

Summary
Sleep reactivity is the trait-like degree to which stress exposure disrupts sleep, resulting in difficulty falling and staying asleep. Individuals with highly reactive sleep systems experience drastic deterioration of sleep when stressed, whereas those with low sleep reactivity proceed largely unperturbed during stress. Research shows that genetics, familial history of insomnia, female gender and environmental stress influence how the sleep system responds to stress. Further work has identified neurobiological underpinnings for sleep reactivity involving disrupted cortical networks and dysregulation in the autonomic nervous system and hypothalamic-pituitary-adrenal axis. Sleep reactivity is most pathologically and clinically pertinent when in excess, such that high sleep reactivity predicts risk for future insomnia disorder, with early evidence suggesting high sleep reactivity corresponds to severe insomnia phenotypes (sleep onset insomnia and short sleep insomnia). High sleep reactivity is also linked to risk of shift-work disorder, depression and anxiety. Importantly, stress-related worry and rumination may exploit sensitive sleep systems, thereby augmenting the pathogenicity of sleep reactivity. With the development of cost-effective assessment of sleep reactivity, we can now identify individuals at risk of future insomnia, shift-work disorder and mental illness, thus identifying a target population for preventive intervention. Given that insomniacs with high sleep reactivity tend to present with severe insomnia phenotypes, patient sleep reactivity may inform triaging to different levels of treatment. Future research on sleep reactivity is needed to clarify its neurobiology, characterize its long-term prospective associations with insomnia and shift-work disorder phenotypes, and establish its prognostic value for mental illness and other non-sleep disorders.
Keywords: Ford insomnia response to stress test, mental health, preventive treatment
1 |. INTRODUCTION
Insomnia disorder affects 7% of adults in the EU (Wittchen et al., 2011) and 9–20% of adults in the USA (Morin, LeBlanc, Daley, Gregoire, & Merette, 2006; Roth et al., 2011), with incidence rates at 7–10% in the USA (LeBlanc et al., 2009; Morin et al., 2006; Singareddy et al., 2012). Upwards of 37% of adults in the UK complain of insomnia (Morphy, Dunn, Lewis, Boardman, & Croft, 2007), whereas 19% of adults in France report the disorder (Leger, Guilleminault, Dreyfus, Delahaye, & Paillard, 2000). Indeed, the insomnia epidemic is reflected by high prevalence rates across the globe (Cho et al., 2009; Kim, Uchiyama, Okawa, Liu, & Ogihara, 2000; Panda et al., 2012; Xiang et al., 2008). Insomnia disorder has devastating effects on psychological (Baglioni et al., 2011; Riemann & Voderholzer, 2003) and cardiometabolic health (Palagini et al., 2013; Vgontzas, Fernandez-Mendoza, Liao, & Bixler, 2013) and heavily burdens the economy (Daley, Morin, LeBlanc, Grégoire, & Savard, 2009). Although pharmacological and cognitive-behavioural treatments for insomnia have proven efficacy and may limit morbidity (Drake, Roehrs, & Roth, 2003; Koffel, Koffel, & Gehrman, 2015; Okajima, Komada, & Inoue, 2011; Riemann & Perlis, 2009; Roehrs & Roth, 2012; Smith et al., 2002), relapse rates are high (Morin et al., 2009) and the field has struggled to develop preventive interventions. Disrupting the progression of insomnia is naturally difficult while its pathophysiology remains enigmatic, but recent advances using stress-diathesis models of insomnia have offered critical insights into its disease processes.
The stress response is highly complex, as highlighted by identical stressors of equal potency eliciting different reactions across individuals (Ellis, Jackson, & Boyce, 2006; Pacak & Palkovits, 2001; Selye, 1985). Indeed, the differential resilience of various psychoneurobiological systems to the ravages of stress exposure moderates vulnerability to many medical and psychological illnesses (Kobasa, Maddi, & Courington, 1981; Pacak & Palkovits, 2001; Selye, 1985). In other words, stress-related pathologies often manifest in accordance with the constitutionally weak systems being overwhelmed by pathogenic stress. It is thus crucial to identify and characterize underlying mechanisms of vulnerable systems. One such process is the degree to which a given amount of stress (cognitive, physiological, etc.) disrupts the sleep system, which has been described as sleep reactivity (Drake, Pillai, & Roth, 2013). A burgeoning body of research points to sleep reactivity as a key aetiological vulnerability to insomnia and possibly other sleep and stress-related disorders. In this review, we discuss the concept of sleep reactivity, its aetiological role in sleep–wake disorders and other areas of disease, its neurobiological bases, and relationship with cognitive-emotional processes.
2 |. SEARCH STRATEGY AND SELECTION CRITERIA
A literature search was conducted for all articles published before and including December 2017 using the search terms ‘sleep reactivity’, ‘stress-related sleep disturbance’ and ‘stress’, along with ‘sleep’ and ‘Ford insomnia response to stress test’ in the PubMed and Google Scholar databases. Additional references were identified by reviewing manuscript reference sections and publications by leading experts in the field of stress and insomnia.
3 |. STRESS AND SLEEP
Stress-diathesis theories constitute the most empirically robust aetiological models of many forms of pathology, including depression, post-traumatic stress disorder, schizophrenia, cardiovascular disease, infectious disease (Hammen, 2005; Hill, 2001; McKeever & Huff, 2003; Ordovas et al., 2002; Walker & Diforio, 1997) and, of course, insomnia and other sleep and circadian pathologies (Drake & Wright, 2011; Morin, Rodrigue, & Ivers, 2003; Riemann et al., 2015; Singareddy et al., 2012). The literature on stress and sleep is as vast as it is rich, and its comprehensive analysis deserves its own review. Here, we broadly describe the stress–sleep relationship to serve as a backdrop for a more in-depth review of sleep reactivity. For more thorough examinations of stress and sleep, please refer to reviews on stress dysregulation in sleep (Basta, Chrousos, Vela-Bueno, & Vgontzas, 2007; Buckley & Schatzberg, 2005; Meerlo, Sgoifo, & Suchecki, 2008; Steiger, 2002) and the neurobiology of insomnia (Feige et al., 2013; Palagini, Biber, & Riemann, 2014; Riemann et al., 2010, 2015) and short sleep insomnia (Vgontzas et al., 2013), as well as investigations on structural brain abnormalities in insomnia (Riemann et al., 2007; Stoffers et al., 2012, 2013, 2014; Van Someren et al., 2013).
Overall, the literature presents a dynamic and complex relationship between stress and sleep, but one that is unequivocally dysbiotic, wherein exposure to stressful events (such as major life events and daily hassles) impairs normal sleep function (Bastien, Vallieres, & Morin, 2004; Friedman, Brooks, Bliwise, Yesavage, & Wicks, 1995; Hall et al., 2000, 2007, 2008; Healey et al., 1981; LeBlanc et al., 2009; Mezick et al., 2009; Morin et al., 2003; Pillai, Roth, Mullins, & Drake, 2013). Even so, given the complicated inner workings of stress regulation, individuals do not exhibit uniform sleep responses to stress, either in manifestation or severity (Bonnet & Arand, 2003; Drake, Richardson, Roehrs, Scofield, & Roth, 2004). Indeed, evidence roundly rejects a uniform dose–response relationship across individuals (Drake et al., 2013; Morin et al., 2003; Pillai et al., 2013; Sadeh, Keinan, & Daon, 2004). Rather, the impact of a given dose of stress on sleep includes a differential sleep response across individuals (Bonnet & Arand, 2003). Myriad factors, including (but not limited to) event appraisal (Morin et al., 2003), stressor chronicity (Pillai et al., 2013), coping behaviour (Morin et al., 2003; Pillai et al., 2013; Sadeh et al., 2004), cognitive-emotional and psychological defects (Chung, Chang, Yang, Kuo, & Hsu, 2009; Fortunato & Harsh, 2006; Morin et al., 2003; Pillai & Drake, 2014; Pillai et al., 2013; Williams & Moroz, 2009; Zoccola, Dickerson, & Lam, 2009), and substance misuse (Pillai et al., 2013), impact the sleep response to stress (see Figure 1). However, it was unknown until relatively recently whether a sleep-specific trait vulnerability, potentially distinct from these cognitive, emotional and behavioural processes, represented an individual predisposition to insomnia from a broad array of triggers, including several of those listed above.
FIGURE 1.

Conceptual diagram depicting moderators of the effect of stress on sleep disturbance. Negative event appraisal, greater stressor chronicity, poor coping, greater substance misuse and negative cognitive-emotional factors increase the risk of and severity of sleep disturbance following a stressor
It has long been held that insomnia begins with an organic predisposition towards poor sleep and wake-promoting hyperarousal (Borkovec, 1982). Indeed, insomnia disorder is often triggered by stressful life events, and physiologically and cognitive-emotionally induced hyperarousal can interfere with sleep and lead to chronic insomnia in a subset of the population (Drake et al., 2013; Ellis, Gehrman, Espie, Riemann, & Perlis, 2012; Singareddy et al., 2012). Although predisposition to insomnia may include broad cognitive-emotional underpinnings, a highly specific and independent physiological vulnerability to exaggerated sleep responses to stress may be equally or perhaps even more so germane to insomnia aetiology and course.
The substantial differences across individuals in risk of insomnia following stress exposure highlight the moderating influence of endogenous neurobiological vulnerabilities. An important goal of current sleep research is thus the identification of premorbid vulnerabilities to insomnia, and the delineation of their interaction with environmental triggers. Individuals with insomnia exhibit exaggerated neurobiological and cognitive-emotional reactivity to stress (Baglioni, Spiegelhalder, Lombardo, & Riemann, 2010; Drake & Roth, 2006; Drake et al., 2004; Fernández-Mendoza et al., 2010; Morin et al., 2003; Riemann et al., 2010). Until recently it was unclear whether this exaggerated stress reactivity had a sleep-specific component (see Figure 2) and, if so, whether this exaggerated reactivity preexisted or was a consequence of insomnia disorder (Palagini et al., 2014). Although growing evidence suggests that stress dysregulation typically preexists and even contributes to insomnia development, it had been unclear until recently whether this dysregulation produces a uniquely sleep-specific vulnerability or rather a broad cognitive-emotional response that can impact a myriad of stress-linked physiological systems, such as sleep regulation, the autonomic nervous system, cardiovascular regulation and gastrointestinal function (Drake et al., 2013; Fernández-Mendoza et al., 2010). In recent years, research has identified that a sleep-specific component of stress reactivity exists, and the degree to which stress reactivity manifests in the sleep system has been referred to as situational insomnia (Bonnet & Arand, 2003) and, now more commonly, sleep reactivity (Drake et al., 2004).
FIGURE 2.
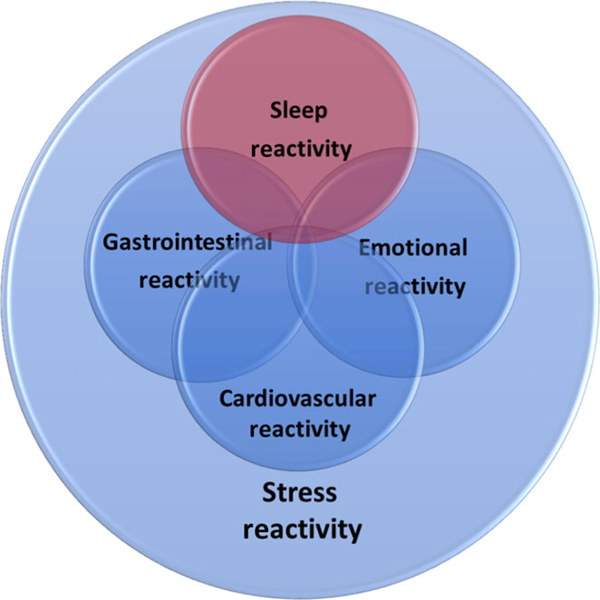
Theorized relation of sleep reactivity to other components of stress reactivity
4 |. SLEEP REACTIVITY: A CONCEPTUALIZATION
A vignette: Adrian and Addison are both laid off from work. There are presumably similar psychosocial contexts, stressor potency and cognitive reactions for both former employees, but even so Adrian begins having significant difficulty falling and staying asleep at night. In contrast, Addison has a couple of mildly disturbed nights, but overall continues to sleep reasonably well. Here, Addison’s largely unperturbed sleep reflects a robust sleep system resilient to the saprogenic cognitive and non-sleep effects of stress (e.g. increased heart rate, gastrointestinal disturbance, etc.). By comparison, the system regulating sleep–wake function in Adrian is weaker and thereby predisposed to exaggerated disruptions to its homeostatic pattern. How this develops into chronic insomnia disorder can involve additional responses and vulnerabilities (cognitive, behavioural, etc.), but it is this initial sleep-specific vulnerability to acute sleep disruption that characterizes Adrian’s high level of sleep reactivity and risk of insomnia disorder.
In its most simple conceptualization, sleep reactivity is the degree to which a stressor (broadly defined) disrupts sleep; behaviourally, sleep reactivity is the degree to which individuals exhibit acute sleep-disruptive responses to stress. Other terms used to describe sleep reactivity include sleep system sensitivity, stress reactivity of the sleep system and vulnerability to stress-related insomnia. It is important to emphasize here that sleep reactivity is a normal phenomenon, as all individuals experience some degree of difficulty sleeping when faced with certain sleep challenges. Even mild challenges, including unfamiliar sleep environments, small circadian shifts and ill-timed low-dose stimulants, produce transient sleep difficulties in many individuals (Bonnet & Arand, 2003).
Individuals vary greatly in the degree to which they experience stress-related sleep problems (Bonnet & Arand, 2003; Drake et al., 2004; Jarrin, Chen, Ivers, & Morin, 2014). Highly reactive sleepers experience drastic deterioration of sleep when stressed, whereas those with low sleep reactivity can proceed largely unperturbed during stress. Moreover, sleep reactivity exhibits trait-like characteristics such that individuals’ sleep responses to stress are consistent over time and across a variety of stressful stimuli (e.g. caffeine, and change in bedtime or sleep environment) (Bonnet & Arand, 2003; Drake et al., 2004; Jarrin, Chen, Ivers, Drake, & Morin, 2016). By extension, sleep reactivity is most pathologically and ergo clinically pertinent when in excess, as observed in those with exaggerated and non-retractable sleep disturbances during or even following the removal of stress (Drake et al., 2013; Kalmbach, Pillai, Arnedt, & Drake, 2016b).
5 |. ROOTS IN SITUATIONAL INSOMNIA
Situational insomnia refers to poor sleep in response to specific stressful circumstances, but of course somnological responses to uniform stressors differ across individuals, thus reflecting varying degrees of sensitivity of the sleep system (Bonnet & Arand, 2003; Drake et al., 2004). Insomnia researchers proposed that exaggerated disruptions in sleep in response to stress, even prior to any history of sleep morbidity, may be a marker for vulnerability to insomnia disorder (Bonnet & Arand, 2003; Drake et al., 2004).
