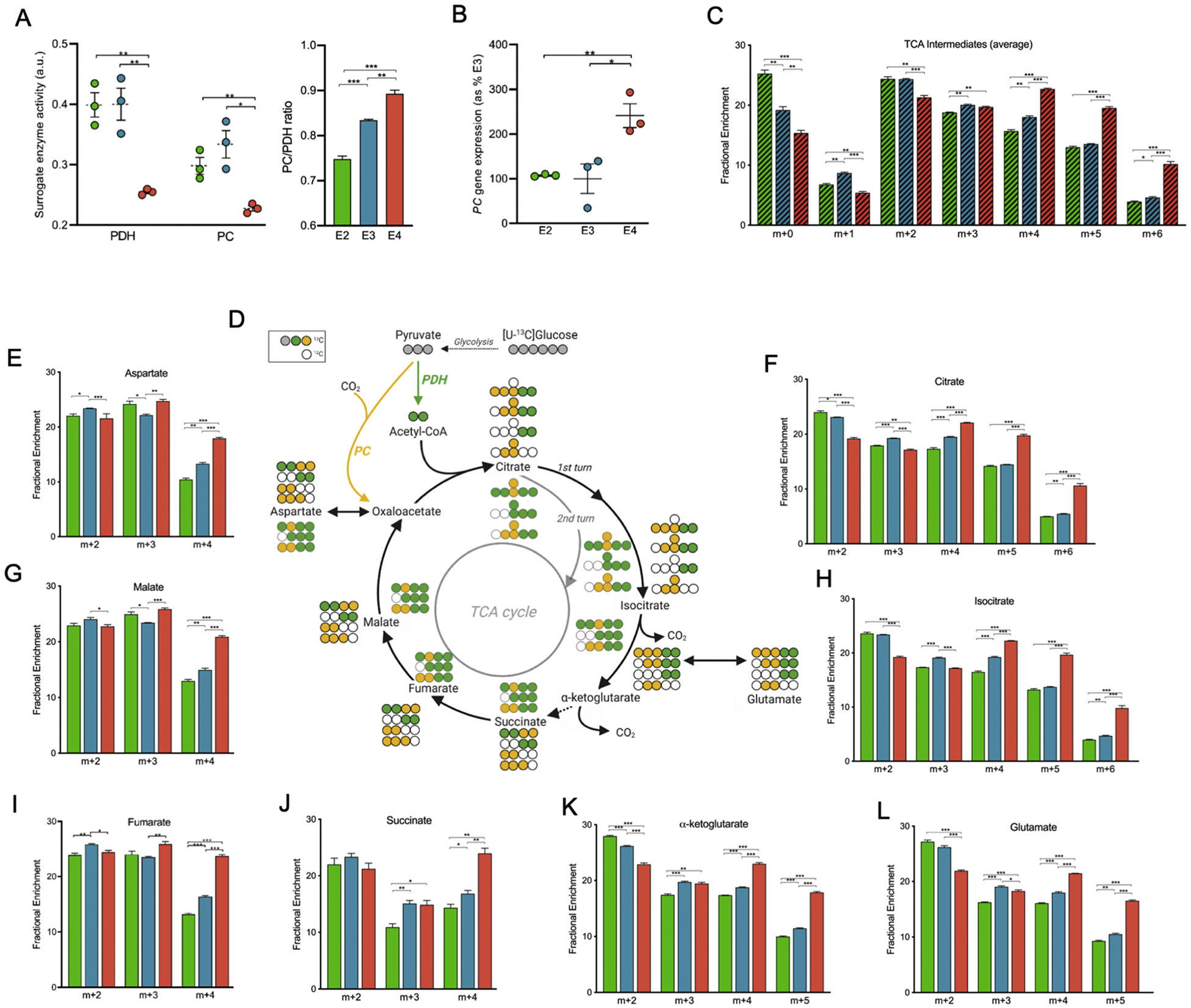
A) Decreased activity of TCA cycle entry enzymes pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC)Relative enzyme activity of PDH and PC; ratio of PC/PDH. (PC enzyme activity is estimated based on the ratio of m+3 citrate relative to m+3 pyruvate. PDH enzyme activity is estimated based on the ratio of m+2 citrate relative to m+3 pyruvate.)
B) Increased PC/PDH ratio mRNA gene expression. Gene expression of PC mRNA as percentage of E3
C) Isotopic distribution pattern reveals greater incorporation of glucose-derived carbon into TCA cycle in E4, relative to E2 or E3. The average isotopic distribution of TCA cycle intermediates (shown individually in E-L).
D) Glucose-derived carbon tracing through TCA cycle. Not all possible labeled metabolites are shown. Carbons are color coded based on the pathway and/or enzyme the carbon was provided from (Grey circles: 13C; yellow: 13C from PC; green circles: 13C from PDH; white circle: non-labeled 12C; black outlined circles: first round of TCA; grey outlined circles: second round of TCA).
E-L) Fractional enrichments in the isotopologues of aspartate, citrate, malate, isocitrate, fumarate, succinate, alpha-ketoglutarate, and glutamate highlighting differences among apoE isoforms. The x-axis denotes isotopic distribution per isotopologue shown.
Data analyzed by t-test (PDH and PC) and multiple t-tests (fractional enrichment). Values shown are mean +/− SEM (n=3). * p < 0.05; **p < 0.01; *** p < 0.001.
A general overview of the TCA cycle can be obtained from averaging the fractional enrichments of the entire TCA intermediates. This yields a distribution of all isotopologues and offers insight to global patterns of the TCA cycle. Higher fractions of m+2 isotopologues in E2 and E3 astrocytes, likely due to increased PDH activity, suggests 13C input undergoes a single turn of the TCA cycle before being released as 13CO2 (Figure 4C). Multiple turns of the TCA cycle would steadily feed more 13C into the TCA cycle, sustaining the 13CO2 lost to oxidation. This would result in an increase of the more enriched isotopologues, as demonstrated by the m+4, m+5, and m+6 fractions of E4 (Figure 4C–L). Also referred to as reverse Krebs cycle, a reductive TCA cycle operates in the reverse, or non-oxidative direction to replenish TCA substrates lost to cataplerosis (Verschueren et al., 2019). Cataplerosis is evidenced in E4 astrocytes by increased enrichment of amino acids synthesized from the TCA cycle, aspartate (m+4) and glutamate (m+4 and m+5) (Figure 4E,L). The decreased labeling in m+4, m+5 and m+6 isotopologues of E2 and E3, relative to E4, could result from glutamine import and subsequent oxidation via glutaminolysis, which would contribute unlabeled carbon atoms into TCA intermediates.
