

Abstract
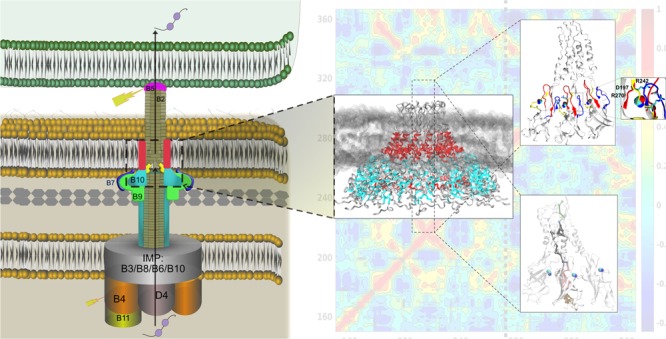
Type IV secretion systems are large nanomachines assembled across the bacterial cell envelope for effector translocation and conjugation. VirB10 traverses the inner and outer membranes, sensing cellular signals for coordinating the conformational switch for pilus biogenesis and/or secretion. Mutations uncoupling secretion from pilus biogenesis were identified in Agrobacterium tumefaciens VirB10 including a gating defect mutation G272R that made VirB10 unresponsive to intracellular ATP, causing unregulated secretion of VirE2 in a contact-independent manner. Comparative long-timescale molecular dynamics of the wild type and G272R mutant of the A. tumefaciens VirB10CTD tetradecamer reveals how the G272R mutation locks the oligomer in a rigid conformation by swapping the ionic interactions between the loops from the β-barrel close to the inner leaflet of the outer membrane. This electrostatic switching changes the allosteric communication pathway from the extracellular loop to the base of the barrel, suggesting that the local conformational dynamics in the loops can gate information across VirB10.
Introduction
Bacterial type IV secretion (T4S) systems are large versatile molecular machines that span the entire bacterial cell envelope coordinating translocation of a diverse set of macromolecules to prokaryotic and eukaryotic cells (recent reviews1−3). Conjugation machineries and effector translocation systems are the main subfamilies of T4S systems. They are the main virulence factors in various pathogenic bacteria affecting plants, animals, and humans. Some examples of T4S systems as virulence factors for pathogenesis include Agrobacterium tumefaciens that causes crown-gall disease, Helicobacter pylori for the transport of oncoprotein CagA leading to various gastrointestinal diseases, facilitating DNA uptake for genomic plasticity and diversity in H. pylori, and DNA release in Neisseria gonorrhea to name a few.4 The best studied T4S systems are the conjugation-related type IVa secretion system exemplified by the VirB/D4 system in A. tumefaciens. The building blocks of the T4S system include the type IV coupling protein (VirD4) that couples substrate recruitment to the secretion channel, the energizing components (VirB4 and VirB11), the core channel components (inner membrane: VirB3, VirB6, VirB8, and VirB10; and outer membrane core complex: VirB7, VirB9, and VirB10), the hydrolase (VirB1), and the pilus components (VirB2 (major) and VirB5 (minor)).
The structural architecture of the T4S system has come to light in the last decade, revealing how the various components come together to span the bilayer. The negative stain electron microscopy (nsEM) structure of the TrwR388 T4S system (from plasmid R388) VirB3–VirB10 complex revealed the membrane spanning structure of the secretion channel composed of the outer membrane core complex (OMCC) joined to the inner membrane complex (IMC) by a thin stalk.5 The IMC has a platform made of 12 copies of VirB3, VirB5, and VirB8 proteins with 24 copies of the VirB6 protein with 14 copies of the VirB10 cytoplasmic transmembrane (TM) region connected to two barrel-like structures of the hexameric cytoplasmic ATPase. The OMCC is composed of 14 copies of VirB7, VirB9, and VirB10 proteins, a conserved architecture seen in various OMCC structures including Escherichia coli conjugation plasmids pKM101 and R388, A. tumefaciens VirB/D4, Legionella pneumophila Dot/Icm, and the H. pylori Cag T4S system.6−9 The OMCC has an outer layer (O-layer) formed by C-terminal domains of VirB9 and VirB10 with lipoprotein VirB7 and an inner layer (I-layer) formed by the N-terminal domains of VirB9 and VirB10. The high-resolution crystal structure of the tetradecameric outer membrane complex (OMC) of the T4S system (O-layer) from the E. coli conjugation plasmid pKM101 revealed the atomistic details of the unusual helical outer membrane pore.10 The O-layer has a main body and a cap. The cap forms the outer membrane pore composed of two helix bundles of VirB10 proteins described as the antenna projections (APs), and the basal body is entirely lined on the interior by VirB10 and surrounded on the exterior by VirB9 and VirB7. This structure established that the VirB10 protein formed the outer membrane pore and was the first protein known to traverse both the inner and outer membrane—apt to be the scaffold protein of the T4S system. The recent high-resolution Cryo-TEM structures of the T4S system OMCC from bacteria-killing Xanthomonas citri and the H. pylori Cag T4S system further confirm the conservation of this architecture.9,11
The crystal structure of TraF/VirB1010 in the O-layer reveals an N-terminal helical extended lever arm that wraps around neighboring subunits followed by an atypical splayed open β-barrel with loops bulging out between the β-strands in the inter-subunit interface and an antenna projection (AP) emanating out of the barrel forming a two-helix bundle (α2 helix–APL loop−α3 helix; APL: antenna projection loop) wherein the α2 helix forms the OM helical pore in the OMC structure. The VirB10 protein is the sensor protein that coordinates the conformational changes required for function based on intracellular signals and the energy state of the ATPases.12 Seminal experiments by Cascales and Christie12 showed VirB10 to adopt alternate protease-susceptible or protease-resistant conformations in response to the intracellular energy state of the VirD4 and VirB11 ATPases. Energy poisons arsenate and protonophore CCCP rendered VirB10 unable to adopt the protease-susceptible conformation. Recent in situ Cryo-ET structures at various stages of assembly for mating-competence and pilus biogenesis of the F plasmid conjugation T4S system showed a structural transition between the body and cap of the OMC (30 to 90°) at the inner leaflet of the OM to accommodate the growing pilus.13 Although we have a structural snapshot of the VirB10 oligomer in the OMC and structural snapshots for larger secretion channel complexes, the molecular mechanism of action or how the ATP state is conveyed by VirB10 in the OMC is still not known.
