

Abstract
Introduction
Autologous T-cells transduced to express a chimeric antigen receptor (CAR) directed against CD19 elicit high response rates in relapsed or refractory (r/r) B-cell non-Hodgkin lymphoma (B-NHL). However, r/r B-NHL remissions are durable in fewer than half of recipients of second-generation CAR T-cells. Third-generation (3G) CARs employ two costimulatory domains, resulting in improved CAR T-cell efficacy in vitro and in animal models in vivo. This investigator-initiated, phase I dose escalation trial, termed ENABLE, will investigate the safety and preliminary efficacy of WZTL-002, comprising autologous T-cells expressing a 3G anti-CD19 CAR incorporating the intracellular signalling domains of CD28 and Toll-like receptor 2 (TLR2) for the treatment of r/r B-NHL.
Methods and analysis
Eligible participants will be adults with r/r B-NHL including diffuse large B-cell lymphoma and its variants, follicular lymphoma, transformed follicular lymphoma and mantle cell lymphoma. Participants must have satisfactory organ function, and lack other curative options. Autologous T-cells will be obtained by leukapheresis. Following WZTL-002 manufacture and product release, participants will receive lymphodepleting chemotherapy comprising intravenous fludarabine and cyclophosphamide. A single dose of WZTL-002 will be administered intravenously 2 days later. Targeted assessments for cytokine release syndrome and immune cell effector-associated neurotoxicity syndrome, graded by the American Society Transplantation and Cellular Therapy criteria, will be made. A modified 3+3 dose escalation scheme is planned starting at 5×104 CAR T-cells/kg with a maximum dose of 1×106 CAR T-cells/kg. The primary outcome of this trial is safety of WZTL-002. Secondary outcomes include feasibility of WZTL-002 manufacture and preliminary measures of efficacy.
Ethics and dissemination
Ethical approval for the study was granted by the New Zealand Health and Disability Ethics Committee (reference 19/STH/69) on 23 June 2019 for Protocol V.1.2. Trial results will be reported in a peer-reviewed journal, and results presented at scientific conferences or meetings.
Trial registration number
Keywords: chimeric antigen receptor, CD19 Antigen, non-hodgkin lymphoma, B-Cell Lymphoma, clinical trial protocol
Strengths and limitations of this study.
Uses a new third-generation (3G) anti-CD19 chimeric antigen receptor (CAR) construct incorporating both CD28 and Toll-like receptor 2 (TLR2) costimulatory domains.
Establishes feasibility of T-cell harvest, CAR T-cell manufacture and treatment delivery at a New Zealand centre.
Employs consensus grading systems for cytokine release syndrome and immune cell effector-associated neurotoxicity syndrome.
Dose escalation and dosing steps similar to those employed in other 3G anti-CD19 CAR T-cell trials.
Small sample size, inclusion of several B-cell non-Hodgkin’s lymphoma subtypes and dose escalation design means that efficacy and exploratory outcomes will be descriptive only.
Introduction
Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma
Non-Hodgkin lymphoma (NHL) is the seventh most common malignancy worldwide, accounting for over 200 000 deaths annually.1 Over 90% of NHLs stem from the B-cell lineage (B-NHL) and can be divided into aggressive and indolent forms.2 While aggressive subtypes of B-NHL, exemplified by diffuse large B-cell lymphoma (DLBCL), are often cured with chemoimmunotherapy, around 20% are either refractory to treatment or will relapse.3–5 For most indolent B-NHL subtypes, such as follicular lymphoma (FL), relapses after chemoimmunotherapy are the norm, and while allogeneic stem cell transplantation is curative for some patients, its use is limited by significant short-term and long-term toxicities and by the need to identify a matched haematopoietic stem cell donor.
Autologous T-cells transduced to express a chimeric antigen receptor (CAR) specific for the B-cell antigen CD19 can lyse B-NHL cells.6 Two such ‘CAR T-cell therapies’ have been licensed, incorporating a single intracellular costimulatory domain derived from either CD28 (axicabtagene ciloleucel) or 4-1BB (tisagenlecleucel). CAR T-cell therapies lead to impressive response rates in those with relapsed or refractory (r/r) DLBCL7 8 and with indolent B-NHL subtypes.9 However, only 35%–40% of recipients of currently licensed CAR T-cells for DLBCL remain free of progression for longer than 12 months, a lack of complete metabolic response by 6 months being a major predictor of CAR T-cell treatment failure.10 Anti-CD19 CAR T-cell therapies that exhibit improved early complete metabolic response rates and long-term disease-free survival rates could fulfil an unmet need in r/r B-NHL.7 8
Third-generation CAR T-cells
One way of enhancing CAR T-cell efficacy is to incorporate a second intracellular costimulatory domain within the CAR, generating the so-called ‘third-generation’ (3G) CAR T-cells.11 This can lead to improved CAR T-cell proliferation, cytotoxicity and persistence in vivo.12 13 Most 3G CAR T-cells in registered clinical trials combine a costimulatory domain derived from an immunoglobulin (Ig) superfamily member (such as CD28 or ICOS) alongside one derived from a tumour necrosis factor receptor superfamily member (such as 41BB or OX40) (see table 1).11 Potential benefits of 3G CAR constructs over second-generation (2G) CARs have been demonstrated in preclinical studies.14–16 For example, Zhao et al reported that 3G CARs containing both CD28 and 41BB costimulatory domains led to greater expansion of CD4+ and CD8+ T-cells, along with improved B-cell acute lymphoblastic leukaemia (B-ALL) tumour regression in xenograft models.15 However, it is not yet clear whether 3G CAR T-cells offer improved clinical efficacy.