3.4. E4 astrocytes show increased purine and pyrimidine synthesis
The synthesis of nucleotides from glucose utilizes PPP products to generate the five membered sugar ring attached to nucleotides. The stepwise APOE-associated increases in 13C labeling of R5P (E2<E3<E4) (Figure 5D) may suggest differences in nucleotide biosynthesis, as R5P serves as a critical precursor to both purine and pyrimidine ribonucleotide synthesis. To determine the effect of APOE on the rate of nucleotide biosynthesis, we next examined the patterns of [U-13C] glucose-derived 13C in various purine and pyrimidine nucleotides.
Figure 5. E4 astrocytes show increased de novo synthesis of nucleotides from of glucose -derived carbon.
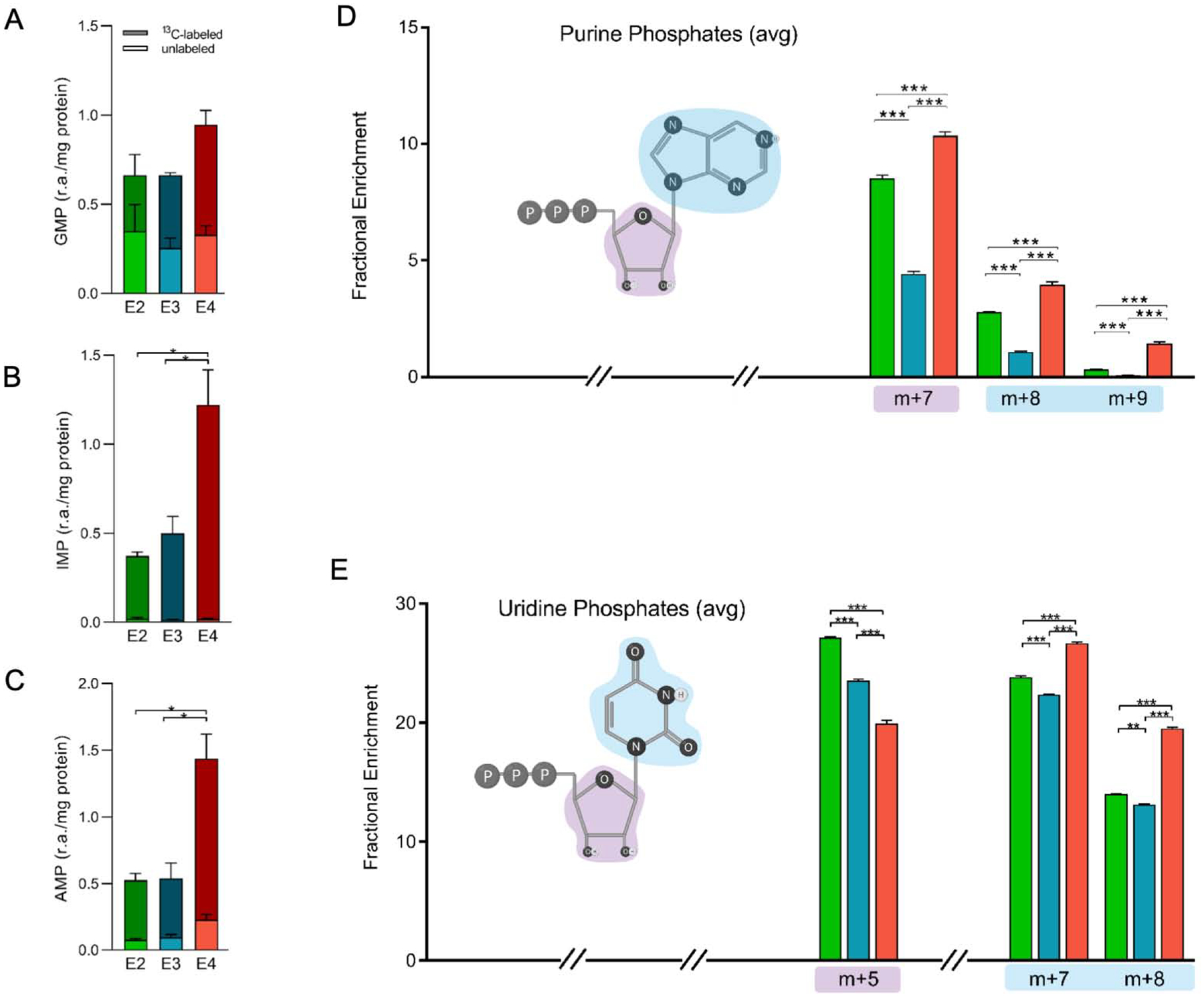
A-C) Comparison of total labeled isotopologues (m+1 + m+2 + … m+n) and unlabeled (m+0) of GMP, IMP and AMP. The y-axis denotes relative abundance of each purine phosphate normalized to cell amount (protein).
D-E) Fractional enrichment of D) purine phosphates (AMP, ADP, AMP, GMP, GDP, GTP) and E) uridine phosphates (UMP, UDP, UTP) highlighting structural components attributable to isotopic distribution. Purple and blue “clouds” denote contributions of various carbon positions to isotopologues shown (ex. purple, pentose ring). The x-axis denotes the percentage of total 13C distribution for each m+n fraction (select isotopologues shown).
Data analyzed by multiple t-tests. Values shown are mean +/− SEM (n=3). * p < 0.05; **p < 0.01; *** p < 0.001.
