

Abstract
Antimicrobial peptides (AMPs), an essential component of innate immunity, are very important resources for human therapeutics to counter the current threat of drug resistance. We have previously established that one such AMP, α-melanocyte stimulating hormone (α-MSH), an endogenous neuropeptide, and its derivatives have potent antimicrobial activity against Staphylococcus aureus, including methicillin-resistant S. aureus (MRSA). However, the immense potential of α-MSH for therapeutic development against staphylococcal infections is marred by its reduced efficacy in the presence of standard microbiological culture medium. To overcome this issue, in this study, we designed a series of five novel analogues of the C-terminal fragment of α-MSH, i.e., α-MSH(6–13), by replacing uncharged and less hydrophobic residues with tryptophan and arginine to increase the hydrophobicity and cationic charge of the peptide, respectively. While all of the peptides showed a preferential interaction with negatively charged phospholipid vesicles, the most hydrophobic and cationic peptide, i.e., Ana-5, exhibited the highest activity against S. aureus cells while maintaining cell selectivity. Moreover, Ana-5 could retain its activity even in complex media like the Mueller Hinton broth and displayed rapid bactericidal activity in the presence of serum. Ana-5 also caused rapid bacterial membrane depolarization, permeabilization, and cell lysis and was able to bind to polyanionic plasmid DNA suggesting a possible dual mode of action of the peptide. Importantly, Ana-5 was able to eradicate intracellular S. aureus in fibroblast cells similar to conventional antibiotics. Collectively, in the present study, we obtained a potent α-MSH-based analogue with excellent staphylocidal potency in microbial growth medium and ex vivo efficacy, which may translate into therapeutic application.
Introduction
Staphylococcus aureus is a major etiological agent of skin, soft tissue, device-related infections as well as threatening diseases such as bacteremia, pneumonia, and infective endocarditis contributing significantly to morbidity and mortality in nosocomial infections.1,2 Over the last few decades, S. aureus has equipped itself with multiple drug resistance (MDR) and a plethora of virulence factors that allow this deadly pathogen to avoid immune clearance and thrive inside human hosts.3 In the face of escalating MDR, membrane disruptive cationic antimicrobial peptides (CAMPs) with nonspecific multitargeting potential have emerged as lucrative candidates. CAMPs are ubiquitous, naturally occurring host defense peptides (11–60 residues) serving as the first line of defense in almost all life forms.4 A number of peptide hormones and neuropeptides that share cationic and amphipathic structures have been shown to modulate immune response, thereby protecting the host against microbial invasion and infection.5 Several neuropeptides such as α-melanocyte stimulating hormone (α-MSH), substance P, and neuropeptide Y show direct anti-infective properties regulated through spatial specificity and delivery at the site of infection.6,7 α-MSH is an endogenous trideca-neuropeptide (Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2) produced through cleavage of precursor polypeptide pro-opiomelanocortin (POMC). It primarily induces melanogenesis and also exerts antipyretic, anti-inflammatory, neuroprotective, and immunomodulatory effects by interacting with various melanocortin receptors.8,9
Over the past decade, our group has established the rapid and potent activity of α-MSH and its shorter C-terminal fragments against various clinical strains of S. aureus, including methicillin-resistant S. aureus (MRSA).10−12 We also demonstrated that α-MSH primarily acts through membrane permeabilization and depolarization in S. aureus and also hampers DNA replication and protein synthesis without compromising cell selectivity.11,13 Encouragingly, α-MSH showed synergistic activity with different classes of antibiotics and effective in vivo activity against S. aureus.13,14 Further, we showed that α-MSH has a better affinity toward anionic membrane environment, and therefore the de novo α-MSH designed analogues with enhanced cationic charge promoted the staphylocidal efficacy.15,16 In our previous study, we also demonstrated the C-terminal region of α-MSH, i.e., α-MSH(6–13), to be equally potent against S. aureus as the entire peptide.11 However, α-MSH-based peptides have shown little antimicrobial activity in standard bacterial growth media like Mueller Hinton broth (MHB).10−17 It might be due to the complex formed by these cationic analogues with components present in MHB like carbohydrates and proteins.18,19 Therefore, in this study, we aimed to design analogues that could overcome the barrier of α-MSH inactivity in media. Several natural antimicrobial peptides, e.g., cathelicidins (indolicidin and tritrpticin), lactoferricin B, and defensin, and successful synthetic CAMPs have shown potent antimicrobial activity due to the presence of Arg and Trp residues or domains.20 Being shorter in length, the analogues of α-MSH(6–13) may also cost less for pharmaceutical production. Keeping these points in mind, we selected this truncated region of α-MSH to design novel analogues and modified the pharmacophore region His6-Phe7-Arg8-Trp9 to diminish their melanogenic activity and other downstream effects. Thus, we designed a series of five novel analogues by increasing the cationic charge and hydrophobicity of the peptides by substituting Arg and Trp residues in the primary sequence of α-MSH(6–13), respectively. To understand the basis of antibacterial activity of the designed analogues, we first evaluated their secondary structure and interaction with artificial bacterial mimic lipid vesicles by employing circular dichroism (CD) spectroscopy, dynamic light scattering (DLS), and Trp fluorescence studies. Next, we determined the antibacterial efficacy of the designed analogues against S. aureus cells and their toxicity toward mammalian cells. The active analogues were then investigated for their mode of action using membrane perturbation assay, lipid binding assay, and DNA gel retardation assay. Among them, the most active analogue was further assessed for the peptide-membrane interaction by employing flow cytometric and microscopic tools. Finally, the potency of the most active analogue was evaluated under ex vivo conditions against intracellular S. aureus.
Results
Peptide Design
Based on the mounting repertoire of biologically active α-MSH-based peptides, we designed five novel analogues of α-MSH by incorporating basic (Arg) and hydrophobic (Trp, Phe) residues in the sequence of α-MSH(6–13), as shown in Table 1. Arg and Trp/Phe are known to enhance the ability of the peptide to interact with and insert into the bacterial membrane, respectively.21 Therefore, maintaining the charge (+2) of the parent peptide, in Ana-1, we replaced Phe7 with Trp residue to alter its hydrophobicity (Table 1). Next, in Ana-2, the Gly10 residue of Ana-1 was replaced with Trp to enhance its hydrophobicity further. As expected, the reversed-phase high performance liquid chromatography (RP-HPLC) retention time data indicated no change and an ∼11% increase in % hydrophobicity of Ana-1 and Ana-2 relative to the parent peptide, respectively (Table 1). Further, in both Ana-1 and Ana-2, His6 residue was replaced with Arg to increase their cationic charge to +3, giving rise to Ana-3 and Ana-4, respectively. Interestingly, it has also been reported that replacement of Pro12 with Phe/Tyr in α-MSH(6–13) affords conformational stabilization through aromatic interactions, which in turn improves the antifungal activity of the peptide.22 Keeping this in mind, we designed Ana-5 by replacing the Pro12 residue with Phe in Ana-4. Ana-5, therefore, showed maximum charge (+3) and hydrophobicity (∼20% increase from parent peptide) among the designed peptides (Table 1). The retention time of the analogues was ascertained through RP-HPLC for all peptides.
Table 1. Sequence and Physicochemical Properties of α-MSH(6–13) and the Designed Analogues.
| mass (Da) |
|||||
|---|---|---|---|---|---|
| peptide | sequence | Calc. | Obs. | charge | % Ha |
| α-MSH(6–13) | Ac-H6-F7-R8-W9-G10-K11-P12-V13-CONH2 | 1067.25 | 1067.27 | +2 | 46.4 |
| Ana-1 | Ac-H6-W7-R8-W9-G10-K11-P12-V13-CONH2 | 1106.29 | 1106.88 | +2 | 46.4 |
| Ana-2 | Ac-H6-W7-R8-W9-W10-K11-P12-V13-CONH2 | 1235.43 | 1235.60 | +2 | 51.7 |
| Ana-3 | Ac-R6-W7-R8-W9-G10-K11-P12-V13-CONH2 | 1125.33 | 1125.98 | +3 | 46.9 |
| Ana-4 | Ac-R6-W7-R8-W9-W10-K11-P12-V13-CONH2 | 1254.49 | 1254.52 | +3 | 51.3 |
| Ana-5 | Ac-R6-W7-R8-W9-W10-K11-F12-V13-CONH2 | 1304.55 | 1304.58 | +3 | 55.6 |
% hydrophobicity calculated on the basis of percentage of acetonitrile at RP-HPLC elution.