In a landmark study on stress response and sleep, Bonnet and Arand (2003) investigated the consistency of situational insomnia across several stressful conditions in individuals without any history of sleep or circadian pathology. The authors leveraged the well-established first night effects of polysomnography (PSG), characterized as sleep difficulty when initially exposed to overnight PSG monitoring during an adaptation night. In this study, subjects underwent a PSG adaptation night, and PSG-defined sleep efficiency (time asleep/time in bed) was used to divide the sample into good sleepers (top quartile in efficiency) and those with ‘situational insomnia’ (bottom quartile). Confirming that the insomnia was indeed situational, sleep efficiency in the situational insomnia group increased into the normal range on a second night of PSG monitoring (i.e. after the so-called first night effects have worn off). Nevertheless, subjects in the situational insomnia group exhibited lower sleep efficiency than good sleepers during subsequent nights in response to circadian phase advance (lights out 3 or 6 hr prior to habitual bedtime) and administration of 400 mg of caffeine 30 min prior to lights out. The authors pointed to reduced parasympathetic activity as a potential autonomic marker of situational insomnia. These findings suggest that stress reactivity within the sleep system is consistent across various situational stressors for a segment of historically good sleepers, and that stress-related sleep disturbances may have neurobiological underpinnings in autonomic dysregulation. Bonnet and Arand’s (2003) early work led to further research on the aetiological role of stress reactivity of the sleep system in sleep pathology; namely, insomnia disorder.
6 |. MEASURING SLEEP REACTIVITY
Given the normal distribution of insomnia in the population (Beattie, Espie, Kyle, & Biello, 2015) and its polygenic basis (Gehrman, Byrne, Gillespie, & Martin, 2011; Hammerschlag et al., 2017; Lane et al., 2017; Lind & Gehrman, 2016), individual differences in vulnerability to sleep disturbance and insomnia are likely to constitute a continuum from outright resilience to vulnerability to acute/transient sleep disturbance through overt chronic insomnia disorder. Although sleep profiles on PSG adaptation nights successfully identify individuals with high sleep reactivity, this method of assessment is hindered by exorbitant resource demands. This method is further limited to PSG-naïve individuals, and first night effects may not generalize to the myriad of potential naturalistic stressors. To address these assessment concerns, a self-report instrument was developed and validated to allow the assessment of sleep reactivity in the broad population to facilitate prospective studies of aetiology and clinical relevance.
The Ford Insomnia Response to Stress Test (FIRST) (Drake et al., 2004) is a self-report instrument designed to assess sleep reactivity. For its creation, four insomnia experts submitted a total of 27 items assessing stress-related sleep disturbance. A total of 104 subjects were asked to rate the likelihood of experiencing difficulty sleeping (1 = not likely, 2 = somewhat likely, 3 = moderately likely, 4 = very likely) in response to a number of both broad and highly specific hypothetical stressors, such as ‘Before an important meeting the next day’ or ‘After a stressful experience in the evening’ (see Drake et al., 2004 for the full instrument). Exploratory factor analysis identified a single-factor structure for the FIRST, comprised of nine items accounting for 25.7% of the variance among all 27 items. This single-factor nine-item structure has also been validated for use in early pregnancy (Gelaye et al., 2016) and translated into Spanish (Gelaye et al., 2016), Japanese (Nakajima et al., 2014), European Portuguese (Marques, Allen Gomes, Drake, Roth, & De Azevedo, 2018), French (Chen et al., 2015), German (Dieck, Helbig, Drake, & Backhaus, 2017) and Italian (Palagini et al., 2016).
7 |. SLEEP REACTIVITY MODERATES THE IMPACT OF STRESS ON SLEEP PHYSIOLOGY
7.1 |. Experimental studies
To validate the FIRST, our team tested whether self-reported sleep reactivity corresponded to objectively measured sleep reactivity (i.e. physiological changes in sleep following stress; Drake et al., 2004). In PSG-naïve normal sleepers, we compared PSG-based sleep parameters on an adaptation night between individuals with high FIRST scores and those with low FIRST scores (median split; Drake et al., 2004). Consistent with PSG first-night effects reflecting high sleep reactivity (Bonnet & Arand, 2003), we found that highly reactive sleepers, per self-report, exhibited lower PSG-based sleep efficiency (81% versus 89%), longer latency to stage 1 sleep (23 min versus 9 min) and longer latency to persistent sleep (36 min versus 16 min) than low-reactivity sleepers. Moreover, individuals with high FIRST scores showed increased daytime arousal per prolonged sleep latency on the Multiple Sleep Latency Test (MSLT) compared to individuals with low FIRST scores (13 min versus 11 min). These elevated latencies on MSLT suggest that physiological hyperarousal may override homeostatic sleep pressure in reactive individuals even during the day, and even despite disturbed sleep during the preceding night.
Importantly, these data were obtained in a sample of normal sleepers without evidence of poor sleep prior to the laboratory challenge. Thus, the insomnia-like characteristics seen in those with high FIRST scores were a departure from the habitual good sleep these individuals typically experienced. Furthermore, as the PSG adaptation night is an affectively-neutral stimulus, the exaggerated first-night effects among highly reactive sleepers are unlikely to be accompanied by significant anxiety or elevated cognitive responses, and therefore are likely to be sleep specific.
A recent study by the Morin group in Canada supported these findings by showing that higher FIRST scores were associated with longer self-reported sleep latency, higher somatic arousal and elevated heart rate in a sample of 45 good sleepers (Chen et al., 2015). In a subsample of 14 subjects, the authors examined whether self-reported sleep reactivity on the FIRST was associated with PSG-defined sleep disturbances. Results showed that highly reactive sleepers had lower sleep efficiency, longer sleep latency and more total wake time on a PSG adaptation night than non-reactive sleepers, thus further validating this conceptualization of sleep reactivity across laboratories and countries.
Self-reported sleep reactivity has also been shown to generalize broadly to various controlled stressors, including objective measures of sleep response to pharmacological challenge (Drake, Jefferson, Roehrs, & Roth, 2006). Eleven highly reactive good sleepers and 10 good sleepers with low reactivity (median split) underwent 3 nights of PSG: (a) adaptation night, (b) control night and (c) caffeine night (3 mg/kg caffeine at 1 hr prior to lights out). Highly reactive good sleepers took 47 min longer to fall asleep on the caffeine night than on the control night (65 min versus 18 min). Further, their sleep latency on the caffeine night was 40 min longer than that of the low-reactive sleepers on the caffeine night (65 min versus 25 min). Although all subjects were insomnia free, the severely prolonged sleep latency exhibited by highly reactive sleepers in response to this pharmacological sleep challenge is similar to the latency reported by chronic insomniacs (Walsh et al., 2000). It is important to highlight here that sleep latency did not differ between these two groups on the control night (17.5 ± 14.3 min versus 18.2 ± 18.8 min), indicating that high and low-reactive sleepers do not differ in sleep quality in the absence of stress. Not only do these data complement Bonnet & Arand, 2003 caffeine findings (Bonnet & Arand, 2003), but they also show that highly reactive sleepers can be identified a priori utilizing a validated self-report measure (i.e. FIRST).
7.2 |. Observational studies
Much of the knowledge on sleep reactivity to this point had been gained from laboratory studies utilizing neutrally valenced non-psychosocial stressors (i.e. first night effects, phase advance and stimulant administration).1 Although this emphasizes the independence of this construct from cognitive components of stress reactivity, Petersen et al. extended this work to include psychosocial and mental stress by examining differences in PSG responses to naturally occurring work stress between good sleepers with high and those with low sleep reactivity (above versus below a FIRST mean item score of 2.5, corresponding to FIRST ≥23 for high reactivity; Petersen, Kecklund, D’onofrio, Nilsson, & Akerstedt, 2013). PSG showed that highly reactive sleepers exhibited more sleep stage transitions and nocturnal arousals than low-reactive sleepers during high stress. These profiles suggest that highly reactive sleepers exhibit much more microlevel sleep fragmentation than low-reactivity sleepers when stressed, even in the absence of macrolevel differences in sleep efficiency.
Curiously, rapid eye movement (REM) sleep duration interacted with stress exposure (Petersen et al., 2013). Among highly reactive sleepers, REM sleep duration decreased in response to stress (low to high stress, 119 to 92 min), whereas no such decrease was observed for low-reactive sleepers when stressed (low to high stress, 91 to 97 min, non-significant). Importantly, the highly reactive sleepers exhibited decreased REM sleep and increased nocturnal arousals (particularly during REM) in response to stress, which have been proposed as sleep physiological markers of chronic insomnia (Riemann et al., 2012). Laboratory studies using animal models show that manipulated stress exposure leads to REM sleep instability in rats (i.e. short and fragmented REM sleep), especially among those with high stress reactivity (Cano, Mochizuki, & Saper, 2008; Fenzl et al., 2011; Pawlyk, Morrison, Ross, & Brennan, 2008; Revel, Gottowik, Gatti, Wettstein, & Moreau, 2009). Findings on REM sleep instability and stress dysregulation in humans, however, have been more mixed (Akerstedt, Kecklund, & Axelsson, 2007; Buckley & Schatzberg, 2005; Pillai & Drake, 2014; Vandekerckhove & Cluydts, 2010). Even so, in the case of REM decreasing during stress among highly reactive sleepers, it is possible that Petersen et al. (2013) captured stress-induced REM instability in this insomnia-vulnerable group. Although REM sleep duration did not differ between the sleep-reactivity groups under high stress, a striking observation was the long REM sleep duration among highly reactive sleepers during low stress. By extension, it is possible that a vulnerability to REM change (or REM instability) during stress is a marker for insomnia. Although the association of REM instability with sleep reactivity is consistent with the animal models of stress reactivity noted above, this isolated finding is difficult to interpret as other studies have not revealed stress-related REM abnormalities associated with high sleep reactivity (Drake et al., 2004, 2006).
Taken together, early experimental and observation studies indicated that normal sleepers, even without a history of sleep issues, can differ greatly in their sleep responses to stress. Those with high sleep reactivity had difficulty falling and staying asleep and had more microlevel sleep fragmentation during stress than those with low sleep reactivity. The question remained, however: do these highly sleep-reactive ‘normal sleepers’ have a greater risk of developing insomnia disorder in the future?
8 |. THE PATHOGENICITY OF SLEEP REACTIVITY: INSOMNIA
8.1 |. Insomnia incidence and chronicity
Early research on sleep reactivity confirmed that certain individuals are predisposed to exaggerated sleep responses to a wide range of environmental, pharmacological and psychosocial stressors. The implications of this research centred on the potential role of high trait sleep reactivity as a diathesis for sleep disorders, such that insomnia may be conceptualized as the somnological fallout and detritus of a constitutionally weak sleep system in stress-potentiated manifest.
In 2014, two prospective studies supported the role of high premorbid sleep reactivity in vulnerability to insomnia (Drake et al., 2013; Jarrin et al., 2014). In a sample of 1449 good sleepers, Jarrin et al. found that individuals with high sleep reactivity were nearly 60% more likely to develop insomnia symptoms and were twice as likely to develop chronic insomnia over the next 2 years compared with low-reactive sleepers (Jarrin et al., 2014). Importantly, the relationship between premorbid sleep reactivity and insomnia risk was independent of sleep history, depressive symptoms, and stress exposure and impact. Further, sleep reactivity predicted future insomnia while controlling for arousal predisposition, supporting sleep reactivity as a sleep-specific vulnerability that is distinct from general trait hyperarousability.
In the Evolution of Pathways to Insomnia Cohort (EPIC) study, our team investigated whether sleep reactivity predicted the incidence of insomnia disorder in a sample of 2,316 individuals without a history of DSM-IV (American Psychiatric Association, 2000) insomnia or major depression (Drake et al., 2013). The results showed that higher premorbid sleep reactivity increased the risk of developing DSM-IV insomnia disorder over the next year. Critically, this study offered the first empirical evidence showing that stress exposure exploits the fragility of highly reactive sleep systems. Firstly, an interaction between sleep reactivity and stress exposure approached significance (p = 0.06), suggesting that major life events may exploit sensitive sleep systems in highly reactive sleepers, thereby triggering the onset of insomnia. However, as noted in our overview of stress and sleep above, our appraisals of and reactions to stress often influence the sleep response (Morin et al., 2003; Pillai & Drake, 2014; Pillai et al., 2013; Sadeh et al., 2004). Here, our team showed that cognitive and emotional reactions to stress (i.e. rumination) interact with sleep reactivity to amplify the risk of developing insomnia disorder (see Figure 3; Drake et al., 2013). This interaction shows the unique and synergistic contribution to insomnia disorder of the ‘cognitive-emotional’ (appraisal, rumination, coping, etc.) and ‘sleep reactivity’-related responses to stress. That is, individuals with both corrosive psychological reactions to stress and a sensitive sleep system are at especially high risk of developing insomnia disorder. The finding that rumination intensifies the pathogenicity of stress in those with fragile sleep systems constituted the first evidence emphasizing the importance of psychological processes in the risk relationship between sleep reactivity and insomnia development.
FIGURE 3.
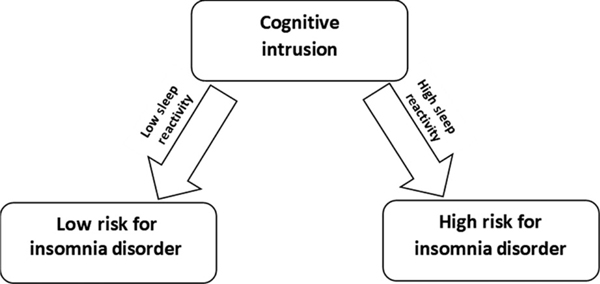
The role of sleep reactivity in the relationship between cognitive intrusion (i.e. rumination) and risk of insomnia disorder. In response to the same degree of cognitive intrusion, persons with greater sleep reactivity are more likely to develop insomnia disorder
A follow-up report from the EPIC study examined the ability of the FIRST to identify good sleepers predisposed to incident insomnia and to identify clinically relevant cut-points for the measure (Kalmbach, Pillai, Arnedt, & Drake, 2016b). In a sample of 2,892 individuals without a history of DSM-IV (American Psychiatric Association, 2000) insomnia disorder or major depression, cut-offs for the FIRST were established using ROC curve analysis to identify good sleepers at elevated risk of incident insomnia disorder. Where prior studies had used sample medians to split subjects into high versus low sleep-reactivity groups, this study statistically derived normative cut-offs for elevated risk for 1-year incidence of insomnia in a healthy sample.