The carbon atoms for de novo biosynthesis of purines come from glycine, bicarbonate, and the methyl donor formyltetrahydrofolate (fTHF) (Villa et al., 2019). Once generated, inosine monophosphate (IMP) can then be converted to guanosine or adenine phosphates. While guanosine monophosphate levels were not significantly different between genotypes, the amount of both 13C-labeled IMP and 13C-labeled adenosine monophosphate (AMP) were significantly higher in E4 compared to E2 or E3 astrocytes (Figure 5A–C). This substantial increase in 13C-labeled IMP and AMP suggests that compared to E2 and E3 astrocytes, cells expressing E4 are diverting significantly more glucose toward de novo purine nucleotide biosynthesis.
For a broad look at purine and pyrimidine biosynthesis, we analyzed the average 13C labeling patterns for adenine phosphates (AMP, ADP, ATP) and uridine phosphates (UMP, UDP and UTP) (Figure 5D–E). Synthesis was increased in E4 astrocytes relative to E2 and E3 as evidenced by greater m+7, m+8, and m+9 fractions in purine phosphates and m+7 and m+8 fractions in uridine phosphates. These fractions correspond to the nucleotide base where either m+2, m+3, or m+4 aspartate was utilized, the latter of which is also highest in E4 astrocytes (Figure 5D). Conversely, the m+5 isotopologue of the uridine phosphates was highest in E2, followed by E3 and then E4 astrocytes (Figure 5E). This fraction denotes labeling in the ribose sugar, suggesting that the majority of uridine phosphates in E2, and to a lesser extent E3, contain only a newly synthesized ribose ring along with a recycled or salvaged base. Pyrimidines with deoxyribose sugar rings (dUMP, dTMP, and dTDP) display a similar pattern of increased m+5 fractions in E2 and E3, suggesting that of the labeled isotopologues the majority contain only a labeled ribose. In contrast, the labeling pattern in E4 astrocytes indicates an increase in newly synthesized bases demonstrated by greater fractions of m+7, m+8 and m+9 relative to E2 or E3 (Figure 5D–E), suggestive of an E4-associated upregulation of nucleotide biosynthetic pathways.
3.5. E4 astrocytes show increased biosynthesis of glutathione, lipid precursors, NADH, and UDP hexosamines.
Amino acids generated by the TCA cycle are utilized in a variety of biosynthetic pathways. For example, glutathione is synthesized using TCA-derived glutamate, cysteine and glycine (derived from serine via glycolytic intermediate 3PG). Interestingly, the fractional labeling pattern of glutathione m+1 through m+5 mirrors that of glutamate, where E2 is highest in m+2 and E4 higher in fractions m+4 and m+5 (Figure 6A). Additionally, the m+6 and m+7 fractions, which are indicative of 13C contributions from glycine, are also increased in E4 astrocytes compared to E2 and E3 (Figure 6A). The absence of fractions m+8, m+9 and m+10 suggest that in all three genotypes cysteine utilized for glutathione biosynthesis is not derived from glucose. However, the prominent m+6 and m+7 fractions seen in E4 suggest a much higher flux of glucose out of glycolysis, where 3PG is used to synthesize serine and subsequently glycine.
Figure 6. Increased de novo biosynthesis of glutathione, NADH, phospholipids, and UDP-hexoses biosynthesis in E4 astrocytes.
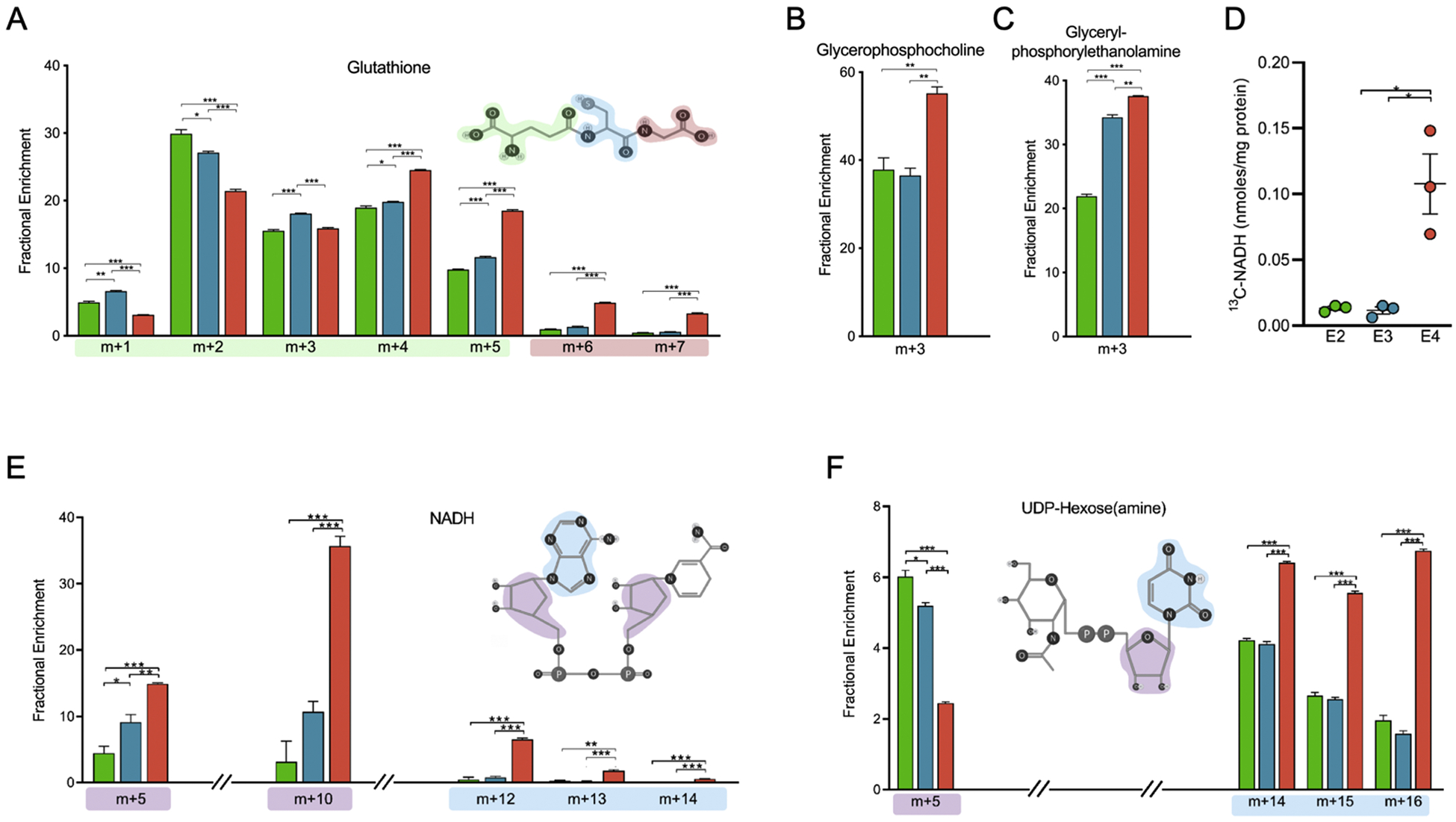
A) Fractional enrichment of selected isotopologues of glutathione highlighting structural components attributable to isotopic distribution. Red, blue, or green outline denotes contributions of carbon positions to isotopologues shown.