Table 1.
Other third-generation anti-CD19 CAR T-cell trials registered on ClinicalTrials.gov
| Study ClinicalTrials.gov ID |
B-cell malignancy subtypes | CAR generation | Study phase | Lymphodepletion | Study start date | Results published (yes/no) |
| NCT02963038 | B-ALL+B-NHL | 3G | I+II | Not specified | June 2016 | No |
| NCT03068416 | B-ALL+B-NHL | 3G | II | Not specified | September 2017 | No |
|
NCT02132624 (see the Discussion section in paper) |
B-NHL | 3G | I | Flu 25 mg/m2×3 days, Cy 500 mg/m2×3 days |
April 2014 | Yes* |
| NCT03146533 | B-NHL | 3G | I+II | Flu 30 mg/m2×3 days Cy 800 mg/m2×3 days |
April 2017 | No |
|
NCT01853631 (see the Discussion section in paper) |
B-ALL+B-NHL | 3G and 2G† | I | Flu 30 mg/m2×3 days, Cy 500 mg/m2×3 days |
February 2014 | Yes‡ |
| NCT03676504 | B-ALL+B-NHL | 3G | I+II | Flu 30 mg/m2×3 days, Cy 500 mg/m2×3 days |
September 2018 | No |
| NCT02822326 | B-ALL | 3G | I | Flu 25 mg/m2×3 days Cy 300 mg/m2×3 days |
January 2016 | No |
As at 16/09/2019.
*See Enblad et al. 23
†Co-infused with CD28 containing second-generation CAR and CD28+41BB containing third-generation CAR.
‡See Ramos et al. 13
B-ALL, B-cell acute lymphoblastic leukaemia; B-NHL, B-cell non-Hodgkin's lymphoma; CAR, chimeric antigen receptor; Cy, cyclophosphamide; Flu, fludarabine.
Activated T-cells express TLRs, particularly TLR2, a pattern recognition receptor that recognises bacterial cell wall components.17 18 Ligation of TLR2 enhances Akt and Erk1/Erk2 phosphorylation in response to T-cell receptor (TCR) stimulation, enhancing TCR-induced cytokine production and proliferation.19 T-cell intrinsic TLR2 signalling lowers the T-cell activation threshold in response to costimulatory signals received from antigen presenting cells and enables the generation of functional memory CD8 T-cells in response to T-cell activation.20 21
Third-generation CAR T-cells incorporating the Toll/interleukin-1 receptor (TIR) domain from TLR2, which mediates the intracellular signalling of TLR2, show improved anti-tumour activity compared with 2G CAR T-cells both in vitro and in vivo.16 The safety and efficacy of a 3G CAR T-cell product combining CD28 and TLR2 TIR costimulatory domains has been explored in a phase I clinical trial in B-cell acute lymphoblastic leukaemia (B-ALL) (ClinicalTrials.gov reference NCT02822326), in which clinical responses were observed, including among participants with extramedullary B-ALL tumours.22
We have modified the manufacture of 3G anti-CD19 CAR T-cells incorporating CD28 and TLR2 TIR costimulatory domains to employ a 3G self-inactivating lentiviral vector for T-cell transduction and to adopt process modifications designed to meet local Good Manufacturing Practice (GMP) requirements. We plan a phase I dose escalation trial to assess the safety of this product, WZTL-002, for the treatment of r/r B-NHL.
Methods
Study design
This investigator-initiated open-label phase I dose escalation trial is named ENABLE: Engaging Toll-like Receptor Signalling for B-cell Lymphoma Chimeric Antigen Receptor Therapy (ClinicalTrial.gov number: NCT04049513). The ENABLE trial aims to assess the safety of WZTL-002, comprising autologous anti-CD19 3G CAR-T cells incorporating CD28 and TLR2 TIR costimulatory domains, for the treatment of r/r B-NHL. The sponsor is the Malaghan Institute of Medical Research (MIMR), and the trial is conducted in collaboration with Wellington Zhaotai Therapies Limited (WZTL). The study site is Wellington Hospital, Capital & Coast District Health Board, New Zealand.
Key inclusion and exclusion criteria are presented in box 1. In addition to the inclusion and exclusion criteria presented in box 1, immunosuppressive therapies, with the exception of lymphodepleting chemotherapy, must be avoided during the week before WZTL-002 administration (72 hours for systemic corticosteroids). Prior autologous and allogeneic stem cell recipients are eligible to participate in the study. Structure of the CAR employed in WZTL-002 is presented in figure 1, and the protocol schema in figure 2.
Box 1. Inclusion and exclusion criteria for ENABLE.
Inclusion criteria
Age 16–75 years (inclusive).
Biopsy-proven relapsed or treatment refractory B-cell non-Hodgkin's lymphoma of the following subtypes per WHO classification: diffuse large B-cell lymphoma (DLBCL) and its variants, primary mediastinal B-cell lymphoma (PMBCL), transformed follicular lymphoma (tFL), follicular lymphoma (FL) and mantle cell lymphoma (MCL).
Requirement for treatment in the opinion of the investigator.
No other curative treatments available, or not suitable due to patient or disease characteristics or lack of stem cell donor.
Malignancy documented to express CD19 based on flow cytometric or immunohistochemical staining.
Provision of written informed consent for this study.
Life expectancy from non-lymphoma-related causes of >12 months.
European Cooperative Oncology Group (ECOG) performance status of 0 to 2 inclusive.
Adequate haematologic function, defined by neutrophils ≥1.0×109/L and platelets ≥50×109/L.
-
No serious cardiac, pulmonary, hepatic or renal disease.