Analyses revealed an optimal cut-off of FIRST ≥ 18, which will be referred to henceforth as the high-risk cut-point. The high-risk cut-point of FIRST ≥ 18 had 62% sensitivity and 67% specificity for predicting 1-year incidence of insomnia, and 15.7% of these individuals developed insomnia disorder within a year, nearly double the annual incidence rate of 9.1% for the full sample. However, because preventive efforts may benefit from a more sensitive test, we examined additional potential cut-points indicating the risk of insomnia with greater sensitivity. Results showed that a FIRST score of 16 or higher conferred the second optimal cut-point in our good-sleeping, non-depressed sample, as appropriate for preventative interventions. This moderate-risk cut-point of FIRST ≥ 16 had 77% sensitivity and 50% specificity for predicting insomnia onset 1 year later. Among those who scored FIRST ≥ 16 a full 13.2% developed insomnia disorder within a year, compared with the incidence rate of 9.1% for the entire sample.
Comparison of the cut-off values showed that the highly reactive sleepers (FIRST ≥ 18) were more than twice as likely to develop insomnia than those who scored in the moderate 16–17 range (16.1% versus 7.7% incidence; relative risk = 2.09). Even so, we would like to emphasize that neither cut-off should be considered universally preferable. Selection of cut-offs should be driven by research and clinical needs regarding sensitivity and specificity. Future research may elucidate the prognostic benefits associated with each respective cut-off.
8.2 |. Sleep onset insomnia
Heterogeneity in insomnia symptoms and phenotypes is likely to reflect correspondingly heterogeneous aetiological factors underlying the disorder (Benjamins et al., 2016; Pillai, Roth, & Drake, 2014; Roth, Roehrs, & Pies, 2007; Vgontzas et al., 2013). Efforts have thus explored whether insomnia phenotypes (e.g. sleep onset versus sleep maintenance) differ between individuals with high versus low sleep reactivity. Data from the EPIC study suggest that insomniacs with high premorbid sleep reactivity are two to three times more likely to have sleep onset insomnia complaints than non-reactive sleepers (Kalmbach, Pillai, Arnedt, & Drake, 2016b). Highly reactive insomniacs estimated their habitual sleep latency to be nearly twice as long as that estimated by non-reactive sleepers at disease onset (65–68 min versus 37–44 min). These findings lend important ecological validity to laboratory studies that show highly reactive sleepers struggle to fall asleep during stress (Drake et al., 2004, 2006). Further, prolonged sleep latency among highly reactive sleepers is consistent with prior evidence showing that rumination (a) is a cardinal feature of pre-sleep arousal (Baglioni et al., 2010; Harvey, 2000, 2002a; Pillai & Drake, 2014; Riemann et al., 2010, 2012), (b) is associated with sleep reactivity (Drake et al., 2013; Fernández-Mendoza et al., 2010), and (c) potentiates the effects of stress on the sleep system (Drake et al., 2013). Together, these findings suggest that one mechanism by which sleep reactivity initiates and perpetuates insomnia disorder may involve the cognitive-emotional processes during the pre-sleep period.
8.3 |. Short sleep insomnia phenotype
Short sleep insomnia is an especially severe and treatment-resistant phenotype of insomnia disorder (Bathgate, Edinger, & Krystal, 2017; Vgontzas et al., 2013). Although reference-standard classification of the short sleep phenotype is recommended via PSG (e.g. <6 hr), results from a recent study in the EPIC cohort showed that findings in insomniacs with self-reported habitual short sleep are consistent with the extant literature on insomnia with objectively measured short sleep (Kalmbach, Pillai, Arnedt, & Drake, 2016a). Underlying the aetiology of short sleep insomnia is severely dysfunctional neurobiological regulation of stress (Vgontzas et al., 2013). Despite foundations of stress dysregulation in short sleep insomnia and sleep reactivity, no study to date has examined whether sleep reactivity differs between insomniacs with short and normal sleep duration.
Thus for the purposes of this review, we revisited the data from our 2016 report on DSM-5 insomnia disorder and short sleep (Kalmbach, Pillai, Arnedt, & Drake, 2016a). We conducted post hoc analyses to test whether sleep reactivity was associated with the short sleep phenotype of insomnia in a large community sample of nonobese US adults aged 18–69 years. Specifically, we cross-sectionally compared sleep reactivity across 1,684 good sleepers with normal sleep duration, 294 insomniacs with normal sleep duration and 284 short-sleeping insomniacs (see Kalmbach, Pillai, Arnedt, & Drake, 2016a for full study details).
Short-sleeping insomniacs reported slightly higher levels of sleep reactivity than insomniacs with normal sleep duration (FIRST scores, 23.49 ± 6.16 versus 22.44 ± 5.95, t = 2.08, p < 0.04, Cohen’s d = 0.17). We then used dummy coded logistic regression to compare odds of having high sleep reactivity (FIRST ≥ 18) for insomniacs with short and normal sleep duration compared with good sleepers as the reference group, while controlling for gender effects. Unsurprisingly, both insomnia groups were more likely to have high sleep reactivity than good sleepers. Short-sleeping insomniacs had a striking 10-fold greater odds of having high sleep reactivity (OR = 9.89, 95% CI = 7.15–13.68, p < 0.001) than good sleepers, whereas insomniacs with normal sleep duration had 6–7-fold greater odds of high sleep reactivity than good sleepers (OR = 6.73, 95% CI = 5.02–9.03, p < 0.001). A follow-up logistic regression directly comparing insomnia phenotypes showed that short-sleeping insomniacs may be 49% more likely to have high sleep reactivity than insomniacs with normal sleep duration, but this finding was a statistical trend (OR = 1.49, 95% CI = 0.99–2.26, p < 0.06).
These cross-sectional findings indicate that the sleep system may be more fragile in short-sleeping insomniacs than in their longer-sleeping insomniac counterparts. However, prospective investigations are needed to characterize sleep reactivity prior to developing insomnia with short versus normal PSG sleep duration; this is especially crucial as insomnia development and stress exposure can sensitize the sleep system (i.e. increase the reactivity of the sleep system), as discussed below. Only then can we determine whether exceptionally high sleep reactivity predicts the onset of short sleep insomnia and/or develops concurrently with the disorder. Given the burgeoning literature suggesting that objectively measured short sleep in insomnia is a better indicator of biological severity and comorbidity than subjectively estimated sleep (Bathgate, Edinger, Wyatt, & Krystal, 2015; Fernandez-Mendoza et al., 2011; Vgontzas et al., 2013), future research on sleep reactivity and short sleep insomnia would greatly benefit from objective measurement of sleep duration.
8.4 |. Pathogenicity of sleep reactivity versus subthreshold insomnia
Transient difficulties in falling and staying asleep can mark a prodromal state leading to chronic insomnia (Singareddy et al., 2012). Given that sleep reactivity is associated with transient sleep problems and subthreshold insomnia (Bonnet & Arand, 2003; Drake et al., 2004), it is important for aetiological research to disentangle the influences of sleep reactivity versus acute sleep disturbance.
Jarrin et al. (2014) provided the first evidence of independence between sleep reactivity and subthreshold insomnia. In a prospective investigation, sleep reactivity predicted future insomnia, independent of prior difficulties in sleeping. More importantly, sleep reactivity predicted future subthreshold insomnia symptoms, suggesting that self-reported sleep reactivity is independent from and is even predictive of future subthreshold or transient insomnia complaints. Moreover, insomnia symptoms fluctuate and are thereby limited as a prognostic marker of insomnia disorder by their very symptomatic ebbs and flows, whereas sleep reactivity is a non-state-dependent trait-like marker of insomnia vulnerability (Drake, Friedman, Wright, & Roth, 2011; Drake, Scofield, & Roth, 2008).
In addition to predicting the degree of the transient sleep response to stress, among those with subthreshold insomnia, trait sleep reactivity influences whether sleep difficulties remain transient or become syndromal and rather independent from acute stress exposure (see Figure 4). Within individuals who reported nocturnal symptoms of insomnia but did not meet diagnostic criteria as a result of not experiencing daytime impairment, future insomnia disorder was two to three times more likely to develop in highly reactive poor sleepers than in non-reactive poor sleepers (Kalmbach, Pillai, Arnedt, & Drake, 2016b). Although it is quite normal to experience transient sleep difficulties (Ohayon, 2002), individuals with sensitive sleep systems may be especially vulnerable to minor disruptions in sleep function that spin out of control and evolve into something vicious and pathological.
FIGURE 4.
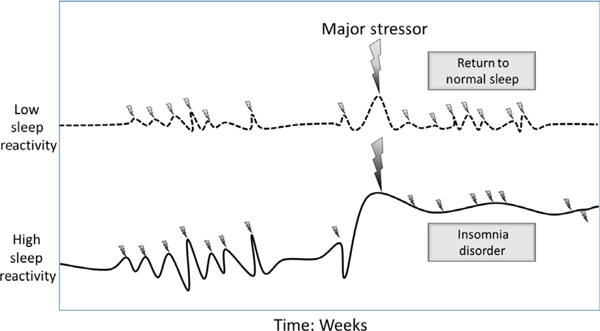
The role of sleep reactivity in the impact of stress on sleep. Although stress disturbs sleep in persons with both low and high sleep reactivity, those with high sleep reactivity experience relatively greater sleep disturbance. Moreover, whereas persons with low sleep reactivity return to normal sleep following a normal stressor, those with high sleep reactivity are less likely to experience such a reprieve, and are thus more likely to develop insomnia disorder
8.5 |. Pathogenicity of sleep reactivity versus family history
Family history of insomnia has been long viewed as a reliable indicator of insomnia risk, with maternal history as arguably the most robust predictor (Bastien & Morin, 2000; Dauvilliers et al., 2005; Singareddy et al., 2012). With maternal history as a reference standard prognostic marker, we compared the risk of insomnia between sleep reactivity and self-reported parental insomnia history in the EPIC cohort (Kalmbach, Pillai, Arnedt, & Drake, 2016b). Notably, history of paternal insomnia was not associated with risk of insomnia. However, in support of prior work, good sleepers with maternal histories of insomnia were at elevated risk of insomnia. Even so, sleep reactivity, as measured by the FIRST, was a stronger predictor of incident insomnia than maternal history. Specifically, the effect sizes for the FIRST clinical cut-points (moderate risk, ≥16; high risk, ≥18) were twice as large as the effect sizes associated with subject-reported history of maternal insomnia.
It is important to note here that more than one-third of our sample indicated not knowing whether or not their biological mother or father had a history of insomnia. This is a serious limitation that critically restricts the utility of solely relying on patient- or subject-reported biological family history as an indicator of risk of insomnia. Further, substantial method error is likely to be involved when individuals report on family history of clinical sleep disorders, as most individuals are not trained to identify these disorders and may not be privy to family members’ health histories.
8.6 |. Sensitization of the sleep system
Evidence widely supports trait-like characteristics for sleep reactivity. Specifically, individuals’ sleep responses to stress exposure are fairly consistent across stressful conditions (Bonnet & Arand, 2003; Drake et al., 2004), and self-reported sleep reactivity is stable over weeks (Drake et al., 2004) and even months (Jarrin et al., 2016). Even so and perhaps contrary to some conventional thought, trait factors are not necessarily immutable and may even alter under certain conditions. Several researchers have emphasized that exposure to stress and even to insomnia itself causes changes in the regulation of stress and sleep (Meerlo et al., 2008; Palagini et al., 2014; Riemann et al., 2010). As a result, the conditions under which the initial episode of insomnia develops may differ from the conditions that perpetuate its course or give rise to recurrent episodes. By extension, this framework offers insight into the robustness of personal history of insomnia as a predictor of future sleep and mood pathology (Baglioni et al., 2011; Fernandez-Mendoza et al., 2012; Riemann & Voderholzer, 2003; Singareddy et al., 2012), even following insomnia remission (Fernandez-Mendoza et al., 2012); that is, stress exposure and disease development fundamentally and functionally change the very ways our minds and bodies regulate sleep and stress.
In the EPIC cohort, we investigated whether sleep reactivity changed in response to insomnia development and exposure to major life stress (Kalmbach, Pillai, Arnedt, Anderson, & Drake, 2016). Specifically, we followed 262 subjects from the community who were insomnia and depression free at baseline but went on to develop DSM-IV insomnia disorder 1 year later. We found that sleep reactivity increased substantially in insomniacs with low baseline sleep reactivity, such that 41 of 60 were reclassified at insomnia onset as having elevated sleep reactivity (FIRST ≥ 16). In addition, exposure to major life stress predicted greater increases in sleep reactivity between baseline and insomnia onset; this finding is consistent with prior research showing that stress exposure erodes the ability to regulate stress and creates a neurotoxic environment, a process demonstrated in both human studies and animal models (Lupien, Mcewen, Gunnar, & Heim, 2009; Palagini et al., 2014). Importantly, sleep reactivity did not return to pre-insomnia baseline levels even after disease remission, indicating that stress exposure and insomnia development have a scarring effect on the sleep system resulting in sensitization, which cannot be attributed to mere state-dependent fluctuations in sleep reactivity. By comparison, insomniacs with high premorbid sleep reactivity did not show any significant changes in sleep reactivity, irrespective of an acute (1 year, then remission) or chronic (2 years) course for insomnia.
These findings strongly support sensitization of the sleep system in response to stress exposure. It is important to emphasize that insomnia itself has been conceptualized as a stressor that ravages systems regulating stress and sleep (Palagini et al., 2014). Importantly, sleep system sensitization is consistent with models of stress sensitization and neurotoxicity in depression. These models propose that aetiological factors evolve in response to development of stress and depression, such that chronic stress and additional episodes of depression increase the risk of recurrence and chronicity by sensitizing the stress-response system (Kendler, Thornton, & Gardner, 2000; Lupien et al., 2009; Monroe & Harkness, 2005). Stress sensitization effects are most pronounced in individuals at low genetic risk of pathology, whereas individuals with high genetic risk have been described as pre-kindled; that is, those with high genetic risk are born sensitized to the disorder (in line with the vulnerability hypothesis; Lupien et al., 2009), whereas those with low genetic risk become sensitized over time if strong environmental stressors wear down psycho-neurobiological stress systems. Sleep system sensitization very closely mirrors stress sensitization in depression, such that the preponderance of individuals who develop the disease (either insomnia or depression) carry a high genetic load and may be described as pre-kindled (i.e. inherited fragile sleep and stress systems), whereas a smaller proportion of individuals with low genetic risk go on to develop insomnia nevertheless and in the process undergo critical changes as a result of pathogenic stress corroding sleep-regulating and stress-regulating systems.
9. |. THE PATHOGENICITY OF SLEEP REACTIVITY: SHIFT WORK DISORDER
Disruptions in sleep–wake patterns can reflect normative adjustment to shift work and resultant circadian misalignment (Drake & Wright, 2011; Schwartz & Roth, 2006). However, some workers are unable to sufficiently adjust to shift-work schedules and are at risk of developing shift work disorder (SWD). SWD is marked by chronic and severe sleep–wake disturbances in response to the misalignment between work-enforced sleep schedules and endogenous timing of the circadian rhythm. The cardinal features of SWD are sleep disturbance/insomnia complaints during the sleep period and/or excessive sleepiness during the wake period in accordance with the mismatch between internal rhythmicity and external demands (American Academy of Sleep Medicine, 2014).