B) Total 13C-labeled NADH. The y-axis denotes total NADH containing at least one 13C atom (sum of m+1, m+2 … m+10).
C-D) Fractional enrichment of (C) Glycerophosphocholine and (D) Glycerylphosphorylethanolamine
E) Fractional enrichment of selected isotopologues of NADH highlighting structural components attributable to isotopic distribution. Blue and purple outlines denote contributions of carbon positions to isotopologues shown.
F) Average fractional enrichment of selected isotopologues of UDP-hexoses (UDP-glucose, UDP-galactose, UDP-N-acetylglucose, and UDP-N-acetylgalactose highlighting structural components attributable to isotopic distribution. Blue and purple outlines denote contributions of carbon positions to isotopologues shown.
X-axes denote percentage of total isotopic distribution per isotopologue shown. Data analyzed by multiple t-tests. Values shown are mean +/− SEM (n=3). * p < 0.05; *** p < 0.001.
Glycerophosphocholine (GPC) and glycerylphosphorylethanolamine (GPE) are found in the brain and involved in the phospholipid biosynthesis pathway (Klein, 2000). The higher m+3 fractions of both GPC and GPE in E4 astrocytes suggests increased activity of the phospholipid biosynthesis pathway relative to E2 or E3 (Figure 6B–C).
The total amount of labeled NADH was higher in E4 astrocytes relative to E2 or E3 (Figure 6D). NADH labeling in m+5 and m+10 are increased in E4 and provides indication of 13C enrichment from labeled ribose (Figure 6E). The m+12, m+13, and m+14 fractions in NADH are also increased in E4 and indicate labeling in the purine ring (Figure 6E). The nicotinamide ring shows no labeling as it is only synthesized in astrocytes from essential amino acids (Harlan et al., 2016).
Glucose can be diverted to the hexosamine biosynthetic pathway (HBP), which culminates in the production of the amino sugars essential for the biosynthesis of glycosaminoglycans, proteoglycans, and glycolipids. To measure glucose flux into the HBP, we analyzed the average fractional enrichment patterns of multiple UDP-hexosamines (UDP-glucose, UDP-galactose, UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine). The observed increases in the unlabeled m+0 fractions in E2 and E3 astrocytes suggests a higher rate of glycolysis, which has been shown to decrease glucose-derived 13C incorporation into HBP (Gibb et al., 2017) (Figure 6F). Conversely, the increased enrichment in m+14, m+15, and m+16 fractions seen in E4 astrocytes is due to labeling in the nucleotide base and requires contributions from 13C labeled aspartate (generated from TCA intermediates) during pyrimidine biosynthesis. Together, the labeling patterns in metabolites containing nucleotides in E4 astrocytes show increased 13C enrichment in the nucleotide bases, both pyrimidines and purines, and their downstream products.
4. Discussion
Changes in glucose metabolism have been characterized in E4 carriers for decades, and several cellular mechanisms have been described to potentially explain these phenomena. This includes impaired insulin receptor signaling, altered expression of glucose transporters and mitochondrial proteins, and mitochondrial dysfunction due to E4 proteolysis (Dose et al., 2016). However, the majority of these E4-mediated detrimental effects on metabolic function have been demonstrated in whole brain tissue or neurons, and thus provide an important but incomplete picture of the metabolic landscape influenced by APOE, especially in astrocytes (the main source of APOE synthesis in the brain) (Chang et al., 2005; Huang et al., 2001; Nakamura et al., 2009; Xu et al., 2006).
In the current study, we aimed to determine the precise effects of APOE on glucose utilization in astrocytes. To do so, we traced 13C atoms from [U-13C] glucose through multiple central carbon metabolic pathways in astrocytes expressing human E2, E3, or E4. The approach identified multiple APOE isoform-dependent alterations, including differences in glucose flux through glycolysis, PPP, and glucose utilization within the TCA cycle. Specifically, we observed an E4-associated shift in glucose entry into the TCA cycle, and stepwise APOE effects (E2<E3<E4) on the rate of glucose cycling through the non-oxidative PPP and on glucose flux through pathways of de novo nucleotide biosynthesis.
The differences observed in the labeling patterns of glycolytic intermediates containing six carbon atoms (F1,6BP, F6P and G6P) suggests that alternative mechanisms are contributing to glycolytic activity. E2 astrocytes displaying the highest m+6 fractions in six-carbon glycolytic intermediates and m+3 fractions in three-carbon intermediates suggests greater glucose flux directly through glycolysis relative to E3 and E4. Conversely, E4 astrocytes appear to cycle a higher fraction of glucose through the non-oxidative PPP and then back into glycolysis via gluconeogenesis. This cycling is evidenced by higher fractional labeling in the m+3 and m+5 fractions of glycolytic intermediates F1,6BP, F6P and G6P, respectively.