Serum bilirubin <2.5 times upper limit of normal (ULN).
Estimated creatinine clearance ≥50 mL/min using the modified Cockroft Gault estimation or as assessed by direct measurement.
Cardiac ejection fraction ≥50% as determined by echocardiogram or MUGA scan.
Oxygen saturations >92% on room air.
Diffuse capacity of the lungs for carbon monoxide (DLCO) or carbon monoxide transfer coefficient (KCO), forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) are all ≥50% of predicted by spirometry after correcting for haemoglobin and/or volume on lung function testing.
Exclusion criteria
Confirmed active or prior central nervous system (CNS) involvement by lymphoma. In patients with a clinical suspicion of CNS disease, lumbar puncture and MRI brain must be performed.
Active CNS pathology including: epilepsy, seizure within the preceding year, aphasia, paresis, stroke, dementia, psychosis within the preceding year, severe brain injury, Parkinson disease or cerebellar disease.
Richter syndrome.
Active autoimmune disease requiring systemic immunosuppression.
Prior solid organ transplantation.
Allogeneic stem cell transplantation within the preceding 3 months or still requiring systemic immunosuppression.
Current grade II–IV acute graft vs host disease (GVHD), any prior grade IV acute GVHD, or current moderate or severe chronic GVHD.
Need for systemic corticosteroids to treat a condition other than B-NHL at a daily dose of ≥10 mg prednisone (or equivalent).
Peripheral blood lymphocytes <0.5×109/L as assessed by complete blood count.
Peripheral blood CD3+ T-cells <350/μL as assessed by lymphocyte subset analysis.
Pregnant or lactating female.
Women of childbearing potential who are not willing to use highly effective methods of contraception during study participation and for at least 1 year after WZTL-002 administration.
Men who are not willing to use highly effective methods of contraception during study participation and for at least 1 year after WZTL-002 administration.
Men who have a pregnant partner and are not willing to use a condom while performing sexual activity during study participation and for at least 3 months after WZTL-002 administration.
Subjects with known sensitivity to immunoglobulin or to components of the investigational product (IP).
History of active malignancy other than B-cell malignancy within 2 years prior to enrolment, with the exception of: adequately treated in situ carcinoma of the cervix; adequately treated basal cell carcinoma (BCC) or localised squamous cell carcinoma (SCC) of the skin; other localised malignancy surgically resected (or radically treated with another treatment modality) with curative intent.
Current or prior HIV infection.
Vaccination with a live virus within the preceding 4 weeks.
Treatment with a purine analogue within the preceding 4 weeks.
Treatment with alemtuzumab within the preceding 12 weeks.
Prior gene therapy, including prior anti-CD19 chimeric antigen receptor T-cell therapy.
Receipt of an investigational medicine within another clinical trial within the preceding 4 weeks.
Inadequately controlled systemic infection.
Serologic status reflecting active viral hepatitis B or any history of hepatitis C infection as follows:
Presence of hepatitis B surface antigen (HBsAg) or hepatitis B core antibody (HBcAb). Patients with presence of HBcAb, but absence of HBsAg, are eligible if hepatitis B virus (HBV) DNA is undetectable (<20 IU), and if they are willing to receive appropriate anti-viral prophylaxis.
Presence of hepatitis C virus (HCV) antibody.
Presence of New York Heart Association (NYHA) class 2 or higher cardiac symptoms not related to lymphoma.
Significant concomitant illnesses which would in the investigator’s opinion make the patient an unsuitable candidate for the trial.
Subjects who have diminished capacity or any circumstance that would prohibit them from understanding and providing informed consent in accordance with ICH-GCP (International Conference on Harmonisation, Good Clinical Practice).
Subject does not provide consent to enrol onto International Cellular Therapy Registry.
Figure 1.
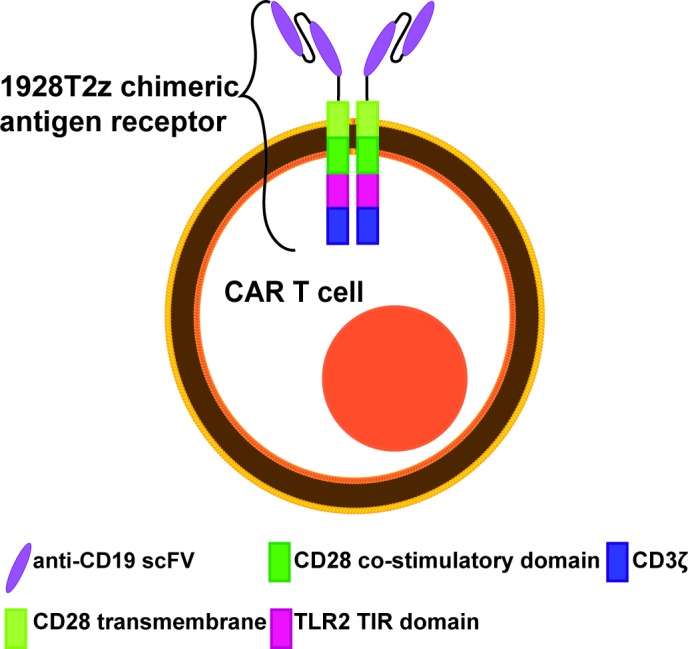
Diagrammatic representation of WZTL-002 anti-CD19 third-generation chimeric antigen receptor (CAR) T-cell illustrating the costimulatory domains and components of the CAR.
Figure 2.

