

Neuronal diversity provides the spinal cord with the functional flexibility required to perform complex motor tasks. Spinal neurons arise during early embryonic development with the establishment of spatially and molecularly discrete progenitor domains that give rise to distinct, but highly heterogeneous, postmitotic interneuron (IN) populations. Our previous studies have shown that Sim1-expressing V3 INs, originating from the p3 progenitor domain, are anatomically and physiologically divergent.
Keywords: Spinal cord, V3 interneurons, neurogenesis timing, interneuron subpopulations, neuronal differentiation, Sim1 transcription factor
Abstract
Neuronal diversity provides the spinal cord with the functional flexibility required to perform complex motor tasks. Spinal neurons arise during early embryonic development with the establishment of spatially and molecularly discrete progenitor domains that give rise to distinct, but highly heterogeneous, postmitotic interneuron (IN) populations. Our previous studies have shown that Sim1-expressing V3 INs, originating from the p3 progenitor domain, are anatomically and physiologically divergent. However, the developmental logic guiding V3 subpopulation diversity remains elusive. In specific cases of other IN classes, neurogenesis timing can play a role in determining the ultimate fates and unique characteristics of distinctive subpopulations. To examine whether neurogenesis timing contributes to V3 diversity, we systematically investigated the temporal neurogenesis profiles of V3 INs in the mouse spinal cord. Our work uncovered that V3 INs were organized into either early-born [embryonic day 9.5 (E9.5) to E10.5] or late-born (E11.5–E12.5) neurogenic waves. Early-born V3 INs displayed both ascending and descending commissural projections and clustered into subgroups across dorsoventral spinal laminae. In contrast, late-born V3 INs became fate-restricted to ventral laminae and displayed mostly descending and local commissural projections and uniform membrane properties. Furthermore, we found that the postmitotic transcription factor, Sim1, although expressed in all V3 INs, exclusively regulated the dorsal clustering and electrophysiological diversification of early-born, but not late-born, V3 INs, which indicates that neurogenesis timing may enable newborn V3 INs to interact with different postmitotic differentiation pathways. Thus, our work demonstrates neurogenesis timing as a developmental mechanism underlying the postmitotic differentiation of V3 INs into distinct subpopulation assemblies.
SIGNIFICANCE STATEMENT Interneuron (IN) diversity empowers the spinal cord with the computation flexibility required to perform appropriate sensorimotor control. As such, uncovering the developmental logic guiding spinal IN diversity is fundamental to understanding the development of movement. In our current work, through a focus on the cardinal spinal V3 IN population, we investigated the role of neurogenesis timing on IN diversity. We uncovered that V3 INs are organized into early-born [embryonic day 9.5 (E9.5) to E10.5] or late-born (E11.5–E12.5) neurogenic waves, where late-born V3 INs display increasingly restricted subpopulation fates. Next, to better understand the consequences of V3 neurogenesis timing, we investigated the time-dependent functions of the Sim1 transcription factor, which is expressed in postmitotic V3 INs. Interestingly, Sim1 exclusively regulated the diversification of early-born, but not late-born, V3 INs. Thus, our current work indicates neurogenesis timing can modulate the functions of early postmitotic transcription factors and, thus, subpopulation fate specifications.
Introduction
As mitotic progenitor cells undergo their final division phase, they exit the cell cycle, becoming postmitotic neurons. The timing of the cell cycle exit of a neuron, marking its genesis, plays a vital role in determining its terminal fate specification via the dynamic exposure of intrinsic and extrinsic factors (Syed et al., 2017; Holguera and Desplan, 2018). Within layered neural tissues, such as the neocortex and retina, distinct neuronal populations differentiate into anatomically organized rows stacked across a single spatial axis. Neurogenesis timing plays an essential role in ordering postmitotic neurons into distinct layers from common progenitor domains (Bassett and Wallace, 2012; Sultan and Shi, 2018; Kawaguchi, 2019). Within other neural tissues, such as the ventral spinal cord and brainstem, postmitotic neurons do not form distinctive anatomically layered structures but instead are organized into clustered and/or mosaic arrangements across multiple anatomical axes. The role of neurogenesis timing in ordering such complex neuronal diversity from common progenitor domains remains largely unknown.
Interneurons (INs) within the spinal cord form highly heterogeneous circuits, which are crucial for varied sensorimotor functions (Goulding, 2009; Kiehn, 2016; Deska-Gauthier and Zhang, 2019). Spinal IN diversity emerges during early embryonic development with the establishment of spatially and molecularly discrete progenitor domains along the dorsoventral axis (Jessell, 2000). As progenitor cells become postmitotic, they express distinct transcription factors, display unique axon projection profiles, and migrate to final mediolateral and dorsoventral locations within specific spinal laminae. Recent work, however, has revealed vast IN subpopulation diversity within cardinal classes born from the same progenitor domains (Bikoff et al., 2016; Gosgnach et al., 2017; Hayashi et al., 2018). Interestingly, it was suggested that some of these IN subpopulations are generated at specific embryonic time points. For example, Renshaw cells are the earliest born neurons among all V1 INs (Benito-Gonzalez and Alvarez, 2012; Stam et al., 2012). Yet, the full extent of the role temporal factors play in ordering IN diversity between lineage-related subpopulations remains obscure. In addition, to what extent neurogenesis timing can influence early postmitotic transcription factor functions, and thus, subpopulation fate specifications, remains unclear.
V3 INs are a major cardinal class of spinal INs. V3 INs are glutamatergic, display predominantly commissural axon projections, and are necessary for robust locomotor output (Zhang et al., 2008). V3 INs emerge from the most-ventral, Nkx2.2+ p3 progenitor domain, as marked by postmitotic expression of the Sim1 transcription factor. During early embryonic stages, postmitotic V3 INs form divergent migratory streams and begin to express diverse transcription factor combinations (Francius et al., 2013). By postnatal stages, clusters of V3 INs assemble from ventral to deep dorsal horn laminae. These spatially separate V3 IN subpopulations are also morphologically and electrophysiologically heterogeneous (Borowska et al., 2013, 2015). Interestingly, the Sim1 transcription factor—although expressed in all V3 INs—exclusively regulates the laminar clustering of dorsal and intermediate V3 IN subpopulations (Blacklaws et al., 2015). However, the factors influencing the subpopulation-specific functions of Sim1 have remained unknown until now.
In the current study, we have begun to address the developmental logic guiding divergent V3 IN differentiation through investigating the role of p3–V3 neurogenesis timing. First, we investigated the neurogenesis timing of anatomically distinct V3 IN subsets. Second, we investigated the functional capacity of the Sim1 transcription factor in promoting V3 IN diversity across distinct neurogenesis times. Anatomically discrete V3 INs were temporally organized into either early- or late-neurogenesis waves. Early-born [embryonic day 9.5 (E9.5) to E10.5] V3 INs were a mixture of ascending and descending commissural INs (CINs), which formed discrete clusters distributed in specific laminae of both ventral and deep dorsal horns. Late-born (E11.5–E12.5) V3 INs were mostly descending commissural INs, which predominantly clustered within ventral laminae. Further, the Sim1 transcription factor exclusively regulated the dorsal clustering and electrophysiological diversification of early-born, but not late-born, V3 INs. Thus, our work indicates that the temporal control of V3 neurogenesis may have a dynamic interaction with the postmitotic function of Sim1. As a result, Sim1 promotes the physiological and anatomical diversification of distinctly early-born V3 INs.
Materials and Methods
Mouse strains.