In the late stages of glycolysis, the decreases observed in glycolytic end products lactate and pyruvate at m+3 in E2 astrocytes suggest that either i) non-glycolytic sources of pyruvate and/or lactate (as the two are actively interconverted) are contributing to metabolite pools, or ii) that E2 astrocytes have a much higher reserve pool of pyruvate and/or lactate. The first is more likely given that the media was supplemented with unlabeled pyruvate and that 24 hours has proven sufficient to establish isotopic steady state of glycolysis during in vitro tracer experiments (Fan et al., 2011). Thus, E4 astrocytes appear to have increased glucose flux into oxygen-independent glycolysis, as evidenced by the significantly higher fraction of m+3 lactate. However, it should be noted that the decreased m+3 in pyruvate and lactate relative to other glycolytic intermediates may instead reflect extracellular pyruvate uptake, as the media was supplemented with sodium pyruvate thus contributing a source of 12C atoms. Therefore, an alternative explanation for the observed labeling patterns in pyruvate and lactate is that E4 astrocytes take up and/or utilize less of this extracellular 12C pyruvate compared to E2 and E3 astrocytes.
The increased m+5 fractions supports entry of PPP intermediates into glycolysis and are likely derived from ribose-5-phosphate. Recycling PPP intermediates through glycolysis to once again enter the PPP might occur in conditions requiring more NADPH, such as during lipid and nucleic acid biosynthesis, which is increased in certain brain regions of E4 mice (Nuriel et al., 2017). NADPH is also utilized in glutathione recycling, a major pathway for mitigating oxidative stress (McBean, 2018). Under conditions of oxidative stress astrocytes have been shown to increase PPP activity (Garcia-Nogales et al., 1999). AD brains exhibit increased oxidative damage that correlates with E4 (Shea et al., 2002). AD brains with presence of at least one E4 allele also display increased glutathione peroxidase activity, a process that would drive the need for NADPH production to reduce oxidized glutathione (Ramassamy et al., 1999). The switch to a reductive TCA cycle as opposed to oxidative, results in the generation of less reactive oxygen species. Perhaps E4 astrocytes initiate a switch to a reductive TCA cycle in order to mitigate an environment of increased oxidative stress (Glodzik-Sobanska et al., 2009; Montine et al., 1998; Ramassamy et al., 1999; Ramassamy et al., 2000).
When undergoing oxidative metabolism in the TCA cycle, glutamine can be used to generate ATP via conversion to alpha-ketoglutarate and subsequently succinate. Using non-glucose derived glutamine would contribute higher unlabeled fractions, seen in E2 and to a lesser extent E3. Mice expressing human E4 alleles had decreased brain glutaminase levels, suggesting altered glutamate-glutamine cycle (Dumanis et al., 2013). Our results showing increased labeling of TCA intermediates in E4 astrocytes further indicate lower glutaminolysis activity, as glutamine oxidation would contribute a higher unlabeled fraction. Reductive carboxylation of glucose – or alternatively, multiple turns of the TCA cycle – would generate more enriched isotopologues, as evidenced in E4 astrocytes. Also referred to as reverse Krebs cycle, a reductive TCA cycle operates in the reverse, or non-oxidative direction to replenish TCA substrates lost to cataplerosis, a potential explanation for the observed patterns of TCA labeling in E4 astrocytes (Verschueren et al., 2019). Increased TCA cycle activity in E4 might result from a shift toward oxidative phosphorylation, which in turn could contribute to elevated oxidative stress (Guo et al., 2013). Thus, undergoing greater oxidative phosphorylation could be deleterious in E4 cells, given their decreased ability to mitigate oxidative stress (Jofre-Monseny et al., 2008). Oxidative phosphorylation is increased with age, and a predisposition of E4 to undergo more oxidative phosphorylation may exacerbate oxidative damage contributing to AD onset (Harris et al., 2014).
Decreased de novo synthesis of nucleotides from glucose observed in E2, and to a lesser extent E3, could be due to higher activity of purine and pyrimidine salvage pathways, thereby mitigating the need to synthesize new nucleotides. The increased conversion of serine to glycine, as denoted by higher m+6 and m+7 fractions of glutathione in E4 astrocytes, generates a methyl group that can be utilized in nucleotide biosynthesis. Together, the labeling patterns in metabolites containing nucleotides in E4 astrocytes show increased 13C enrichment in the nucleotide bases, in both pyrimidines and purines. This may suggest either altered nucleotide salvage ability, an increased demand for nucleotides, or a combination of the two. When taken together with labeling patterns from TCA cycle intermediates and increased labeling in acetylated metabolites, this may suggest an E4-mediated shift toward a cataplerotic TCA cycle, in which substrates are utilized for biosynthesis rather than driving oxidative phosphorylation.
Our results highlighting APOE-specific alterations in glucose flux into the PPP and purine biosynthesis pathways in astrocytes are particularly interesting given that these two pathways are linked to both E4 status and AD. Purine metabolism is dysregulated in the brains of individuals with mild cognitive impairment (MCI) and AD (Ansoleaga et al., 2015; Kaddurah-Daouk et al., 2013; Sims et al., 1998), and postmortem analyses of AD brain tissue also show alterations in PPP metabolites (Orešič et al., 2011; Palmer, 1999). Additionally, an untargeted, integrated ‘omics analysis of hippocampal tissue from mice expressing human E3 or E4 revealed that the top two pathways altered were PPP and purine metabolism (Johnson et al., 2017a). The PPP plays a critical role in managing cellular redox through regeneration of NADPH, and dysregulation of this pathway may contribute to the increased oxidative stress associated with both AD and E4. The PPP also metabolizes glucose to generate the five-carbon precursors for a variety of biosynthetic reactions, including purines and NADH biosynthesis. Finally, as the purine adenosine is a critical building block for ATP generation, deficits in purine biosynthesis may provide a link to help support the bioenergetic failure hypothesis of AD (Wilkins et al., 2014). The current study provides new insight into the specific changes in glucose flux within these pathways that may contribute to the differences in total metabolite concentrations reported in E4 and AD brains (Johnson et al., 2017a; Nuriel et al., 2017).