9.1 |. Incidence after transitioning to rotating shifts
Understandably, much of the work on SWD aetiology and symptomatology has focused largely on circadian misalignment. By extension, well-characterized risk factors for SWD centre on circadianrelated and chronotype-related influences, such as morning preference (early chronotype) and genetic variants known to segregate with SWD and other circadian pathologies (Archer et al., 2003; Dijk & Archer, 2010; Hilliker, Muehlbach, Schweitzer, & Walsh, 1992; Kalmbach et al., 2016). However, as noted above, insomnia complaints are a key feature of SWD (American Academy of Sleep Medicine, 2014), but nevertheless insomnia risk factors have received little attention in SWD development. Given the evidence showing that individuals with high sleep reactivity are prone to stress-related sleep disturbance in response to circadian challenges (Bonnet & Arand, 2003), we evaluated whether high versus low reactive sleepers differed in their response to shift-work-related changes in sleep–waking timing.
Using the transition to rotating shift work as a naturalistic exposure to enforced circadian misalignment, we conducted the only study we know of to date that focuses on sleep reactivity as a vulnerability to circadian rhythm sleep disorders. In a sample of 96 healthy individuals from the EPIC study, we prospectively investigated premorbid sleep reactivity as a vulnerability to incident SWD in response to starting a rotating work schedule (Kalmbach, Pillai, Chen, Arnedt, & Drake, 2015). We observed an overall incidence rate of 18.8% (n = 18/96) for SWD after transitioning to rotating shifts. Notably, 88.9% (n = 16/18) of those with SWD had elevated sleep reactivity prior to starting shift work. In fact, rates of SWD were nearly seven times higher among highly reactive sleepers than non-reactive sleepers (incidence rates, 30.8% versus 4.5%). Consistent with prior findings on prolonged sleep latency among highly reactive sleepers (Drake et al., 2004; Kalmbach, Pillai, Arnedt, & Drake, 2016b), here we found that shift workers with SWD (89% highly reactive) reported sleep latency nearly twice as long as normal-sleeping shift workers (54 min versus 27 min). Not only do these findings extend the diathetic role of sleep reactivity from insomnia to SWD, thereby identifying a trait characteristic to screen for individuals at risk of SWD, but also these findings lend ecological support for previous studies of heightened sensitivity to circadian challenges in highly reactive sleepers (Bonnet & Arand, 2003). Furthermore, caffeine is commonly used by shift workers to combat nocturnal sleepiness and improve vocational function (Åkerstedt & Wright, 2009). Given the high rates of sleep reactivity among individuals who develop SWD (Kalmbach et al., 2015), coupled with the exaggerated sleep responses of highly reactive sleepers to caffeine (Drake et al., 2006), these data suggest that highly reactive sleepers could potentially increase their risk of sleep disturbance and SWD when relying on caffeine and other stimulants to combat fatigue and sleepiness during shift work.
9.2 |. Insomnia versus sleepy versus mixed phenotypes
Three phenotypes of SWD exist consistently with diagnostic criteria: (a) the insomnia phenotype (insomnia during the sleep period, but normal wakefulness during the wake period), (b) the sleepy phenotype (excessive sleepiness during wake periods, no insomnia during the sleep period) and (c) mixed (Gumenyuk, Belcher, Drake, & Roth, 2015). Because of under-representation of non-reactive SWD workers in our sample (n = 2/18), we were unable to empirically compare SWD symptoms between subgroups of reactive and non-reactive SWD sufferers. Consequently, we can only mix extrapolation with conservative speculation to posit that high sleep reactivity is likely to be a particularly germane vulnerability for the insomnia and perhaps mixed phenotypes of SWD, rather than the sleepy phenotype, especially as prior evidence suggests that highly reactive sleepers exhibit wake-promoting hyperarousal during wake periods even after disrupted sleep (Drake et al., 2004). By extension, the aetiology and ergo preventive interventions for the SWD insomnia and mixed phenotypes (with heightened stress-reactivity of the sleep system as a key vulnerability) may differ greatly from those associated with the SWD sleepy phenotype.
10 |. THE PATHOGENICITY OF SLEEP REACTIVITY: PSYCHIATRIC ILLNESS
10.1 |. Depression
Four studies support two non-mutually-exclusive conceptualizations of the risk relationship between sleep reactivity and depression: (a) excessive sleep reactivity directly increases the risk of depression and (b) the pathogenic effects of sleep reactivity on depression are largely mediated by sleep disturbances.
A report from the EPIC study in 2013 first examined sleep reactivity as a diathesis of clinical depression and provided support for both direct and indirect influences of sleep reactivity on incident depression (Drake et al., 2013). Among 2,316 US adults without history of DSM-IV insomnia disorder or major depression, higher levels of premorbid sleep reactivity were associated with risk of incident depression 2 years later (OR = 1.67, p < 0.01). However, a significant proportion of sleep reactivity’s influence on development of depression at 2 years post-baseline was mediated by incident insomnia disorder at 1 year post-baseline.
Findings from the Colorado Longitudinal Twin Study and Community Twin Study provided further support for both direct and indirect aetiological influences of sleep reactivity on depression (Vargas, Friedman, & Drake, 2015). In 2,250 young US adults, sleep reactivity was significantly and positively associated with concurrent symptoms of depression. Further, the relationship between sleep reactivity and depression was partly mediated by concurrent insomnia. Although these findings are methodologically limited by their cross-sectional nature, these findings add support for direct and indirect relationships between sleep reactivity and depression and are consistent with prospective evidence (Drake et al., 2013).
In a follow-up report from the EPIC study on longitudinal changes in sleep reactivity coinciding with insomnia development (i.e. sleep system sensitization, as described above; Kalmbach, Pillai, Arnedt, Anderson et al., 2016), we found that premorbid sleep reactivity at baseline predicted depression severity at 2 years post-baseline. Perhaps more interestingly, however, there was a subgroup of insomniacs who did not have high sleep reactivity prior to developing insomnia. For these individuals with low premorbid sleep reactivity, we found that stress exposure and insomnia development caused increases in sleep reactivity, suggesting that exposure to stress sensitizes the sleep system, particularly among those without this premorbid vulnerability. Moreover, our evidence suggested that sleep system sensitization is directly associated with severity of depression at the onset of insomnia disorder and even a year later; that is, greater increases in sleep reactivity that occur in response to stress and insomnia development result in greater severity of depression during insomnia (Kalmbach, Pillai, Arnedt, Anderson et al., 2016).
Although prior studies have examined the role of insomnia in the risk relationship between sleep reactivity and depression, one report from the EPIC study has examined SWD as a mediator between sleep reactivity and depression (Kalmbach et al., 2015). In this previously described prospective study of 96 rotating shift workers, highly reactive sleepers reported greater depressive symptoms after transitioning to shift work, but this relationship was largely mediated by incident SWD (Kalmbach et al., 2015).
10.2 |. Anxiety
Three studies have investigated the relationship between sleep reactivity and anxiety (two prospective). In a sample of 338 Japanese adults, sleep reactivity (measured by the FIRST) was associated with trait anxiety in both good sleepers and insomniacs (Nakajima et al., 2014). Among 262 adults from the EPIC study who developed insomnia disorder 1 year after baseline, premorbid sleep reactivity was significantly associated with anxiety symptoms at 1 year follow-up (Kalmbach, Pillai, Arnedt, Anderson et al., 2016). Further, and again mirroring depression results, sleep system sensitization (i.e. increases in sleep reactivity associated with stress exposure) was associated with anxiety severity both at insomnia onset and a year later. In another report from the EPIC study on SWD, premorbid sleep reactivity was not directly associated with anxiety symptoms when individuals transitioned to rotating shift work (Kalmbach et al., 2015). However, and again similar to depression, there was an indirect effect wherein greater premorbid sleep reactivity predicted higher shift work-related anxiety, but this effect was mediated by the onset of SWD.
11 |. THE NEUROBIOLOGY OF SLEEP REACTIVITY
Although evidence for sleep reactivity as a phenomenon and diathesis for stress-related illnesses is unequivocally clear, its mechanistic foundations remain mysterious. Growing evidence points to the neurobiological and potentially psychological underpinnings that contribute to individual differences in sleep system sensitivity. Here, we discuss some identified bases and correlates as well as peripheral psychological factors of sleep reactivity that may contribute to its pathogenicity.
11.1 |. Neurobiological bases
Given the heritability of sleep–wake characteristics and insomnia (Gehrman, Byrne et al., 2011; Gehrman, Meltzer et al., 2011; Hammerschlag et al., 2017; Lane et al., 2017; Lind & Gehrman, 2016; Pack, Keenan, Byrne, & Gehrman, 2016; Palagini et al., 2014), and prognostic value of familial insomnia (Bastien & Morin, 2000; Dauvilliers et al., 2005; Kalmbach, Pillai, Arnedt, Anderson et al., 2016; Singareddy et al., 2012), it is not surprising that early evidence points to a substantial genetic influence on sleep reactivity. The first data to support heritability examined familial aggregation for self-reported sleep reactivity on the FIRST among 23 insomnia-free sibling pairs (Drake et al., 2008). Analyses estimated that 37.2% of sleep reactivity is accounted for by familial aggregation after controlling for age, gender, work schedules and psychiatric history. In 2011, heritability estimates for sleep reactivity were 29% for women and 43% for men using a sample of 988 monozygotic and 1,086 dizygotic twins from the Colorado Longitudinal Twin Study and Community Twin Study. Notably, few sex differences in the genetic architecture of sleep reactivity were observed, and genetic contribution was similar for men and women. Rather, the sex difference in heritability estimates differed largely because of a greater environmental influence on sleep reactivity for women. It is critical to highlight here that this study showed that women report substantially higher levels of sleep reactivity than men, despite similar genetic architecture. In 2014, the sleep team at Penn State provided corroborating evidence in the most thorough examination of sleep reactivity heritability in a family study (Fernandez-Mendoza, Shaffer et al., 2014). From 135 nuclear families (including one mother, one father and one progeny), sleep reactivity heritability was estimated at 29%, and progeny of highly reactive parents had 3–7-fold odds for also having high sleep reactivity. Taken together, these three studies offer strong empirical support for the genetic inheritance of sleep reactivity.
Importantly, genetic architecture probably only tells a portion of the story, as epigenetics (i.e. the expression of genes) is likely to contribute a large part to sleep reactivity. For a thorough review on epigenetics in sleep and insomnia, please refer to Palagini et al.’s (2014) exploration of the topic. These authors emphasized that stress can alter gene expression and brain plasticity. They posited, by extension, that stress and insomnia onset (as a stressor itself) may perpetuate the course of insomnia via these neurobiological changes in systems regulating the stress-response and sleep function. This theory is consistent with evidence supporting a substantial environmental influence on sleep reactivity (Brummett et al., 2007; Drake et al., 2011). Data showing that stress exposure increases sleep system sensitivity among insomniacs with low premorbid reactivity (Kalmbach, Pillai, Arnedt, Anderson et al., 2016) may reflect changes in epigenetically controlled regulation of stress and sleep. As sensitization of the sleep system is not reversed after insomnia remission (Kalmbach, Pillai, Arnedt, Anderson et al., 2016), the sensitization process is consistent with a scarring effect on the sleep system.
Overall, little research has been conducted on the neurobiological underpinnings of sleep reactivity. Even so, potential mechanisms can be gleaned from what we know about the neurobiology of sleep disturbance and insomnia (please refer to reviews by Baglioni et al., 2010; Basta et al., 2007; Bonnet & Arand, 2010; Buysse et al., 2012; Harvey et al., 2014; Palagini et al., 2014; Riemann et al., 2010, 2015; and Vgontzas et al., 2013 for in-depth analysis in this area). Briefly, increased activity in the cortex (Fernandez-Mendoza et al., 2016), cerebrum (Nofzinger et al., 2004), HPA axis (Buckley & Schatzberg, 2005; Fernandez-Mendoza, Vgontzas et al., 2014; Riemann et al., 2010; Vgontzas et al., 2001, 2013), sympathetic nervous system (Vgontzas et al., 2013) and amygdala (Baglioni et al., 2014) have been observed in poor sleepers. However, given the effects of sleep loss and comorbid disorders on neurobiological networks, it is important to characterize the integrity of these brain structures prior to morbidity. That is, if we can better understand the neurobiology underlying exaggerated sensitivity in the sleep system prior to disease development, then we will better understand the transdiagnostic neurobiology complicit in the development of sleep disorders following stress and their impact on mental illness and other areas of health and function. We will discuss the next steps in this area of research below in the Future Directions section.
11.2 |. Related psychological factors
Fernandez-Mendoza and colleagues conducted the most comprehensive investigation to date on the cognitive and emotional processes related to sleep reactivity (Fernández-Mendoza et al., 2010). In a sample of adults with good sleeping, the authors showed that sleep reactivity (FIRST scores) was positively associated with indices of cognitive and emotional hyperarousal, including pre-sleep cognitive and somatic arousal, rumination, perceived stress, arousability, neuroticism and emotion-oriented coping. These data suggest that sleep reactivity and cognitive-emotional reactivity are two distinct yet interrelated responses to stress. It is important to emphasize here that sleep reactivity and cognitive-emotional reactivity may share a bidirectional relationship. For instance, ruminating on a stressor may activate wake-promoting processes complicit in stress-related sleep disturbance, whereas difficulty sleeping when stressed serves as a breeding ground for ruminative thinking, given that the inability to fall asleep creates a period of unstructured and socially isolated time in bed.
Not only is sensitivity of the sleep system related to cognitive and emotional processes, but early evidence suggests that the two forms of stress reactance may interact to compound the risk of disease. Cognitive-emotional arousal in the form of a trait-like tendency to worry and ruminate exploits the fragility of sensitive sleep systems, and highly reactive sleepers are especially vulnerable to insomnia when they engage in rumination during high stress (Drake et al., 2013; see Figure 5). Given that rumination is particularly prevalent and insomniogenic during the pre-sleep period (Craske et al., 2002; Harvey, 2000, 2002a, 2002b; Pillai & Drake, 2014), the synergistic effects of high sleep reactivity and rumination are likely to most evidently manifest as prolonged sleep latency (Drake et al., 2004; Kalmbach, Pillai, Arnedt, & Drake, 2016b). That is, highly reactive sleepers are especially vulnerable to difficulty falling asleep when ruminating on stressors compared to those individuals with low sleep reactivity.
FIGURE 5.