Sim1Cre mice (Zhang et al., 2008; Goulding Laboratory, Salk Institute) were crossed with TdTomato (TdTom) Ai14 conditional reporter mice (Jackson Laboratory) to generate Sim1Cre;Rosa.lsl.tdTom mice (Zhang et al., 2008; Blacklaws et al., 2015). These mice were used for fate mapping Sim1+ V3 interneurons through embryonic and postnatal stages. Sim1 knock-out (KO) mice were generated by crossing Sim1Cre;Rosa.lsl.tdTom mice with Sim1taulacz mice (Goulding Laboratory, Salk Institute) to produce Sim1Cre/taulacz;Rosa.lsl.tdTom mice (Blacklaws et al., 2015). All procedures were performed in accordance with the Canadian Council on Animal Care and approved by the University Committee on Laboratory Animals at Dalhousie University.
Spinal cord tissue dissection, processing, and sectioning.
Spinal cords were obtained at embryonic (E10.5, E11.5, E12.5, E14.5) and postnatal [postnatal day 0 (P0)] stages. For embryonic ages, the dates of fertilization were identified by the presence of a vaginal plug. The morning of vaginal plug discovery was defined as E0.5. Before embryonic dissection, pregnant mothers were anesthetized via intraperitoneal injections of a ketamine (60 mg/kg) and xylazine (12 mg/kg) mixture. Once a mouse no longer responded to the pedal reflex, it was decapitated and its embryos removed via cesarean section. Embryonic and postnatal mice were killed via decapitation, and spinal cords were subsequently dissected in oxygenated Ringer's solution (111 mm NaCl, 3.08 mm KCl, 11 mm glucose, 25 mm NaHCO3, 1.25 mm MgSO4, 2.52 mm CaCl2, 1.18 mm KH2PO4, pH 7.4). Spinal cords were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS at 4°C for varying times (E10.5 for 15 min; E11.5 for 20 min; E12.5 for 25 min; E14.5 for 35 min; P0 for 1 h). Following fixation, spinal cords were washed in PBS at 4°C overnight and subsequently cryoprotected in 30% sucrose in PBS at 4°C overnight. Cryoprotected spinal cords were embedded in Fisher Healthcare O.C.T compound (Thermo Fisher Scientific) and flash frozen at −55°C in a mixture of dry ice and ethanol. Frozen lower thoracic (T) and higher lumbar (L; T12–L3) spinal cord segments were sectioned transversely using a cryostat (Leica CM1950). Embryonic spinal cords were sectioned at 14 μm and postnatal spinal cords were sectioned at 30 μm onto Fisherbrand Superfrost Plus Microscope Slides (Thermo Fisher Scientific).
5-Ethynyl-2′-deoxyuridine pulse labeling of V3 IN neurogenesis profiles.
Ethynyl-2′-deoxyuridine (EdU; Thermo Fisher Scientific) was dissolved in saline solution to make 6 mg of EdU/1 ml of saline solution. Pregnant Sim1Cre;Rosa.lsl.tdTom mice were injected with EdU solution (6 mg/ml) according to their body weight (50 μl/10 g) at gestational stages E10.5, E11.5, and E12.5. Pregnant Sim1Cre;Rosa.lsl.tdTom mice injected at E9.5 were pulsed with 12 mg/ml EdU solution. EdU pulsed spinal cords were collected at E12.5, E14.5, or P0. Mice pulsed and analyzed at E12.5 were first pulsed with EdU followed by a 2 h window before spinal cord dissection. Fluorescent labeling of DNA-incorporated EdU was achieved using a Click-iT EdU Alexa Fluor 647 Imaging Kit (Thermo Fisher Scientific). To determine the neurogenesis profiles of anatomically distinct V3 IN subsets, fluorescent labeling of EdU was performed following immunohistochemistry and retrograde tract tracing protocols.
Retrograde tract tracing with biotin-conjugated dextran amine.
Axonal projection patterns of P0 V3 INs were determined via retrograde biotin-conjugated dextran amine (BDA) tracing of isolated Sim1Cre;Rosa.lsl.tdTom spinal cords. Immediately following spinal cord dissection, 3000 molecular weight, lysine-fixable Invitrogen BDA (Thermo Fisher Scientific) was inserted into one half of the high lumbar spinal cord approximately between the L1 and L2 segments. Injected spinal cords were incubated overnight in Ringer's solution (111 mm NaCl, 3.08 mm KCl, 11 mm glucose, 25 mm NaHCO3, 1.25 mm MgSO4 2.52 mm CaCl2, 1.18 mm KH2PO4, pH 7.4) bubbled with 95% O2/5% CO2 at 20°C. Following incubation, spinal cords underwent the aforementioned processing and sectioning protocols. These tract-tracing procedures were previously described in greater detail (Nissen et al., 2005; Blacklaws et al., 2015). Fluorescent labeling of BDA was performed in concert with immunohistochemistry and Click-iT EdU labeling of spinal cord sections. Alexa Fluor 405-conjugated streptavidin (Thermo Fisher Scientific), Alexa Fluor 488-conjugated streptavidin (Thermo Fisher Scientific), or Alexa Fluor 647-conjugated streptavidin (Thermo Fisher Scientific) was diluted at 1:500 and added to secondary antibody solutions.
Immunohistochemistry.
Mounted spinal cord sections were first incubated in PBS containing 0.1% Triton X-100 (PBS-T) for three consecutive washes of 5 min each. Subsequently, spinal cord sections were incubated in 0.1% PBS solution containing primary antibodies and 10% Invitrogen heat-inactivated horse serum (Thermo Fisher Scientific) overnight at 4°C. Primary antibodies (Abs) used were mouse anti-nkx2.2 (1:100; catalog #74.5A5, Developmental Studies Hybridoma Bank) and rabbit anti-DsRed (1:2000; catalog #632496, Clontech). Following primary Ab incubation, spinal cord sections were washed with 0.1% PBS-T for 15 min (3 × 5 min fresh solution) and subsequently incubated in 0.1% PBS solution containing secondary Abs (Alexa Fluor-conjugated streptavidin when relevant) and 10% Invitrogen heat-inactivated horse serum (Thermo Fisher Scientific) for 2 h at 4°C. Secondary Abs used were Alexa Fluor 488 donkey anti-mouse IgG (1:500; catalog #715-545-150, Jackson ImmunoResearch) and Alexa Fluor 594 donkey anti-rabbit IgG (1:500; catalog #711-585-152, Jackson ImmunoResearch). When applicable, secondary antibody incubation was followed by Click-iT EdU Alexa Fluor 647 fluorescent labeling (Thermo Fisher Scientific) and Hoechst 33342 solution (Thermo Fisher Scientific) staining. Last, spinal cord sections were washed in PBS for 15 min (3 × 5 min fresh solution) and coverslipped with Dako fluorescent mounting medium (Agilent).
Image capture and cell position analysis.
Fluorescent micrographs of spinal cord sections were captured using a Zeiss LSM 710 upright confocal microscope with ZEN 2009 Microscope and Imaging Software. Cell numbers and laminar positions were quantified using ImageJ and MATLAB. Using the ImageJ Cell Counter Plugin, x,y coordinates of individual cell bodies as well as the maximum and minimum x,y coordinates of corresponding spinal cord outlines were denoted. At E12.5, a total of three spinal cords were processed and analyzed for each EdU pulse time and genotype (E9.5, n = 3 animals; E10.5, n = 3 animals; E11.5, n = 3 animals; E12.5, n = 3 animals) for both Sim1 control and KO. At E14.5, a total of three spinal cords were processed and analyzed for each EdU pulse time (E9.5, n = 3; E10.5, n = 3; E11.5, n = 3; E12.5, n = 3). For each animal, 10 randomly chosen 14 μm transverse sections were analyzed within approximate lower thoracic and higher lumbar segments per animal. At P0, four spinal cords were processed and analyzed for each EdU pulse time (E9.5, n = 4; E10.5, n = 4; E11.5, n = 4; E12.5, n = 4) for both Sim1 control and KO. For each animal, a total of nine randomly chosen 30 μm transverse sections were analyzed for high lumbar (L1–L3) segments per animal. Cell body laminar distribution and cell body density contour plots were subsequently constructed using “grid-data” and “contour” functions in MATLAB. In brief, within each section, cell body x,y positions were normalized against the maximum and minimum hemicord x,y coordinates. Heat maps were then constructed to display pooled cell body densities across the mediolateral and dorsoventral axes at P0.