Secondary Structure of Designed Peptides
CD spectra of the designed peptides were acquired in a 5 mM sodium phosphate buffer (PB) and bacterial membrane mimic DMPC/DMPG (7:3, w/w) and DMPC/DMPG (1:1, w/w) small unilamellar vesicles (SUVs). In this study, we also used Trp-Arg rich indolicidin as a control due to the availability of its detailed structural characterization in different environments in the literature.23 As the designed sequences are short and contain multiple numbers of aromatic side chains (Trp and Phe residues), there might be a strong influence of aromatic side chains on the peptide backbone signals.23
In buffer, spectra of all of the peptides were consistent with an unordered/turn conformation except Ana-5, which showed a tendency toward structuring with double minima at 205 and 227 nm (Figure 1a). Further, in both the bacterial mimic SUVs (7:3, w/w and 1:1, w/w, DMPC/DMPG),16,24 Ana-5 showed a very prominent minimum at ∼230 nm coupled with a maximum at 212 nm. This enhancement in the minimum at ∼230 nm in Ana-5 in the presence of lipids indicates further induction of ordered conformation. Similarly, the appearance of a double minima was also observed for Ana-2 and Ana-4 (Trp-Trp domain-containing peptides like Ana-5) in the presence of negatively charged lipid vesicles, whereas Ana-1 and Ana-3 did not show any ordered structure in the same environment. Similar to earlier reports, indolicidin, a standard Trp-Trp domain-containing peptide, exhibited a double minima at 207 and ∼230 nm coupled with a positive peak at ∼220 nm (characteristic of a turn conformation) in the presence of both studied SUVs but not in buffer.25 The CD spectra of the designed analogues containing a Trp-Trp domain, i.e., Ana-2, Ana-4, and Ana-5, as well as the standard peptide indolicidin are governed through exciton coupling between bulky indole rings and the formation of intramolecular H-bonding effect upon partitioning into bacterial membrane mimic vesicles.26
Figure 1.

Structural characterization of the designed peptides. (a) Circular dichroism spectra of α-MSH(6–13) and its analogues in 5 mM PB (blue), DMPC/DMPG (7:3, w/w) SUVs (green), and DMPC/DMPG (1:1, w/w) SUVs (pink). (i) α-MSH(6–13), (ii) Ana-1, (iii) Ana-2, (iv) Ana-3, (v) Ana-4, (vi) Ana-5, and (vii) indolicidin, at peptide and lipid concentrations of 35 and 1453 μM, respectively (L/P molar ratio, 41.5:1). The spectrum of each peptide represents an average of three scans and was plotted as mean residue ellipticity (in deg cm2 dmol–1) against wavelength (nm). (b) Dynamic light scattering profile expressed as relative intensity vs radius (nm) for DMPC/DMPG (1:1, w/w) SUVs alone (yellow), peptides dissolved in water (blue), and with added DMPC/DMPG (1:1, w/w) SUVs (pink). Size distribution of the peptides (i) Ana-2, (ii) Ana-4, and (iii) Ana-5, at peptide and lipid concentrations of 35 and 1453 μM, respectively (L/P molar ratio of 41.5:1). Experiments were performed twice, and representative spectra are shown here.
Further, as our peptides Ana-2, Ana-4, and Ana-5 exhibited enhanced hydrophobicity (Table 1) and ordered structure (Figure 1a) in lipid environment compared to the parent peptide and other analogues, we tried to ascertain that these peptides did not form aggregates, as more hydrophobic peptides have been previously shown to have greater tendency for aggregation, which may adversely affect their activity.27,28 For this, we determined the hydrodynamic size of the peptides in water and the presence of DMPC/DMPG (1:1, w/w) SUVs through DLS. The study revealed the average hydrodynamic radius of Ana-2, Ana-4, and Ana-5 to be 86.9, 86.9, and 62.3 nm, respectively, in water, whereas, the same was 52.8, 62.3, and 62.3 nm, respectively, in the presence of the SUVs (Figure 1b). Though Ana-5 is the most hydrophobic analogue, its lower hydrodynamic radius compared to Ana-2 and Ana-4 in water may indicate that unlike Ana-5, both these analogues have a slight tendency to display aggregation in water. This tendency appears to be lost in a lipid environment as implied by the reduced radius of Ana-2 and Ana-4 in the presence of SUVs. Interestingly, a similar radius of Ana-5 in water, as well as in the presence of SUVs, may suggest that it did not undergo aggregation in both the environments.
Trp Fluorescence Emission Spectrum of the Peptides
We monitored the extent of Trp side-chain insertion of peptides into the bacterial and mammalian membrane mimic vesicles. In buffer (10 mM 2-[tris(hydroxymethyl)-methylamine]-1-ethanesulfonic acid (TES), pH 7.4), all of the peptides showed an emission maximum at 357 nm characteristic of the Trp residue in a free aqueous environment (Table 2 and Figure 2). While in mammalian mimic zwitterionic DMPC SUVs, the emission maximum, as well as the emission intensity of the peptides, remained almost unchanged, in bacterial mimic SUVs, appreciable blue shifts were observed for all of the peptides (9−12 nm) with a concomitant 3- to 6-fold increase in fluorescence intensity. Such preferential interaction with anionic vesicles compared to neutral vesicles has been previously reported for cationic analogues of α-MSH16 and Trp rich peptides.21,29
Table 2. Tryptophan Fluorescence Emission Maxima of α-MSH(6–13) and Its Analogues.
| emission
maxima (in nm) |
|||
|---|---|---|---|
| peptides | TES buffer | DMPC | DMPC/DMPG (7:3, w/w) |
| α-MSH(6–13) | 357 | 357 | 348 |
| Ana-1 | 357 | 357 | 348 |
| Ana-2 | 357 | 357 | 345 |
| Ana-3 | 357 | 357 | 348 |
| Ana-4 | 357 | 357 | 346 |
| Ana-5 | 357 | 354 | 346 |
| indolicidin | 357 | 353 | 346 |
Figure 2.

Tryptophan fluorescence emission spectra of peptides in different milieus: in buffer (blue), DMPC (red), and DMPC/DMPG (7:3, w/w) (green) SUVs. (a) α-MSH(6–13), (b) Ana-1, (c) Ana-2, (d) Ana-3, (e) Ana-4, (f) Ana-5, and (g) indolicidin. Peptide and lipid concentrations were 14.5 and 726 μM, respectively, i.e., L/P molar ratio was 50:1.
Staphylocidal Efficacy of α-MSH(6–13) and Its Designed Peptides in Physiological Buffer
To assess the effect of enhanced charge and hydrophobicity on the relative potency of the designed peptides, we determined their bactericidal efficacy against S. aureus (ATCC 29213). As evident from Figure 3, compared to the parent peptide α-MSH(6–13) at 10 μM concentration, all of the designed analogues apart from Ana-1 showed better killing efficacy. After 2 h of incubation, though α-MSH(6–13) and Ana-1 did not exert any observable effect on the bacterial cells, Ana-3 reduced the viable cell count by 2 ± 1 log. Remarkably, all of the Trp-Trp domain-containing peptides, i.e., Ana-2, Ana-4, and Ana-5, demonstrated bactericidal action against the S. aureus cells and resulted in 3 ± 1, 5 ± 1, and 6 ± 1 log reduction in viability, respectively, under identical conditions. The considerable enhancement in the activity of Ana-4 compared to Ana-2 also highlights the significance of increased charge as both these peptides possessed similar hydrophobicities (Table 1). The standard peptide indolicidin also showed potent activity and caused >5 log reduction in bacterial cell viability.
Figure 3.

Antibacterial activity of α-MSH(6–13) and designed analogues against the mid-log phase of S. aureus. Cell viability was observed after 2 h incubation with 10 μM α-MSH(6–13) and its analogues in 5 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES)-20 mM glucose buffer, pH 7.4. Experiments were done on three different days and presented as mean ± standard deviation (SD) (***P < 0.001).
Minimum Inhibitory Concentration (MIC) of the Designed Peptides
Previously, α-MSH and its synthetic analogues have been shown to exhibit potent antibacterial activity in physiological buffers but not in the standard growth medium, which is a major limitation toward their clinical use.10−17 Thus, in this study, we examined whether the most potent peptides Ana-4 and Ana-5 would retain their activity in standard medium. For this purpose, we employed two microbiological media: (i) a less complex minimal essential medium (MEM) and (ii) a nutrient-rich MHB. Both the analogues that exhibited the highest activity against S. aureus cells in physiological buffer, Ana-4 and Ana-5, also showed potent growth inhibition with MIC values of 11.3–22.7 and 1.4 μM, respectively, against MSSA as well as MRSA in complex MEM (Table 3).
Table 3. MIC of the Potent Analogues against MSSA (ATCC 29213) and MRSA (ATCC 33591) in MHB and MEM Growth Media.
| MIC (in μM) |
||||
|---|---|---|---|---|
| MHB |
MEM |
|||
| peptides | MSSA | MRSA | MSSA | MRSA |
| Ana-4 | >90.8 | >90.8 | 11.3–22.7 | 11.3–22.7 |
| Ana-5 | 11.3 | 11.3 | 1.4 | 1.4 |
| indolicidin | 1.4 | 1.4 | 0.3 | 0.3 |
In the more complex and rich medium, such as MHB, Ana-4 did not show any growth inhibition up to the tested concentration of 90.8 μM. However, Ana-5 remained active even in MHB exhibiting an MIC value of 11.3 μM against both MSSA and MRSA. Thus, the most hydrophobic peptide among the designed series, Ana-5 was also the most potent with the ability to retain its antistaphylococcal activity even in complex growth medium. The standard peptide indolicidin also showed high efficacy with an MIC value of 1.4 μM against both the tested strains in MHB medium, while in the MEM, the MIC value was further reduced to 0.3 μM.