Schema for the enable phase I dose escalation study. Second attempt at cell harvest and WZTL-002 production may be considered at discretion of TMC. Six-month PET scan if first PET scan post WZTL-002 treatment shows partial response. Long-term follow-up through bone marrow transplant clinic and Cellular Therapies Registry enrolment. FluCy, fludarabine and cyclophosphamide IV; PET, positron emission tomography; TMC, trial management committee.
A modified 3+3 dose escalation scheme with four dose steps (5×104; 1×105; 5×105; 1×106 CAR T-cells/kg) is planned. The first dose step is two steps (10-fold) below the recommended phase II dose determined in a phase I trial of a similar product in r/r B-ALL (ClinicalTrials.gov reference NCT02822326), and is similar to that used in two reported clinical trials of 3G CAR T-cell products.13 23 The final dose step was selected because dose-limiting toxicities (DLTs) were observed at this level in r/r B-ALL using a similar product (ClinicalTrials.gov reference NCT02822326) and because, based on preclinical data, the dose of WZTL-002 is expected to be lower than that recommended for the licensed 2G CAR T-cell product axicabtagene ciloleucel (2×106 CAR T-cells/kg). Additional dose steps may be incorporated if recommended by the Data Safety Monitoring Committee (DSMC). The DLT definitions are presented in box 2.
Box 2. Dose-limiting toxicities.
A Dose Limiting Toxicity (DLT) is a toxicity or Adverse Event (AE) occurring during the DLT assessment period (first 21 days after WZTL-002 administration), which is not attributable to a cause unrelated to WZTL-002 (such as underlying lymphoma, concurrent illness or concomitant medications), and meets one of the following criteria:
Grade 4 or greater CRS or ICANS or grade 3 CRS or ICANS that does not resolve to grade 2 or lower within 7 days, both as per the American Society for Transplantation and Cellular Therapy (ASTCT) criteria.
Any adverse event requiring airway intubation (including neurotoxicity requiring intubation for airway protection).
Grade 4 neutropenia that does not resolve to grade 3 or lower within 21 days after WZTL-002 administration.
Platelet transfusion-dependent thrombocytopenia persisting for 21 days or longer after WZTL-002 administration.
-
All grade 4 toxicities and grade 3 toxicities that do not resolve to grade 2 or lower within 7 days, with the exception of the following, which are not automatically considered DLTs:
-
Myelosuppression, including
Neutropenia.
Bacterial infection in the setting of neutropenia with neutrophils<1.0×109/L.
Thrombocytopenia.
Bleeding in the setting of thrombocytopenia with platelets<50×109/L.
Anaemia.
Lymphopenia.
Hypersensitivity reactions occurring within 2 hours of WZTL-002 administration (and considered related to cell administration) that resolve to grade 2 or less within 24 hours.
Asymptomatic biochemical abnormalities that resolve to grade 2 or lower within 7 days.
Hypogammaglobulinaemia.
-
For CRS and ICANS, ASTCT grading criteria will be used. For all other toxicities, CTCAE V.5.0 will be used.
AEs, adverse events; CRS, cytokine release syndrome; ICANS, immune cell effector-associated neurotoxicity syndrome.
Study procedures
All potential participants will be assessed at a lymphoma multidisciplinary team meeting to confirm that no other curative treatment options are available. Following written informed consent and screening, eligible participants will undergo a leukapheresis procedure to harvest autologous peripheral blood mononuclear cells (PBMCs) for WZTL-002 manufacture. Following cell harvest, bridging therapy will be permitted to provide disease control during manufacturing and treatment scheduling, and to reduce lymphoma disease bulk before WZTL-002 administration. Antimicrobial prophylaxis and tumour lysis prophylaxis will be given as per standard of care for patients receiving treatment for haematological malignancies.
Once product release criteria are met, eligibility to proceed to WZTL-002 treatment is confirmed, and following any bridging chemotherapy or radiotherapy, a baseline PET/CT scan will be performed. This will be followed by lymphodepleting chemotherapy comprising intravenous fludarabine (30 mg/m2/day×3 days) and cyclophosphamide (500 mg/m2/day×3 days). WZTL-002 will be administered following two chemotherapy-free days as a slow intravenous push. Participants will be monitored as an inpatient for 14 days, using both regular observations and specific CRS and neurotoxicity assessments, including the Immune Effector-cell Encephalopathy (ICE) score, at least twice daily.24 Daily assessment will continue until 21 days after WZTL-002 administration. To inform treatment of the next participant, assessment of DLTs will be undertaken 21 days after WZTL-002 administration (see box 2).
Response assessment will be by PET/CT scan 3 months after WZTL-002 administration using the Deauville 5-point scoring system, and response to treatment will be assigned as either complete response (CR), partial response (PR), stable disease (SD) or progressive disease (PD), according to 2014 Lugano response criteria for lymphoma.25 A further PET/CT scan will be performed at 6 months for those with partial response at the 3-month timepoint. Additional imaging to assess or confirm treatment response, to investigate toxicities or to seek potential disease progression may be carried out at any time, as clinically indicated. Trial follow-up will take place at 3 monthly intervals until 1 year, 6 monthly intervals until 2 years and annually until 5 years post-WZTL-002 administration. Participants will be registered in the Centre for International Blood and Marrow Transplant Research (CIBMTR) Cellular Therapies Registry and Australasian Bone Marrow Transplant Recipient Registry (ABMTRR) in order to capture low incidence or late treatment-related toxicities.
Study aim and outcomes
The overall aim of the ENABLE trial is to assess the safety of 3G autologous anti-CD19 CAR T-cells incorporating CD28 and TLR2 TIR costimulatory domains (WZTL-002) in individuals with r/r B-NHL.