Patch-clamp recording and intrinsic properties analysis.
P0 Sim1 control and Sim1 KO mice were decapitated, and their higher lumbar spinal cords (T13–L3) were dissected in ice-cold oxygenated modified sucrose Ringer's solution (3.5 mm KCL, 25 mm NaHCO3, 1.2 mm KH2PO4, 1.3 mm MgSO4 1.2 mm CaCl2, 10 mm glucose, 212.5 mm sucrose, 2 mm MgCl2, pH 7.4). Dissected cords were then sectioned into 300–350 μm transverse slices using a VT1200 S vibratome (Leica). Following a >30 min recovery period, slices were transferred into a recording chamber mounted on an Olympus BX51WI microscope and perfused with Ringer's solution (111 mm NaCl, 3.08 mm KCl, 11 mm glucose, 25 mm NaHCO3, 1.25 mm MgSO4 2.52 mm CaCl2, 1.18 mm KH2PO4, pH 7.4) bubbled with 95% O2/5% CO2 at room temperature. V3 INs were identified and visualized through the expression of tdTomato fluorescent protein using a 40× water-immersion objective (numerical aperture, 0.8) with the aid of a Dage-MTI IR-1000 CCD camera. Conventional whole-cell voltage- and current-clamp recordings were obtained using a MultiClamp 700B amplifier (Molecular Devices). Patch-clamp recording pipettes were filled with a recording buffer containing 128 mm K-gluconate, 4 mm NaCl, 0.0001 mm CaCl2, 10 mm HEPES, 1 mm glucose, 5 mm Mg-ATP, and 0.3 mm GTP-Li. To facilitate mapping of recorded neurons, 0.4 mg/ml Lucifer yellow dilithium salt (Sigma-Aldrich) and 1 mg/ml neurobiotin (Vector Laboratories) were added to the pipette solution before recording. All electrophysiological data were obtained by Clampex 10.3 (Molecular Devices) and analyzed with Clampfit 10.3 and Spike2 5.0 software (Cambridge Electronic Design).
Experimental design and statistical analysis.
To compare the temporal pattern of V3 subpopulations with different projection profiles, a total of three spinal cords were processed and analyzed for each EdU pulse time (E9.5, n = 3; E10.5, n = 3; E11.5, n = 3). For each individual spinal cord, sections were analyzed two segments rostral (t13–L1) and two segments caudal (L2–L3) to the injection site. A total of 12 sections (20% of entire cord) were analyzed both rostral to (descending) and caudal to (ascending) the BDA injection site. The percentage of EdU+ descending and ascending V3 INs displayed unequal variances. Thus, statistical significance between descending versus ascending V3 at each EdU pulse time was measured via unpaired t test with Welch's unequal variance correction on Prism (GraphPad).
The comparative analysis of the laminar distribution patterns between Sim1 control and Sim1 KO mice was adopted from Laumonnerie et al. (2015). In brief, normalized hemicord distributions were divided into five rows and five columns, producing 25 discrete bins along the dorsoventral and mediolateral axes. The total number of V3 INs within each bin was compared between Sim1 control and Sim1 KO cords. Statistical significance was measured via the nonparametric two-tailed Mann–Whitney U test on Prism (GraphPad).
A model used to partition V3 INs into distinct electrophysiological categories was previously described (Borowska et al., 2013, 2015). In brief, four electrophysiological attributes [×1, ×2, ×3, ×4) = (F/I slope, membrane capacitance, sag amplitude, and first-spike frequency], obtained from 122 P21 V3 INs, were selected from a set of 21 parameters based on their utility in categorizing cells using a method based on principal component analysis (Borowska et al., 2013). It was found that the sign of the first principal component (PC1) of the four attributes could be used to partition INs into two categories that happened to correspond to cells in either the dorsal or ventral lobes of the spinal cross section (corresponding to the regions above and below the central canal, respectively). The same model was used to compute PC1 for the cells analyzed in this article as follows:
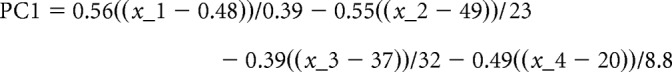 |
where the vector of model coefficients (0.56, 0.55, 0.39, 0.49) is the first empirical orthogonal function of the four attributes, and (0.48, 49, 37, 20) and (0.39, 23, 32, 8.8) are the means and SDs of the four attributes, all computed from the data used by Borowska et al. (2013). Statistical significance differences between dorsal and ventral V3 INs in Sim1 control and Sim1 KO mice were measured using the nonparametric Kruskal–Wallis test.
Results
Anatomically distinct V3 IN subpopulations display distinct temporal neurogenesis patterns
Neurogenesis timing orders postmitotic V3 INs into spatially and temporally distinct migratory streams
As Nkx2.2+ p3 progenitor cells exit their final division cycle, they express the Sim1 transcription factor defining them as postmitotic V3 INs (Zhang et al., 2008; Fig. 1A). Sim1+ V3 INs, indicated by the expression of tdTomato in the lumbar spinal cord of Sim1Cre;Rosa.lsl.tdTom mice, continue to express Nkx2.2 at E10.5 (∼100%, n = 1 animal; Fig. 1Ai), E11.5 (99% V3, n = 4 animals, data not shown; Fig. 1Aii), and E12.5 (95% V3, n = 4 animals; Fig. 1Aiii). During E10.5 and E11.5, V3 INs move laterally from the midline; however, by E12.5, V3 INs begin to form distinct dorsolateral and ventrolateral V3 IN clusters (Fig. 1B).
Figure 1.

Neurogenesis timing orders postmitotic V3 INs into spatially and temporally distinct migratory streams. A, Representative images of Nkx2.2 immunoreactivity and TdTomato+ V3 INs from Sim1Cre;Rosa.lsl.tdTom mice at E10.5 (Ai), E11.5 (Aii), and E12.5 (Aiii). Scale bars: Aiii, i, 10 μm; Aiii, ii, 100 μm. B, Representative image of TdTomato+ V3 INs clustered within presumed dorsolateral and ventrolateral migratory streams at E12.5. Scale bar, 100 μm. C, Representative images of EdU pulses at E9.5 (Ci), E10.5 (Cii), E11.5 (Ciii), and E12.5 (Civ) and subsequent detection EdU+, Nkx2.2+, and Hoechst+ labeling at E12.5. Scale bars: 50 μm; inset, 5 μm. D–H, Hemicord EdU+/Nkx2.2+/Hoechst+ cell position plots (Di–Hi) and mediolateral cell count quantifications (Dii–Hii) at E12.5 from respective E9.5 (Di, Dii; n = 3 animals), E10.5 (Ei, Eii; n = 3 animals), E11.5 (Fi, Fii; n = 3 animals), and E12.5 (Gi, Gii; n = 3 animals) EdU pulse times. Hi, Hii, Combined EdU pulse times at E12.5 (n = 3 animals for each EdU pulse time). Ii, Representative image of EdU+/TdTomato+/Hoechst+ V3 INs at E14.5 pulsed with EdU at E11.5. Scale bars: Ii, 50 μm; inset, 15 μm). Iii–Iiv, E14.5 hemicord EdU+/TdTomato+ cell position plots of E9.5 and E10.5 early-born V3 (Iii, Iiv; n = 3 animals for each EdU pulse time) and E11.5 and E12.5 late-born V3 (Iiii, Iiv, n = 3 animals for each EdU pulse time).