Bacterial Killing Kinetics of the Most Potent Peptide in Growth Media
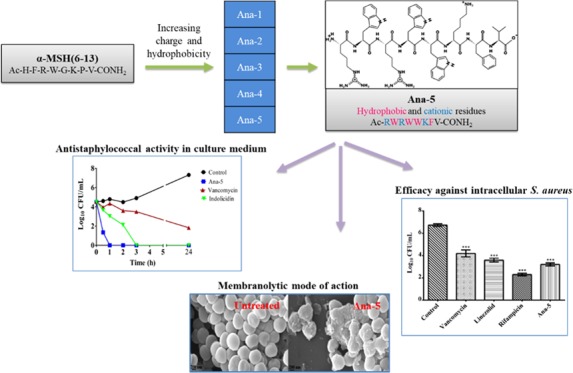
The time dependence of the staphylocidal potency of the most active peptide, Ana-5, was evaluated in MHB medium and compared to the activity of standard comparator peptides indolicidin and vancomycin at 4 × MIC of the peptides. As seen in Figure 4, Ana-5 exhibited potent rapid bactericidal activity, i.e., 3 log reduction, within 30 min of incubation itself and was able to completely eradicate the viable S. aureus cells within only 1 h of incubation. Under similar circumstances, indolicidin and vancomycin showed much slower killing kinetics, as indolicidin required 3 h for the eradication of the cells while vancomycin even after 24 h was only able to cause 3 log reduction and not complete eradication of the bacterial cells. The results observed for indolicidin and vancomycin are in accordance with earlier reports.30,31
Figure 4.
In vitro bacterial killing kinetics of log phase cells of S. aureus in culture medium. The cells were treated with Ana-5, vancomycin, and indolicidin at their 4 × MIC in MHB media for 24 h. The experiments were repeated on three different days, and similar data were obtained. Representative data are shown here.
Serum Stability of the Most Potent Peptide
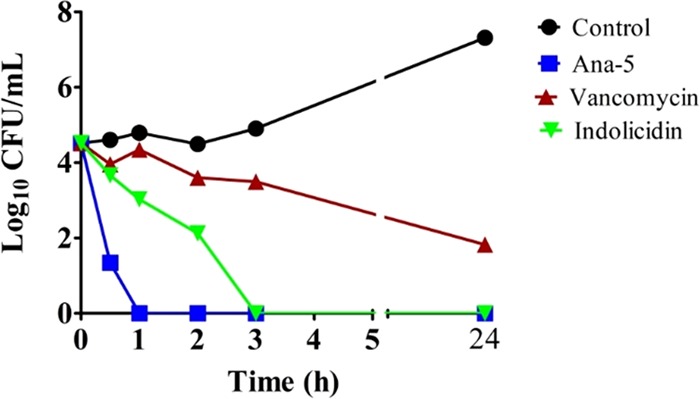
In general, many AMPs are able to exert potent in vitro activity against bacterial cells, but in the presence of a complex biomatrix like serum, i.e., more realistic physiological condition, their antibacterial activity is often inhibited or even completely lost.32 Therefore, next, we determined the activity of Ana-5 in the presence of serum, which is known to diminish the antimicrobial activity of most CAMPs through proteolysis. The bactericidal kinetics of Ana-5, indolicidin, and vancomycin at their 4 × MIC in the presence of MHB with 10% fetal bovine serum (FBS) is shown in Figure 5. Under these conditions, while the comparator peptides indolicidin and vancomycin were able to cause only 3 and 1 log reductions in cell viability, respectively, even after 3 h exposure, Ana-5 exhibited bactericidal action within only 1 h of treatment and completely cleared the bacterial cells in 2 h. Thus, Ana-5 was found to be highly effective against S. aureus cells even under physiologically relevant conditions like the presence of serum, and it was able to maintain its potent bactericidal activity, unlike the standard peptides.
Figure 5.
Antibacterial activity against S. aureus in the presence of 10% serum. Log phase bacterial cells were treated with Ana-5, vancomycin, and indolicidin at their 4 × MIC in MHB media with 10% serum for 3 h. The experiments were repeated on three different days, and similar data were obtained. Representative data are shown here.
Toxicity against Mammalian Cells
Another major limitation toward the translational potential and commercial development of AMPs is their likely toxicity.33 Earlier reports have established α-MSH and its analogues to be nontoxic toward mammalian cells.11,13,16 Therefore, in this study, we also investigated the toxicity of designed peptides toward mammalian cells by determining the % hemolysis of murine red blood cells (RBCs) upon 1 h incubation with various concentrations of the peptides. Encouragingly, the results showed very minimal hemolysis, i.e., <5%, for all designed peptides up to 200 μM, while the standard peptide indolicidin caused 65 and 75% hemolysis at 100 and 200 μM, respectively, under identical conditions (Figure 6a).
Figure 6.

Toxicity studies of α-MSH(6–13) and its analogues against mammalian cells. (a) % Hemolysis of mouse RBCs upon 1 h treatment with different concentrations of the peptides. (b) % Cytotoxicity observed in 3T3 murine fibroblast cell line on treatment with the peptides at 15, 30, and 60 μM concentrations for 2 h. Each assay was done in triplicate on two different days.
Additionally, the cytotoxicity of the designed peptides was also evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay by incubating 3T3 murine fibroblast cell line in serum-free medium with different concentrations of the peptides. As seen in Figure 6b, at 15 and 30 μM concentrations, α-MSH(6–13) and the designed analogues did not exhibit any toxicity, and on increasing the concentration to 60 μM, they showed very minimal toxicity (≤22 ± 4%). At the same concentrations, standard peptide indolicidin exhibited a much higher toxicity, i.e., at 60 μM, it caused 66 ± 4% toxicity toward 3T3 fibroblast cells.
Ability of the Most Potent Peptide to Depolarize the Bacterial Membrane
Following our previous reports, which established a membranolytic mode of action of α-MSH(6–13),11 toward the delineation of the mechanism of staphylocidal action of Ana-5, we determined its effect on the membrane potential of S. aureus cells using the potentiometric dye DiSC3(5). As all of the mechanistic studies have been carried out in buffer, for comparison, we also included Ana-4, another peptide from the designed series with potent activity in the presence of a physiological buffer, and the Trp-Arg rich peptide indolicidin in this study. In Figure 7a, we observed that the addition of 30 μM concentration of the peptides to dye-loaded bacterial cells resulted in instant depolarization of the bacterial membrane, and the fluorescence intensity observed was higher for Ana-5 compared to the parent peptide α-MSH(6–13) as well as Ana-4. The membrane-depolarizing ability of both Ana-4 and Ana-5 was found to be concentration-dependent, and their effect was almost comparable to that of indolicidin (Figure 7b). Of note, unlike Ana-4, Ana-5 was able to dissipate the membrane potential appreciably at a concentration as low as 2 μM. The results of the corresponding killing assay of the dye-loaded cells at 30 μM concentration of the peptides also corroborated with the membrane depolarization study (Figure 7c). It appears that there exists a disparity between the high membrane depolarizing ability of indolicidin in this study (Figure 7a,b) and its slower killing kinetics compared to Ana-5, as shown in Figure 4. This may have resulted due to the difference in the concentration of the peptides used in both these experiments, and as the MIC of indolicidin is much lower compared to Ana-5 (Table 3), it is pertinent to consider that a relatively higher concentration of indolicidin is required to achieve the same order of bactericidal kinetics as Ana-5.30
Figure 7.

Membrane depolarization efficacy of the peptides. (a) Depolarization kinetics measured at a fixed concentration of 30 μM of α-MSH(6–13), Ana-4, Ana-5, and indolicidin for 15 min. (b) Concentration-dependent effect of the peptides on the membrane potential of S. aureus measured by fluorescence intensity of the dye. (c) Corresponding cell viability of the DiSC3(5)-loaded cells after immediate exposure to 30 μM of the peptides in 5 mM HEPES-20 mM glucose buffer, pH 7.4.
Lipid Binding Affinity toward Artificial Bacterial Membrane Mimics
Further, to better understand the enhanced membrane activity of Ana-5 compared to Ana-4 and the parent peptide α-MSH(6–13), we evaluated their relative binding affinity to bacterial mimic liposomes (DMPC/DMPG, 7:3, w/w) and indolicidin was used as a positive control (Figure 8). After the addition of increasing concentrations of bacterial membrane mimetic SUVs to a fixed concentration of the peptide (10 μM) resulted in an increase in the fluorescence intensity, which reached a saturation value, the dissociation constant was calculated by plotting a fraction of bound peptide [(F – F0)/(Fmax – F0)] versus lipid concentration, and the data were fitted to a hyperbolic curve by Origin 8 (2015) software. During fitting of the curve, it was observed that while a hyperbola curve fit the data best for Ana-4, Ana-5, and indolicidin, in the case of α-MSH(6–13), the curve appeared to be sigmoidal. While a hyperbolic curve has been earlier attributed to a noncooperative binding model, a sigmoidal binding curve may suggest that the peptide does not oligomerize upon binding.34,35 The binding constant observed for Ana-4 and Ana-5 was 27 000 and 47 000 M–1, respectively, while for indolicidin, the binding constant observed was 21 000 M–1. Thus, Ana-5 clearly showed better binding to bacterial mimic liposomes compared to Ana-4 and the standard control indolicidin, i.e., 1.7- to 2.2-fold.