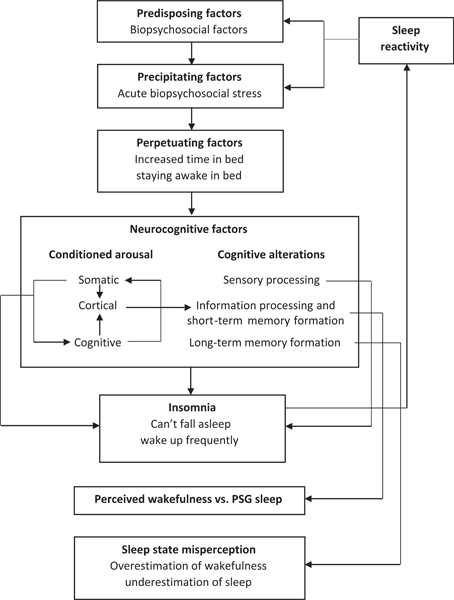
Depiction of the cyclical impact of sleep reactivity in sleep disturbance. Sleep reactivity predisposes individuals to sleep disturbance and subsequent neurocognitive factors following stress, including conditioned arousal and cognitive alterations. These in turn presage the development of insomnia disorder, which increases sleep reactivity, heightening the likelihood of future sleep disturbance. PSG, polysomnography
The effects of pre-sleep cognitive arousal are not limited to sleep initiation. In a recent review, Riemann et al. (2012) reported that pre-sleep cognitive content often reappears later in the night during dreams. Notably, sleep physiology research shows that insomniacs have fragmented REM sleep (Feige et al., 2008, 2013; Riemann et al., 2012), and a recent study showed that a pre-sleep negative affect induction task increased the number of awakenings during REM sleep (Vandekerckhove & Cluydts, 2010). Subjective reports are consistent with these sleep physiology findings, such that insomniacs report REM sleep mentation as more wake-like and emotionally negative than good sleepers (Feige et al., 2018). Riemann’s team presented these findings in the context of highlighting REM instability (i.e. frequent micro- and macro-arousals during REM sleep) as a possible biomarker of insomnia. They proposed that the content of pre-sleep cognitive arousal dominates dream content and increases arousal, thus resulting in fragmentation of REM sleep. Indeed, recent evidence suggests that REM instability reflects interference of overnight resolution of stress, the accumulation of which may contribute to chronic hyperarousal (Wassing et al., 2016). Riemann et al. proposed that REM sleep fragmentation may lead to rebounds in REM pressure (↓ REM latency, ↑ REM density) resembling sleep abnormalities in the transition from acute to chronic insomnia (Ellis, Perlis, Bastien, Gardani, & Espie, 2014) and in depression (Pillai, Kalmbach, & Ciesla, 2011).
Owing to the hallmark of cognitive-emotional arousal in both sleep reactivity and REM instability, it is possible that highly reactive sleepers, even prior to morbidity, show REM instability in their sleep profiles. Moreover, REM instability may explain the reduction in REM and micro-fragmentation of sleep among highly reactive sleepers during stress (Petersen et al., 2013), although it is important to emphasize here that no consistent evidence regarding REM abnormalities in high sleep reactivity yet exists. More laboratory-based PSG studies are needed to clarify the relationship between sleep reactivity and REM sleep architecture during high and low stress. Laboratory studies characterizing baseline and stress-induced REM sleep profiles in high versus low-sleep-reactivity individuals may clarify REM sleep characteristics associated with sleep reactivity and stress.
12 |. FUTURE DIRECTIONS
Sleep reactivity is a relatively young area of research. Here, we propose four key areas for future research in sleep reactivity, including: (a) improved characterization of its neurobiology and relation to cognitive and emotional processes, (b) associations with various insomnia phenotypes, (c) implications for prevention and treatment in behavioural sleep medicine, and (d) prognostic value of sleep reactivity for non-sleep-related disorders.
12.1 |. The neurobiology of sleep reactivity: Next steps
12.1.1 |. Neurobiology next steps: Genetic and genomic science
Although transient sleep difficulties are quite normal on occasion (Ohayon, 2002), individuals with high sleep reactivity appear especially vulnerable to stress-related sleep disturbances spinning out of control and evolving into insomnia disorder. The precise neurobiological system involved and how these aetiological processes occur over time is an important area of further study. Despite heritability studies supporting a genetic basis for sleep reactivity (Drake et al., 2008, 2011), no studies to date have utilized large-scale genomewide association techniques or genetic sequencing to adequately investigate its genetic architecture. This is particularly intriguing given that a premorbid study of sleep reactivity would allow investigation of genetic aspects of insomnia that are independent of the potentially confounding effects of environmental stress exposure and chronic sleep loss associated with the onset of insomnia disorder.
Genomic methods hold promise for providing critical information on the sleep, stress and other unknown systems involved in sleep reactivity. Further, evidence suggests that exposure to stress and insomnia development can sensitize the sleep system, thereby increasing sensitivity to future stress and subsequent pathology (Kalmbach, Pillai, Arnedt, Anderson et al., 2016). These findings are consistent with Palagini and colleagues’ proposal that exposure to stress and insomnia changes the expression of genes involved in sleep and stress regulation. Thus, characterization of these epigenetic mechanisms may provide much needed insights into the genetic influences of sleep reactivity beyond simply its architecture.
12.1.2 |. Neurobiology next steps: Disrupted cortical networks
Research shows that insomnia can result from a conflict between sleep-promoting processes and inappropriate activation of the central nervous system (CNS; Bonnet & Arand, 2010). Elevated encephalographic-defined (EEG) cortical activity when trying to fall asleep and during sleep is a marker of insomnia (Fernandez-Mendoza et al., 2016; Perlis, Merica, Smith, & Giles, 2001) and may also represent a shared mechanism that increases the risk of mental illness among insomniacs (Fernandez-Mendoza et al., 2016). Laboratory research is needed to evaluate whether good sleepers with high versus low sleep reactivity differ in cortical activity prior to and in response to stress exposure. These results can establish whether disrupted cortical networks preexist insomnia development and are indicative of a sensitive sleep system.
Earlier in the review, we stated that sleep reactivity is most pathological and ergo clinically pertinent when in excess. We would like to expand on this statement here to emphasize that individuals with high cognitive-emotional hyperarousal may remain very much ‘under stress’ cognitively and emotionally, long after the elimination of a stressful stimulus. Given the close relationship between cognitive-emotional hyperarousal and sleep reactivity (Fernandez-Mendoza, Shaffer et al., 2014; Fernandez-Mendoza, Vgontzas et al., 2014), and their combined synergistic effects on negative health outcomes (Drake et al., 2013), individuals with high sleep reactivity appear especially vulnerable to the insomniogenic effects of disrupted cortical networks. One potential mechanism may involve stress exposure exploiting the fragility of sensitive sleep systems via cortical hyperactivity or hyperreactivity overriding sleep-promoting processes. Evidence clearly shows that cognitive-emotional and sleep reactivityrelated factors produce disruptions in sleep, and that these constructs are unique but tightly interrelated. Future research using objective markers is needed to tease apart the influences of sleep reactivity and cognitive-emotional reactivity to determine their respective and relative contributions to the development of insomnia and related disorders.
12.1.3 |. Neurobiology next steps: Stress dysregulation
Early evidence also implicates sympathetic nervous system hyperactivation and parasympathetic under-activation in high sleep reactivity (Bonnet & Arand, 2010). Among good sleepers, those with high sleep reactivity exhibit low parasympathetic activity and high sympathetic activity (Bonnet & Arand, 2003). In this study, high versus low reactive sleepers did not differ in mood or personality indices and stress exposures were affectively neutral. Thus, autonomic imbalance in highly reactive sleepers appears largely independent of potential cognitive and emotional influences, at least at this early premorbid stage. Notably, this high sympathetic activity and low parasympathetic activity profile is consistent with what we see in chronic insomniacs in comparison to healthy controls (Bonnet & Arand, 1998).
Adding to this literature, a recent study by Dang-Vu’s team examined parasympathetic activity (high-frequency and low-frequency heart rate variability [HF- and LF-HRV]) in association with stress exposure, worry and sleep (Gouin, Wenzel, Deschenes, & Dang-Vu, 2013). The authors found that low HF-HRV during worry (i.e. blunted parasympathetic activation) was associated with poor sleep quality and corresponded to greater sleep reactivity (measured by stress-induced changes in reported sleep quality). These data indicate that low parasympathetic activity during worry predicts poor sleep and greater sensitivity of the sleep system to stress, thus providing laboratory support for our prior epidemiological finding that rumination augments the pathogenicity of sleep reactivity in insomnia development (Drake et al., 2013). Even so, future work is needed to clarify the role of ANS dysregulation in sleep reactivity, particularly in light of some evidence suggesting that ANS indices may not differ between those with high and low sleep reactivity (Chen, Jarrin, Ivers, & Morin, 2017).
In further support of an underlying neurobiological diathesis for insomnia, Chen et al. compared physiological stress regulation across three groups of adolescents: insomniacs, good sleepers with high sleep reactivity, and good sleepers with low sleep reactivity (Chen et al., 2017). Overall findings suggested that increased hyperarousal at night and greater HPA axis reactivity to acute laboratory stress (Trier Social Stress Test) may constitute trait-like markers for insomnia vulnerability. These data suggest that stress-related HPA reactivity may underlie high sleep reactivity. However, contrary to these data, our team recently identified that good sleepers with high sleep reactivity and familial insomnia risk exhibit a blunted cortisol response to laboratory stress (also the Trier Social Stress Test; Drake, Cheng, Almeida, & Roth, 2017). The role of cortisol as the critical negative feedback component of the stress response suggests that a lack of such a response may reduce an individual’s adaptive reaction to naturalistic stress and is thus a plausible mechanism for the evolution to chronic insomnia (Vgontzas et al., 2001). Taken together, these two recent studies suggest that the sensitivity of the sleep system is associated with HPA dysregulation, while offering very different characterizations of the inner machinations (i.e. an exaggerated versus blunted HPA stress response corresponding to high sleep reactivity). Further research is needed to clarify the relationship between HPA activity and sleep reactivity. Moreover, the causal implications of these unique HPA axis markers of insomnia diathesis remain unknown. Potential next steps include prospectively investigating whether a blunted and/or exaggerated cortisol response to stress associated with high versus low sleep reactivity augurs future insomnia development, and how HPA regulation changes with new versus recurrent disease onset.
Going forward, a notable obstacle to the identification of unique neurobiological bases of sleep reactivity that are independent of general cognitive responses to stress is that the assessment of sleep reactivity generally relies on self-report measures. This method is of course in contrast to PSG-defined first night effects and similar methods (discussed in Roots in Situational Insomnia and Measuring Sleep Reactivity sections earlier in the review), which are limited by high resource demands and dependence on PSG-naïve subjects (for first night effects). Nevertheless, reliance on perceptual evaluation of a stress–sleep response is likely to overrepresent the cognitive component of this relationship. Indeed, it would appear unlikely that any self-report measure of sleep reactivity could fully dissociate sleep system sensitivity from differential cognitive responses to stress. Further explorations of sleep reactivity as a process independent of cognitive responses (i.e. an inherent part of the sleep-response system) will require the development of more sophisticated objective tools for the assessment of sleep reactivity. Although this is undoubtedly a challenge for future studies on sleep reactivity, the recent advancement of cost-effective ambulatory objective sleep measures may play a critical role.
12.1.4 |. Insomnia phenotypes
Insomnia is a heterogeneous disorder, reflecting its multifactorial and complex aetiology. Depending on operationalizations, it is estimated that 62%–77% of insomniacs have high premorbid sleep reactivity, and women are over-represented among highly reactive sleepers and insomniacs. Prolonged sleep latency is a robust hallmark of stress-related sleep disturbance and sleep reactivity (Bonnet & Arand, 2003; Drake et al., 2004; Kalmbach, Pillai, Arnedt, & Drake, 2016b), and implicates pre-sleep arousal as their basis. The sleep-onset insomnia phenotype is associated with poor prognosis as it portends a higher likelihood of persistence than the sleep-maintenance or combined (onset and maintenance) phenotypes at the time of disease onset (Pillai et al., 2014). In addition, preliminary evidence presented in this review suggests that exceptionally high sleep reactivity may underlie the short-sleep insomnia phenotype, which is the most biologically severe and treatment-resistant phenotype (Bathgate et al., 2017; Vgontzas et al., 2013). Recent theories propose that HPA dysregulation, disrupted cortical networks, sympathetic over-activation and parasympathetic under-activation comprise the neurobiological bases of short sleep insomnia (Vgontzas et al., 2013). This neurobiological hyperarousal and stress dysregulation contribute to the high rates of mental illness among short-sleeping insomniacs, even in the absence of poor cognitive-emotional coping (Fernandez-Mendoza et al., 2015; Vgontzas et al., 2013). High sleep reactivity augurs highly severe, persistent and treatment-resistant forms of insomnia. By extension, premorbid sleep reactivity can be used to identify high-risk individuals toward whom preventive efforts can be targeted, whereas sleep reactivity among the already affected may aid in triaging to different levels of treatment.
12.1.5|. Treatment implications: Insomnia
A crucial goal in sleep health is prevention, and a necessary but not sufficient step towards prevention is identifying individuals at risk of disease prior to onset. Highly reactive sleepers represent a critical target group at risk of insomnia, SWD and comorbid mental illness. Occupational research has supported poor sleep as a risk factor for sleep problems, mental illness and vocational impairment across a variety of professions (Baldwin & Daugherty, 2004; Charles et al., 2007; Kalmbach, Arnedt, Song, Guille, & Sen, 2017; Mohr et al., 2003; Olson, Drage, & Auger, 2009; Rajaratnam et al., 2011; Sen et al., 2010, 2013; Vorona, Chen, & Ware, 2009). Thus, health evaluation at the time of employment may involve assessing sleep reactivity. Although evaluating for other sleep parameters would be helpful and identify poor sleepers, research shows that sleep reactivity is a better prognostic indicator for future sleep pathology than poor sleep quality (Kalmbach, Pillai, Arnedt, & Drake, 2016b). This is perhaps attributable to the often-transient nature of sleep problems because an absence of sleep problems at a given moment does not necessarily reflect a healthy sleep system, nor do sleep problems at any one moment necessarily reflect a fragile sleep system. Highly reactive sleepers transitioning to shift work or stressful and demanding fields may benefit from the knowledge that they have a high risk of sleep problems, which can help them detect early signs of insomnia or SWD. Moreover, these at-risk individuals can be provided with sleep information or even low-intensity stepped-care approaches designed to help mitigate the effects of stress and shift work on the sleep system (Drake, 2015). Given the cognitive-emotional component of sleep reactivity, guidance and promotion of adaptive coping strategies for stress may lessen the risk of disease.