To determine whether neurogenesis timing orders postmitotic V3 INs into potential early embryonic migratory streams, we pulsed pregnant mice with EdU at E9.5, E10.5, E11.5, or E12.5. We then mapped the neurogenesis timing of V3 INs by combining EdU detection with Nkx2.2 immunolabeling in E12.5 lumbar spinal segments (Fig. 1Ci–Civ). Interestingly, at E12.5, both dorsolateral and ventrolateral V3 IN migratory streams were exclusively composed of early-born V3 INs [those pulsed at E9.5 (Fig. 1D; n = 3 animals) and E10.5 (Fig. 1E; n = 3 animals)]. Neither V3 INs pulsed at E11.5 (Fig. 1F; n = 3 animals) nor those pulsed at E12.5 (Fig. 1G; n = 3 animals) entered the migratory streams at E12.5.
Although early-born V3 INs exclusively formed embryonic migratory streams at E12.5, it remained possible for late-born V3 INs to also form postmitotic migratory streams, but at later embryonic time points. To examine this possibility, we repeated EdU pulsing at E9.5, E10.5, E11.5, or E12.5 and quantified EdU+ V3 INs in E14.5 Sim1Cre;Rosa.lsl.tdTom spinal cords (Fig. 1I). By E14.5, early-born (E9.5 and E10.5, n = 3 animals for each EdU pulse time) V3 INs distributed away from the p3 progenitor domain in both dorsal and lateral directions (Fig. 1Iii,Iiv). Interestingly, late-born E11.5 EdU+ V3 INs also displayed assemblies away from the p3 progenitor domain (n = 3 animals), dominantly restricted in a lateral direction (Fig. 1Iiii,Iiv). However, late-born E12.5 EdU+ V3 INs (n = 3 animals) displayed almost no sign of migration and predominantly clustered in the ventromedial spinal cord (Fig. 1Iiii,Iiv).
Together, neurogenesis timing ordered V3 INs into temporally and spatially distinct postmitotic migratory streams. Between E11.5 and E12.5, early-born (E9.5 and E10.5) V3 INs formed dorsolateral and ventrolateral migratory streams. Subsequently, between E13.5 and E14.5, late-born (E11.5) V3 INs formed a predominantly ventrolateral migratory stream, whereas E12.5 EdU+ V3 INs did not display substantial migration away from the p3 progenitor.
Dorsoventral V3 IN clusters display distinct neurogenesis windows in the postnatal high lumbar spinal cord
Our previous studies (Blacklaws et al., 2015) and current work have shown that subsets of V3 INs cluster across dorsoventral and mediolateral axes in low thoracic and high lumbar (L1–L3) spinal cords postnatally (Fig. 2A). These laminarly discrete dorsal and ventral V3 IN clusters display distinct axon projection profiles, intrinsic membrane properties, and dendritic morphologies (Borowska et al., 2013, 2015; Blacklaws et al., 2015). Thus, we next asked whether neurogenesis timing underlies V3 postnatal heterogeneity. To begin, we assayed the neurogenetic timing of V3 laminar clusters in P0 high lumbar (L1–L3) spinal cords via preemptive EdU pulsing (Fig. 2Bi–Biv). We then plotted EdU+ V3 INs across spinal laminae (Fig. 2Ci–Gi) and quantified their distributions across dorsoventral and mediolateral spinal axes (Fig. 2Cii–Gii; n = 4 animals for each EdU pulse time).
Figure 2.

Dorsoventral V3 IN clusters display a distinct neurogenesis windows in the postnatal high lumbar spinal cord. A, Representative image of TdTomato+ V3 INs separated within distinct dorsal, intermediate, and ventral laminar clusters in the high lumbar (L3) spinal cord of a P0 Sim1Cre;Rosa.lsl.tdTom mouse. Scale bar, 100 μm. Bi–Biv, Representative images of EdU+/TdTomato+/Hoechst+ V3 INs at P0 pulsed at E9.5 (Bi), E10.5 (Bii), E11.5 (Biii), and E12.5 (Biv) time points. Scale bars, 100 and 10 μm. C–G, Hemicord EdU+/TdTomato+ cell position plots (Ci–Gi), dorsoventral cell count quantifications (Cii–Gii), and ventral V3 mediolateral cell count quantifications (Ciii–Giii) at P0 from E9.5 (Ci–Ciii, n = 4 animals), E10.5 (Di–Diii, n = 4 animals), E11.5 (Ei–Eiii, n = 4 animals), and E12.5 (Fi–Fiii, n = 4 animals) EdU pulse times. Gi–Giii, Combined EdU pulse times (n = 4 animals for each EdU pulse time).
Dorsal and intermediate V3 INs displayed a restricted neurogenesis window consisting mostly of early-born (E9.5–E10.5) neurogenesis times (Fig. 2C,D). These results are in accordance with early embryonic V3 dorsolateral and ventrolateral migratory streams being composed of early-born V3 INs (Fig. 1). In contrast to dorsal and intermediate V3 IN clusters, ventral V3 INs displayed a wide neurogenesis window spanning E9.5 to E12.5 (Fig. 2C–G). However, whereas E9.5 to E11.5 EdU+ V3 INs spanned mediolateral locations within the ventral V3 IN cluster (Fig. 2Ciii–Eiii), E12.5 EdU+ V3 INs localized almost exclusively within ventromedial locations (Fig. 2Fiii). Thus, whereas dorsal and intermediate high lumbar V3 INs displayed a narrow neurogenesis window, mostly between E9.5 and E10.5, ventral V3 INs displayed a wide neurogenesis window ranging between E9.5 and E12.5.
Commissural descending and ascending V3 INs display distinct neurogenesis windows
Previously, we showed that dorsal and intermediate V3 INs project almost exclusively ascending axons, whereas ventral V3 INs display a mixture of both ascending and descending axon projections (Blacklaws et al., 2015). To identify the axon projection profiles and neurogenesis times of V3 INs, we used retrograde BDA tracing of Sim1Cre;Rosa.lsl.tdTom P0 spinal cords that were preemptively pulsed with EdU at E9.5, E10.5, E11.5, or E12.5 time points (Fig. 3Ai,Aii). BDA+ V3 INs were then grouped as either V3 ascending or descending CINs, depending on whether they were labeled caudal or rostral to the BDA injection site, respectively.
Figure 3.