Figure 8.

Binding isotherms derived by plotting fractions of the membrane-bound peptide (F – F0)/(Fmax – F0) as a function of the concentration of the lipid. The Trp fluorescence intensity at 295 nm was recorded by titrating 10 μM of the peptides with increasing molar concentrations of SUVs in 10 mM TES buffer (pH 7.4). Data were fitted (OriginLab Corp.) with the hyperbolic saturation curve for DMPC/DMPG (7:3, w/w) in the case of Ana-4 (pink, ●), Ana-5 (green, ▲), and indolicidin (red, ▼), whereas for the parent peptide α-MSH(6–13) (blue, ■), due to weak initial binding, the data points could not be fitted with the same. The data acquired are presented (mean ± SD) after background subtraction and dilution correction.
Bacterial Membrane Permeabilization and Morphological Changes
The membrane damaging ability of Ana-5 was further validated through membrane permeabilization assay and visualization of the damaged bacterial cell through scanning electron microscopy (SEM). The extent of membrane permeabilization of S. aureus cells in the presence of Ana-5 was determined through calcein leakage study by incubating the calcein-loaded cells with 30 μM concentration of the peptide for 2 h. As evident from the histogram of calcein-loaded cells (Figure 9a), compared to the parent peptide α-MSH(6–13), exposure to Ana-5 caused a large shift in the dye fluorescence from higher to lower values, indicating the release of calcein from the bacterial cells with damaged membrane.11,30 The percentage reduction in the observed fluorescence for Ana-5-treated cells was 74 ± 9% (P < 0.01, compared to α-MSH(6–13)) (Figure 9b). The corresponding killing assay also performed at 30 μM concentration of the peptides (Figure 9c) corroborated with the membrane damage as the percentage survival of S. aureus cells treated with Ana-5 was 3 ± 1%, whereas 30 ± 4% of the cells were still viable after α-MSH(6–13) treatment.
Figure 9.

S. aureus membrane perturbation by Ana-5. Membrane permeabilization measured by the percentage of calcein leakage from calcein-AM-loaded 106 CFU mL–1 bacterial cells after 2 h treatment with α-MSH(6–13) and Ana-5. (a) Histograms (from left to right) for calcein-loaded mid-log phase cells that were (i) untreated, (ii) treated with α-MSH(6–13), and (iii) treated with Ana-5. (b) % Calcein leakage from S. aureus cells after treatment with α-MSH(6–13) and Ana-5 at 30 μM concentration. (c) Corresponding % survival of cells after treatment with the same concentration of peptides for 2 h (**P < 0.01, ***P < 0.001). (d) Scanning electron micrographs of S. aureus: (i) untreated control, treated with 50 μM of (ii) α-MSH(6–13) and (iii) Ana-5 at 50 000× magnification.
The morphological changes in the staphylococcal membrane upon treatment with Ana-5 and α-MSH(6–13) were also visualized through SEM (Figure 9d). The untreated control showed cells with an intact membrane, whereas bacterial cells treated with 50 μM of Ana-5 or α-MSH(6–13) for 2 h exhibited membrane perturbation. The peptide treatments resulted in damaged cells with membrane abnormalities, including the collapse of the cell membrane and oozing out of cellular content. However, compared to α-MSH(6–13), the extent of S. aureus membrane disruption was much more pronounced in the case of Ana-5-treated cells.
DNA Binding Affinity of the Most Potent Peptide
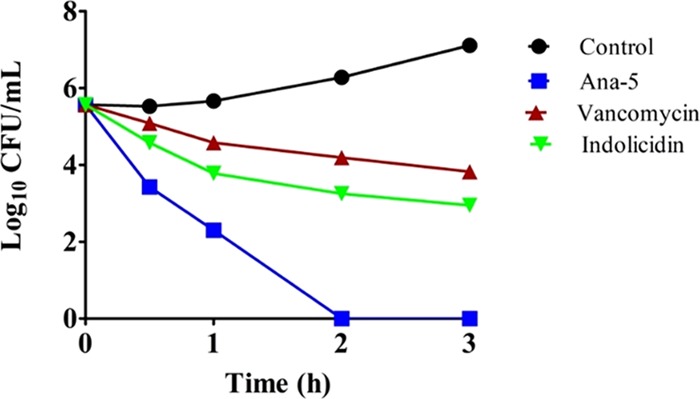
We further investigated whether apart from the bacterial membrane, Ana-5 had another target, i.e., an intracellular target like DNA. For this, we evaluated the ability of the two potent peptides, Ana-4 and Ana-5, to bind to plasmid DNA through electrophoretic mobility shift assay by incubating different concentrations of peptides with plasmid DNA (pBluescriptII SK(+)) for 1 h and measuring the inhibition of DNA migration in 1% (w/v) agarose gel. The results showed that although Ana-4 and Ana-5 have similar cationic charge, the latter bound more strongly to polyanionic plasmid DNA as Ana-5 completely retarded DNA migration at a concentration ≥25 μM, whereas Ana-4 inhibited the migration of DNA at much higher concentrations, i.e., ≥100 μM (Figure 10). Thus, at comparable concentrations, Ana-5 exhibited better DNA binding ability than Ana-4. This suggests that apart from the interaction of cationic residues with phosphodiester bonds, the aromatic residues of Ana-5 might also have stacked between the nucleotide bases of each DNA strand.36 Indolicidin, the standard peptide, was able to inhibit DNA migration at a concentration of 12.5 μM.
Figure 10.
Binding of Ana-4, Ana-5, and indolicidin with plasmid DNA assessed through gel retardation assay. Peptide interaction with plasmid DNA (pBluescriptII SK(+)) was determined via inhibition of DNA migration in 1% (w/v) agarose gel. Different peptide concentrations were incubated with 100 ng of plasmid DNA for 1 h. Lane 1 contains plasmid DNA, and lanes 2–5, 6–9, and 10–13 contain Ana-4, Ana-5, and indolicidin, respectively. The four lanes of each peptide are in decreasing order of peptide concentration, i.e., 100, 50, 25, and 12.5 μM, respectively.
Killing of Intracellular S. aureus in a Coculture Model by Ana-5
During infection, the uptake of S. aureus cells by neutrophils (phagocytic cells) often results in an incomplete clearance of the pathogen, leading to the dissemination of the bacteria to nonphagocytic host cells, including epithelial cells, fibroblasts, osteoblasts, endothelial cells, and keratinocytes.3,37 Therefore, considering the importance of the capability of antimicrobial agents to eradicate internalized bacterial cells, we evaluated the ability of Ana-5 to clear intracellular S. aureus infection and compared it to the activity of conventional antibiotics like rifampicin and the glycopeptide vancomycin. For this, the murine fibroblast cell line was infected with S. aureus cells at a multiplicity of infection (MOI) 10:1, followed by treatment with antibiotics and Ana-5 at their 5 × MIC (Figure 11). While rifampicin exhibited the most effective intracellular killing causing ∼4 log reduction in the viable cell count compared to the untreated control, Ana-5 also resulted in ≥3 log reduction, which was similar to the effect of linezolid and even better than vancomycin, which remains a drug of choice for the treatment of critical MRSA infections and caused ≥2 log reduction. Thus, the results established the ability of Ana-5 to target intracellular S. aureus with similar efficacy to clinically relevant antibiotics and AMPs.
Figure 11.

Ex vivo efficacy of Ana-5, vancomycin, linezolid, and rifampicin at their 5 × MIC in a coculture model of S. aureus and 3T3 murine fibroblast cell line at MOI 10:1. The log reduction of intracellular S. aureus cells upon 24 h incubation with Ana-5 and the antibiotics is shown here. Data were acquired on three different days in duplicates and represent mean ± SD (***P < 0.001).
Discussion
Considering the increasing burden of antibiotic resistance in bacteria and other microorganisms, cationic AMPs have been extensively explored for the last 3 decades to find AMPs with potential as effective therapeutics. Over the years of investigative studies with respect to its antimicrobial potential, α-MSH, with its widespread pleiotropic expression and ability to regulate biological and physiological response in an organism, has emerged as an effective AMP against S. aureus. We established the staphylocidal potency of α-MSH and its C-terminal fragments and designed α-MSH-based analogues with enhanced cationicity against S. aureus, including MRSA strains.11,16 However, a major limitation of these peptides is their inactivation in the presence of bacterial growth medium. Recently, a study by Grieco et al. in 2013 described analogues of NDP-MSH, a biologically active analogue of α-MSH with broad-spectrum activity against microorganisms in standard culture media, but they also displayed a detrimental effect against mammalian cell line.17
Toward overcoming the constraint of inactivity in culture media, we systematically designed novel analogues of α-MSH(6–13) by increasing its cationic charge and hydrophobicity through the incorporation of Arg, Trp, and Phe residues. Arg residues were incorporated as they have guanidine group, which facilitates perturbation of the headgroup region in the lipid bilayer through its bidentate functionality while also engaging in hydrogen bond formation with negatively charged phosphate groups.21 Similarly, we incorporated aromatic hydrophobic residues Trp and Phe as they can partition into the membrane interface and lead to membrane perturbation.38 RP-HPLC of the designed analogues revealed that the increase in percentage hydrophobicity was in accordance with substituting hydrophobic residues Trp and Phe. The analysis of the CD data demonstrated that all of the designed peptides remained unstructured in buffer solution except the most hydrophobic analogue, Ana-5, which not only included a Trp9-Trp10 domain but also possessed a Phe12 residue. Previous studies have shown that the presence of aromatic residue (Phe12/Tyr12) in the 10–13 turn region of α-MSH(6–13) stabilizes the π-stacking interactions, which are more prominent in the presence of multiple Trp residues and may be responsible for the stability of the native fold of Ana-5.22,39 Additionally, all of the designed analogues exhibited a similar ability to interact with artificial membranes as there were no appreciable differences in the blue shift of the emission maximum of the peptides in the presence of bacterial membrane mimic SUVs. A similar result was observed by Cho et al. in 2012, in which the designed analogues of a CAMP, pleurocidin, despite substitution with hydrophobic amino acids such as Trp and Phe, showed a similar blue shift in a membrane mimetic environment.40 Additionally, the designed analogues did not show any considerable interaction with mammalian membrane mimic SUVs, suggesting that the peptides possessed negligible toxicity, which was later confirmed through the hemolytic and cytotoxicity assays.