During the biogenesis of the T4S system, two terminal assemblies take place—the pilus polymerization and the formation of an active translocation channel. Mutational studies have been carried out for the A. tumefaciens VirB10 protein (AtVirB10) to understand domain requirements that support these terminal structures—substrate transport (Tra) and pilus biogenesis (Pil).7,14−16 T4S systems lacking surface-exposed features such as the TraC/VirB5 pilus tip or deletion of the OMCC cap (deletion of the AP between residues 282–335 in AtVirB10 or 307–355 in TraF) revealed a phenotype with no discernible pilus but supported substrate release including pilin monomers, uncoupling the two functions—substrate secretion and pilus assembly (Tra+ Pil–).7 The α2 helix and α3 helix and not the APL are important for pilus biogenesis. A gating defect mutation (G272R)15 was identified that also blocked pilus biogenesis but did not affect substrate transfer to target cells. This mutation also rendered VirB10 unresponsive to the cellular energy state, adopting the alternative protease-susceptible conformation even in the energy-depleted state, allowing the unregulated release of VirE2, independent of host cell contact. This gating defect mutation also increased the sensitivity of the mutant to vancomycin and SDS but no leakage of periplasmic components. Some of the other mutations that attenuated substrate transfer but not pilus biogenesis (Traatt Pil+) included a V243C mutation in one of the loops close to the groove in the splayed β-barrel, Q295C in the α2 helix, N218C in the flap toward the base of the barrel, and T173C in the 310 α-helix connecting the base of the barrel to the extended arm.14 The C-terminal end of VirB10 (CT) has a conserved RDLDF motif, deletion of which abrogates both functions (Tra– Pil–).7,14
In an attempt to visualize the structural transitions that VirB10CTD might adopt in the O-layer to support secretion and/or pilus biogenesis, we embarked on an unbiased molecular dynamics study to compare the dynamics of the wild-type VirB10CTD protein with the constitutively open-secretion competent conformation adopted by the ATP-insensitive gating defect mutant. To the best of our knowledge, this is the first time that long-timescale molecular dynamics has been employed to study the conformational dynamics of the outer membrane protein oligomers in bacterial secretion systems, including the T4S system. This is mainly due to the computational challenges posed by the large oligomeric states of the outer membrane proteins (14-mer in T4S system). This would be one of the first studies to simulate the 328 kDa outer membrane protein oligomer in a lipid bilayer to trap the conformational states otherwise difficult to visualize in experimental techniques. Differences between the wild-type VirB10CTD oligomer and the gating defect ATP-insensitive mutant G272R VirB10CTD oligomer could highlight regions important for coordinating the ATP state sensed by VirB10 in the OMC and for supporting the different terminal structures for secretion and pilus biogenesis.
We have successfully modeled the A. tumefaciens VirB10CTD tetradecamer in the outer membrane complex using the pKM101 OMC structure as the template. We chose to simulate the A. tumefaciens VirB10 to correlate with the mutation studies directly. We also built the APL loop that was cleaved in the crystal structure between the two-helix bundle, one of which spans the outer membrane. We used the EM structure as a reference for guidance and docked the outer membrane complex by guiding the loop in the extra density. Some of the extra density also is contributed by the detergent micelles.
Results
Modeling of Wild Type (wt-AtVirB10CTD) and G272R Mutant (mt-AtVirB10CTD)
E. coli conjugation plasmid pKM101-encoded TraF and A. tumefaciens VirB10 (AtVirB10) sequences share 40% sequence identity. The homology model of wt-AtVirB10CTD was generated in two steps: first by aligning the E. coli TraF/VirB10CTD and AtVirB10CTD sequences followed by extracting structural features from the E. coli TraF/VirB10CTD template (PDB ID 3JQO). The model (Figure 1) was generated and the loop between α2 and α3 helices in the antenna projection, which was missing in the high-resolution structures of OMC, was built and refined to fit the OMCC nsEM structure.6 Each VirB10CTD has an N-terminal extended arm followed by a 7-stranded β-barrel and an antenna projection made up of a helix–loop–helix motif jutting out from the barrel to form the outer membrane pore. The β-barrel is atypical as it is splayed open at the base and has loops between β strands bulging out into the inter-subunit interface. We refer to these loops as L1 (between β1 and β2 strands: residues 192–201), L2 (between β3 and β4 strands: residues 231–246), L3 (between β6a and β6b strands that lead to α2 OM-TM helix (residues 266–275)), and a flap region between β2b and β3a (residues 210–220) (Figure 1C, secondary structure labels are in accordance with the pKM101 OMC structure (3JQO)10). The L3 loop features the conserved GxxGxxG (G269xxG272xxG275 in AtVirB10CTD) motif in the VirB10 protein. The tetradecameric model was generated with symmetry operation switched on, and the final Cα RMSD between the template and the model was 0.68 Å. The mt-AtVirB10CTD tetradecamer was modeled by mutating G272 to R272 in the wt-AtVirB10CTD tetradecamer using an in silico site-directed mutagenesis method implemented in the ICM-Pro suite. The calculated free energy change in protein stability showed a decrease in ΔΔG value (−0.4), suggesting that the mutation at this position was tolerated and did not destabilize the protein fold.
Figure 1.
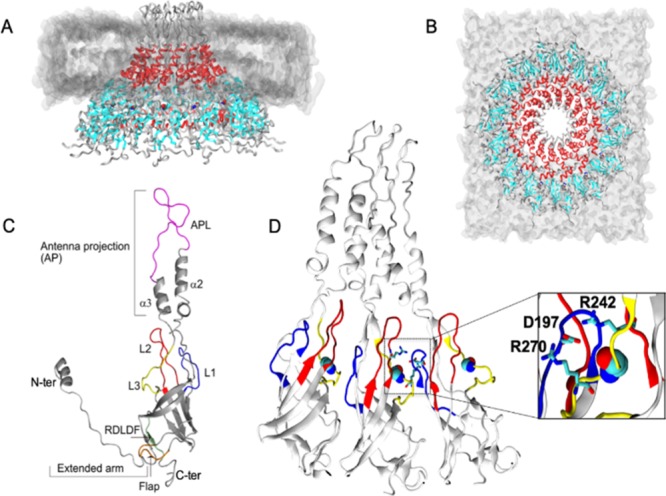
(A, B) Side and top view of the tetradecamer wt-AtVirB10CTD system embedded in a dimyristoylphosphatidylcholine (DMPC) bilayer, illustrated in gray. The β-barrel is colored cyan, and the α2 and α3 helices are colored red. (C) Details highlighting different structural elements within a monomer subunit. Loop 1 (L1; blue), loop 2 (L2; red), loop 3 (L3; yellow), and antenna projection loop (APL; magenta) are noteworthy. (D) Conformation of the L1, L2, and L3 loops and the ion pair interaction (inset) formed between R242–D197–R270. The relative position of G272 (space fill) is also illustrated.