Commissural descending and ascending V3 INs display distinct neurogenesis windows. Ai, Aii, V3 neurogenesis profiles were determined by injecting EdU into pregnant Sim1Cre;Rosa.lsl.tdTom mice at E9.5, E10.5, E11.5, or E12.5. Ai, To classify V3 INs as ascending or descending, hemicord BDA injections were inserted between L1 and L2 spinal cord segments at P0. Aii, Representative image of EdU pulsed at E11.5 and subsequent hemicord BDA injection and EdU detection of descending V3 INs in the t13 spinal cord segment at P0. Scale bars: Aii, 100 μm; inset, 20 μm. Bi–Biii, Hemicord cell position plots of BDA+ and EdU+ V3 INs contralateral to the BDA injection [E9.5, n = 3 animals (Bi); E10.5, n = 3 animals (Bii); E11.5, n = 3 animals (Biii)]. EdU+/BDA+ V3 INs located rostral to the BDA injection site are classified as descending (dark green), and cells caudal to the site are classified as ascending (light green). Biv, Quantification of the percentage of total descending and ascending V3 CINs born at E9.5, E10.5, and E11.5 (n = 3 animals for each EdU pulse time; *p < 0.05, unpaired t test with Welch's correction). Error bars, SD. C, EdU+ V3 INs pulsed at E12.5 displayed no descending or ascending projections. To capture locally projecting V3 CINs, we adapted our tract-tracing procedure by inserting BDA just lateral to the ventral commissure. Under these conditions, E12.5 EdU+ V3 INs were labeled as local CIN V3 at P0. Scale bars: C, 100 μm; insets, 10 μm). Di–Div, Hemicord cell position plots of total BDA+ and EdU+ commissural INs [E9.5, n = 3 animals (Di); E10.5, n = 3 animals (Dii); E11.5, n = 3 animals (Diii); E12.5, n = 3 animals (Div)]. Edu+/BDA+ V3 INs located rostral to the BDA injection site are classified as descending (black), and cells caudal to the site are classified as ascending (gray). Dv, Quantification of the total number of descending and ascending CINs born at E9.5, E10.5, E11.5, and E12.5 (n = 3 animals for each EdU pulse time; *p < 0.05, unpaired t test with Welch's correction). Error bars, SD. E, Summary of neurogenesis timeline of V3 IN projection profiles. ns = nonsignificant.
Both ascending and descending V3 CINs spanning dorsoventral spinal laminae were labeled with E9.5 and E10.5 EdU pulsing (Fig. 3Bi,Bii). However, at these early neurogenesis time points, ascending V3 INs had a higher portion of their total cell numbers labeled relative to descending V3 INs (Fig. 3Biv; n = 3 animals for each EdU time point). In contrast, E11.5 EdU+ V3 INs were almost exclusively descending CINs (Fig. 3Biii,Biv; n = 3 animals for each EdU time point). Interestingly, E12.5 EdU+ V3 INs displayed a complete absence of both ascending and descending axon projections at P0 (data not shown). We reasoned that E12.5 EdU+ V3 INs may not project to adjacent segments, but rather project within restricted local segments. To test this, we inserted BDA just lateral to the ventral midline on one half of the spinal cord and analyzed cells directly contralateral to the injection site. Indeed, the E12.5 EdU+/BDA+ V3 INs remained restricted to the local segment, thus identifying these late-dividing V3 INs as locally projecting V3 CINs (Fig. 3C).
Together, in the higher lumbar spinal cord, a major wave of V3 neurogenesis occurs between E9.5 and E10.5. These early-born V3 INs diversify into heterogeneous clusters across dorsoventral and mediolateral spinal axes composed of V3 ascending and descending CINs. Subsequently, two other distinct V3 IN subsets born at E11.5 and E12.5 are generated. Those V3 INs born at E11.5 define a group of ventral localized V3 descending CINs, whereas those V3 INs born at E12.5 define a ventromedial population that contains local segmental projections (Fig. 3E). Thus, V3 neurogenesis timing correlates with the postnatal anatomical heterogeneity of V3. Furthermore, this temporal neurogenesis order of V3 IN diversity appears to be p3 lineage specific, as the total number of ascending and descending CINs were equivalent across E9.5–E12.5 neurogenesis times (Fig. 3Di–Dv; n = 3 animals for each EdU pulse time).
Postmitotic Sim1 expression specifically promotes the anatomical and physiological diversification of early-born V3 INs
Sim1 expression is essential for the laminar clustering of early-born, but not late-born, V3 INs
We have previously shown that, within low thoracic and high lumbar spinal segments, Sim1 is necessary for V3 dorsal and intermediate, but not ventral, clustering (Blacklaws et al., 2015). As such, the differential neurogenesis timing of V3 IN subsets along dorsoventral spinal axes prompted us to further investigate whether Sim1 differentially regulates the formation of temporally ordered V3 IN subsets.
We first examined whether Sim1 regulates V3 cell cycle exit as distinct V3 INs undergo neurogenesis. We compared V3 IN neurogenesis profiles in Sim1 control and KO mice via embryonic EdU pulsing. We then analyzed EdU+ V3 distributions at E12.5 as previously described. Sim1 KO V3 INs displayed uniform cell cycle exit relative to wild-type cells across all stages examined (E9.5–E12.5; Fig. 4A,B; n = 4 animals for each EdU pulse time). Next, we analyzed their laminar distributions at P0 (n = 4 animals for each EdU pulse time). In Sim1 KOs, early-born V3 INs (E9.5–E10.5) displayed disorganized laminar distributions across the dorsoventral axis (Fig. 4Ci,Cii,Dii), whereas late-born V3 INs (E11.5–E12.5) maintained organized laminar distributions (Fig. 4Ciii,Civ,Diii). Together, Sim1 did not affect V3 IN cell cycle exit. However, the postmitotic expression of Sim1 drove the dorsoventral migration and laminar clustering of exclusively early-born (E9.5–E10.5), but not late-born (E11.5–E12.5), V3 INs by P0. These results indicate Sim1 may promote dorsal and intermediate laminar clustering within a confined pool of early-born V3 INs.
Figure 4.

Sim1 expression is essential for the laminar clustering of early-born V3 INs. Ai, Representative image of E9.5-pulsed EdU detection (confirmed by Hoechst staining) and Nkx2.2 immunoreactivity in an E12.5 lumbar spinal section of a Sim1Cre/taulacz mouse. Scale bars: Ai, 50 μm, insets, 50 μm). Merged image (Mask) shows a colocalization mask of EdU+/Hoechst+/Nkx2.2+ cells using Imaris software. Aii, Representative image of EdU detection (confirmed with Hoechst staining) in V3 INs (TdTom) in a P0 high lumbar spinal section of a Sim1Cre/taulacz;Rosa.lsl.tdTom mouse. Scale bars: Aii, 100 μm; insets, 50 μm). Bi, Bii, Hemicord cell position plots of EdU+/Hoechst+/Nkx2.2+ cells in E12.5 lumbar segments of Sim1Cre (Bi) and Sim1Cre/taulacz (Bii; n = 3 animals for each pulse age and genotype). Biii, The total number of Nkx2.2+/EdU+ cells from respective pulse times (E9.5, E10.5, E11.5, E12.5) from E12.5 lumbar spinal segments compared between Sim1 control (Sim1Cre) and knock-out (Sim1Cre/taulacz) mice (n = 3 animals for each pulse age and genotype, p value > 0.05, unpaired t test with Welch's correction). Error bars, SD. Ci–Civ, Hemicord cell position heat map plots of EdU+/Hoechst+/tdTom+ cells in P0 higher lumbar (L1–L3) segments of Sim1Cre;Rosa.lsl.tdTom and Sim1Cre/taulacz;Rosa.lsl.tdTom mice pulsed at E9.5 (Ci), E10.5 (Cii), E11.5 (Ciii), and E12.5 (Civ; n = 4 animals for each EdU pulse age and genotype). D, EdU+ V3 hemicord cell position plots of P0 higher lumbar (L1–L3) spinal cord sections were pooled together as either early-born (EdU+: E9.5, E10.5, n = 8 animals total for each genotype) or late-born (EdU+: E11.5, E12.5, n = 8 animals total for each genotype) groups. Five-by-five grids were superimposed over hemicord cell position plots, producing 25 discrete bins with specific medial-lateral and dorsal-ventral positions. EdU+ V3 IN counts within corresponding bins were then compared between Sim1Cre;Rosa.lsl.tdTom (Sim1 control) and Sim1Cre/taulacz;Rosa.lsl.tdTom (Sim1 knock-out) cords for early-born and late-born groups: early-born, Sim1 control (n = 8 animals) vs knock-out (n = 8 animals) mice; late-born, Sim1 control (n = 8 animals) vs knock-out (n = 8 animals) mice. Di–Diii, The average count and SD for Sim1 control (top of bin, average ± SD) and Sim1 knock-out (bottom of bin, average ± SD) cords. Statistical significance between Sim1 control and knock-out counts of the same bin were measured with a two-tailed Mann–Whitney U test. Bins with average counts <1 in both control and knock-out mice were considered nonsignificant. Dii, Diii, The p values are presented as a heat map with white bins indicating no change, blue bins indicating gradations of reduced numbers in Sim1 knock-out mice, and red bins indicating gradations of increased numbers in Sim1 knock-out mice.