Next, as anticipated, in the antistaphylococcal assay, the killing activity increased upon the simultaneous increase in cationicity and hydrophobicity of the designed analogues. From our systematically designed set of analogues, two analogues having the highest charge (+3) and hydrophobicity showed a substantial improvement in activity compared to the parent peptide, further validating that both cationicity and hydrophobicity are required for enhanced antimicrobial activity. The high antistaphylococcal activity of Ana-4 and Ana-5 may also be attributed to the particular cation-π orbital interaction between cationic residues (Arg) and aromatic residues (Trp and Phe), which assist the peptide in inserting deeper into the bacterial membrane.20,41 Importantly, the most hydrophobic analogue of α-MSH(6–13), i.e., Ana-5, was able to retain its antibacterial activity against MSSA and MRSA cells in the presence of rich and complex growth media, which is extremely important for the realistic use of this molecule for therapeutic purpose. This enhanced activity of Ana-5 compared to Ana-4 may be attributed to the substitution of Pro12 of Ana-4 with Phe12 in Ana-5. A similar observation was also reported for [d-Nal7,Phe12]-α-MSH(6–13), an analogue of α-MSH(6–13) designed by Grieco et al., which showed maximum candidacidal activity.42 Apart from this, Ana-5 exhibited rapid bactericidal kinetics against S. aureus cells, which remained almost unchanged in the presence of 10% serum. Low serum stability is often a limiting factor toward the development of AMPs as therapeutic agents, e.g., human serum components like mono/divalent cations and serum proteins strongly inhibit the bactericidal activity of human β-defensin 3.32 Therefore, the potent antistaphylococcal efficacy of Ana-5 in the presence of clinically relevant conditions like the presence of serum further validates its development for therapeutic application. This enhanced potential of Ana-5 may in part be explained by the CD data where Phe12 substitution in Ana-5 stabilized the π-stacking interactions, leading to a more ordered structure.22,39
In this study, we also established that similar to the parent peptide α-MSH(6–13),11 the most potent analogue Ana-5 was membrane active. The mechanistic studies revealed that Ana-5 was not only able to instantly depolarize the staphylococcal membrane but also, compared to the parent peptide, the effect of Ana-5 was much more pronounced, suggesting that membrane depolarization was a lethal event for this active analogue. Similar studies showing the correlation between peptides having augmented cationicity and hydrophobicity and enhanced cytoplasmic membrane depolarization and antibacterial activity have been reported earlier.31 The markedly superior membrane activity of Ana-5 was also evident from its higher affinity to bind to artificial bacterial membrane compared to the other designed analogue as well as the standard indolicidin. Though previous studies reported that enhanced binding of peptides which is governed by nonspecific electrostatic interactions with anionic vesicles depends on their cationic charge,16 the nearly 2-fold better binding of Ana-5 than the less hydrophobic analogue Ana-4, having the same charge, may be attributed to its improved hydrophobicity43 and its ordered conformation in buffer.44 Local structure formation in an aqueous environment in the case of short peptides is considered to be important in their binding properties. During binding, the formation of local structure is often accompanied by the reduction in conformational entropy. Additionally, these local structures, being complementary in shape and electrostatic properties, enable the peptide to bind to the target molecule.45 As such, the ability of Ana-5 to acquire an ordered structure in buffer as well as in the presence of bacterial membrane mimic SUVs might have facilitated its binding with the phospholipid bilayer. The membrane disruptive mode of action of the most potent peptide Ana-5 was further validated through calcein leakage assay and SEM studies, which showed substantial membrane damage by Ana-5 with leakage of entrapped dye and oozing out of cellular content. Apart from membrane disruption, Ana-5 was also able to bind to DNA and inhibit its mobility in a gel retardation assay, suggesting that after penetrating the cell membrane, Ana-5 may further attack an intracellular target like DNA, leading to the death of the staphylococcal cells. Thus, Ana-5 with its dual mode of action may act as a “double-edged sword” against staphylococcal infections and also be less prone to resistance development.
An important consideration while designing antimicrobials against S. aureus is the ability of the pathogen to infect and survive inside mammalian cells, which often results in the moderate activity of the antimicrobials in eliminating intracellular S. aureus probably due to their limited intracellular penetration or reduced stability inside mammalian cells.46 Therefore, we also examined the ex vivo ability of Ana-5 to eradicate intracellular bacterial cells in a coculture model of S. aureus and mammalian host cells. In this study, Ana-5 was found to be either equally or more potent compared to conventional antibiotics, especially the peptide antibiotic vancomycin, which, despite its toxicity and poor bioavailability, continues to serve as the cornerstone for anti-MRSA therapy. The effective intracellular killing shown by rifampicin, which was only slightly more efficacious than Ana-5, has been previously attributed to its capability to cross mammalian cell membrane.47 The high potency of Ana-5 to clear internalized bacterial cells was in line with the previous literature which report that some Trp-Arg peptides readily penetrate and get internalized into mammalian cells due to the electrostatic and bidentate H-bonding of Arg residue with sulfates and hydrophobic interactions of Trp moiety with the sugar rings present on the mammalian cell membrane.48
Conclusions
In light of the reduced effectiveness of antibiotics owing to the current resistance situation, AMPs with their broad-spectrum activity, rapid onset of activity, and relatively low possibility of development of resistance appear as potential therapeutic sources.4 In this study, we described the design of a novel analogue of α-MSH(6–13), Ana-5, with enhanced cationic charge and hydrophobicity, having potent efficacy against the opportunistic pathogen S. aureus. Importantly, not only was Ana-5 able to retain its staphylocidal efficacy even in the presence of complex microbiological medium, thereby overcoming one of the major limitations of α-MSH-based peptides, but it was also active in the presence of serum. Furthermore, in addition to being a membrane-active peptide, the preliminary studies presented here suggest that Ana-5 may also have a secondary intracellular target like DNA unlike conventional antibiotics, which mostly target a single specific cellular activity, and thus it may be less prone to the development of resistance. Remarkably, Ana-5 was also able to significantly clear intracellular bacterial cells in a coculture model of S. aureus with mammalian cells. Therefore, the successful design of Ana-5 in this study provides an impetus for the continued development of α-MSH-based peptides toward combating the menace of antibiotic resistance in S. aureus.
Experimental Section
Materials
Brain heart infusion media (BHI), Mueller Hinton broth (MHB), Dulbecco’s modified Eagle’s medium (DMEM), 2-[tris(hydroxymethyl)-methylamine]-1-ethanesulfonic acid (TES buffer), and [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) were purchased from HiMedia Laboratories, India. 3,3′-Dipropylthiadicarbocyanine iodide (DiSC3(5)), calcein acetoxymethyl ester (calcein-AM), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), and 1,2-dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DMPG) were purchased from Sigma-Aldrich. Trypsin (2.5%), FBS, and MEM were purchased from Gibco, India. Plasmid DNA (pBluescriptII SK(+)) was obtained from Agilent Technologies.
α-MSH(6–13) and its analogues (HPLC purity > 98%) were custom synthesized by BioChain Incorporated, India. Indolicidin was purchased from AnaSpec, Inc. The concentration of peptides was determined by UV absorbance of Trp and Tyr residues at 280 nm, with εTrp = 5690 M–1 cm–1 and εTyr = 1280 M–1 cm–1.
Two American Type Culture Collection (ATCC) S. aureus strains were used in the present study, namely, ATCC 29213 (MSSA) and ATCC 33591 (MRSA). The stock cultures were stored at −80 °C in 15% (v/v) glycerol until subcultured onto BHI agar plate. The S. aureus strains were grown at 37 °C in BHI, MHB, or MEM broth with shaking till mid-log phase and adjusted to an optical density (OD600) of ∼0.5, which corresponds to 108 CFU mL–1 for staphylococcal strains.