Structural Variations between wt-AtVirB10CTD and mt-AtVirB10CTD
Molecular dynamics simulation for 1 μs of both wt-AtVirB10CTD and mt-AtVirB10CTD indicated stable dynamic systems (Figure 2). The N-terminal end of VirB10CTD in the O-layer of the OMCC is quite flexible with an N-terminal extended arm that wraps around neighboring subunits and then continues into the I-layer proline-rich domain. Although the N-terminal arm has some role to play in the stability of the oligomer, its functional relevance for both translocation channel and pilus biogenesis is dispensable in mutational studies.7 Due to the flexibility of the extended arm and it not being the main focus of our study, it was eliminated from further analysis. The residues analyzed ranged from 179–359. The individual monomers in both wt-AtVirB10CTD and mt-AtVirB10CTD showed similar motions in the simulation run. A Cα RMSD plot for the individual subunits showed more significant deviation from the starting structure for wt-AtVirB10CTD (4.0 Å) than for mt-AtVirB10CTD (2.2 Å) (Figure 2A). The RMSDev plot showed RMS coordinate deviations across the backbone for both wt-AtVirB10CTD and mt-AtVirB10CTD with maximum deviation seen in the AP loop in both the wild type and G272R mutant. Deviations in the loops bulging from the β-barrel including L1, L2, L3, and the flap region are more in the wild-type than in the mutant protein, which appears to be quite rigid (Figure 2B). The RMSF plot (Figure 2C) also showed fluctuations of backbone atoms for all the chains of wt-AtVirB10CTD and mt-AtVirB10CTD with maximum fluctuations seen in the extracellular loop in the AP between α2 and α3 helices, which is flexible and missing in the high-resolution structures of the OMC. Peaks in fluctuation can also be noted in the regions surrounding the loops L1, L2, and L3, which are more pronounced in wt-AtVirB10CTD, consistent with the RMSDev plots. The G272 in wt-AtVirB10CTD showed marked fluctuations compared to R272 in mt-AtVirB10CTD, which is stabilized by a salt bridge it forms with the D240 residue in the L2 loop and interacts with S199 of the L1 loop from the neighboring subunit (Figure 5, lower panel). These interactions restrict the loop movements, supporting a more rigid conformation for mt-AtVirB10CTD compared to wt-AtVirB10CTD (Movies S1 and S2).
Figure 2.

Comparison of Cα conformational drifts between the wild type (wt-AtVirB10CTD) and G272R mutant (mt-AtVirB10CTD). (A) Time-dependent root-mean-square deviation (RMSD); (B) root-mean-square deviation (RMSDev) of each residue; (C) dynamical flexibility was characterized with RMSF with the slowest 10 modes (RMSF10). RMSF10 provides better estimates of the intrinsic flexibility of the protein compared to all motions. Faster motions are noisy and depend on temperature, while slower motions are characteristic of protein architecture and sequence. (D) Comparison of dynamical flexibility of loops 1, 2, and 3 between wild type and G272R. Results are shown for individual monomer A (red curves) and an average of all 14 monomers (black curves). The dotted lines in A, B, and C represent RMSD, RMSDev per residue, and RMSF of each individual monomer, respectively, whereas the bold line is the average of all the subunits.
Figure 5.

Sequence alignment of VirB10CTD shown for the conjugation plasmid pKM101 T4S system (TraF), bacteria-killing effector translocator X. citri T4S system (VirB10), H. pylori Cag T4S system (CagY), and A. tumefaciens VirB10 using ENDscript/ESPript44 and HMM logo for loops L1, L2, and L3 displayed below the sequence using Skylign.45 The lower panel shows the conserved salt bridge triad in the inter-subunit interface for the above three systems and the corresponding salt bridge triad in the wild-type A. tumefaciens VirB10CTD oligomer (R242:D197:R270). Also shown for the wild type are other interacting residues involved in switching ionic interactions in the mutant G272R VirB10CTD. Loops L1 in blue, L2 in red, and L3 in yellow, and conserved G272 and corresponding middle Gly in the GxxGxxG motif in loop L3 are shown as spheres.
The aggregated root-mean-square fluctuations for the top 10 quasi-harmonic modes (RMSF10) were used to characterize protein flexibility. It is well known that the slowest 10 modes contribute to the majority of fluctuations in proteins (>80%) and the use of RMSF10, instead of all modes, removes the faster stochastic motions of the protein, allowing focus on intrinsic dynamics of proteins.17 The single mutation G272R causes significant changes in conformational/dynamical flexibility of the entire complex, particularly for loop L2 and loop L3. Figure 2D shows that the dynamical flexibility is changed for the mutant for all three loops. For loop L2 (Figure 2D: top-right panel), the averaged dynamical flexibility for all 14 monomers is the highest in the region 236–239, which is decreased due to a single mutation in loop L3. For a monomer, the dynamical flexibility is slightly decreased for the entire loop L2. The results are also similar for loop L3 (Figure 2D: bottom panel). However, the case for loop L1 is reversed (Figure 2D: top-left panel) as the mutant is slightly more flexible than the wild type.