The primary outcome is safety profile of WZTL-002, determined by the number and severity of adverse events (AEs) assessed by CTCAE v5.0, except for CRS and ICANS, which will be assessed by American Society Transplantation and Cellular Therapy (ASTCT) consensus grading criteria.24
Secondary outcomes are as follows:
Feasibility of WZTL-002 manufacture, as determined by the proportion of enrolled participants undergoing at least one study leukapheresis procedure that receive WZTL-002.
Overall response rate (ORR) as determined by CR plus PR 3 months after WZTL-002 administration.
Cumulative CR rate 6 months after WZTL-002 administration.
Relapse-free survival (RFS) for participants treated with WZTL-002 over a period of 24 months after WZTL-002 administration.
Overall survival (OS) for participants treated with WZTL-002 over a period of 24 months after WZTL-002 administration.
The recommended phase II dose of WZTL-002 for the treatment of patients with r/r B-NHL.
Exploratory outcomes are as follows:
Kinetics and persistence of WZTL-002 following administration, determined by peripheral blood PCR for the CAR transgene.
Extent and duration of B-cell aplasia, determined by peripheral blood flow cytometry and serum IgG concentration.
Serum cytokine profile following WZTL-002 administration.
Phenotype of the WZTL-002 CAR T-cell product before administration, and of circulating CAR T-cells following administration.
Manufacture of WZTL-002
WZTL-002 will be manufactured in the Clinical Human Immunology Laboratory at the Malaghan Institute of Medical Research in Wellington, New Zealand, which is licensed by Medsafe, the New Zealand Medicines and Medical Devices Safety Authority. Briefly, peripheral blood mononuclear cells (PBMCs) are isolated from the leukapheresis using a density gradient medium. T-cells are selected and activated using immunomagnetic CD3/CD28 microbeads and genetically modified using a 3G self-inactivating non-replication competent lentiviral vector (manufactured in-house and tested according to EMEA guidelines). After washing to remove lentiviral vector and microbeads, CAR T-cells are expanded in a GMP-grade medium supplemented with interleukin (IL)-2 and human serum for 7 days. The CAR T-cell product is harvested and formulated in a cryopreservation medium containing 10% dimethyl sulfoxide (DMSO). Release criteria for the CAR T-cell product include product sterility, identity, purity and absence of residual lentiviral vector.
Immune monitoring and exploratory endpoints
Kinetics of WZTL-002 will be determined by determining the CAR transgene in patient’s peripheral blood by quantitative PCR before and at 1, 2, 4, 7, 9, 11, 14, 16, 18, 21 and 28 days, at 2, 3, 6, 12 and 24 months, and at 3, 4 and 5 years, after WZTL-002 administration. Serum cytokine profile after WZTL-002 administration will be determined by ELISA (for IL-6) and by cytokine bead array before and at 1, 2, 4, 7, 9, 11, 14, 16, 18, 21 and 28 days after WZTL-002 administration. At each timepoint, 25 mL of blood is taken for study-specific analyses. In subjects with neurotoxicity, cerebrospinal fluid samples may be stored for assessment of cytokine and CAR transgene levels. Depth and duration of B-cell aplasia will be established by peripheral blood flow cytometry and by nephelometric determination of serum IgG concentration, both to inform infection risk among participants and to serve as a surrogate measure for WZTL-002 persistence. The immunophenotype (including CD4, CD8, CD45RA and CD62L) of WZTL-002 CAR T-cells will be determined in the preadministration product, and within participant PBMCs after administration (if CAR T-cells are detectable).
Samples of the CAR T-cell product and recipient PBMCs postadministration (at each protocolised timepoint) will be cryopreserved. The timepoints for exploratory analyses will be selected based on pharmacokinetics determined by PCR for the CAR transgene. The outcomes of exploratory analyses are not expected to be definitive and are included to inform design of a subsequent efficacy trial.
Toxicity management
Based on clinical experience with similar constructs, CRS and ICANS are anticipated toxicities of WZTL-002. Accordingly, a CAR T-cell toxicity (CARTOX) team comprising local intensive care, neurology, haematology, immunology and infectious disease specialists and nursing representatives was formed. This team localised consensus assessment and treatment protocols for CRS and ICANS, and reviewed safety measures and training and competency assessment materials.24 26 A summary of key measures taken to prepare for CRS and ICANS is provided in box 3.
Box 3. Summary of measures taken to prepare for CRS and ICANS.
Formation of CAR T-cell toxicity working group composed of haematologists, neurologist, intensivist, immunologist and haematology nurses.
Attendance of principal investigator, coinvestigators, study nurse, senior haematology nurses and nurse educators at a CAR T-cell toxicities preceptorship day.
Localisation of guidelines for CRS and ICANS identification, management and escalation, and the upload of these to institutional electronic treatment guide.
Education sessions and competency assessments on CAR T-cell toxicities delivered to nursing, patient at risk and ICU teams.
Allowance for bridging treatment between leukapheresis and lymphodepleting chemotherapy to ‘debulk’ or control disease before WZTL-002 administration.
Confirmation that three doses of tocilizumab are on site before WZTL-002 administration.
Levetiracetam 750 mg twice daily to be given for 28 days following WZTL-002 administration for anti-seizure prophylaxis.
Completion of clinical checklist before WZTL-002 administration.
Notification of neurology and intensive care unit teams before WZTL-002 administration.
Scheduled nurse-led CRS and ICANS assessments.
Provision of participant-held wallet card and discharge summary sheet.
CRS, cytokine release syndrome; ICANS, immune cell effector-associated neurotoxicity syndrome.