Preparation of Small Unilamellar Vesicles (SUVs)
Artificial bacterial membrane mimic (DMPC/DMPG, 1:1, w/w and 7:3, w/w) and mammalian membrane mimic (DMPC) SUVs were prepared by probe sonication after direct hydration of phospholipid films, as previously described.15 Briefly, the desired amount of lipids was dissolved in chloroform and methanol, and the solvents were evaporated to dryness under nitrogen flux. After overnight incubation at −20 °C, the dried lipid film was rehydrated in the buffer (either 10 mM TES, pH 7.4 or 5 mM PB, pH 7.4). The suspension was then sonicated using a titanium tip sonicator with burst and halt times of 30 and 10 s, respectively, in an ice bath. Titanium debris was removed by centrifugation at 10 000 rpm for 2 min.
Circular Dichroism (CD) Spectroscopy
CD measurements were performed using an Applied PhotoPhysics Chirascan (Surrey, U.K.) instrument set at a Peltier temperature of 37 °C.15 The spectra of the peptides were acquired in the presence of two different environments, namely, 5 mM PB and bacterial mimic SUVs (DMPC/DMPG, 7:3 and 1:1 w/w). In the far-UV region (190–260 nm), scans were recorded at a step size of 0.2 nm, bandwidth of 1 nm, path length of 2 mm, and time per point of 0.2 s. Three CD spectra scans of peptides were first averaged, then subtracted from their respective medium. The representative spectrum was converted from millidegree to molar ellipticity (deg cm2 dmol–1), as mentioned below and plotted against wavelength.49
Dynamic Light Scattering (DLS)
The hydrodynamic radius and size distribution of peptides were measured by DLS using the instrument Xtal (Spectrosize300, Germany). Samples were filtered using a 0.22 μm syringe filter, and two separate DLS measurements were acquired at 25 °C. Each measurement consisted of 10 subruns with 20 s duration. A representative run was selected and provided. Peptides (35 μM) were dissolved in either water or DMPC/DMPG (1:1, w/w) SUVs (1453 μM).27
Trp Fluorescence of Peptides
All fluorescence measurements were done with quartz cuvettes with 1 cm path length on a Shimadzu RF-5301 spectrofluorimeter. Briefly, peptides were added to either 10 mM TES buffer, pH 7.4, or SUVs solutions at a fixed lipid to peptide ratio. The excitation wavelength was 295 nm, and the emission was recorded from 310 to 450 nm. The excitation and emission slit widths were set at 1.5 and 3 nm, respectively. Each peptide in the lipid spectrum was subtracted with their respective blank, and an emission maximum of peptides in the presence of lipids was compared to peptides in buffer to calculate the blue shift (nm).15,16
In Vitro Bactericidal Assay in Physiological Buffer
To determine the staphylocidal activity of peptides, S. aureus was grown in BHI broth and resuspended and diluted to the desired cell densities in a 5 mM HEPES-20 mM glucose buffer (pH 7.4), as mentioned elsewhere.11 The bacterial cells were incubated with desired concentrations of the peptides at 37 °C for 2 h. Aliquots were then serially diluted in the buffer, and 15 μL of each dilution was plated in triplicate on BHI agar and incubated overnight at 37 °C. Afterward, the viable cell count was determined by counting the colony forming units (CFU) and comparing it with the untreated control. The mean ± standard deviation (SD) for surviving S. aureus cells was plotted in terms of log10 CFU mL–1.
Minimum Inhibitory Concentration (MIC) Determination
The MIC of peptides was determined by the serial broth microdilution method with slight modifications, as reported previously,30 in MEM and MHB media. Briefly, the peptides were serially 2-fold diluted in 0.01% acetic acid containing 0.2% bovine serum albumin (BSA) in sterile polypropylene 96-well microtiter plates (Corning, Inc.). To 10 μL of the serially diluted peptide, 100 μL of initial bacterial inoculum of 5 × 105 CFU mL–1 in MHB or MEM was added. The medium without cells was considered as a negative control. The microtiter plate was read at 600 nm using an ELISA plate reader (Molecular Devices, Sunnyvale, CA) after overnight incubation with shaking at 180 rpm at 37 °C. The experiments were carried out in duplicate on at least three different days. The lowest concentration of peptide, which completely inhibited the growth of bacteria, was taken as the MIC.
Time-Kill Kinetics of the Peptides
The active peptide in the standard medium was further evaluated for its time-dependent staphylocidal activity in MHB alone or with 10% serum, as described previously.50 Briefly, mid-logarithmic S. aureus cells grown in MHB were adjusted to an OD600 of ∼0.5 and were further diluted to ∼105 CFU mL–1 in MHB to which peptides/antibiotics were added at their 4 × MIC values obtained in MHB and incubated at 37 °C with shaking at 180 rpm. Aliquots were removed at fixed time intervals, diluted in 10 mM PBS (sodium phosphate, 150 mM NaCl, pH 7.4), and 15 μL were plated in triplicate on BHI agar plates. After overnight incubation at 37 °C, bacterial colonies were enumerated and compared to those of the untreated control. To study the effect of serum, bacterial suspension in MHB containing 10% FBS and peptides at 4 × MIC were incubated for 3 h at 37 °C with shaking at 180 rpm. The experiment was repeated on three different days, and representative data from one set are shown.
Hemolytic Activity of the Peptides
The hemolytic activity of the peptides was determined using a protocol as described previously with slight modifications.30,51 Fresh blood from mice was washed twice with 35 mM PBS (pH 7.4) via centrifugation at 1500 rpm for 10 min to remove the plasma and buffy coat. The red blood cells (RBCs) pellet was resuspended to 4% v/v in PBS and 100 μL of this suspension was dispensed into a 96-well plate already containing 100 μL of previously serially diluted peptides in PBS. After 1 h incubation at 37 °C, the plates were centrifuged at 1500 rpm for 10 min and 20 μL of the supernatant was added to 80 μL of PBS in a fresh 96-well plate. Absorbance was measured at 414 nm using an ELISA plate reader (Molecular Devices, Sunnyvale, CA) to determine the hemoglobin release. 0.1% Triton X-100 (v/v) was used as a positive control (100% hemolysis), and the percentage of hemolysis was calculated using the following equation
The experiment was conducted following the “Committee for the Purpose of Control and Supervision of Experiments on Animals” (CPCSEA) guidelines and Institutional Animal Ethics Committee (IAEC-02/2014) of JNU, New Delhi, India.
Measuring Cytotoxicity by MTT Assay
The viability of 3T3 murine fibroblast cells was determined through the MTT assay to evaluate the cytotoxicity of peptides, as described previously.13,16 The cell line was propagated in 24-well plates (∼5 × 105 cells per well) in DMEM supplemented with 10% FBS and antibiotics at 37 °C in 5% CO2. After 24 h, the spent media was aspirated and various concentrations of peptides, i.e., 15, 30, and 60 μM, dissolved in DMEM without serum were added to each well (in triplicate). The medium without peptide and with 2% Triton X-100 was set as negative control and positive control, respectively. The plate was incubated for 2 h at 37 °C. The MTT solution (1 mL, 0.1 mg mL–1) was added to each well, and the plates were again incubated for 2 h at 37 °C in the dark. Subsequently, after removal of the supernatants, 200 μL of dimethyl sulfoxide was then added to each well and kept for 5 min in the incubator to dissolve the formazan crystals formed, and absorbance was measured at 570 nm. The assay was done in triplicate on three different days, and the percentage of cytotoxicity was determined using the following equation
Membrane Depolarization Assay using DiSC3(5) Fluorescent Dye
S. aureus (ATCC 29213) membrane depolarization was monitored through the cationic membrane potential-sensitive fluorescent probe DiSC3(5), as mentioned elsewhere.30,31,51 Briefly, mid-logarithmic S. aureus cells were washed and resuspended in 5 mM HEPES-20 mM glucose buffer (pH 7.4) at an OD600 of 0.05, which corresponds to ∼107 CFU mL–1. To each 1 mL of this bacterial suspension, 2 μM DiSC3(5) was added and incubated for 1 h at room temperature to get a stable reduction in fluorescence intensity. Next, the dye-loaded cells were treated with increasing peptide concentrations from 2 to 40 μM in a quartz cuvette. The increase in fluorescence was monitored after 2 min of peptide incubation in a Shimadzu RF-5301 PC spectrofluorimeter, which was set at λexcitation and λemission of 622 and 670 nm, respectively, with a slit width of 5 nm at 37 °C. The spectrum mode was changed to kinetics mode in the instrument to measure depolarization kinetics for up to 15 min after the addition of peptides at 30 μM concentration. The effect of depolarization on the viability of S. aureus was measured through the corresponding killing assay where the dye-loaded cells were treated with 30 μM peptides, and the aliquots were immediately plated onto BHI agar, which was incubated overnight at 37 °C, and the viable cells were enumerated by counting the colonies.