Pore Analysis of wt-AtVirB10CTD and mt-AtVirB10CTD
Changes in the physical dimensions of the pore in both wt-AtVirB10CTD and mt-AtVirB10CTD were calculated from the MD trajectory. A time-averaged pore radius profile was constructed and then compared to determine the changes brought about as a result of the mutation over the course of the simulation. The mutant appears to exhibit far less structural variation throughout the trajectory (Figure 3). The minimum pore radius decreases from 8–14 Å in wt-AtVirB10CTD to 6.5–7.5 Å in mt-AtVirB10CTD. The variability in the pore dimensions for wt-AtVirB10CTD is the largest in the region between the barrel and AP domains under the inner leaflet of the OM and then the AP loop between α2 and α3. The loop L2 wherein the D240 and a conserved R242 residue exist is situated just under the inner leaflet of the OM and exposed to the interior of the VirB10 channel in the OMC. Any variations in this loop can affect the pore dynamics. The loop L3 where the G272 residue resides continues toward the α2 OM-TM followed by a short β6b strand. mt-AtVirB10CTD locks the L2 and L3 loops due to salt bridge D240:R272, thereby affecting L2 loop flexibility, locking the mutant oligomer in a rigid conformation. However, in both conformations, there was no barrier to permeation of water and ions anywhere along the pore.
Figure 3.

Pore profiles of (A) wt-AtVirB10CTD and (B) mt-AtVirB10CTD from MD simulations. (i) Annotation of the solvent conduction pathway. (ii) Time-averaged radius profile calculated as a function of position, s, along the pore axis. Calculations were done over 2000 frames at an interval of 0.5 ns and drawn as a solid line (standard deviation as a gray band). Residues facing the pore for more than 50% of the simulation time are illustrated as dots and colored based on their hydrophobicity. (iii) Dynamic radius profile highlights the variation in pore dimensions over the course of the simulation. The pore is structured and displayed less variation in the mutant.(iv) Minimum pore radius over simulated time highlights stabilization of the pore in the mutant when compared with the wild type.
Direct Domain Correlations in wt-AtVirB10CTD and mt-AtVirB10CTD
Average dynamical cross-correlation maps (DCCMs) were calculated for wt-AtVirB10CTD and mt-AtVirB10CTD for comparison. A comparison of DCCMs between various subunits of wt-AtVirB10CTD and mt-AtVirB10CTD after 500 ns and 1 μs of simulation showed very little variation, suggesting that the correlations based on 1 μs of trajectories are well converged. Figure 4A,B compares the DCCM for wt-AtVirB10CTD and mt-AtVirB10CTD with a schematic of the structural features of VirB10CTD aligned below. The single mutation (G272R) causes dynamical cross-correlations to be changed across the entire monomer in mt-AtVirB10CTD. Positive correlations (values closer to 1) imply that residues move together in the same direction, and large negative correlations (values closer to −1) imply that residues are correlated but they move in the opposite direction. Intermediate values (closer to 0) indicate that the residues are not dynamically correlated. The wild-type protein shows significant positive correlations throughout the protein (Figure 4A), which are drastically reduced for the G272R mutant (Figure 4B).
Figure 4.

Comparison of dynamical cross-correlation maps for wt-AtVirB10CTD and mt-AtVirB10CTD: (A) Wild-type monomer and (B) G272R monomer. (C) Comparison of correlations between regions 232–246 and 265–273 in wt-AtVirB10CTD and (D) comparison of correlations between regions 232–246 and 265–273 in mt-AtVirB10CTD. White ellipses in (A) and (B) mark the regions of correlations between loops 1, 2, and 3, and the site of mutation is marked by a black vertical line. Note that the maps in (A) and (B) are symmetric. The red regions show positive correlations, and blue regions are negative. From wild type to mutant, the biggest changes are in the disappearance of several positively correlated areas. Network analysis for (E) wild type and (F) mutant calculated over three adjacent subunits. The analysis focused on the effective signal transmission between two ends of the subunit. In the wild type, the community interactions are limited to within the subunits, and at no point, the shortest path passes via G272. In the mutant, there is significant repartitioning of the network. The shortest path runs through the R272–D240 ion pair interaction adopting both, intra- and inter-subunit routes.
In the wild-type protein, notable positive correlation is observed between the loops L1, L2, and L3 and the C-terminal (CT) region beyond the AP loop including the α3 helix of the two-helix bundle in the OM and the β7c strand that features the conserved motif R353DLDF3577,14 important for T4S system function. There is a weak negative coupling of domain movements of the loops L1, L2, and L3 with the α2 helix that forms the TM helix in the OM. However, analysis of the DCCM for mt-AtVirB10CTD has a different correlation pattern. In particular, the correlations between regions 232–246 and 265–273 show remarkable differences (Figure 4C,D). These regions show strongly positively correlated motions in the wild-type protein, which disappears completely with the mutation. However, an area showing an increase in negative correlation emerges with the mutation. Overall, these results indicate that the single mutation causes significant changes in the dynamical flexibility of loops L2 and L3 and dynamical correlations of residues across the entire protein. The different correlation patterns in the two simulations suggest the loops and CT domain are more strongly coupled in wt-AtVirB10CTD, thereby being able to respond to the cellular signals compared to mt-AtVirB10CTD that has different local dynamics between the loops with some negative correlation that might affect response to intracellular signals.
Network Analysis
Dynamical network analysis has been used to describe correlated and/or anticorrelated residue motions in order to identify possible communication pathways between different parts of biomolecular complexes. This method has been successfully applied to study the allosteric coupling between tRNA–protein complexes,18 M2 muscarinic receptors,19 cysteinyl tRNA synthetase,20 GPCRs,21 and long-range signaling in very large multisubunit complexes of the MutS component of the methyl-directed DNA mismatch repair system.22 MD simulation analysis revealed large-scale conformational changes in wt-AtVirB10CTD, while the G272R mutation in mt-AtVirB10CTD restricts these movements. The lateral distance between extreme ends of a monomer between the AP loop residue (G309) at the extracellular end and a residue at the base of the barrel (T216) is 101 Å with residue G272R positioned ∼30 Å from T216. The lockdown of the conformational dynamics as a result of G272R mutation suggested that communication in this system takes place via changes in the local dynamics rather than an induced mechanism. Since AtVirB10CTD is an extremely large protein complex, dynamical networks would be employed to transmit information via pathways connecting residue pairs along the shortest path with the highest pairwise local correlations. We focused our analysis on the functionally most relevant signal propagation between two ends of a subunit and whether residue 272 falls on this effective signal transmission pathway. Previously, such analysis has identified how single mutations can change the dynamical characteristic of entire proteins.23 Analysis of wt-AtVirB10CTD suggested that the communities of network are predominantly limited to interactions within the subunit and rarely make contacts with the adjacent subunit. At no point, the shortest path passes via G272 (Figure 4E). Mutation G272R in mt-AtVirB10CTD leads to a significant repartitioning of the dynamical network (Figure 4F). In the mutant, the shortest path runs through the subunit, passing through the R272 and D240 ion pair. There are two other sets of paths that can be identified. Of these, the major path is via the adjacent subunit, again passing via the R272–D240 ion pair. The minor path follows an intra-subunit route but avoids the R272–D240 ion pair interaction altogether. This variability in the pathways observed in mt-AtVirB10CTD could be an attribute of the rigidification of the complex arising as a result of the lockdown of loops 2 and 3 due to the formation of the R272–D240 ion pair interaction.