Monitoring and data management
A trial management committee including the principal investigator, at least one co-investigator, the study nurse, and a representative of the Clinical Human Immunology Laboratory, will meet at least monthly during study recruitment to review recruitment rates, trial conduct, trial procedures, AEs and serious adverse events (SAEs). An independent Data and Safety Monitoring Committee (DSMC) will include clinicians with experience in early phase T-cell trials and in haemato-oncology. Per the DSMC Charter, the DSMC will meet and review trial accrual, conduct and safety data a minimum of 6-monthly and before each dose step. An independent study will monitor the study and will report to the sponsor.
The study site will hold responsibility for the confidentiality of electronic and paper clinical records held for the study participant. To maintain confidentiality of trial participants, study data or samples sent to collaborating investigators or external contractors for analysis or review will be labelled with study-specific codes and not with patient identifiers. The principal investigator will hold responsibility for ensuring that presentations and publications of the study findings do not contain identifiable information. Laboratory records will be kept for a minimum of 15 years. Clinical data (including Case Report Forms) will be stored securely for a minimum of 15 years.
Statistical analysis
This phase I trial will be analysed using descriptive statistics; no formal hypothesis testing will be undertaken. All participants who commence lymphodepleting chemotherapy will be included in the summaries of the safety outcomes.
Safety outcomes including AEs, SAEs, suspected unexpected serious adverse reactions, CRS, ICANS and DLTs will be individually listed by dose group and summarised as the frequency of events and percentages of individuals experiencing each event type. Each event summary will include details of the timing, grade and outcome of the event. Response outcomes including ORR (defined as CR rate plus PR rate) and CR rate will be individually listed and summarised as frequencies (%) by dose group. The survival outcomes, RFS and OS will also be summarised as frequencies (%) at 24 months and the times to events will be individually listed and may be summarised with Kaplan-Meier curves for individual dose groups if sample sizes permit. Associations between safety outcomes and presenting features will be explored in a qualitative manner.
The study sample size will depend on DLTs observed during dose escalation and is estimated at 12 participants, with at least three participants treated at each dose step. If no DLTs are observed, escalation to the next dose step may occur (see box 2 for DLT definition). If a DLT is observed in one of the first three participants treated at a specific dose step, a further three participants should be treated at that dose. If two or more participants develop a DLT at a specific dose step, escalation to the next dose step should not occur, indicating that the maximum tolerated dose has been reached. The DMSC will meet before each proposed dose escalation and may recommend de-escalation to a lower dose level, protocol modification or for more participants to be treated at that dose step, based on available safety and/or efficacy data.
Ethics
The study will be performed in accordance with the principles of the International Conference on Harmonisation Guidelines on Good Clinical Practise (ICH-GCP) (Step 4, dated 10 June 1996) that have their origins in the Declaration of Helsinki.27
The trial has been approved by the New Zealand Health and Disability Ethics Committee (reference 19/STH/69) and has been endorsed by Research Advisory Group Māori at Capital & Coast District Health Board, which is mandated to provide consultation for cultural appropriateness of clinical research conducted within the region (reference RAG-M #662).
Patient and public involvement
The study protocol was developed after discussion at a blood cancer patient forum convened by Leukaemia & Blood Cancer New Zealand, and at meetings of the Lymphoma Network of New Zealand and the NZ Branch of the Haematology Society of Australia and New Zealand. The study protocol and consent form were developed in consultation with Research Advisory Group – Māori, a Māori relationship board, which includes lay representation, to Capital & Coast District Health Board. The participant information and consent form was reviewed by a patient representative with relevant personal experience. The study has been publicised in national media, although due to regulatory and logistical considerations, referrals must come from a relevant specialist rather than directly from potential participants. Study results will be presented in the lay media and in scientific journals. The patient information and consent form includes an option to request a lay summary of the study results.
Data dissemination
Participants will be given the option to receive a summary of the trial results. Trial results will be published in a peer-reviewed journal after completion of the trial.
Discussion
This manuscript describes the protocol for ENABLE, an investigator-led phase I dose escalation trial evaluating a new 3G autologous anti-CD19 CAR T-cell product (WZTL-002) for the treatment of individuals with r/r B-NHL. The primary outcome is safety, which will be assessed by determining the number and severity of AEs. Secondary outcomes will assess feasibility, efficacy and recommended WZTL-002 dose for subsequent efficacy trials.
As well as resulting in improved cytotoxicity against target cells, the incorporation of a second costimulatory domain can enhance CAR T-cell proliferation and cytokine production.11 16 Thus, while 3G CAR T-cell products have the potential for increased efficacy, there is also the potential for an increased risk of toxicities, including CRS and ICANS, compared with 2G products. Accordingly, CRS and ICANS were identified as events of special interest early during trial development. The risk of both toxicities may relate to CAR T-cell dose and to disease burden.28 29 Therefore, to mitigate CRS and ICANS risks, conservative starting and maximum WZTL-002 doses have been selected, based on clinical experience using similar CAR T-cell products. Targeted CRS and ICANS assessments will be performed, and a comprehensive risk mitigation plan has been developed, including the development of institutional policies and protocols, documented staff training, intensive care escalation plans and on-site tocilizumab availability. Use of the ASTCT international consensus CRS and ICANS grading system will facilitate the comparison of toxicity rates with other anti-CD19 CAR T-cell trials conducted internationally.24
The use of ‘bridging’ chemotherapy and radiotherapy between enrolment and WZTL-002 administration is permitted. This will facilitate WZTL-002 treatment scheduling during dose escalation and may allow reduction of disease burden before WZTL-002 therapy, potentially reducing CRS and ICANS risk.30 To mitigate the impact of bridging therapy on efficacy assessments, baseline PET-CT scans will be conducted after completing bridging therapy and before starting lymphodepleting chemotherapy and WZTL-002 administration.