Membrane Permeabilization Assay using Calcein-AM Dye
The extent of S. aureus (ATCC 33591) membrane permeabilization upon exposure to the active peptide was measured through flow cytometry (BD FACSverse, San Jose, CA), as mentioned elsewhere.11,30,51 Briefly, mid-logarithmic S. aureus cells were adjusted to an OD600 value of 1.0 (∼109 CFU mL–1) in 10 mM PBS, pH 7.4. These cells were incubated with calcein-AM (2 μg mL–1) supplemented with 10% MHB at 37 °C with shaking at 180 rpm for 2 h. The cells were washed once with PBS, diluted to 106 CFU mL–1, and incubated with 30 μM of the peptides for 2 h at 37 °C in the dark. After 2 h, 10 000 cells were acquired for each treated and untreated sample using a flow cytometer to determine the extent of release of the preloaded fluorophore calcein and the cells were gated using unloaded cells. The experiment was conducted on three different days, and the percentage of calcein leakage was plotted as mean ± SD. The corresponding viability of calcein-loaded S. aureus was also determined by the drop plate method after an incubation of 2 h at 37 °C.
Measurements of Binding of the Peptide with Lipid through Fluorescence Titration
Binding of the peptides to bacterial membrane mimic SUVs (DMPC/DMPG, 7:3, w/w) was assessed through monitoring the changes in intrinsic Trp fluorescence intensity of the peptides. The lipids were titrated by adding SUVs suspension to a fixed peptide concentration (10 μM) and varying the lipid to peptide ratio from 3.3 to 75.6. Fluorescence spectra were measured with excitation at 295 nm and a slit width of 3 nm. The spectra were corrected by subtracting the blank spectra (without peptide) to account for light scattering. The fraction of peptide bound to the lipid vesicles, i.e., (F – F0)/(Fmax – F0), where F is the peptide fluorescence intensity in the presence of lipid, F0 is its intensity in buffer alone, and Fmax is its intensity in the presence of the highest lipid concentration tested, was plotted against the lipid concentrations to determine the binding constant.16 The Origin 8 (2015) software was used to fit the obtained data to a hyperbolic curve.
Scanning Electron Microscopy (SEM)
For this experiment, a procedure, as described previously, was followed with slight modifications.11,16 Briefly, mid-log phase S. aureus (ATCC 29213) cells were grown in BHI broth, washed thrice, and resuspended in 10 mM PBS, pH 7.4, to remove any traces of media. OD600 was adjusted to 1.0 (∼109 CFU mL–1), and 50 μM concentration of the peptides was added to the cells in PBS (pH 7.4) for 2 h. After incubation, the cells were pelleted down at 6000 rpm for 10 min and washed with 10 mM PB (pH 7.4) thrice to remove any salt traces and fixed overnight in 2.5% (v/v) glutaraldehyde at 4 °C. The next day, fixative was removed by washing with PB (pH 7.4), the samples were dehydrated in a series of graded ethanol solutions (30–100%), and dried in a vacuum desiccator. The specimens were then viewed via a scanning electron microscope (EVO 40; Carl Zeiss, Germany) after coating them with 20 nm gold particles using an automatic sputter coater (Polaron OM-SC7640).
Gel Retardation Assay
The electrophoretic mobility shift assay was performed by incubating different concentrations of peptides with plasmid DNA (pBluescriptII SK(+)) for 1 h at room temperature, as described previously.30 Briefly, reaction mixtures containing 100 ng of plasmid DNA with increasing concentrations of peptide in 20 μL of binding buffer (5% glycerol, 10 mM Tris-HCl, pH 8.0, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol, 20 mM KCl, and 50 μg mL–1 BSA) were incubated at 37 °C for 1 h. Next, 4 μL of native loading buffer (10% Ficoll 400, 10 mM Tris-HCl, pH 7.5, 50 mM EDTA, 0.25% bromophenol blue, and 0.25% xylene cyanol) was added to this reaction mixture and a 20 μL aliquot of this mix was then subjected to 1% agarose gel electrophoresis in 0.5× Tris-borate-EDTA buffer (45 mM Tris-borate and 1 mM EDTA, pH 8.0). The gel was visualized under a UV transilluminator.
Inhibition of Intracellular Staphylococcal Infection by Peptides in Mammalian Cells by Coculture Method
Infection of 3T3 fibroblast cells with S. aureus was done as described previously with slight modifications.37,46 Initially, a monolayer of the mammalian cells was grown in DMEM supplemented with 10% FBS and 1× anti-anti at 37 °C, 5% CO2. Nearly 106 cells (semiconfluence) in a 24-well tissue culture plate were preincubated in DMEM for 24 h. Mid-logarithmic phase cells (∼107 CFU mL–1) were used to infect 3T3 cells at MOI 10:1 (i.e., the ratio of S. aureus to fibroblast cells) in DMEM with 10% FBS. The infected monolayers were incubated for 2 h at 37 °C at 5% CO2. The medium was aspirated, and subsequently, fresh DMEM with 10% FBS containing 100 μg mL–1 of gentamycin was added to kill the extracellular bacteria for 2 h. The coculture was washed with PBS and treated with peptide or antibiotics in DMEM with 10% FBS for 16 h at 37 °C. The cells were maintained at 4 μg mL–1 of gentamycin to prevent extracellular bacteria growth.52 The cells were then lysed by adding 100 μL of 0.1% Triton X-100 in PBS. The bacterial suspensions were diluted and plated on BHI agar to quantify the number of viable intracellular bacteria by CFU count.
Statistical Analysis
Data were analyzed using one-way analysis of variance, and the significance of the experiment among the groups was determined by Bonferroni’s post hoc test using GraphPad Prism 8 software. Data are expressed as mean ± standard deviation (SD) for at least three different measurements.
Acknowledgments
This study was supported by grants from Department of Science and Technology (DST-SERB, EMR/2016/001708), Department of Biotechnology (DBT, BT/PR27737/MED/29/1265/2018), DST-PURSE (PAC-JNU-DST-PURSE-462 (Phase-II)), and University Grants Commission (UGC) (UPE-II, JNU) to K.M. J.S., S.M., and S.J. are thankful to funding agencies ICMR and UGC, and to Dr. D. S. Kothari for their research fellowships, respectively. The authors also acknowledge DBT for funding AIRF (BT/PR3130/INF/22/139/2011) in JNU for instrumentation facilities. They thank Dr. Ruchita Pal and Dr. Manish in AIRF, JNU, for their help in SEM experiment and CD experiments, respectively. The authors thank Prof. Rupesh Chaturvedi and his research scholar Rohit Tiwari (School of Biotechnology, JNU) for providing biosafety level-II (BSL-2) facility and assisting in mammalian cell culture technique, respectively.
Glossary
Abbreviations
- α-MSH
α-melanocyte stimulating hormone
- MSSA
methicillin-sensitive Staphylococcus aureus
- MRSA
methicillin-resistant S. aureus
- CAMP
cationic antimicrobial peptide
- POMC
pro-opiomelanocortin
- RP-HPLC
reversed-phase high performance liquid chromatography
- TFE
2,2,2-trifluoroethanol
- MIC
minimum inhibitory concentration
- SUVs
small unilamellar vesicles
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DPMG
1,2-dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol)
Author Contributions
† J.S. and S.M. contributed equally to this work.
Author Contributions
S.J. and K.M. conceived and designed the peptides. J.S. performed the experiments. J.S., S.J., and K.M. analyzed the data. J.S., S.M., S.J., and K.M. wrote the manuscript.
The authors declare no competing financial interest.