Sim1 expression is essential for the electrophysiological diversification of V3 IN subpopulations across the dorsoventral axis
In addition to their spatial partition, by P0, dorsal and ventral V3 IN clusters display distinct electrophysiological properties in the high lumbar (L1–L3) spinal cord (Borowska et al., 2013, 2015). Because early-born V3 dorsoventral clustering is perturbed in Sim1 KO animals, we examined whether Sim1 is also necessary for the physiological separation of dorsoventral V3 INs at P0.
To assess electrophysiological properties, we conducted whole-cell patch-clamp recordings of V3 INs in slices from Sim1 wild-type and Sim1 KO spinal cords at P0 (Fig. 5A). In our previous study, we generated a computational model that used four electrophysiological properties [the slope of frequency–current plot (F–I slope), membrane capacitance (Cm), sag voltage amplitude, and first-spike frequency) to calculate the PC1 score for each V3 IN. We demonstrated that respective PC1 scores could be used to separate V3 INs into dorsal or ventral subpopulations at different ages (Borowska et al., 2013, 2015), which was also observed in P0 wild types in our current study (Fig. 5D). However, in Sim1 KO mice, PC1 scores were no longer significantly different between ventral V3 (V3v; 1.12 ± 0.82, n = 23) and dorsal V3 [V3d; 1.00 ± 0.69, n = 17; not significant (ns), p value > 0.05; Fig. 5E). Further assessment of the individual attributes used in the model revealed that all four attributes (F–I slope, Cm, sag voltage amplitude, first-spike frequency) were significantly different between V3d and V3v INs in Sim1 wild-type cords (Fig. 5Bi–Biv), whereas in Sim1 KO mice, neither first-spike frequency (Fig. 5Ci) nor sag amplitude (Fig. 5Cii) was statistically significant between ventral and dorsal V3 INs. Thus, Sim1 is required for both the correct laminar clustering and electrophysiological diversification of early-born dorsal V3 INs in the high lumbar (L1–L3) spinal cord.
Figure 5.

Sim1 expression is essential for the electrophysiological diversification of dorsal V3 IN subsets in the higher lumbar spinal cord. Ai, Spatial criteria for recorded V3 INs as either dorsal or ventral. Aii, Aiii, Representative current-clamp traces from Sim1 control (Aii) and Sim1 knock-out (Aiii) V3d and V3v INs responding to 1 s suprathreshold (left) and subthreshold (right) 40 pA current injections. First-spike frequencies and sag voltages are indicated on traces. Bi–Civ, First-spike frequency (Bi, Ci), sag amplitude (Bii, Cii), F–I slope (Biii, Ciii), and Cm (Biv, Civ) comparisons between V3v and V3d V3 INs in Sim1 control (B) and knock-out mice (C; each point represents an individual cell; *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed Mann–Whitney U test). Error bars, SD. D, E, Contour plots of V3 IN PC1 scores across the mediolateral and dorsoventral axis of higher lumbar (L1–L3) P0 Sim1 control (D) and Sim1 knock-out (E) spinal cords. Color bars indicate PC1 scores from high (yellow) to low (blue). Dashed lines indicate separation of V3d and V3v INs above and below the central canal, respectively. Statistical significance between PC1 scores of dorsal (D) and ventral (V) V3 INs in Sim1 control and KO mice was determined using the Kruskal–Wallis test (D, Sim1 control V3d, n = 25, V3v, n = 33, ***p < 0.001; E, Sim1 knock-out V3d, n = 23, V3v, n = 17; ns, p > 0.05).
Discussion
In the current study, we have systematically investigated the neurogenesis patterns of spinal V3 INs, uncovering significant temporal components contributing to the spatial, anatomical, and physiological diversification of V3 INs. Our data have revealed that early-born V3 INs (E9.5–E10.5) cluster across dorsoventral spinal laminae, displaying both ascending and descending commissural axon projections within low thoracic and high lumbar segments (Fig. 6Ai). In contrast, late-born V3 INs (E11.5–E12.5) cluster mostly within ventral spinal laminae, displaying descending and local commissural axon projections (Fig. 6Aii). Furthermore, we uncovered that the V3-defining Sim1 transcription factor, although expressed in all V3 INs, exclusively regulated the anatomical and electrophysiological clustering of early-born dorsal V3 INs (Fig. 6B). This latter result indicates that the sequential generation of V3 INs may regulate terminal subpopulation characteristics through temporally restricted postmitotic differentiation pathways. Thus, we propose that the timing of neurogenesis serves as a crucial developmental strategy in ordering V3 subpopulation fates across the dorsoventral spinal axis.
Figure 6.

The temporal neurogenesis ordering of postmitotic V3 IN diversity. Postnatal V3 INs display distinct temporal neurogenesis patterns across dorsoventral and mediolateral low thoracic (t12–t13) and high lumbar (L1–L3) spinal segments. Ai, Early-born (E9.5–E10.5) V3 INs assemble within ventral, intermediate, and dorsal V3 IN laminar clusters and display both ascending and descending commissural axon projections. Aii, In contrast, late-born V3 INs (E11.5–E12.5) are fate-restricted to ventral laminar clusters and descending or local commissural axon projection profiles. Bi, Bii, In Sim1 knock-out spinal cords, early-born V3 INs display disorganized dorsoventral laminar clustering and electrophysiological diversification (Bi), whereas late-born V3 INs display unchanged ventral clustering and electrophysiological properties (Bii).
V3 neurogenesis timing and postnatal subpopulation diversification
During early embryonic development, gradient morphogen expression profiles along the neural tube determine the spatial motif of progenitor domains that generate diverse populations of spinal INs (Jessell, 2000; Goulding, 2009). However, this spatial arrangement is not able to account for the extensive subpopulations that have recently been discovered within respective cardinal spinal IN classes. Temporal regulation of spinal IN neurogenesis has been shown to play crucial roles in the formation of dorsal Lbx1+ INs (John et al., 2005); V0 INs (Satou et al., 2012); V1 Renshaw cells (Benito-Gonzalez and Alvarez, 2012; Stam et al., 2012); and most recently, Gata2/3+ CSF-contacting neurons (CSF-cNs; Petracca et al., 2016) and V2a INs (Hayashi et al., 2018). In our current work, by systematically tracing V3 IN neurogenesis, we further demonstrate the contribution of temporal factors in the formation of highly diversified spinal IN subpopulations.