Discussion
The T4S system functions in two modes: (a) the pilus biogenesis mode also described as a mate-seeking mode and (b) the translocation mode for secretion also described as the mating mode.2,7 T4S systems are versatile in the diverse array of substrates they can transport, and in the translocation mode, it has been suggested that there could be two further conformational states—one for transporting protein substrates that later switches to an ssDNA substrate translocation mode in conjugation machineries.2 The inner membrane complex and the outer membrane core complex assemble initially to form the core assembly across the cell membrane. ATPases associate at the cytoplasmic side and power the release of a pilin–phospholipid complex from the inner membrane to initiate pilus biogenesis within the T4S core assembly to form the mate-seeking complex. In response to intra and extracellular signals on host–cell contact, structural transitions take place in the T4S assembly to switch the pilus biogenesis assembly to a translocation or mating complex.
The VirB10 protein provides the scaffold for the assembly of the T4S system during biogenesis and responds to the intracellular ATP state and substrate recruitment by undergoing a conformational change, transmitting this information essential in both terminal functions—pilus biogenesis and secretion. VirB10 inserts into both the inner and outer membranes, thereby being uniquely positioned to channel information across the inner membrane complex (IMC) and OMCC in the T4S system. High-resolution structures of diverse outer membrane complexes from both a conjugation system such as the E. coli pKM101 plasmid and an effector translocation system such as the X. citri and H. pylori Cag T4S system show a conserved architecture with VirB10 forming the central channel in the outer layer of the OMCC. However, the structural transitions that could respond to intracellular or extracellular signals and changes in conformational dynamics required to bring about function are poorly understood. The substrate translocation path for ssDNA chalked out using the TrIP assay for VirB proteins that make contact with the substrate identified VirB9 and VirB2 subunits but not VirB10 in the O-layer,12 leaving the puzzle for what forms the translocation path in different modes unsolved. Mutation studies to understand domain contribution of VirB10 protein to specific functions provided the first hints toward mechanistic understanding. Uncoupling mutations that support secretion in the absence of surface-exposed pilus were identified that included the AP region of VirB10 (VirB10: Δ282–335) revealing a role of the distal end of the OMCC in regulating pilus biogenesis and a G272R mutation in the OMCC chamber close to the AP that spans the OM. We performed extended molecular dynamics simulations of both wt-AtVirB10CTD and G272R mt-AtVirB10CTD to capture the structural transitions that the VirB10 CTD oligomer can undergo and the difference that might explain the phenotype seen in mt-AtVirB10CTD. These transitions can point toward the structural mechanism that senses cellular signals to bring about the switching required for various functional modes. The ATP-insensitive open complex of mutant G272R showed a more rigid conformation throughout the dynamic simulation run compared to the wild-type oligomer, consistent with the lack of conformational flexibility required for sensing the intracellular energy state. RMSDev and RMSF plots showed maximum fluctuations in the AP loop for both wt-AtVirB10CTD and mt-AtVirB10CTD followed by noticeable deviations in the loop regions of VirB10 in the inter-subunit interface including the L1, L2, L3, and flap regions for wt-AtVirB10CTD. The AP loops are extracellular and flexible in all structures of the OMC solved so far. They can tolerate insertions and deletions for both secretion and pilus biogenesis functions, suggesting a more structural role for this flexible loop. This suggests that the conformational flexibility in the loops bulging out from the barrel domain might coordinate information in the wild-type protein to relay function across the tetradecamer. In order to further investigate the role of these loops, we compared the inter-subunit interface of VirB10CTD in high-resolution structures that are available for OMC for diverse systems—conjugative plasmid pKM101 and effector translocators—X. citri and H. pylori Cag T4S systems (Figure 5).
What is striking in all known structures is a conserved salt bridge triad in the inter-subunit interface formed between L2 and L3 loops of one subunit with the L1 loop of the neighboring subunit. A conserved arginine (R263 in E. coli TraF, R257 in X.citri VirB10, and R1754 in H. pylori CagY) in loop L2 forms a salt bridge with a charged residue in the L3 loop (N288 in E. coli TraF, D281 in X. citri VirB10, and E1783 in H. pylori CagY), which has the conserved GxxGxxG motif supported by an ionic interaction with a charged residue from loop L1 of a neighboring subunit (E216 in E. coli TraF, R211 in X. citri VirB10, and K1705 in H. pylori CagY).
The equilibrium model of the wild-type VirB10CTD tetradecamer from A. tumefaciens and the structures at the end of 500 ns and 1 μs simulation show a similar salt bridge triad coordinated by the corresponding conserved arginine R242 in loop L2 with R270 in the loop L3 and D197 in loop L1 of the neighboring subunit. The salt bridge triad can coordinate the movements of these loops, consistent with the positive correlations noted for these loops in the DCCM maps. Although no mutations of the residues in this triad have been carried out previously, mutation of V243C in the L2 loop attenuated substrate secretion, suggesting a role for this loop in T4S function. The DCCM data also showed the coupling of the loops with the C-terminal region beyond the AP loops, including the conserved R353DLDF357 region, which has been shown to be essential for both secretion and pilus biogenesis. The conserved RDLDF region and the flap region are in close proximity to the L1, L2, and L3 loop salt bridge triad in the inter-subunit interface (Figure 1C).