The reduction in glucose uptake observed in E4 astrocytes may suggest an important role for this cell type in contributing to the cerebral glucose hypometabolism in E4 carriers observed with PET imaging (Yao et al., 2015). Intriguingly, it may also not only contribute to the reduction in glucose flux through glycolysis but may also drive the increased recycling of PPP intermediates seen in these cells. Perhaps E4 astrocytes re-appropriate metabolic intermediates in order to supply substrates for the PPP to facilitate NADPH and biosynthesis processes.
Our results suggest that E4 astrocytes flux more 13C from glucose to lactate than E2 and E3 astrocytes. This is interesting for a number of reasons. First, studies show that greater expression of lactate-producing enzymes are correlated with increased memory in mice, and increased lactate concentrations during neural activity may help explain the increased cognitive function and alterations in regional brain activation patterns via fMRI reported in young and middle-aged E4 carriers (Dennis et al., 2010; Evans et al., 2014; Filippini et al., 2009; Harris et al., 2016). A preference for lactate production in E4 astrocytes would also facilitate more oxygen-independent glycolysis, which while potentially beneficial for brain function and memory, could contribute to cognitive decline later in life due to age-related impairments in lactate processing (Harris et al., 2016). Interestingly, oxygen-dependent glycolysis decreases with age and may also be altered in AD (Goyal et al., 2017; Vlassenko and Raichle, 2015). Second, E4 astrocytes were recently shown to carry aberrant endosomes that are highly acidified, leading to down regulation of Na+ /H+ exchanger 6 and subsequent impairments in membrane receptor trafficking (Prasad and Rao, 2018). Apart from endosomal pH, cytoplasmic pH of E4 astrocytes was also shown to be lower, though non-significantly so. We hypothesize that E4 astrocytes may be decreasing their pH based on increased lactate synthesis from cytoplasmic glycolysis. This, in part, may be mitigated by endosomal H+ sinks that sequester H+ from the cytoplasm to maintain physiologic pH. This endosomal acidification then reduces trafficking and surface expression of receptors such as LRP1, a key receptor for amyloid-β which is known to be decreased in E4 astrocytes (Prasad and Rao, 2018). Additionally, lower pH in itself is considered a post-translational structural modifier of protein conformation (Schonichen et al., 2013). It has been reported that stability, aggregation, and lipidation of APOE depends strongly on pH (Garai et al., 2011). Specifically, lower pH drives self-aggregation and increases lipid binding. E4 astrocytes display both increased aggregation of intracellular APOE and increased intracellular lipid content (Farmer et al., 2019; Rawat et al., 2019). Addition of unlabeled lactate to a 13C-glucose treatment decreases the incorporation of isotopic label into fatty acids by up to 40%, indicating that lactate is a major carbon source and carbon competitor for fatty acid biogenesis (Liu et al., 2017).
Here, we show that glycerophosphocholine and glyceryl-phosphorylethanolamine are enriched in glucose derived carbons in E4 astrocytes. Furthermore, our group has shown an increased lipid droplet phenotype in E4 astrocytes, supporting the idea that E4 astrocytes may be managing increased cellular lactate via lipogenic means (Farmer et al., 2019). Increased synthesis of GPC observed in E4 astrocytes could result from elevated phosphatidylcholine hydrolysis, a process mediated by phospholipase A2. The ratios of GPC to choline and GPE to ethanolamine were increased certain AD brain regions also known to be associated with glucose hypometabolism (Nitsch et al., 1992). Interestingly, GPC is increased in both cerebrospinal fluid and brains of AD patients compared to age-matched controls (Valenzuela and Sachdev, 2001; Walter et al., 2004). Amyloid β peptides have been shown to activate cPLA2, the subtype likely responsible for phosphatidylcholine hydrolysis in the brain, while the breakdown product GPC was shown to promote β-amyloid aggregation (Kanfer et al., 1998; Klunk et al., 1997).
Although our study provides new detail and insight into APOE-directed changes in astrocyte metabolism, it has several limitations. First, the model of choice – an in vitro system immortalized murine astrocyte – may limit extrapolation of our results. While focusing on one cell type in vitro limits confounding variables – those very confounders (i.e. the multiple cell types and intricate interplay and interdependence in vivo) are critical to the ultimate translation of these findings to the E4 and AD human brain. Second, while we also focused on a 24 hour timepoint in order to reach a steady state of 13C incorporation, this extended timeline may cause us to miss other potential APOE effects in pathways like glycolysis that occur very quickly. Finally, our study focused solely on 13C-labeled glucose as an energy source. Although astrocytes are thought to primarily rely on glucose for ATP production, they are known to metabolize numerous other substrates for energy production (fatty acids, glutamine, and glycogen-derived glucose) (Hertz et al., 2007). Contributions from these non-glucose sources could be contributing to the unlabeled fractions of pyruvate and lactate. For example, the higher m+3 fraction in G6P seen in E4 astrocytes implicates gluconeogenesis and could be attributed to either lactate and/or pyruvate being utilized to generate G6P and F6P, or contributions from non-oxidative PPP products. Future studies such as astrocyte-neuron cocultures to elucidate cell-to-cell metabolic interactions, the effect of insulin signaling on glucose flux, in vivo tracer studies in human APOE mice, and 13C-tracer analyses in isogenic human APOE iPSCs may help to address some of these limitations and provide additional insight into the effects of APOE on cerebral metabolism and its ultimate relevance to the pathophysiology of AD.
Conclusions
APOE genotype is differentially modulating glucose metabolism in astrocytes. E2 and E3 have an increased flux through early glycolysis and are utilizing carbon for oxidative metabolism in the TCA cycle. E4 astrocytes have a more active cataplerotic TCA cycle, and increased glucose flux into lactate as well as into the PPP, which may ultimately fuel biosynthesis pathways. Additionally, E4 has increased de novo glutathione synthesis, which may drive the alterations in both PPP and TCA cycle.