Evaluating sleep reactivity in patients with insomnia may have value in triaging to different levels of intervention. Stepped-care approaches have been recommended for cognitive-behavioural therapy for insomnia (CBTI; Espie, 2009), and early evidence supports the scalability of CBTI for insomnia (Manber, Simpson, & Bootzin, 2015). Insomniacs with low sleep reactivity may have less severe forms of the disorder that may be amenable to less intensive treatment modalities such as self-guided digital/internet-based delivery of CBTI, which has been shown to effectively alleviate symptoms of insomnia, anxiety and depression (Blom et al., 2015; Espie, Hames, & Mckinstry, 2013; Feuerstein et al., 2017; Lancee, Van Straten, Morina, Kaldo, & Kamphuis, 2016; Pillai et al., 2015; Zachariae, Lyby, & Ritterband, 2016). Highly reactive insomniacs, however, may benefit more from intensive and highly personalized cognitive-behavioural treatment provided by clinical experts. Given the vulnerability to wake-promoting arousal among highly reactive sleepers (Drake et al., 2004), insomniacs with high sleep reactivity may benefit from adjunct pharmacotherapy to augment cognitive-behavioural interventions. Overall treatment response and long-term outcomes may also be enhanced by greater focus on maladaptive reactions to stress, including maladaptive coping and dysfunctional beliefs about sleep. Even after insomnia remission, evidence suggests that sleep reactivity does not decrease into the healthy range (Kalmbach, Pillai, Arnedt, Anderson et al., 2016). Thus, even after treatment success, remitted insomniacs with elevated sleep reactivity should continue to adopt ongoing preventive efforts.
12.2 |. Treatment implications: Positive airway pressure treatment for sleep-disordered breathing
Obstructive sleep apnea (OSA) is the most common form of sleep-disordered breathing. OSA is caused by obstructions of the upper airway, resulting in shallowed and interrupted breathing during sleep (American Academy of Sleep Medicine, 2014). The most widely supported and prescribed treatment for OSA is positive airway pressure (PAP) therapy (Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine, 2009), which involves a mobile respiratory device to provide compressed air to support the airway during sleep via a facial mask. Individuals undergoing PAP therapy vary greatly in their ability to tolerate wearing a mask or breathing with constant in-flow of air pressure during sleep. Indeed, intolerance of the mask and/or air pressure is the largest barrier to successful OSA treatment with PAP therapy. Future research is needed to determine whether highly reactive sleepers are more likely to experience insomnia as a result of PAP therapy intolerance. If high sleep reactivity portends difficulty tolerating PAP therapy, then highly reactive sleepers may be good candidates for (a) early psychological intervention to promote PAP tolerance, habituation and desensitization or (b) alternative treatment methods for OSA, such as oral appliances, when applicable.
12.3 |. Prognostic value of sleep reactivity for non-sleep stress-related disorders
Recent evidence suggests that the pathogenicity of a highly sensitive sleep system is not limited exclusively to sleep and circadian disorders. Depression and anxiety are more severe at insomnia onset among those with highly sensitive sleep systems (Kalmbach, Pillai, Arnedt, Anderson et al., 2016). More to the point, evidence also shows that high sleep reactivity directly increases the risk of mental illness, although some data suggest that this relationship is partly mediated by developing sleep pathology (Drake et al., 2013; Kalmbach et al., 2015; Kalmbach, Pillai, Arnedt, Anderson et al., 2016; Vargas et al., 2015). Even so, these findings offer strong early support for sleep reactivity as a trait that indicates vulnerability to mood and anxiety disorders.
A potential next step for sleep reactivity research may involve evaluating its prognostic value for non-sleep stress-related disorders outside of mental illness. A burgeoning literature shows that stress has harmful effects on the cardiovascular, metabolic and gastrointestinal systems (Brotman, Golden, & Wittstein, 2007; Mulle & Vaccarino, 2013; Rosmond & Björntorp, 2000). It is possible that individuals with sensitive sleep systems are more vulnerable to the pathogenic effects of stress on the cardiometabolic systems, suggesting a potential direct link between high sleep reactivity and cardiometabolic dysregulation. Further, given the high rates of cardiovascular disease and metabolic disorders among insomniacs with short sleep duration (Vgontzas et al., 2013), any links between sleep reactivity and cardiometabolic risk may be partly mediated through the development of short sleep and insomnia.
Exploration of a potential relationship between sleep reactivity and cardiovascular reactivity lends itself to a variety of innovative laboratory experiments. High cardiovascular reactivity among individuals with high sleep reactivity would support the transdiagnostic validity of both sleep reactivity and cardiovascular reactivity, and possibly implicate cardiovascular dysregulation in sleep pathology. Such evidence would be consistent with recent findings of genetic overlap in insomnia, mental illness and cardiometabolic disorders (Hammerschlag et al., 2017; Lane et al., 2017), thus supporting insomnia and cardiometabolic disorders as manifestations of overlapping aetiological factors (Kalmbach, Pillai, Arnedt, & Drake, 2016a; Vgontzas et al., 2013). Long-term prospective epidemiological studies examining premorbid sleep reactivity as a predictor of future risk of cardiometabolic disorders would be needed to confirm whether the sensitivity of the sleep system has any prognostic value for this related area of disease.
ACKNOWLEDGEMENTS
Christopher Drake’s effort was supported by an NINR Grant (R01 NR013959) and an NIMH Grant (R56 MH115150). David Kalmbach’s effort was supported by an NHLBI Grant (T32 HL110952) awarded to Dr Allan I. Pack.
Funding information: NHLBI, Grant/Award Number: T32, HL110952
Footnotes
Perhaps ironically, items on the FIRST largely pertain to psychosocial stressors, many of which are negatively valenced (e.g. ‘stressful experience’, ‘bad news’, ‘bad day at work’, ‘after an argument’, etc.). That self-reported sleep disturbance response to psychosocial stressors corresponds to objectively measured sleep responses to non-psychosocial and pharmacological challenges suggests generalizability across a broad range of potential stressors.
CONFLICT OF INTEREST
CLD has received research support from Merck & Co., Eisai Co., Aladdin Dreamer, Jazz, Actelion and Teva, and has served on speakers’ bureau for Merck & Co. DAK and JRA report no conflicts of interest.
REFERENCES
- Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. (2009). Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. Journal of Clinical Sleep Medicine, 5, 263–276. [PMC free article] [PubMed] [Google Scholar]
- Åkerstedt T, Kecklund G, & Axelsson J (2007). Impaired sleep after bedtime stress and worries. Biological Psychology, 76, 170–173. [DOI] [PubMed] [Google Scholar]
- Åkerstedt T, & Wright KP (2009). Sleep Loss and Fatigue in Shift Work and Shift Work Disorder. Sleep Medicine Clinics, 4, 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Academy of Sleep Medicine. (2014). International classification of sleep disorders—third edition (ICSD-3). AASM Resource Library; [Online], Darien, IL, United States. [Google Scholar]
- American Psychiatric Association. (2000). Diagnostic and statistical manual-text revision (DSM-IV-TRim, 2000). Washington, DC, United States: American Psychiatric Association. [Google Scholar]
- Archer SN, Robilliard DL, Skene DJ, Smits M, Williams A, Arendt J, & von Schantz M (2003). A length polymorphism in the circadian clock gene Per3 is linked to delayed sleep phase syndrome and extreme diurnal preference. Sleep, 26, 413–415. [DOI] [PubMed] [Google Scholar]
- Baglioni C, Battagliese G, Feige B, Spiegelhalder K, Nissen C, Voderholzer U, ... Riemann D (2011). Insomnia as a predictor of depression: A meta-analytic evaluation of longitudinal epidemiological studies. Journal of Affective Disorders, 135, 10–19. [DOI] [PubMed] [Google Scholar]
- Baglioni C, Spiegelhalder K, Lombardo C, & Riemann D (2010). Sleep and emotions: A focus on insomnia. Sleep Medicine Reviews, 14, 227–238. [DOI] [PubMed] [Google Scholar]
- Baglioni C, Spiegelhalder K, Regen W, Feige B, Nissen C, Lombardo C, ... Riemann D (2014). Insomnia disorder is associated with increased amygdala reactivity to insomnia-related stimuli. Sleep, 37, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin D Jr, & Daugherty SR (2004). Sleep deprivation and fatigue in residency training: Results of a national survey of first-and secondyear residents. Sleep, 27, 217–223. [DOI] [PubMed] [Google Scholar]
- Basta M, Chrousos GP, Vela-Bueno A, & Vgontzas AN (2007). Chronic insomnia and the stress system. Sleep Medicine Clinics, 2, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien CH, & Morin CM (2000). Familial incidence of insomnia. Journal of Sleep Research, 9, 49–54. [DOI] [PubMed] [Google Scholar]
- Bastien CH, Vallieres A, & Morin CM (2004). Precipitating factors of insomnia. Behavioral Sleep Medicine, 2, 50–62. [DOI] [PubMed] [Google Scholar]
- Bathgate CJ, Edinger JD, & Krystal AD (2017). Insomnia patients with objective short sleep duration have a blunted response to cognitive behavioral therapy for insomnia. Sleep, 40, 10.1093/sleep/zsw012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathgate CJ, Edinger JD, Wyatt JK, & Krystal AD (2015). Objective but Not Subjective Short Sleep Duration Associated with Increased Risk for Hypertension in Individuals with Insomnia. Sleep, 39, 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie L, Espie CA, Kyle SD, & Biello SM (2015). How are normal sleeping controls selected? A systematic review of cross-sectional insomnia studies and a standardized method to select healthy controls for sleep research. Sleep Medicine, 16, 669–677. [DOI] [PubMed] [Google Scholar]
- Benjamins JS, Migliorati F, Dekker K, , Wassing R, Moens S, Blanken TF, ... Van Someren EJW (2017). Insomnia heterogeneity: Characteristics to consider for data-driven multivariate subtyping. Sleep Medicine Reviews, 36, 71–81. [DOI] [PubMed] [Google Scholar]
- Blom K, Tillgren HT, Wiklund T, Danlycke E, Forssén M, Söder-ström A, ... Kaldo V (2015). Internet-vs. group-delivered cognitive behavior therapy for insomnia: A randomized controlled non-inferiority trial. Behaviour Research and Therapy, 70, 47–55. [DOI] [PubMed] [Google Scholar]
- Bonnet M, & Arand D (1998). Heart rate variability in insomniacs and matched normal sleepers. Psychosomatic Medicine, 60, 610–615. [DOI] [PubMed] [Google Scholar]
- Bonnet MH, & Arand DL (2003). Situational insomnia: Consistency, predictors, and outcomes. Sleep, 26, 1029–1036. [DOI] [PubMed] [Google Scholar]
- Bonnet MH, & Arand DL (2010). Hyperarousal and insomnia: State of the science. Sleep Medicine Reviews, 14, 9–15. [DOI] [PubMed] [Google Scholar]
- Borkovec TD (1982). Insomnia. Journal of Consulting and Clinical Psychology, 50, 880–895. [DOI] [PubMed] [Google Scholar]
- Brotman DJ, Golden SH, & Wittstein IS (2007). The cardiovascular toll of stress. The Lancet, 370, 1089–1100. [DOI] [PubMed] [Google Scholar]
- Brummett BH, Krystal AD, Ashley-Koch A, Kuhn CM, Züchner S, Siegler IC, ... Williams RB (2007). Sleep quality varies as a function of 5-HTTLPR genotype and stress. Psychosomatic Medicine, 69, 621–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley TM, & Schatzberg AF (2005). On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: Normal HPA axis activity and circadian rhythm, exemplary sleep disorders. The Journal of Clinical Endocrinology & Metabolism, 90, 3106–3114. [DOI] [PubMed] [Google Scholar]
- Buysse DJ, Germain A, Hall M, Monk TH, & Nofzinger EA (2012). A neurobiological model of insomnia. Drug Discovery Today: Disease Models, 8, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Mochizuki T, & Saper CB (2008). Neural circuitry of stress-induced insomnia in rats. Journal of Neuroscience, 28, 10167–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles LE, Burchfiel CM, Fekedulegn D, Vila B, Hartley TA, Slaven J, ... Violanti JM (2007). Shift work and sleep: The Buffalo Police health study. Policing: An International Journal of Police Strategies & Management, 30, 215–227. [Google Scholar]
- Chen IY, Jarrin DC, Ivers H, & Morin CM (2017). Investigating psychological and physiological responses to the Trier social stress test in young adults with insomnia: A preliminary study. Sleep Medicine, 40, 11–22. [DOI] [PubMed] [Google Scholar]
- Chen I, Jarrin D, Rochefort A, Lamy M, Ivers H, & Morin C (2015). Validation of the French version of the Ford insomnia response to stress test and the association between sleep reactivity and hyperarousal. Sleep Medicine, 16, S238. [Google Scholar]
- Cho YW, Shin WC, Yun CH, Hong SB, Kim J, & Earley CJ (2009). Epidemiology of insomnia in Korean adults: Prevalence and associated factors. Journal of Clinical Neurology, 5, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MH, Chang FM, Yang CC, Kuo TB, & Hsu N (2009). Sleep quality and morningness–eveningness of shift nurses. Journal of Clinical Nursing, 18, 279–284. [DOI] [PubMed] [Google Scholar]
- Craske MG, Lang AJ, Rowe M, DeCola JP, Simmons J, Mann C, ... Bystritsky A (2002). Presleep attributions about arousal during sleep: Nocturnal panic. Journal of Abnormal Psychology, 111, 53–62. [DOI] [PubMed] [Google Scholar]
- Daley M, Morin CM, LeBlanc M, Gregoire J-P, & Savard J (2009). The economic burden of insomnia: Direct and indirect costs for individuals with insomnia syndrome, insomnia symptoms, and good sleepers. Sleep, 32, 55–64. [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Morin C, Cervena K, Carlander B, Touchon J, Besset A, & Billiard M (2005). Family studies in insomnia. Journal of Psychosomatic Research, 58, 271–278. [DOI] [PubMed] [Google Scholar]
- Dieck A, Helbig S, Drake CL, & Backhaus J (2017). Validation of the German version of the Ford Insomnia Response to Stress Test. Journal of Sleep Research, 10.1111/jsr.12621 [DOI] [PubMed] [Google Scholar]
- Dijk D-J, & Archer SN (2010). PERIOD3, circadian phenotypes, and sleep homeostasis. Sleep Medicine Reviews, 14, 151–160. [DOI] [PubMed] [Google Scholar]