Equilibrium structures for G272R mt-AtVirB10CTD at 500 ns and 1 μs simulation run time reveal a repositioning of the ionic interactions between the loops affecting their local dynamics (Figure 5). The G272R mutation disrupts the conserved salt bridge formed between R242 with loop L3 and loop L1. Instead, the R272 in loop L3 forms a salt bridge with D240 in loop L2 changing the interloop dynamics. This interaction is further supported by S199 and D197 interactions in loop L1 of the neighboring subunit. Loop L3 is locked down toward the barrel by interactions between R270 with D266 and E267, further affecting the dynamics of this loop. DCCM maps for G272R mt-AtVirB10CTD show the disappearance of the positive correlation between L2 and L3 loops and instead show the emergence of a negative correlation in the region surrounding R270 in loop L3 and R242 in loop L2, uncoupling these loops in the mutant. Analysis of intrinsic dynamics using aggregated root-mean-square fluctuations for the top 10 quasi-harmonic modes (RMSF10) also showed significant changes in conformational flexibility of loops L2 and L3 in G272R mt-AtVirB10CTD and slightly increased flexibility in loop L1, in contrast to the wild-type protein. Reduced flexibility in loops L2 and L3 and uncoupling between the L1, L2, and L3 loops lead to the rigidification of the entire VirB10CTD oligomer in the mutant.
We employed network analysis to identify the changes in the communication pathway in the wild type versus G272R mutant to understand how the swapping of electrostatic interactions between the loops affects the local dynamics leading to large-scale changes in conformational flexibility. We looked for the shortest communication path with the highest pairwise correlation between the extracellular APL loop to the periplasmic end facing the cytoplasm at the base of the O-layer for the extremely large AtVirB10CTD protein complex and looked for loop L3 that features the conserved GxxG272xxG motif in this path. At no point did the shortest path in wt-AtVirB10CTD pass through G272, and the communities of networks were limited to intra-subunit interactions. However, mt-AtVirB10CTD features the R272–D240 ion pair in its shortest communication path with a few alternate paths through neighboring subunits. The different dynamic pathways in the wild-type and mutant proteins suggest that the conformational switching in the VirB10 oligomer is brought about by changes in local dynamics rather than induced mechanisms.
The VirB10 protein in the T4S channel needs to undergo a conformational switch in response to intracellular and extracellular signals to switch the assembly from pilus biogenesis to the secretion mode. Uncoupling mutations have shown that secretion can take place in the absence of a substrate-exposed pilus, although pilin monomers are expressed. Our studies suggest that this conformational switch is regulated by the local dynamics employed between the loops bulging from the atypical β-barrel and any local changes can cause large-scale conformational changes as seen in the G272R mutant dynamics. The swapping of the electrostatic ion pairs between the loops L1, L2, and L3 can affect local dynamics sufficient to change the communication paths across the complex and detach the communication link between the various regions of the VirB10 protein. This disengagement leads to the rigidification of the mutant protein, making it unable to respond to intracellular signals required for T4S function. However, in situ, where many other proteins, signaling molecules, and host factors are present, an induced mechanism (or a combination of both) could also drive VirB10 conformational switching.
Computational Methods
Since there is no high-resolution structure of VirB10 from A. tumefaciens, we constructed a homology model based on the E. coli plasmid pKM101 TraF/VirB10 structure as a template. The crystal structure of the E. coli outer membrane complex of a type IV secretion (T4S) system was downloaded from Protein Data Bank (PDB ID3JQO). The E. coli T4S system OMC structure is a complex of three proteins, namely VirB7, VirB9CTD, and VirB10CTD. From the complex, the coordinates of VirB7 and VirB9 were removed, and only VirB10CTD was used for further studies. The core of VirB10CTD comprises 14 symmetric subunits, each comprising an N-terminal arm, a 7-sheet atypical β-barrel, and two helices—α2 and α3. There is a 23 amino acid loop that connects α2 and α3 helices, which is missing from the E. coli VirB10 crystal structure.
Modeling
The sequence of A. tumefaciens VirB10 was downloaded from UniProt (P05359) and aligned with the E. coli pKM101 TraF/VirB10 using Clustal Omega.24 The homology model was built based on a 40% sequence identity over 217 residues with E. coli TraF/VirB10. The 23 residues between α2 and α3 helices were built as a loop. The model was generated using Modeller v9.2125 with symmetry operation switched on to generate a tetradecameric assembly. This enables each subunit to be an exact copy of the others. The loop between α2 and α3 was refined until a conformation was obtained, which was able to fit the electron density maps of the OMCC EM structures.6,26 The stereochemical parameters were checked using PROCHECK27 and PROSA.28 The final model was chosen based on the low-energy function and low Cα RMSD overlap between the template and the model. Several rounds of minimization were performed to relieve any steric clashes between the side chains. The final Cα between the template and the model was 0.68 Å.
The central cavity running through the core of the protein was hydrated via the use of a grid-based cavity solvation method as implemented in VOIDOO and FLOOD,29 resulting in a total of 100 initial pore water molecules. The model and the pore waters were then imported in the Schrodinger Maestro suite for pre-processing and protonation state assignment using the protein preparation wizard. The Orientation of Proteins in Membrane (OPM) database was used to identify the spatial arrangement of VirB10 with respect to a preformed dimyristoylphosphatidylcholine (DMPC) lipid bilayer. The DMPC bilayer was chosen as it represented a bacterial model bilayer. A system builder was used to embed the protein in the bilayer, and solvent was placed at either ends. The system was neutralized using counterions, which were added by replacing the solvent molecules. Amberff99SB-ILDN30 was used to describe the protein interactions in the system, with explicit TIP3P water molecules. The final system was a simulation cell consisting of 3038 protein residues, 738 DMPC lipids, 84,301 water molecules, and 70 counterions, totaling 385,570 atoms in a box of size 160 × 160 × 130 Å3.
Simulations
The simulations were run using Desmond3.6.31 A 50 ns NPT equilibration was carried out followed by 1 μs of production run using the NVT ensemble. Production simulations were initiated from the final snapshot of the corresponding equilibration simulation. All bond lengths of hydrogen atoms were constrained using M-SHAKE.32 An r-RESPA integrator was used with a time step of 2 fs for the short-range bonded and non-bonded interactions, and long-range non-bonded interactions were computed every 6 fs.33 The mutant VirB10 was generated by replacing Gly272 with Arg272, and ΔΔG calculated using an in silico site directed mutagenesis method as implemented in ICM-Pro suite.34 The mutant VirB10 system was simulated using the same protocol as the wild type. The simulations were run in parallel on a single GPU 1080Ti workstation at a rate of ∼15 ns/day.