Highlights (3–5 bullet points of novelty/significant findings).
Stable isotope tracing reveals APOE-specific changes in astrocyte glucose utilization
APOE alters glucose entry into the TCA cycle
E4 astrocytes increase glucose flux into PPP and de novo biosynthesis pathways
Acknowledgements
We thank Drs. Ja Brandon and Tim Scott for their assistance. We thank Drs. Richard Higashi, Andrew Lane, Teresa Fan and the staff at the Resource Facility for Stable Isotope Resolved Metabolomics (RCSIRM) at the University of Kentucky for their support and technical expertise.
Funding
This work was made possible by a Pilot and Feasibility Grant through the NIH Common Fund Metabolomics Program, and was also supported by funding from National Institute on Aging 1R01AG060056-01, R01AG060056 02 and COBRE P20 GM103527 - LAJ; 1T32AG057461-01 - HCW; F30AG06342201A1 BCF. COBRE P20 GM121327, American Cancer Society institutional research grant #16-182-28, and St Baldrick’s Foundation Career Development Award, and funding from the University of Kentucky Markey Cancer Center – RCS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests statement: none
References
- Alberini CM, et al. , 2018. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia. 66, 1244–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves TC, et al. , 2015. Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab. 22, 936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansoleaga B, et al. , 2015. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol Aging. 36, 68–80. [DOI] [PubMed] [Google Scholar]
- Bak LK, et al. , 2018. Astrocytic glycogen metabolism in the healthy and diseased brain. J Biol Chem. 293, 7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP, 2002. Alzheimer’s disease and Alzheimer’s dementia: distinct but overlapping entities. Neurobiol Aging. 23, 1077–84. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR, 2007. Astrocyte glycogen and brain energy metabolism. Glia. 55, 1263–71. [DOI] [PubMed] [Google Scholar]
- Buescher JM, et al. , 2015. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Halliwell B, 2019. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience. 20, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell F, et al. , 2016. Modulation of glucose metabolism and metabolic connectivity by β-amyloid. J Cereb Blood Flow Metab. 36, 2058–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, et al. , 2005. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 102, 18694–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GC, et al. , 2010. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 75, 1976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, et al. , 1994. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 7, 180–4. [DOI] [PubMed] [Google Scholar]
- Courtney KD, et al. , 2018. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 28, 793–800.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis NA, et al. , 2010. Temporal lobe functional activity and connectivity in young adult APOE varepsilon4 carriers. Alzheimers Dement. 6, 303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, et al. , 2005. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 65, 404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dose J, et al. , 2016. APOE genotype and stress response - a mini review. Lipids Health Dis. 15, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, et al. , 2013. APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J Neurochem. 124, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S, et al. , 2014. Cognitive and neural signatures of the APOE E4 allele in mid-aged adults. Neurobiol Aging. 35, 1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, et al. , 2011. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 7, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, et al. , 2012. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol Ther. 133, 366–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, et al. , 2016. Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator β-glucan in a two-case ex vivo non-small-cell lung cancer study. Cold Spring Harb Mol Case Stud. 2, a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer BC, et al. , 2019. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, et al. , 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 278, 1349–56. [PubMed] [Google Scholar]
- Fiebig C, et al. , 2019. Mitochondrial Dysfunction in Astrocytes Impairs the Generation of Reactive Astrocytes and Enhances Neuronal Cell Death in the Cortex Upon Photothrombotic Lesion. Front Mol Neurosci. 12, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N, et al. , 2009. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 106, 7209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai K, et al. , 2011. Self-association and stability of the ApoE isoforms at low pH: implications for ApoE-lipid interactions. Biochemistry. 50, 6356–64. [DOI] [PubMed] [Google Scholar]
- Garatachea N, et al. , 2014. ApoE gene and exceptional longevity: Insights from three independent cohorts. Exp Gerontol. 53, 16–23. [DOI] [PubMed] [Google Scholar]
- Garcia-Nogales P, et al. , 1999. Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J Neurochem. 72, 1750–8. [DOI] [PubMed] [Google Scholar]
- Gibb AA, et al. , 2017. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J. 474, 2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodzik-Sobanska L, et al. , 2009. The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer’s disease. Neurobiol Aging. 30, 672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonneaud J, et al. , 2016. Relative effect of APOE epsilon4 on neuroimaging biomarker changes across the lifespan. Neurology. 87, 1696–1703. [DOI] [PubMed] [Google Scholar]
- Goyal MS, et al. , 2017. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 26, 353–360.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, et al. , 1986. Stability of metabolic and neuropsychological asymmetries in dementia of the Alzheimer type. Neurology. 36, 1390–2. [DOI] [PubMed] [Google Scholar]
- Guo C, et al. , 2013. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 8, 2003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan BA, et al. , 2016. Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1). J Biol Chem. 291, 10836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, et al. , 2014. Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology. 15, 559–77. [DOI] [PubMed] [Google Scholar]
- Harris RA, et al. , 2016. Aerobic Glycolysis in the Frontal Cortex Correlates with Memory Performance in Wild-Type Mice But Not the APP/PS1 Mouse Model of Cerebral Amyloidosis. The Journal of Neuroscience. 36, 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxby JV, et al. , 1990. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch Neurol. 47, 753–60. [DOI] [PubMed] [Google Scholar]
- Herholz K, et al. , 2002. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 17, 302–16. [DOI] [PubMed] [Google Scholar]
- Hertz L, et al. , 2007. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 27, 219–49. [DOI] [PubMed] [Google Scholar]
- Higashi RM, et al. , 2014. Stable isotope-labeled tracers for metabolic pathway elucidation by GC-MS and FT-MS. Methods Mol Biol. 1198, 147–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, et al. , 2001. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 98, 8838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mahley RW, 2014. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 72 Pt A, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, et al. , 2012. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 32, 18227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jofre-Monseny L, et al. , 2008. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 52, 131–45. [DOI] [PubMed] [Google Scholar]