- Drake CL (2015). The promise of digital CBT-I. Sleep, 39, 13–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CL, Cheng P, Almeida DM, & Roth T (2017). Familial risk for insomnia is associated with abnormal cortisol response to stress. Sleep, 40, 10.1093/sleep/zsx143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CL, Friedman NP, Wright KP Jr, & Roth T (2011). Sleep reactivity and insomnia: Genetic and environmental influences. Sleep, 34, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CL, Jefferson C, Roehrs T, & Roth T (2006). Stress-related sleep disturbance and polysomnographic response to caffeine. Sleep Medicine, 7, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CL, Pillai V, & Roth T (2013). Stress and sleep reactivity: A prospective investigation of the stress-diathesis model of insomnia. Sleep, 37, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C, Richardson G, Roehrs T, Scofield H, & Roth T (2004). Vulnerability to stress-related sleep disturbance and hyperarousal. Sleep, 27, 285–291. [DOI] [PubMed] [Google Scholar]
- Drake CL, Roehrs T, & Roth T (2003). Insomnia causes, consequences, and therapeutics: An overview. Depression and Anxiety, 18, 163–176. [DOI] [PubMed] [Google Scholar]
- Drake CL, & Roth T (2006). Predisposition in the evolution of insomnia: Evidence, potential mechanisms, and future directions. Sleep Medicine Clinics, 1, 333–349. [Google Scholar]
- Drake CL, Scofield H, & Roth T (2008). Vulnerability to insomnia: The role of familial aggregation. Sleep Medicine, 9, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C, & Wright KP Jr (2011). Shift work, shift-work disorder, and jet lag In: Kryger MH, Roth T & Dement WC (Eds.), Principles and practice of sleep medicine (pp. 784–798). St. Louis, MO: Elsevier. [Google Scholar]
- Ellis JG, Gehrman P, Espie CA, Riemann D, & Perlis ML (2012). Acute insomnia: Current conceptualizations and future directions. Sleep Medicine Reviews, 16, 5–14. [DOI] [PubMed] [Google Scholar]
- Ellis BJ, Jackson JJ, & Boyce WT (2006). The stress response systems: Universality and adaptive individual differences. Developmental Review, 26, 175–212. [Google Scholar]
- Ellis JG, Perlis ML, Bastien CH, Gardani M, & Espie CA (2014). The natural history of insomnia: Acute insomnia and first-onset depression. Sleep, 37, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espie CA (2009). “Stepped care”: A health technology solution for delivering cognitive behavioral therapy as a first line insomnia treatment. Sleep, 32, 1549–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espie CA, Hames P, & Mckinstry B (2013). Use of the internet and mobile media for delivery of cognitive behavioral insomnia therapy. Sleep Medicine Clinics, 8, 407–419. [Google Scholar]
- Feige B, Al-Shajlawi A, Nissen C, Voderholzer U, Hornyak M, Spiegelhalder K, ... Riemann D (2008). Does REM sleep contribute to subjective wake time in primary insomnia? A comparison of polysomnographic and subjective sleep in 100 patients. Journal of Sleep Research, 17, 180–190. [DOI] [PubMed] [Google Scholar]
- Feige B, Baglioni C, Spiegelhalder K, Hirscher V, Nissen C, & Riemann D (2013). The microstructure of sleep in primary insomnia: An overview and extension. International Journal of Psychophysiology, 89, 171–180. [DOI] [PubMed] [Google Scholar]
- Feige B, Nanovska S, Baglioni C, Bier B, Cabrera L, Diemers S, ... Riemann D (2018). Insomnia—perchance a dream? Results from a NREM/REM sleep awakening study in good sleepers and patients with insomnia. Sleep, 10.1093/sleep/zsy032 [DOI] [PubMed] [Google Scholar]
- Fenzl T, Touma C, Romanowski CP, Ruschel J, Holsboer F, Landgraf R, ... Yassouridis A (2011). Sleep disturbances in highly stress reactive mice: Modeling endophenotypes of major depression. BMC Neuroscience, 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Calhoun SL, Bixler EO, Karataraki M, Liao D, Vela-Bueno A, ... Vgontzas AN (2011). Sleep misperception and chronic insomnia in the general population: The role of objective sleep duration and psychological profiles. Psychosomatic Medicine, 73, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Li Y, Vgontzas AN, Fang J, Gaines J, Calhoun SL, ... Bixler EO (2016). Insomnia is Associated with Cortical Hyperarousal as Early as Adolescence. Sleep, 39, 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Shaffer ML, Olavarrieta-Bernardino S, Vgontzas AN, Calhoun SL, Bixler EO, & Vela-Bueno A (2014). Cognitive–emotional hyperarousal in the offspring of parents vulnerable to insomnia: A nuclear family study. Journal of Sleep Research, 23, 489–498. [DOI] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Shea S, Vgontzas AN, Calhoun SL, Liao D, & Bixler EO (2015). Insomnia and incident depression: Role of objective sleep duration and natural history. Journal of Sleep Research, 24, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Vela-Bueno A, Vgontzas AN, Ramos-Platon MJ, Olavarrieta-Bernardino S, Bixler EO, & De la Cruz-Troca JJ (2010). Cognitive-emotional hyperarousal as a premorbid characteristic of individuals vulnerable to insomnia. Psychosomatic Medicine, 72, 397–403. [DOI] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Vgontzas AN, Bixler EO, Singareddy R, Shaffer ML, Calhoun SL, ... Liao D (2012). Clinical and polysomnographic predictors of the natural history of poor sleep in the general population. Sleep, 35, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mendoza J, Vgontzas AN, Calhoun SL, Vgontzas A, Tsaoussoglou M, Gaines J, ... Bixler EO (2014). Insomnia symptoms, objective sleep duration and hypothalamic-pituitary-adrenal activity in children. European Journal of Clinical Investigation, 44, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein S, Hodges SE, Keenaghan B, Bessette A, Forselius E, & Morgan PT (2017). Computerized Cognitive Behavioral Therapy for Insomnia in a Community Health Setting. Journal of Clinical Sleep Medicine, 13, 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato VJ, & Harsh J (2006). Stress and sleep quality: The moderating role of negative affectivity. Personality and Individual Differences, 41, 825–836. [Google Scholar]
- Friedman L, Brooks JO, Bliwise DL, Yesavage JA, & Wicks DS (1995). Perceptions of life stress and chronic insomnia in older adults. Psychology and Aging, 10, 352–357. [DOI] [PubMed] [Google Scholar]
- Gehrman PR, Byrne E, Gillespie N, & Martin NG (2011). Genetics of insomnia. Sleep Medicine Clinics, 6, 191–202. [Google Scholar]
- Gehrman PR, Meltzer LJ, Moore M, Pack AI, Perlis ML, Eaves LJ, & Silberg JL (2011). Heritability of insomnia symptoms in youth and their relationship to depression and anxiety. Sleep, 34, 1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelaye B, Zhong Q-Y, Barrios YV, Redline S, Drake CL, & Williams MA (2016). Psychometric Evaluation of the Ford Insomnia Response to Stress Test (FIRST) in Early Pregnancy. Journal of Clinical Sleep Medicine, 12, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin J, Wenzel K, Deschenes S, & Dang-Vu T (2013). Heart rate variability predicts sleep efficiency. Sleep Medicine, 14, e142. [Google Scholar]
- Gumenyuk V, Belcher R, Drake CL, & Roth T (2015). Differential sleep, sleepiness, and neurophysiology in the insomnia phenotypes of shift work disorder. Sleep, 38, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Buysse DJ, Nofzinger EA, Reynolds CF 3rd, Thompson W, Mazumdar S, & Monk TH (2008). Financial strain is a significant correlate of sleep continuity disturbances in late-life. Biological Psychology, 77, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Buysse DJ, Nowell PD, Nofzinger EA, Houck P, Reynolds CF 3rd, & Kupfer DJ (2000). Symptoms of stress and depression as correlates of sleep in primary insomnia. Psychosomatic Medicine, 62, 227–230. [DOI] [PubMed] [Google Scholar]
- Hall M, Thayer JF, Germain A, Moul D, Vasko R, Puhl M, ... Buysse DJ (2007). Psychological stress is associated with heightened physiological arousal during NREM sleep in primary insomnia. Behavioral Sleep Medicine, 5, 178–193. [DOI] [PubMed] [Google Scholar]
- Hammen C (2005). Stress and depression. Annual Review of Clinical Psychology, 1, 293–319. [DOI] [PubMed] [Google Scholar]
- Hammerschlag AR, Stringer S, De Leeuw CA, Sniekers S, Taskesen E, Watanabe K, ... Posthuma D (2017). Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nature Genetics, 49, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AG (2000). Pre-sleep cognitive activity: A comparison of sleep-onset insomniacs and good sleepers. British Journal of Clinical Psychology, 39, 275–286. [DOI] [PubMed] [Google Scholar]
- Harvey AG (2002a). A cognitive model of insomnia. Behaviour Research and Therapy, 40, 869–893. [DOI] [PubMed] [Google Scholar]
- Harvey AG (2002b). Trouble in bed: The role of pre-sleep worry and intrusions in the maintenance of insomnia. Journal of Cognitive Psychotherapy, 16, 161–177. [Google Scholar]
- Harvey CJ, Gehrman P, & Espie CA (2014). Who is predisposed to insomnia: A review of familial aggregation, stress-reactivity, personality and coping style. Sleep Medicine Reviews, 18, 237–247. [DOI] [PubMed] [Google Scholar]
- Healey SE, Kales A, Monroe LJ, Bixler EO, Chamberlin K, & Soldatos CR (1981). Onset of Insomnia: Role of Life-Stress Events. Psychosomatic Medicine, 43, 439–451. [DOI] [PubMed] [Google Scholar]
- Hill AV (2001). The genomics and genetics of human infectious disease susceptibility. Annual Review of Genomics and Human Genetics, 2, 373–400. [DOI] [PubMed] [Google Scholar]
- Hilliker N, Muehlbach M, Schweitzer P, & Walsh J (1992). Sleepiness/alertness on a simulated night shift schedule and morningnesseveningness tendency. Sleep, 15, 430–433. [DOI] [PubMed] [Google Scholar]
- Jarrin DC, Chen IY, Ivers H, Drake CL, & Morin CM (2016). Temporal stability of the ford insomnia response to stress test (first). Journal of Clinical Sleep Medicine, 12, 1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrin DC, Chen IY, Ivers H, & Morin CM (2014). The role of vulnerability in stress-related insomnia, social support and coping styles on incidence and persistence of insomnia. Journal of Sleep Research, 23, 681–688. [DOI] [PubMed] [Google Scholar]
- Kalmbach DA, Arnedt JT, Song PX, Guille C, & Sen S (2017). Sleep disturbance and short sleep as risk factors for depression and perceived medical errors in first-year residents. Sleep, 40, 10.1093/sleep/zsw073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach DA, Pillai V, Arnedt JT, Anderson JR, & Drake CL (2016). Sleep system sensitization: Evidence for changing roles of etiological factors in insomnia. Sleep Medicine, 21, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach DA, Pillai V, Arnedt JT, & Drake CL (2016a). DSM-5 insomnia and short sleep: Comorbidity landscape and racial disparities. Sleep, 39, 2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach DA, Pillai V, Arnedt JT, & Drake CL (2016b). Identifying at-risk individuals for insomnia using the Ford Insomnia Response to Stress Test. Sleep, 39, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach DA, Pillai V, Chen P, Arnedt JT, & Drake C (2015). Shift work disorder, depression, and anxiety in the transition to rotating shifts: The role of sleep reactivity. Sleep Medicine, 16, 1532–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach DA, Schneider LD, Cheung J, Bertrand SJ, Kariharan T, Pack AI, & Gehrman PR (2016). Genetic basis of chronotype in humans: Insights from three landmark GWAS. Sleep, 40, Zsw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Thornton LM, & Gardner CO (2000). Stressful life events and previous episodes in the etiology of major depression in women: An evaluation of the “kindling” hypothesis. American Journal of Psychiatry, 157, 1243–1251. [DOI] [PubMed] [Google Scholar]
- Kim K, Uchiyama M, Okawa M, Liu X, & Ogihara R (2000). An epidemiological study of insomnia among the Japanese general population. Sleep, 23, 41–47. [PubMed] [Google Scholar]
- Kobasa SC, Maddi SR, & Courington S (1981). Personality and constitution as mediators in the stress-illness relationship. Journal of Health and Social Behavior, 22, 368–378. [PubMed] [Google Scholar]
- Koffel EA, Koffel JB, & Gehrman PR (2015). A meta-analysis of group cognitive behavioral therapy for insomnia. Sleep Medicine Reviews, 19, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancee J, Van Straten A, Morina N, Kaldo V, & Kamphuis JH (2016). Guided online or face-to-face cognitive behavioral treatment for insomnia: A randomized wait-list controlled trial. Sleep, 39, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JM, Liang J, Vlasac I, Anderson SG, Bechtold DA, Bowden J, ... Saxena R (2017). Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nature Genetics, 49, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc M, Merette C, Savard J, Ivers H, Baillargeon L, & Morin CM (2009). Incidence and risk factors of insomnia in a population-based sample. Sleep, 32, 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger D, Guilleminault C, Dreyfus JP, Delahaye C, & Paillard M (2000). Prevalence of insomnia in a survey of 12 778 adults in France. Journal of Sleep Research, 9, 35–42. [DOI] [PubMed] [Google Scholar]
- Lind MJ, & Gehrman PR (2016). Genetic pathways to insomnia. Brain Sciences, 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, Mcewen BS, Gunnar MR, & Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nature Reviews Neuroscience, 10, 434–445. [DOI] [PubMed] [Google Scholar]
- Manber R, Simpson NS, & Bootzin RR (2015). A step towards stepped care: Delivery of CBT-I with reduced clinician time. Sleep Medicine Reviews, 19, 3–5. [DOI] [PubMed] [Google Scholar]
- Marques DR, Allen Gomes A, Drake CL, Roth T, & De Azevedo MHP (2018). Assessing stress-induced sleep reactivity in college students: The European Portuguese version of the Ford insomnia response to stress test (FIRST). Behavioral Sleep Medicine, 16, 337–346. 10.1093/sleep/zsw073 [DOI] [PubMed] [Google Scholar]
- McKeever VM, & Huff ME (2003). A diathesis-stress model of post-traumatic stress disorder: Ecological, biological, and residual stress pathways. Review of General Psychology, 7, 237–250. [Google Scholar]
- Meerlo P, Sgoifo A, & Suchecki D (2008). Restricted and disrupted sleep: Effects on autonomic function, neuroendocrine stress systems and stress responsivity. Sleep Medicine Reviews, 12, 197–210. [DOI] [PubMed] [Google Scholar]