Analysis
To reduce the noise from the simulations, the incomplete N-terminal arm was trimmed by removing the first 29 residues, resulting in the VirB10 core retaining 200–386 residues. VirB10 exhibits tetradecameric symmetry; thus, we used three consecutive subunits (A B C) for further analysis. The simulations were analyzed using the VMD suite and the Gromacs suite, while the structural figures were generated using VMD, ICM-Pro, and PyMol software.34−36
Hole Plot
The time-dependent behavior of the pore was calculated using the channel annotation package CHAP (www.channotation.org).37 The trajectories were aligned along the z axis following which each frame is then analyzed using the CHAP program. The program calculates summary statistics (mean, standard deviation, minimum, and maximum) over time for each point in the pore profile. The s coordinate follows the pathway of the pore. CHAP calculates the hydrophobicity profile according to a scale proposed by Wimley and White38 and rescales between −1 (hydrophilic) to +1 (hydrophobic). The pores were visualized using VMD software.35
RMSF10 Calculations for Protein Flexibility/Dynamics
The aggregated root-mean-square fluctuations for the top 10 quasi-harmonic modes (RMSF10) were used to characterize protein flexibility. It is well known that the slowest 10 modes contribute to the majority of fluctuations in proteins (>80%) and the use of RMSF10, instead of all modes, removes the faster stochastic motions of the protein, allowing focus on intrinsic dynamics of proteins.17 These calculations were performed using the ptraj analysis program.39 As a full protein oligomer comprises over 3000 protein residues, for computational reasons, the quasi-harmonic analysis was performed for three monomers at a time (monomer and its two adjoining neighbors). The results for all 14 sets of trimers were collected. For each calculation, 2001 frames from MD simulations were used, and the coordinates were aligned to the first frame to remove rotation and translation before the calculation.
Dynamic Cross-correlation Matrix (DCCM)
The dynamic cross-correlations for Cα–Cα were calculated using the ptraj analysis program.39 These calculations were also performed for a set of trimers in a similar way to RMSF10 calculations, and the results were plotted and visualized using MATLAB (www.mathworks.com).
Network Analysis
Communication networks within the wild type and the mutant were identified using NetworkView.18,40 The trajectories from the molecular dynamics simulations were used to construct the dynamic networks. The system was simplified by representing each residue as a single Cα node. A local contact matrix was generated after extracting all conformations from a simulation run. Two nodes (excluding neighboring nodes) are in contact if they are within a distance of 4.5 Å for more than 75% of the simulation time. The interdependence between nodes is weighted by correlation and represented as a connecting edge. The dynamic cross-correlational maps (DCCM) generated have the ability to identify highly correlated or anticorrelated nodes. However, to compute communication pathways, it is useful to construct a matrix (C) where small values indicate highly correlated or anticorrelated motions. This can be functionalized by wij = −log(|Cij|) where wij can be thought of as a distance in the functionalized correlation space between node–node pairs i and j. The DCC maps for network analysis were calculated using the program Carma.41 The shortest (optimal) path between two nodes was obtained using the Floyd–Warshall algorithm.42 The number of optimal paths that cross one edge is termed as betweenness of the edge (node). The length of a path is the sum of the edge weights between the consecutive nodes along this path. Suboptimal paths within a certain limit (offset) between the two nodes were also determined in addition to the optimal path. The number of suboptimal paths shows the path degeneracy. Communities or disjoined subnetworks were calculated using the Girvan–Newman algorithm43 where nodes have stronger and more connections to other nodes within the same community than they have to those outside the community.
Acknowledgments
We thank Aravindan Ilangovan for discussion and useful comments on the manuscript. S.H. would like to thank Paul Barchilon (www.barchilonceramics.com) for providing the 7-fold geometric pattern image.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03313.
Figures S1: multiple sequence alignments of VirB10 sequences from diverse T4S systems; Figure S2: network communities for wild-type and G272R mutant AtVirB10; and Figure S3: stereochemical parameters check of the A. tumefaciens VirB10 model (PDF)
Movie S1: wild type (MPG)
Movie S2: mutant G272R (MPG)
Author Present Address
# P.K.A.: Department of Physiological Sciences, Oklahoma State University, Stillwater, Oklahoma 74078, United States
Author Contributions
V.C.D. and S.H. conceived the project, S.H. carried out the modeling and molecular dynamics runs, S.H., V.C.D., J.C., J.S.P., B.I., and P.A. performed the trajectory analysis, S.H., V.C.D., and P.A. wrote the main text, and all authors edited the manuscript.
S.H. is supported by NIH grant RO1AI063517. V.C.D. is supported by an institution start up grant from the Queen Mary University of London. P.K.A. is supported by NIH grant GM105978.
The authors declare no competing financial interest.