The selection of eligible lymphoma subtypes was based on evidence of efficacy from other clinical trials evaluating anti-CD19 CAR T-cell therapies in this population.7–9 31 32 Limitations of this trial include its small size, inclusion of several B-NHL subtypes and dose escalation design, as a result of which efficacy and exploratory outcomes will be descriptive only. In particular, secondary outcomes assessing WZTL-002 efficacy (ORR, CR rate, RFS and OS) will be preliminary, and are included to help inform the design of future phase II trials. Similarly, the exploratory outcomes, which explore WZTL-002 kinetics, phenotype, serum cytokines and B-cell aplasia, are intended to inform outcome measure selection for future larger trials.
The published clinical experience of 3G anti-CD19 CAR T-cells for the treatment of r/r B-NHL is limited, with the final results of only two other early-phase trials published, to our knowledge.13 23 Enblad et al treated 11 patients with r/r B-NHL or chronic lymphocytic leukaemia with 3G anti-CD19 CAR T-cells combining CD28 and 41BB costimulatory domains, in a phase I dose escalation study.23 Of the 11 treated participants, 4 did not receive lymphodepletion before CAR T-cell administration. The dose range of 3G anti-CD19 CAR T-cells administered this study was 2×107–2×108 cells/m2 (approximately equivalent to 5×105–5×106 CAR T-cells/kg). A response to treatment was observed in four participants (36%), all of whom reached CR.23 Severe CRS was reported in two participants (18%), and severe neurotoxicity in one (9%).
Ramos et al reported results of a phase I anti-CD19 CAR T-cell trial involving simultaneous administration of autologous 2G (CD28 only) and 3G (4-1BB plus CD28) anti-CD19 CAR T-cell products to participants with r/r B-NHL.13 This dose escalation study treated 11 participants with active lymphoma and 5 in remission after autologous stem cell transplant (ASCT). All participants with active lymphoma received lymphodepletion with cyclophosphamide and fludarabine before CAR T-cell infusion, whereas no further lymphodepletion was given to those post ASCT. The dose range of total CAR T-cells administered on this study (2G+3G CAR T-cells in 1:1 ratio) was 5×104–1×106 CAR T-cells/kg. Six of 11 with active lymphoma (54%) responded, three (27%) reaching CR. All five recipients of CAR T-cells after ASCT remained in CR at least 9 months after CAR T-cell administration. No cases of severe CRS, and only one of severe neurotoxicity, were reported.13 Ramos et al found that the 3G anti-CD19 CARs showed superior in vivo expansion and persisted longer than their 2G counterparts, although the relative contribution of the 2G and 3G CAR T-cells to anti-tumour efficacy and to toxicity could not be assessed with this study design.13
In conclusion, published phase I trials suggest that manufacture of 3G CAR T-cells is feasible and do not yet indicate that CRS and ICANS rates are higher than for 2G products. Moreover, the Ramos et al study indicates that 3G CAR T-cells can exhibit improved proliferation and persistence in humans compared with 2G counterparts. However, because of the small number of reported 3G CAR T-cell recipients, and the likely suboptimal CAR T-cell dosing in the early cohorts of these dose escalation studies, conclusions cannot be drawn about the relative efficacy and safety of 3G compared with 2G CAR T-cells.13 23 Other 3G anti-CD19 CAR T-cell trials in patients with r/r B-NHL are underway (table 1). As well as adding to the clinical experience of 3G anti-CD19 CAR T-cell therapies for the treatment of B-NHL, the ENABLE trial will inform the clinical safety and potential utility of a new intracellular TLR2 costimulatory domain within CAR T-cells.
Supplementary Material
Acknowledgments
The authors would like to thank Wellington Zhaotai Therapies Limited (WZTL) for providing the construct used to generate WZTL-002 for use in this trial and for supporting WZTL-002 development and manufacture. The authors would like to thank Research Advisory Group – Māori, the Māori relationship board to Capital and Coast District Health Board for their input into study design. The authors would like also to acknowledge a number of volunteers for providing feedback on the study protocol, particularly Julie Jones, Marina Dzhelali and David Downs.
Footnotes
Twitter: @PipG05
Contributors: RW conceived the clinical trial. RW and PG designed and wrote the study protocol. ND, PG and RW drafted the manuscript. RW, PG, ND, GG, BM, EB, BA, TP, TO, CF, DR, CMB and IH provided input into the study protocol. PL conceived the TLR2 costimulatory domain construct, and provided background, preclinical and clinical data regarding its use. All authors reviewed the study manuscript. RW, TP, PG and TO will conduct study procedures. RW is the principal investigator.
Funding: The Malaghan Institute of Medical Research (MIMR) is the sponsor of this investigator-initiated trial. Delegated duties will be assigned to Capital and Coast District Health Board and its employees by means of the site clinical trial agreement, and to the study monitor by means of a monitor agreement. The study is funded by philanthropic support to MIMR, an independent biomedical research institute and registered charity. A private company, Wellington Zhaotai Therapies Limited (WZTL), provided the rights to use the 1928T2z construct, the source plasmids for vector production, and contributed to MIMR costs for the production of WZTL-002. WZTL is not involved in study design, conduct or reporting, which are responsibilities of the principal investigator.