- Mezick EJ, Matthews KA, Hall M, Kamarck TW, Buysse DJ, Owens JF, & Reis SE (2009). Intra-individual variability in sleep duration and fragmentation: Associations with stress. Psychoneuroendocrinology, 34, 1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr D, Vedantham K, Neylan T, Metzler TJ, Best S, & Marmar CR (2003). The mediating effects of sleep in the relationship between traumatic stress and health symptoms in urban police officers. Psychosomatic Medicine, 65, 485–489. [DOI] [PubMed] [Google Scholar]
- Monroe SM, & Harkness KL (2005). Life stress, the” kindling” hypothesis, and the recurrence of depression: Considerations from a life stress perspective. Psychological Review, 112, 417–445. [DOI] [PubMed] [Google Scholar]
- Morin CM, Bélanger L, LeBlanc M, Ivers H, Savard J, Espie CA, ... Grégoire JP (2009). The natural history of insomnia: A population-based 3-year longitudinal study. Archives of Internal Medicine, 169, 447–453. [DOI] [PubMed] [Google Scholar]
- Morin CM, LeBlanc M, Daley M, Gregoire J, & Merette C (2006). Epidemiology of insomnia: Prevalence, self-help treatments, consultations, and determinants of help-seeking behaviors. Sleep Medicine, 7, 123–130. [DOI] [PubMed] [Google Scholar]
- Morin CM, Rodrigue S, & Ivers H (2003). Role of stress, arousal, and coping skills in primary insomnia. Psychosomatic Medicine, 65, 259–267. [DOI] [PubMed] [Google Scholar]
- Morphy H, Dunn KM, Lewis M, Boardman HF, & Croft PR (2007). Epidemiology of insomnia: A longitudinal study in a UK population. Sleep, 30, 274–280. [PubMed] [Google Scholar]
- Mulle JG, & Vaccarino V (2013). Cardiovascular disease, psychosocial factors, and genetics: The case of depression. Progress in Cardiovascular Diseases, 55, 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Okajima I, Sasai T, Kobayashi M, Furudate N, Drake CL, ... Inoue Y (2014). Validation of the Japanese version of the Ford Insomnia Response to Stress Test and the association of sleep reactivity with trait anxiety and insomnia. Sleep Medicine, 15, 196–202. [DOI] [PubMed] [Google Scholar]
- Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, & Kupfer DJ (2004). Functional neuroimaging evidence for hyperarousal in insomnia. American Journal of Psychiatry, 161, 2126–2128. [DOI] [PubMed] [Google Scholar]
- Ohayon MM (2002). Epidemiology of insomnia: What we know and what we still need to learn. Sleep Medicine Reviews, 6, 97–111. [DOI] [PubMed] [Google Scholar]
- Okajima I, Komada Y, & Inoue Y (2011). A meta-analysis on the treatment effectiveness of cognitive behavioral therapy for primary insomnia. Sleep and Biological Rhythms, 9, 24–34. [Google Scholar]
- Olson EJ, Drage LA, & Auger RR (2009). Sleep deprivation, physician performance, and patient safety. CHEST Journal, 136, 1389–1396. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Corella D, Demissie S, Cupples LA, Couture P, Coltell O, ... Tucker KL (2002). Dietary fat intake determines the effect of a common polymorphism in the hepatic lipase gene promoter on high-density lipoprotein metabolism. Circulation, 106, 2315–2321. [DOI] [PubMed] [Google Scholar]
- Pacak K, & Palkovits M (2001). Stressor specificity of central neuroendocrine responses: Implications for stress-related disorders. Endocrine Reviews, 22, 502–548. [DOI] [PubMed] [Google Scholar]
- Pack AI, Keenan BT, Byrne EM, & Gehrman PR (2016). Genetics and genomic basis of sleep disorders in humans In: Kryger MH, Roth T & Dement WC (Eds.), Principles and practice of sleep medicine, 6th ed (322–339). St. Louis, MO: Elsevier. [Google Scholar]
- Palagini L, Biber K, & Riemann D (2014). The genetics of insomnia–Evidence for epigenetic mechanisms? Sleep Medicine Reviews, 18, 225–235. [DOI] [PubMed] [Google Scholar]
- Palagini L, Bruno RM, Paolo T, Caccavale L, Gronchi A, Mauri M, ... Drake CL (2016). Association between stress-related sleep reactivity and metacognitive beliefs about sleep in insomnia disorder: Preliminary results. Behavioral Sleep Medicine, 14, 636–649. [DOI] [PubMed] [Google Scholar]
- Palagini L, Maria Bruno R, Gemignani A, Baglioni C, Ghiadoni L, & Riemann D (2013). Sleep loss and hypertension: A systematic review. Current Pharmaceutical Design, 19, 2409–2419. [DOI] [PubMed] [Google Scholar]
- Panda S, Taly AB, Sinha S, Gururaj G, Girish N, & Nagaraja D (2012). Sleep-related disorders among a healthy population in South India. Neurology India, 60, 68. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Morrison AR, Ross RJ, & Brennan FX (2008). Stressinduced changes in sleep in rodents: Models and mechanisms. Neuroscience & Biobehavioral Reviews, 32, 99–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlis ML, Merica H, Smith MT, & Giles DE (2001). Beta EEG activity and insomnia. Sleep Medicine Reviews, 5, 365–376. [DOI] [PubMed] [Google Scholar]
- Petersen H, Kecklund G, D’onofrio P, Nilsson J, & Åkerstedt T (2013). Stress vulnerability and the effects of moderate daily stress on sleep polysomnography and subjective sleepiness. Journal of Sleep Research, 22, 50–57. [DOI] [PubMed] [Google Scholar]
- Pillai V, Anderson JR, Cheng P, Bazan L, Bostock S, Espie CA, ... Drake CL (2015). The anxiolytic effects of cognitive behavior therapy for insomnia: Preliminary results from a web-delivered protocol. Journal of Sleep Medicine & Disorders, 2, 1017. [PMC free article] [PubMed] [Google Scholar]
- Pillai V, & Drake C (2014). Sleep and repetitive thought: The role of rumination and worry in sleep disturbance In Feldner M, & Babson K (Eds.), Sleep and affect: Assessment, theory, & clinical implications (pp. 201–226). London: Elsevier. [Google Scholar]
- Pillai V, Kalmbach DA, & Ciesla JA (2011). A meta-analysis of electroencephalographic sleep in depression: Evidence for genetic biomarkers. Biological Psychiatry, 70, 912–919. [DOI] [PubMed] [Google Scholar]
- Pillai V, Roth T, & Drake C (2014). The nature of stable insomnia phenotypes. Sleep, 38, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai V, Roth T, Mullins HM, & Drake CL (2013). Moderators and mediators of the relationship between stress and insomnia: Stressor chronicity, cognitive intrusion, and coping. Sleep, 37, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaratnam SM, Barger LK, Lockley SW, Shea SA, Wang W, Landrigan CP, ... Czeisler CA (2011). Harvard Work Hours, Health and Safety Group. Sleep disorders, health, and safety in police officers. JAMA, 306, 2567–2578. [DOI] [PubMed] [Google Scholar]
- Revel FG, Gottowik J, Gatti S, Wettstein JG, & Moreau J-L (2009). Rodent models of insomnia: A review of experimental procedures that induce sleep disturbances. Neuroscience & Biobehavioral Reviews, 33, 874–899. [DOI] [PubMed] [Google Scholar]
- Riemann D, Nissen C, Palagini L, Otte A, Perlis ML, & Spiegelhalder K (2015). The neurobiology, investigation, and treatment of chronic insomnia. The Lancet Neurology, 14, 547–558. [DOI] [PubMed] [Google Scholar]
- Riemann D, & Perlis ML (2009). The treatments of chronic insomnia: A review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Medicine Reviews, 13, 205–214. [DOI] [PubMed] [Google Scholar]
- Riemann D, Spiegelhalder K, Feige B, Voderholzer U, Berger M, Perlis M, & Nissen C (2010). The hyperarousal model of insomnia: A review of the concept and its evidence. Sleep Medicine Reviews, 14, 19–31. [DOI] [PubMed] [Google Scholar]
- Riemann D, Spiegelhalder K, Nissen C, Hirscher V, Baglioni C, & Feige B (2012). REM sleep instability-a new pathway for insomnia? Pharmacopsychiatry, 45, 167. [DOI] [PubMed] [Google Scholar]
- Riemann D, & Voderholzer U (2003). Primary insomnia: A risk factor to develop depression? Journal of Affective Disorders, 76, 255–259. [DOI] [PubMed] [Google Scholar]
- Riemann D, Voderholzer U, Spiegelhalder K, Hornyak M, Buysse DJ, Nissen C, ... Feige B (2007). Chronic insomnia and MRI-measured hippocampal volumes: A pilot study. Sleep, 30, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrs T, & Roth T (2012). Insomnia pharmacotherapy. Neurotherapeutics: the Journal of the American Society for Experimental NeuroTherapeutics, 9, 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmond R, & Björntorp P (2000). The hypothalamic–pituitary–adrenal axis activity as a predictor of cardiovascular disease, type 2 diabetes and stroke. Journal of Internal Medicine, 247, 188–197. [DOI] [PubMed] [Google Scholar]
- Roth T, Coulouvrat C, Hajak G, Lakoma MD, Sampson NA, Shahly V, ... Kessler RC (2011). Prevalence and perceived health associated with insomnia based on DSM-IV-TR; international statistical classification of diseases and related health problems, tenth revision; and research diagnostic criteria/international classification of sleep disorders, criteria: Results from the America insomnia survey. Biological Psychiatry, 69, 592–600. [DOI] [PubMed] [Google Scholar]
- Roth T, Roehrs T, & Pies R (2007). Insomnia: Pathophysiology and implications for treatment. Sleep Medicine Reviews, 11, 71–79. [DOI] [PubMed] [Google Scholar]
- Sadeh A, Keinan G, & Daon K (2004). Effects of stress on sleep: The moderating role of coping style. Health Psychology, 23, 542–545. [DOI] [PubMed] [Google Scholar]
- Schwartz JR, & Roth T (2006). Shift Work Sleep Disorder. Drugs, 66, 2357–2370. [DOI] [PubMed] [Google Scholar]
- Selye H (1985). The nature of stress. Basal Facts, 7, 3–11. [PubMed] [Google Scholar]
- Sen S, Kranzler HR, Didwania AK, Schwartz AC, Amarnath S, Kolars JC, ... Guille C (2013). Effects of the 2011 duty hour reforms on interns and their patients: A prospective longitudinal cohort study. JAMA Internal Medicine, 173, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Kranzler HR, Krystal JH, Speller H, Chan G, Gelernter J, & Guille C (2010). A prospective cohort study investigating factors associated with depression during medical internship. Archives of General Psychiatry, 67, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singareddy R, Vgontzas AN, Fernandez-Mendoza J, Liao D, Calhoun S, Shaffer ML, & Bixler EO (2012). Risk factors for incident chronic insomnia: A general population prospective study. Sleep Medicine, 13, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Perlis ML, Park A, Smith MS, Pennington J, Giles DE, & Buysse DJ (2002). Comparative meta-analysis of pharmacotherapy and behavior therapy for persistent insomnia. American Journal of Psychiatry, 159, 5–11. [DOI] [PubMed] [Google Scholar]
- Steiger A (2002). Sleep and the hypothalamo–pituitary–adrenocortical system. Sleep Medicine Reviews, 6, 125–138. [DOI] [PubMed] [Google Scholar]
- Stoffers D, Altena E, Van Der Werf YD, Sanz-Arigita EJ, Voorn TA, Astill RG, ... Van Someren EJ (2013). The caudate: A key node in the neuronal network imbalance of insomnia? Brain, 137, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers D, Moens S, Benjamins J, van Tol MJ, Penninx BW, Veltman DJ, ... Van Someren EJ (2012). Orbitofrontal gray matter relates to early morning awakening: A neural correlate of insomnia complaints? Frontiers in Neurology, 3, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers D, Van Tol M, Penninx B, Veltman D, Van Der Wee N, & Van Someren E (2014). Differential brain structural correlates of insomnia severity in affective disorders. Journal of Sleep Research, 23, 11. [Google Scholar]
- Van Someren EJW, Altena E, Ramauter JR, Stoffers D, Benjamins JS, Moens S, ... Van Der Werf YD (2013). Imaging causes and consequences of insomnia and sleep complaints In Nofzinger E, Maquet P, & Thorpy MJ (Eds.), Neuroimaging of Sleep and Sleep Disorders (pp. 187–196). Cambridge, UK: Cambridge University Press. [Google Scholar]
- Vandekerckhove M, & Cluydts R (2010). The emotional brain and sleep: An intimate relationship. Sleep Medicine Reviews, 14, 219–226. [DOI] [PubMed] [Google Scholar]
- Vargas I, Friedman NP, & Drake CL (2015). Vulnerability to stress-related sleep disturbance and insomnia: Investigating the link with comorbid depressive symptoms. Translational Issues in Psychological Science, 1, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vgontzas AN, Bixler EO, Lin H-M, Prolo P, Mastorakos G, Vela-Bueno A, ... Chrousos GP (2001). Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: Clinical implications. The Journal of Clinical Endocrinology & Metabolism, 86, 3787–3794. [DOI] [PubMed] [Google Scholar]
- Vgontzas AN, Fernandez-Mendoza J, Liao D, & Bixler EO (2013). Insomnia with objective short sleep duration: The most biologically severe phenotype of the disorder. Sleep Medicine Reviews, 17, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorona RD, Chen IA, & Ware JC (2009). Physicians and sleep deprivation. Sleep Medicine Clinics, 4, 527–540. [Google Scholar]
- Walker EF, & Diforio D (1997). Schizophrenia: A neural diathesis-stress model. Psychological Review, 104, 667–685. [DOI] [PubMed] [Google Scholar]
- Walsh JK, Vogel GW, Scharf M, Erman M, William Erwin C, Schweitzer PK, ... Roth T (2000). A five week, polysomnographic assessment of zaleplon 10 mg for the treatment of primary insomnia. Sleep Medicine, 1, 41–49. [DOI] [PubMed] [Google Scholar]
- Wassing R, Benjamins JS, Dekker K, Moens S, Spiegelhalder K, Feige B, ... Van Someren EJ (2016). Slow dissolving of emotional distress contributes to hyperarousal. Proceedings of the National Academy of Sciences of the United States of America, 113, 2538–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PG, & Moroz TL (2009). Personality vulnerability to stress-related sleep disruption: Pathways to adverse mental and physical health outcomes. Personality and Individual Differences, 46, 598–603. [Google Scholar]
- Wittchen H-U, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jönsson B, ... Steinhausen HC (2011). The size and burden of mental disorders and other disorders of the brain in Europe 2010. European Neuropsychopharmacology, 21, 655–679. [DOI] [PubMed] [Google Scholar]
- Xiang Y-T, Ma X, Cai Z-J, Li SR, Xiang YQ, Guo HL, ... Ungvari GS (2008). The prevalence of insomnia, its sociodemographic and clinical correlates, and treatment in rural and urban regions of Beijing, China: A general population-based survey. Sleep, 31, 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae R, Lyby MS, & Ritterband L (2016). M. and O’toole, M. S. Efficacy of internet-delivered cognitive-behavioral therapy for insomnia–A systematic review and meta-analysis of randomized controlled trials. Sleep Medicine Reviews, 30, 1–10. [DOI] [PubMed] [Google Scholar]
- Zoccola PM, Dickerson SS, & Lam S (2009). Rumination predicts longer sleep onset latency after an acute psychosocial stressor. Psychosomatic Medicine, 71, 771–775. [DOI] [PubMed] [Google Scholar]