- Johnson LA, et al. , 2017a. Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci Rep. 7, 43701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, et al. , 2017b. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab. 271678x17746186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R, et al. , 2013. Alterations in metabolic pathways and networks in Alzheimer’s disease. Translational Psychiatry. 3, e244–e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfer JN, et al. , 1998. Phospholipases as mediators of amyloid beta peptide neurotoxicity: an early event contributing to neurodegeneration characteristic of Alzheimer’s disease. Neurosci Lett. 257, 93–6. [DOI] [PubMed] [Google Scholar]
- Klein J, 2000. Membrane breakdown in acute and chronic neurodegeneration: focus on choline-containing phospholipids. J Neural Transm (Vienna). 107, 1027–63. [DOI] [PubMed] [Google Scholar]
- Klunk WE, et al. , 1997. Aggregation of beta-amyloid peptide is promoted by membrane phospholipid metabolites elevated in Alzheimer’s disease brain. J Neurochem. 69, 266–72. [DOI] [PubMed] [Google Scholar]
- Lane AN, et al. , 2008. Isotopomer-based metabolomic analysis by NMR and mass spectrometry. Methods Cell Biol. 84, 541–88. [DOI] [PubMed] [Google Scholar]
- Lin AL, et al. , 2017. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab. 37, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, et al. , 2013. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 9, 106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, et al. , 2017. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 26, 719–737.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I, 2015. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 86, 883–901. [DOI] [PubMed] [Google Scholar]
- McBean GJ, 2018. Astrocyte Antioxidant Systems. Antioxidants (Basel, Switzerland). 7, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoshima S, et al. , 1997. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 42, 85–94. [DOI] [PubMed] [Google Scholar]
- Montine KS, et al. , 1998. Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. J Neuropathol Exp Neurol. 57, 415–25. [DOI] [PubMed] [Google Scholar]
- Morikawa M, et al. , 2005. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis. 19, 66–76. [DOI] [PubMed] [Google Scholar]
- Moseley HN, 2010. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinformatics. 11, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, et al. , 2009. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch RM, et al. , 1992. Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci U S A. 89, 1671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriel T, et al. , 2017. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun. 8, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orešič M, et al. , 2011. Metabolome in progression to Alzheimer’s disease. Transl Psychiatry. 1, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, 1999. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J Neural Transm (Vienna). 106, 317–28. [DOI] [PubMed] [Google Scholar]
- Pellerin L, et al. , 1998. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 20, 291–9. [DOI] [PubMed] [Google Scholar]
- Prasad H, Rao R, 2018. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci U S A. 115, E6640–e6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, et al. , 2004. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 25, 641–50. [DOI] [PubMed] [Google Scholar]
- Ramassamy C, et al. , 1999. Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein E genotype. Free Radic Biol Med. 27, 544–53. [DOI] [PubMed] [Google Scholar]
- Ramassamy C, et al. , 2000. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis. 7, 23–37. [DOI] [PubMed] [Google Scholar]
- Rawat V, et al. , 2019. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J Neurosci. 39, 9611–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. , 2001. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A. 98, 3334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. , 2004. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 101, 284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonichen A, et al. , 2013. Considering protonation as a posttranslational modification regulating protein structure and function. Annu Rev Biophys. 42, 289–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, et al. , 2002. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic Biol Med. 33, 1115–20. [DOI] [PubMed] [Google Scholar]
- Simpson IA, et al. , 2007. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 27, 1766–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims B, et al. , 1998. Elevated adenosine monophosphate deaminase activity in Alzheimer’s disease brain. Neurobiol Aging. 19, 385–91. [DOI] [PubMed] [Google Scholar]
- Small GW, et al. , 2000. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 97, 6037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RC, et al. , 2017. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat Commun. 8, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuminello ER, Han SD, 2011. The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. Int J Alzheimers Dis. 2011, 726197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela MJ, Sachdev P, 2001. Magnetic resonance spectroscopy in AD. Neurology. 56, 592–8. [DOI] [PubMed] [Google Scholar]
- Venzi M, et al. , 2017. Differential Effect of APOE Alleles on Brain Glucose Metabolism in Targeted Replacement Mice: An [(18)F]FDG-μPET Study. J Alzheimers Dis Rep. 1, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren KHG, et al. , 2019. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature. 568, 571–575. [DOI] [PubMed] [Google Scholar]
- Villa E, et al. , 2019. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel). 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Raichle ME, 2015. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin Transl Imaging. 3, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A, et al. , 2004. Glycerophosphocholine is elevated in cerebrospinal fluid of Alzheimer patients. Neurobiol Aging. 25, 1299–303. [DOI] [PubMed] [Google Scholar]
- Wensaas AJ, et al. , 2007. Cell-based multiwell assays for the detection of substrate accumulation and oxidation. J Lipid Res. 48, 961–7. [DOI] [PubMed] [Google Scholar]
- Wilkins HM, et al. , 2014. Bioenergetic dysfunction and inflammation in Alzheimer’s disease: a possible connection. Front Aging Neurosci. 6, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins JM, Trushina E, 2017. Application of Metabolomics in Alzheimer’s Disease. Front Neurol. 8, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, et al. , 2002. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry. 73, 672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart HA, et al. , 2006. Increased brain activation during working memory in cognitively intact adults with the APOE epsilon4 allele. Am J Psychiatry. 163, 1603–10. [DOI] [PubMed] [Google Scholar]
- Xu J, et al. , 2016. Graded perturbations of metabolism in multiple regions of human brain in Alzheimer’s disease: Snapshot of a pervasive metabolic disorder. Biochimica et biophysica acta. 1862, 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, et al. , 2006. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 26, 4985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, et al. , 2015. A FDG-PET Study of Metabolic Networks in Apolipoprotein E ε4 Allele Carriers. PLoS One. 10, e0132300. [DOI] [PMC free article] [PubMed] [Google Scholar]