In mice, V3 INs are generated from the most ventral progenitor domain, p3, between E9.5 and E12.5. Although, currently, no molecular factors have been fully characterized to define V3 subpopulations, previous studies from us and others have demonstrated high heterogeneity of V3 INs. Here, we further revealed that anatomically and physiologically distinct V3 IN subpopulations displayed distinct temporal neurogenesis patterns. By mapping all of the V3 INs generated between E9.5 and 12.5, we found that early-born V3 INs (E9.5–E10.5) formed early embryonic (E12.5) migratory streams and, by P0, clustered across separate laminar locations. Late-born V3 INs (E11.5–E12.5) remained close to their original position, the ventral and medioventral region of the spinal cord (particularly those born at E12.5). Early-born V3 INs traveled further away from their progenitor region, a trait shared by other spinal INs. Renshaw cells, which are one of the earliest-born V1 IN subpopulations, migrate to the furthest edge of the ventral horn, whereas most other V1 INs distribute across lamina VII (Benito-Gonzalez and Alvarez, 2012). In the case of a late-born population (>e14.5), Gata2/3+ CSF-cN neurons were only found around the central canal (Petracca et al., 2016). Such a migration trend may be different from what was found for motor neurons, which is more similar to cortical differentiation, where late-born motor neurons migrate through early-born motor neurons in an inside-out order (Dewitz et al., 2018). The mechanisms that direct the migrations of spinal INs are not well known. Nonetheless, our results indicate that the timing of neurogenesis is crucial for the spatial distribution of spinal INs to reach the correct positions in forming diverse neural circuits.
Another interesting finding from our study is that, in addition to their diverse laminar locations, early-born V3 INs (E9.5–E10.5) display mixed commissural ascending or descending axon projections, although our current results cannot rule out that a small amount of early-born V3 INs may be bifurcating (Nissen et al., 2005). In contrast, late-born V3 INs have much more restricted projection profiles. V3 INs born at E11.5 almost exclusively projected descending commissural axons, and those born at E12.5 projected exclusively local commissural axons. Few studies have systematically investigated differential projection profiles within individual spinal IN populations. In our study, it appears such time-dependent acquisition of projection profiles might be V3 IN specific, because the total number of spinal INs did not share this trend. This suggests a possible V3 lineage-specific temporal development strategy.
The results described above also indicate that, in comparison with late-born V3 INs, early-born V3 INs displayed more diverse topological distributions, anatomical characteristics, and electrophysiological properties, leading to more potential subpopulations (Fig. 6A). Such progressive restriction of postmitotic IN fates from a common progenitor domain has recently been demonstrated in the cerebral cortex. Sultan and Shi (2018) revealed that successive divisions of radial glial progenitors generate distinct groups of inhibitory INs. Interestingly, they also showed that postmitotic INs could be generally categorized into either early- or late-born IN groups, where the physiologically and morphologically distinct chandelier cells were exclusively born at later time points. The unique neurogenesis timing of the small and distinct chandelier cell population raises the possibility that neurogenesis timing could be playing even more of a role in ordering the differentiation of distinct subpopulations within the spinal V3 cardinal class. In addition to the anatomical and physiological criteria we have used to separate V3 subpopulations here, it is possible that further molecularly and/or activity-dependent V3 subpopulations emerge during distinct neurogenesis times. Thus, to further uncover the full extent of neurogenesis timing and V3 diversity, it will be necessary to more precisely uncover molecularly and functionally distinct V3 IN subpopulations in the future. Together, our results support the idea that differential waves of neurogenesis serve as a developmental strategy to guide the diversification of unique IN subpopulations across anatomical spinal axes.
Sim1 differentially promotes the diversification of early-born V3 INs
As progenitor cells exit from the cell cycle, they are exposed to various intrinsic and extrinsic factors, promoting their terminal fate specifications. For example, Stam et al. (2012) showed that the expression of Onecut1 and Onecut2 transcription factors during the first wave of V1 IN neurogenesis is a key step in the Renshaw cell differentiation program. In addition, the development of Renshaw cells is also dependent on the presence of the forkhead transcription factor Foxd3. The molecular mechanisms that regulate the temporal differentiation of V3 IN subpopulations are still not clear. However, we found that Sim1 differentially promotes the specification of certain characteristics of V3 IN subpopulations confined within distinct windows of neurogenesis.
Our previous work showed that the loss of Sim1 expression did not change the identity of V3 INs but was crucial for proper V3 IN migration and axon projections (Blacklaws et al., 2015). Interestingly, we further demonstrated here that Sim1 regulates the anatomical and physiological specification of early-born (E9.5–E10.5), but not late-born (E11.5–E12.5), V3 INs. This result strongly indicates that the timing of neurogenesis enables V3 INs to engage in different and specific postmitotic molecular programs. At least one of these molecular programs may involve Sim1, which plays different functional roles across distinct neurogenesis times. We would also like to emphasize that even though Sim1 may not regulate the clustering and electrophysiological diversification of late-born V3 INs, it may play other functional roles in those subpopulations. Nevertheless, our current data indicate that V3 INs may inherit unique transcription factor expression profiles depending on their neurogenesis time, as has been shown in the retina (Clark et al., 2019) and cerebral cortex (Mayer et al., 2018; Zhong et al., 2018). Indeed, Delile et al. (2019) recently took advantage of single-cell mRNA sequencing across early embryonic spinal stages, revealing temporally distinct gene expression dynamics across p3–V3 neurogenesis. This may modulate the differential regulatory control of Sim1 between sequentially generated V3 IN subpopulations. Furthermore, studies from supraspinal regions have revealed that Sim1 forms distinct heterodimer complexes and plays unique developmental roles in different CNS tissues (Michaud et al., 1998; Caqueret et al., 2005; Marion et al., 2005; Xu and Fan, 2007; Osterberg et al., 2011; Schweitzer et al., 2013; Blacklaws et al., 2015). Sim1 expression levels may also differ among distinct subpopulations, as has recently been demonstrated for Chx10 expression in anatomically distinct V2a IN subpopulations (Hayashi et al., 2018). Last, variations in Sim1/DNA binding in V3 IN subpopulations could modulate Sim1 transcriptional activity (Deplancke et al., 2016). Going forward, it will be important to uncover how the temporal components of V3 neurogenesis interact with postmitotic Sim1 expression to diversify V3 IN subpopulations across the anatomical axes of the spinal cord.
Conclusions
Together, through investigation of the p3–V3 spinal IN lineage, we have uncovered a significant temporal component to thee postmitotic anatomical and physiological diversification of V3. With progressive divisions of p3 progenitor cells, postmitotic V3 INs become increasingly fate-restricted to ventral laminae, descending and local commissural axon projections, and uniform membrane properties. Furthermore, we suggest that this could in part be due to the temporal restriction of the postmitotic function of Sim1 in early-born V3 INs. Thus, in addition to layered supraspinal tissues, our current work indicates neurogenesis timing as a developmental mechanism underlying the postmitotic assembly of nonlayered neural circuits in the spinal cord.
Footnotes
The research was funded by the Natural Sciences and Engineering Research Council of Canada (Grant RGPIN/04880). We thank Dr. Martyn Goulding for the gifts of Sim1Cre/+ and Sim1taulacz/+ mice, and Dallas Bennett, Mari Li, and Laura Bennett for technical support. We also thank Han Zhang and Dr. James Fawcett for their helpful comments and feedback.
The authors declare no competing financial interests.