Supplementary Material
References
- Sgro G. G.; Oka G. U.; Souza D. P.; Cenens W.; Bayer-Santos E.; Matsuyama B. Y.; Bueno N. F.; Dos Santos T. R.; Alvarez-Martinez C. E.; Salinas R. K.; Farah C. S. Bacteria-Killing Type IV Secretion Systems. Front. Microbiol. 2019, 10, 1078. 10.3389/fmicb.2019.01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman G. From conjugation to T4S systems in Gram-negative bacteria: a mechanistic biology perspective. EMBO Rep. 2019, 20, e47012 10.15252/embr.201847012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. G.; Christie P. J. The Agrobacterium VirB/VirD4 T4SS: Mechanism and Architecture Defined Through In Vivo Mutagenesis and Chimeric Systems. Curr. Top. Microbiol. Immunol. 2018, 418, 233–260. 10.1007/82_2018_94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. G.; Hu B.; Christie P. J.. Biological and Structural Diversity of Type IV Secretion Systems. Microbiol. Spectr. 2019,7 (), 10.1128/microbiolspec.PSIB-0012-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low H. H.; Gubellini F.; Rivera-Calzada A.; Braun N.; Connery S.; Dujeancourt A.; Lu F.; Redzej A.; Fronzes R.; Orlova E. V.; Waksman G. Structure of a type IV secretion system. Nature 2014, 508, 550–553. 10.1038/nature13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronzes R.; Schäfer E.; Wang L.; Saibil H. R.; Orlova E. V.; Waksman G. Structure of a type IV secretion system core complex. Science 2009, 323, 266–268. 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J. E.; Costa T. R. D.; Patel R. S.; Gonzalez-Rivera C.; Sarkar M. K.; Orlova E. V.; Waksman G.; Christie P. J. Use of chimeric type IV secretion systems to define contributions of outer membrane subassemblies for contact-dependent translocation. Mol. Microbiol. 2017, 105, 273–293. 10.1111/mmi.13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T.; Koike M.; Bui X. T.; Higaki S.; Aizawa S.-i.; Nagai H. Native structure of a type IV secretion system core complex essential for Legionella pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11804–11809. 10.1073/pnas.1404506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. M.; Sheedlo M. J.; Campbell A. M.; Sawhney N.; Frick-Cheng A. E.; Lacy D. B.; Cover T. L.; Ohi M. D. Structure of the Helicobacter pylori Cag type IV secretion system. Elife 2019, 8, e47644. 10.7554/eLife.47644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V.; Fronzes R.; Duquerroy S.; Cronin N.; Navaza J.; Waksman G. Structure of the outer membrane complex of a type IV secretion system. Nature 2009, 462, 1011–1015. 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgro G. G.; Costa T. R. D.; Cenens W.; Souza D. P.; Cassago A.; Coutinho de Oliveira L.; Salinas R. K.; Portugal R. V.; Farah C. S.; Waksman G. Cryo-EM structure of the bacteria-killing type IV secretion system core complex from Xanthomonas citri. Nat. Microbiol. 2018, 3, 1429–1440. 10.1038/s41564-018-0262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E.; Christie P. J. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17228–17233. 10.1073/pnas.0405843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Khara P.; Christie P. J. Structural bases for F plasmid conjugation and F pilus biogenesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 14222–14227. 10.1073/pnas.1904428116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski S. J.; Kerr J. E.; Garza I.; Krishnamoorthy V.; Bayliss R.; Waksman G.; Christie P. J. Agrobacterium VirB10 domain requirements for type IV secretion and T pilus biogenesis. Mol. Microbiol. 2009, 71, 779–794. 10.1111/j.1365-2958.2008.06565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta L. M.; Kerr J. E.; Cascales E.; Giuliano M. E.; Bailey M. E.; McKay C.; Chandran V.; Waksman G.; Christie P. J. An Agrobacterium VirB10 mutation conferring a type IV secretion system gating defect. J. Bacteriol. 2011, 193, 2566–2574. 10.1128/JB.00038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza I.; Christie P. J. A putative transmembrane leucine zipper of agrobacterium VirB10 is essential for t-pilus biogenesis but not type IV secretion. J. Bacteriol. 2013, 195, 3022–3034. 10.1128/JB.00287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A.; Agarwal P. K. Computational identification of slow conformational fluctuations in proteins. J. Phys. Chem. B 2009, 113, 16669–16680. 10.1021/jp9077213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical networks in tRNA: protein complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6620–6625. 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Nichols S. E.; Gasper P. M.; Metzger V. T.; McCammon J. A. Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10982–10987. 10.1073/pnas.1309755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Sakaguchi R.; Liu C.; Vishveshwara S.; Hou Y.-M. Allosteric Communication in Cysteinyl tRNA Synthetase a network of direct and indirect readout. J. Biol. Chem. 2011, 286, 37721–37731. 10.1074/jbc.M111.246702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.; Vaidehi N. Differences in allosteric communication pipelines in the inactive and active states of a GPCR. Biophys. J. 2014, 107, 422–434. 10.1016/j.bpj.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Francis J.; Sharma M.; Law S. M.; Predeus A. V.; Feig M. Long-range signaling in MutS and MSH homologs via switching of dynamic communication pathways. PLoS Comput. Biol. 2016, 12, e1005159 10.1371/journal.pcbi.1005159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Singh P.; Gabriel G.; Lin J.; Agarwal P. K.; Cheatum C.; Amnon Kohen A.. Establishing the role of dynamic network of coupled motions in human dihydrofolate reductase. Biochemistry (Under Review) 2019.
- Sievers F.; Higgins D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- Šali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Rivera-Calzada A.; Fronzes R.; Savva C. G.; Chandran V.; Lian P. W.; Laeremans T.; Pardon E.; Steyaert J.; Remaut H.; Waksman G.; Orlova E. V. Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J. 2013, 32, 1195–1204. 10.1038/emboj.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 178–185. 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct., Funct., Bioinf. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.; Salmon J. K.; Shan Y.; Shaw D. E.. Scalable algorithms for molecular dynamics simulations on commodity clusters. In SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; IEEE: 2006; pp 43–43.
- Kräutler V.; Van Gunsteren W. F.; Hünenberger P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. . [DOI] [Google Scholar]
- Tuckerman M.; Berne B. J.; Martyna G. J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. 10.1063/1.463137. [DOI] [Google Scholar]
- Abagyan R.; Totrov M.; Kuznetsov D. ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. 10.1002/jcc.540150503. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.PyMOL molecular viewer: Updates and refinements. In Abstracts of Papers of the American Chemical Society; American Chemical Society; 1155 16TH ST, NW, Washington, DC 20036 USA: 2009. [Google Scholar]
- Klesse G.; Rao S.; Sansom M. S. P.; Tucker S. J. CHAP: A Versatile Tool for the Structural and Functional Annotation of Ion Channel Pores. J. Mol. Biol. 2019, 431, 3353–3365. 10.1016/j.jmb.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842. 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Eargle J.; Luthey-Schulten Z. NetworkView: 3D display and analysis of protein· RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykos N. M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- Floyd R. W. Algorithm 97: shortest path. Commun. ACM 1962, 5, 345. 10.1145/367766.368168. [DOI] [Google Scholar]
- Girvan M.; Newman M. E. J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7821–7826. 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P.; Courcelle E. ENDscript: a workflow to display sequence and structure information. Bioinformatics 2002, 18, 767–768. 10.1093/bioinformatics/18.5.767. [DOI] [PubMed] [Google Scholar]
- Wheeler T. J.; Clements J.; Finn R. D. Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinf. 2014, 15, 7. 10.1186/1471-2105-15-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.