Competing interests: Trial principal investigator, RW, and co-investigator, PG, are employees of the Malaghan Institute of Medical Research, a charitable research institute and study sponsor. The other co-investigators have no competing interests to declare. PL has proprietary interest in the intellectual property of the 1928T2z construct. CB is co-Founder and Scientific Advisory Board Member of Mana Therapeutics is on the Advisory Board of Cellectis, has Stock ownership in Torque Therapeutics and Neximmune and is a Board Member of Caballeta Bio.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and National cancer incidence, mortality, years of life lost, years lived with disability, and Disability-Adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017;3:524–48. 10.1001/jamaoncol.2016.5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health organization subtypes. CA Cancer J Clin 2016;66:443–59. 10.3322/caac.21357 [DOI] [PubMed] [Google Scholar]
- 3. Molina TJ, Canioni D, Copie-Bergman C, et al. Young patients with non-germinal center B-cell-like diffuse large B-cell lymphoma benefit from intensified chemotherapy with ACVBP plus rituximab compared with CHOP plus rituximab: analysis of data from the Groupe d'Etudes des Lymphomes de l'Adulte/lymphoma study association phase III trial LNH 03-2B. J Clin Oncol 2014;32:3996–4003. 10.1200/JCO.2013.54.9493 [DOI] [PubMed] [Google Scholar]
- 4. Récher C, Coiffier B, Haioun C, et al. Intensified chemotherapy with ACVBP plus rituximab versus standard CHOP plus rituximab for the treatment of diffuse large B-cell lymphoma (LNH03-2B): an open-label randomised phase 3 trial. The Lancet 2011;378:1858–67. 10.1016/S0140-6736(11)61040-4 [DOI] [PubMed] [Google Scholar]
- 5. Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure—what to do? Hematology 2016;2016:366–78. 10.1182/asheducation-2016.1.366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benmebarek M-R, Karches C, Cadilha B, et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 2019;20:1283 10.3390/ijms20061283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44. 10.1056/NEJMoa1707447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med 2019;380:45-56 10.1056/NEJMoa1804980 [DOI] [PubMed] [Google Scholar]
- 9. Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med 2017;377:2545–54. 10.1056/NEJMoa1708566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Locke FL, Ghobadi A, Jacobson CA, et al. Long-Term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 2019;20:31–42. 10.1016/S1470-2045(18)30864-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weinkove R, George P, Dasyam N, et al. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol 2019;8:e1049 10.1002/cti2.1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karlsson H, Svensson E, Gigg C, et al. Evaluation of intracellular signaling downstream chimeric antigen receptors. PLoS One 2015;10:e0144787 10.1371/journal.pone.0144787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramos CA, Rouce R, Robertson CS, et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin's Lymphomas. Mol Ther 2018;26:2727–37. 10.1016/j.ymthe.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A 2009;106:3360–5. 10.1073/pnas.0813101106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 2015;28:415–28. 10.1016/j.ccell.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai Y, Weng J, Wei X, et al. Toll-Like receptor 2 costimulation potentiates the antitumor efficacy of CAR T cells. Leukemia 2018;32:801–8. 10.1038/leu.2017.249 [DOI] [PubMed] [Google Scholar]
- 17. Komai-Koma M, Jones L, Ogg GS, et al. Tlr2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A 2004;101:3029–34. 10.1073/pnas.0400171101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res 2009;45:25–36. 10.1007/s12026-009-8113-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chapman NM, Bilal MY, Cruz-Orcutt N, et al. Distinct signaling pathways regulate TLR2 co-stimulatory function in human T cells. Cell Signal 2013;25:639–50. 10.1016/j.cellsig.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cottalorda A, Mercier BC, Mbitikon-Kobo FM, et al. TLR2 engagement on memory CD8 + T cells improves their cytokine-mediated proliferation and IFN-γ secretion in the absence of Ag. Eur J Immunol 2009;39:2673–81. 10.1002/eji.200939627 [DOI] [PubMed] [Google Scholar]
- 21. Mercier BC, Cottalorda A, Coupet C-A, et al. Tlr2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. The Journal of Immunology 2009;182:1860–7. 10.4049/jimmunol.0801167 [DOI] [PubMed] [Google Scholar]
- 22. Weng J, Lai P, Qin L, et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J Hematol Oncol 2018;11:25 10.1186/s13045-018-0572-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Enblad G, Karlsson H, Gammelgård G, et al. A phase I/IIa trial using CD19-Targeted third-generation CAR T cells for lymphoma and leukemia. Clinical Cancer Research 2018;24:6185–94. 10.1158/1078-0432.CCR-18-0426 [DOI] [PubMed] [Google Scholar]
- 24. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019;25:625–38. 10.1016/j.bbmt.2018.12.758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. JCO 2014;32:3059–67. 10.1200/JCO.2013.54.8800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neelapu SS, Tummala S, Kebriaei P, et al. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit 'ALL'. Nat Rev Clin Oncol 2018;15:218 10.1038/nrclinonc.2018.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Declaration of Helsinki Assembly tWMAG. Edinburgh, Scotland: World Medical Association, Inc, 2008. [Google Scholar]
- 28. Hay KA, Hanafi L-A, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017;130:2295–306. 10.1182/blood-2017-06-793141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hay KA. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br J Haematol 2018;183:364–74. 10.1111/bjh.15644 [DOI] [PubMed] [Google Scholar]
- 30. Brudno JN, Kochenderfer JN. Recent advances in car T-cell toxicity: mechanisms, manifestations and management. Blood Rev 2019;34:45–55. 10.1016/j.blre.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Turtle CJ, Hanafi L-A, Berger C, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016;8:355ra116 10.1126/scitranslmed.aaf8621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. JCO 2017;35:1803–13. 10.1200/JCO.2016.71.3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.