References
- Bassett EA, Wallace VA (2012) Cell fate determination in the vertebrate retina. Trends Neurosci 35:565–573. 10.1016/j.tins.2012.05.004 [DOI] [PubMed] [Google Scholar]
- Benito-Gonzalez A, Alvarez FJ (2012) Renshaw cells and Ia inhibitory interneurons are generated at different times from p1 progenitors and differentiate shortly after exiting the cell cycle. J Neurosci 32:1156–1170. 10.1523/JNEUROSCI.3630-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikoff JB, Gabitto MI, Rivard AF, Drobac E, Machado TA, Miri A, Brenner-Morton S, Famojure E, Diaz C, Alvarez FJ, Mentis GZ, Jessell TM (2016) Spinal inhibitory interneuron diversity delineates variant motor microcircuits. Cell 165:207–219. 10.1016/j.cell.2016.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklaws J, Deska-Gauthier D, Jones CT, Petracca YL, Liu M, Zhang H, Fawcett JP, Glover JC, Lanuza GM, Zhang Y (2015) Sim1 is required for the migration and axonal projections of V3 interneurons in the developing mouse spinal cord. Dev Neurobiol 75:1003–1017. 10.1002/dneu.22266 [DOI] [PubMed] [Google Scholar]
- Borowska J, Jones CT, Zhang H, Blacklaws J, Goulding M, Zhang Y (2013) Functional subpopulations of V3 interneurons in the mature mouse spinal cord. J Neurosci 33:18553–18565. 10.1523/JNEUROSCI.2005-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowska J, Jones CT, Deska-Gauthier D, Zhang Y (2015) V3 interneuron subpopulations in the mouse spinal cord undergo distinctive postnatal maturation processes. Neuroscience 295:221–228. 10.1016/j.neuroscience.2015.03.024 [DOI] [PubMed] [Google Scholar]
- Caqueret A, Coumailleau P, Michaud JL (2005) Regionalization of the anterior hypothalamus in the chick embryo. Dev Dyn 233:652–658. 10.1002/dvdy.20372 [DOI] [PubMed] [Google Scholar]
- Clark BS, Stein-O'Brien GL, Shiau F, Cannon GH, Davis-Marcisak E, Sherman T, Santiago CP, Hoang TV, Rajaii F, James-Esposito RE, Gronostajski RM, Fertig EJ, Goff LA, Blackshaw S (2019) Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron 102:1111–1126.e5. 10.1016/j.neuron.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delile J, Rayon T, Melchionda M, Edwards A, Briscoe J, Sagner A (2019) Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Development 146:dev173807. 10.1242/dev.173807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Alpern D, Gardeux V (2016) The genetics of transcription factor DNA binding variation. Cell 166:538–554. 10.1016/j.cell.2016.07.012 [DOI] [PubMed] [Google Scholar]
- Deska-Gauthier D, Zhang Y (2019) The functional diversity of spinal interneurons and locomotor control. Curr Opin Physiol 8:99–108. 10.1016/j.cophys.2019.01.005 [DOI] [Google Scholar]
- Dewitz C, Pimpinella S, Hackel P, Akalin A, Jessell TM, Zampieri N (2018) Nuclear organization in the spinal cord depends on motor neuron lamination orchestrated by catenin and afadin function. Cell Rep 22:1681–1694. 10.1016/j.celrep.2018.01.059 [DOI] [PubMed] [Google Scholar]
- Francius C, Harris A, Rucchin V, Hendricks TJ, Stam FJ, Barber M, Kurek D, Grosveld FG, Pierani A, Goulding M, Clotman F (2013) Identification of multiple subsets of ventral interneurons and differential distribution along the rostrocaudal axis of the developing spinal cord. Plos One 8:e70325–e70325. 10.1371/journal.pone.0070325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosgnach S, Bikoff JB, Dougherty KJ, El Manira A, Lanuza GM, Zhang Y (2017) Delineating the diversity of spinal interneurons in locomotor circuits. J Neurosci 37:10835–10841. 10.1523/JNEUROSCI.1829-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M. (2009) Circuits controlling vertebrate locomotion: moving in a new direction. Nat Rev Neurosci 10:507–518. 10.1038/nrn2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Hinckley CA, Driscoll SP, Moore NJ, Levine AJ, Hilde KL, Sharma K, Pfaff SL (2018) Graded arrays of spinal and supraspinal V2a interneuron subtypes underlie forelimb and hindlimb motor control. Neuron 97:869–884.e5. 10.1016/j.neuron.2018.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holguera I, Desplan C (2018) Neuronal specification in space and time. Science 362:176–180. 10.1126/science.aas9435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM. (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet 1:20–29. 10.1038/35049541 [DOI] [PubMed] [Google Scholar]
- John A, Wildner H, Britsch S (2005) The homeodomain transcription factor Gbx1 identifies a subpopulation of late-born GABAergic interneurons in the developing dorsal spinal cord. Dev Dyn 234:767–771. 10.1002/dvdy.20568 [DOI] [PubMed] [Google Scholar]
- Kawaguchi A. (2019) Temporal patterning of neocortical progenitor cells: how do they know the right time? Neurosci Res 138:3–11. 10.1016/j.neures.2018.09.004 [DOI] [PubMed] [Google Scholar]
- Kiehn O. (2016) Decoding the organization of spinal circuits that control locomotion. Nat Rev Neurosci 17:224–238. 10.1038/nrn.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnerie C, Tong YG, Alstermark H, Wilson SI (2015) Commissural axonal corridors instruct neuronal migration in the mouse spinal cord. Nat Commun 6:7028. 10.1038/ncomms8028 [DOI] [PubMed] [Google Scholar]
- Marion JF, Yang C, Caqueret A, Boucher F, Michaud JL (2005) Sim1 and Sim2 are required for the correct targeting of mammillary body axons. Development 132:5527–5537. 10.1242/dev.02142 [DOI] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, Satija R (2018) Developmental diversification of cortical inhibitory interneurons. Nature 555:457–462. 10.1038/nature25999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JL, Rosenquist T, May NR, Fan CM (1998) Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev 12:3264–3275. 10.1101/gad.12.20.3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen UV, Mochida H, Glover JC (2005) Development of projection-specific interneurons and projection neurons in the embryonic mouse and rat spinal cord. J Comp Neurol 483:30–47. 10.1002/cne.20435 [DOI] [PubMed] [Google Scholar]
- Osterberg N, Wiehle M, Oehlke O, Heidrich S, Xu C, Fan CM, Krieglstein K, Roussa E (2011) Sim1 is a novel regulator in the differentiation of mouse dorsal raphe serotonergic neurons. PLoS One 6:e19239–e19239. 10.1371/journal.pone.0019239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petracca YL, Sartoretti MM, Di Bella DJ, Marin-Burgin A, Carcagno AL, Schinder AF, Lanuza GM (2016) The late and dual origin of cerebrospinal fluid-contacting neurons in the mouse spinal cord. Development 143:880–891. 10.1242/dev.129254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou C, Kimura Y, Higashijima S (2012) Generation of multiple classes of V0 neurons in zebrafish spinal cord: progenitor heterogeneity and temporal control of neuronal diversity. J Neurosci 32:1771–1783. 10.1523/JNEUROSCI.5500-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer J, Löhr H, Bonkowsky JL, Hübscher K, Driever W (2013) Sim1a and Arnt2 contribute to hypothalamo-spinal axon guidance by regulating Robo2 activity via a Robo3-dependent mechanism. Development 140:93–106. 10.1242/dev.087825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam FJ, Hendricks TJ, Zhang J, Geiman EJ, Francius C, Labosky PA, Clotman F, Goulding M (2012) Renshaw cell interneuron specialization is controlled by a temporally restricted transcription factor program. Development 139:179–190. 10.1242/dev.071134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan K, Shi S (2018) Generation of diverse cortical inhibitory interneurons. Wiley Interdiscip Rev Dev Biol 7:e306. 10.1002/wdev.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed MH, Mark B, Doe CQ (2017) Playing well with others: extrinsic cues regulate neural progenitor temporal identity to generate neuronal diversity. Trends Genet 33:933–942. 10.1016/j.tig.2017.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Fan CM (2007) Allocation of paraventricular and supraoptic neurons requires Sim1 function: a role for a Sim1 downstream gene PlexinC1. Mol Endocrinol 21:1234–1245. 10.1210/me.2007-0034 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, Fan CM, Goulding M (2008) V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron 60:84–96. 10.1016/j.neuron.2008.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N, Xu X, Tang F, Zhang J, Qiao J, Wang X (2018) A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555:524–528. 10.1038/nature25980 [DOI] [PubMed] [Google Scholar]