References
- Tong S. Y. C.; Davis J. S.; Eichenberger E.; Holland T. L.; Fowler V. G. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomer L.; Schneewind O.; Missiakas D. Pathogenesis of Staphylococcus aureus bloodstream infections. Annu. Rev. Pathol.: Mech. Dis. 2016, 11, 343–364. 10.1146/annurev-pathol-012615-044351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T. J.; Geoghegan J. A.; Ganesh V. K.; Höök M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49. 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M.; Håkansson J.; Ringstad L.; Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. 10.3389/fcimb.2016.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rey E.; Ganea D.; Delgado M. Neuropeptides: keeping the balance between pathogen immunity and immune tolerance. Curr. Opin. Pharmacol. 2010, 10, 473–481. 10.1016/j.coph.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustyniak D.; Nowak J.; T Lundy F. Direct and indirect antimicrobial activities of neuropeptides and their therapeutic potential. Curr. Protein Pept. Sci. 2012, 13, 723–738. 10.2174/138920312804871139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden K. A.; Guthmiller J. M.; Salzet M.; Zasloff M. The nervous system and innate immunity: the neuropeptide connection. Nat. Immunol. 2005, 6, 558. 10.1038/ni1209. [DOI] [PubMed] [Google Scholar]
- Catania A.; Gatti S.; Colombo G.; Lipton J. M. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol. Rev. 2004, 56, 1–29. 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- Singh M.; Mukhopadhyay K. Alpha-melanocyte stimulating hormone: an emerging anti-inflammatory antimicrobial peptide. BioMed Res. Int. 2014, 874610 10.1155/2014/874610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhuri; Shireen T.; Venugopal S. K.; Ghosh D.; Gadepalli R.; Dhawan B.; Mukhopadhyay K. In vitro antimicrobial activity of alpha-melanocyte stimulating hormone against major human pathogen Staphylococcus aureus. Peptides 2009, 30, 1627–1635. 10.1016/j.peptides.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Singh M.; Mukhopadhyay K. C-terminal amino acids of alpha-melanocyte stimulating hormone are requisite for its antibacterial activity against Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 1920–1929. 10.1128/AAC.00957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shireen T.; Singh M.; Dhawan B.; Mukhopadhyay K. Characterization of cell membrane parameters of clinical isolates of Staphylococcus aureus with varied susceptibility to alpha-melanocyte stimulating hormone. Peptides 2012, 37, 334–339. 10.1016/j.peptides.2012.05.025. [DOI] [PubMed] [Google Scholar]
- Singh M.; Gadepalli R.; Dhawan B.; Mukhopadhyay K. Combination of alpha-melanocyte stimulating hormone with conventional antibiotics against methicillin resistant Staphylococcus aureus. PLoS One 2013, 8, e73815 10.1371/journal.pone.0073815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M.; Shireen T.; Mukhopadhyay K. Susceptibility of clinical isolates of Staphylococcus aureus to alpha-melanocyte stimulating hormone and its in vivo efficacy. Am. J. Clin. Microbiol. Antimicrob. 2018, 1, 1–6. [Google Scholar]
- Shireen T.; Basu A.; Sarkar M.; Mukhopadhyay K. Lipid composition is an important determinant of antimicrobial activity of alpha-melanocyte stimulating hormone. Biophys. Chem. 2015, 196, 33–39. 10.1016/j.bpc.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Singh J.; Joshi S.; Mumtaz S.; Maurya N.; Ghosh I.; Khanna S.; Natarajan V. T.; Mukhopadhyay K. Enhanced cationic charge is a key factor in promoting staphylocidal activity of α-melanocyte stimulating hormone via selective lipid affinity. Sci. Rep. 2016, 6, 31492 10.1038/srep31492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco P.; Carotenuto A.; Auriemma L.; Limatola A.; Di Maro S.; Merlino F.; Mangoni M. L.; Luca V.; Di Grazia A.; Gatti S.; Campiglia P.; et al. Novel α-MSH peptide analogues with broad spectrum antimicrobial activity. PLoS One 2013, 8, e61614 10.1371/journal.pone.0061614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab U.; Gilligan P.; Jaynes J.; Henke D. In vitro activities of designed antimicrobial peptides against multidrug-resistant cystic fibrosis pathogens. Antimicrob. Agents Chemother. 1999, 43, 1435–1440. 10.1128/AAC.43.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J.; Cho Y.; Dinh N. N.; Waring A. J.; Lehrer R. I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. 10.1128/AAC.42.9.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. I.; Prenner E. J.; Vogel H. J. Tryptophan-and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1184–1202. 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Torcato I. M.; Huang Y. H.; Franquelim H. G.; Gaspar D.; Craik D. J.; Castanho M. A.; Henriques S. T. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 944–955. 10.1016/j.bbamem.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Masman M. F.; Somlai C.; Garibotto F. M.; Rodríguez A. M.; de la Iglesia A.; Zacchino S. A.; Penke B.; Enriz R. D. Structure–antifungal activity relationship of His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2 and analogues. Bioorg. Med. Chem. 2008, 16, 4347–4358. 10.1016/j.bmc.2008.02.072. [DOI] [PubMed] [Google Scholar]
- Ladokhin A. S.; Selsted M. E.; White S. H. CD spectra of indolicidin antimicrobial peptides suggest turns, not polyproline helix. Biochemistry 1999, 38, 12313–12319. 10.1021/bi9907936. [DOI] [PubMed] [Google Scholar]
- Cheng J. T.; Hale J. D.; Elliot M.; Hancock R. E.; Straus S. K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2009, 96, 552–565. 10.1016/j.bpj.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrushchenko V. V.; Vogel H. J.; Prenner E. J. Solvent-dependent structure of two tryptophan-rich antimicrobial peptides and their analogs studied by FTIR and CD spectroscopy. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1596–1608. 10.1016/j.bbamem.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Nichols M.; Kuljanin M.; Nategholeslam M.; Hoang T.; Vafaei S.; Tomberli B.; Gray C. G.; DeBruin L.; Jelokhani-Niaraki M. Dynamic turn conformation of a short tryptophan-rich cationic antimicrobial peptide and its interaction with phospholipid membranes. J. Phys. Chem. B 2013, 117, 14697–14708. 10.1021/jp4096985. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Pletzer D.; Haney E. F.; Rahanjam N.; Cheng J. T.; Yue M.; Aljehani W.; Hancock R. E.; Kizhakkedathu J. N.; Straus S. K. Aurein-derived antimicrobial peptides formulated with pegylated phospholipid micelles to target methicillin-resistant Staphylococcus aureus skin infections. ACS Infect. Dis. 2019, 5, 443–453. 10.1021/acsinfecdis.8b00319. [DOI] [PubMed] [Google Scholar]
- Yin L. M.; Edwards M. A.; Li J.; Yip C. M.; Deber C. M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. 10.1074/jbc.M111.303602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanan R.; Li X.; Lim K.; Mohanram H.; Peng L.; Mishra B.; Basu A.; Lee J. M.; Bhattacharjya S.; Leong S. S. J. Design of short membrane selective antimicrobial peptides containing tryptophan and arginine residues for improved activity, salt-resistance, and biocompatibility. Biotechnol. Bioeng. 2014, 111, 37–49. 10.1002/bit.25003. [DOI] [PubMed] [Google Scholar]
- Joshi S.; Mumtaz S.; Singh J.; Pasha S.; Mukhopadhyay K. Novel miniature membrane active lipopeptidomimetics against planktonic and biofilm embedded methicillin-resistant Staphylococcus aureus. Sci. Rep. 2018, 8, 1021 10.1038/s41598-017-17234-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich C. L.; Moyles D.; Beveridge T. J.; Hancock R. E. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob. Agents Chemother. 2000, 44, 2086–2092. 10.1128/AAC.44.8.2086-2092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisetta G.; Di Luca M.; Esin S.; Florio W.; Brancatisano F. L.; Bottai D.; Campa M.; Batoni G. Evaluation of the inhibitory effects of human serum components on bactericidal activity of human beta defensin 3. Peptides 2008, 29, 1–6. 10.1016/j.peptides.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. P. Cationic antimicrobial peptides. BioDrugs 2003, 17, 233–240. 10.2165/00063030-200317040-00002. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski S. C.; Paul L. S.; Lonberg N.; Newport J. W.; McSwiggen J. A.; Von Hippel P. H. Cooperative and noncooperative binding of protein ligands to nucleic acid lattices: experimental approaches to the determination of thermodynamic parameters. Biochemistry 1986, 25, 1226–1240. 10.1021/bi00354a006. [DOI] [PubMed] [Google Scholar]
- Middleton E. R.; Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. K.; Park S. C.; Hahm K. S.; Park Y. Antimicrobial HPA3NT3 peptide analogs: placement of aromatic rings and positive charges are key determinants for cell selectivity and mechanism of action. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 443–454. 10.1016/j.bbamem.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Lehar S. M.; Pillow T.; Xu M.; Staben L.; Kajihara K. K.; Vandlen R.; DePalatis L.; Raab H.; Hazenbos W. L.; Morisaki J. H.; Kim J.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- Saint Jean K. D.; Henderson K. D.; Chrom C. L.; Abiuso L. E.; Renn L. M.; Caputo G. A. Effects of hydrophobic amino acid substitutions on antimicrobial peptide behavior. Probiotics Antimicrob. Proteins 2018, 10, 408–419. 10.1007/s12602-017-9345-z. [DOI] [PubMed] [Google Scholar]
- Carotenuto A.; Saviello M. R.; Auriemma L.; Campiglia P.; Catania A.; Novellino E.; Grieco P. Structure–function relationships and conformational properties of α-MSH (6–13) analogues with candidacidal activity. Chem. Biol. Drug Des. 2007, 69, 68–74. 10.1111/j.1747-0285.2007.00473.x. [DOI] [PubMed] [Google Scholar]
- Cho J.; Choi H.; Lee D. G. Influence of the N-and C-terminal regions of antimicrobial peptide pleurocidin on antibacterial activity. J. Microbiol. Biotechnol. 2012, 22, 1367–1374. 10.4014/jmb.1205.05040. [DOI] [PubMed] [Google Scholar]
- Hao Y.; Yang N.; Wang X.; Teng D.; Mao R.; Wang X.; Li Z.; Wang J. Killing of Staphylococcus aureus and Salmonella enteritidis and neutralization of lipopolysaccharide by 17-residue bovine lactoferricins: improved activity of Trp/Ala-containing molecules. Sci. Rep. 2017, 7, 44278 10.1038/srep44278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco P.; Rossi C.; Colombo G.; Gatti S.; Novellino E.; Lipton J. M.; Catania A. Novel α-melanocyte stimulating hormone peptide analogues with high candidacidal activity. J. Med. Chem. 2003, 46, 850–855. 10.1021/jm0204338. [DOI] [PubMed] [Google Scholar]
- Caesar C. E.; Esbjörner E. K.; Lincoln P.; Nordén B. Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: correlations of structure to cellular uptake. Biochemistry 2006, 45, 7682–7692. 10.1021/bi052095t. [DOI] [PubMed] [Google Scholar]
- Dong W.; Mao X.; Guan Y.; Kang Y.; Shang D. Antimicrobial and anti-inflammatory activities of three chensinin-1 peptides containing mutation of glycine and histidine residues. Sci. Rep. 2017, 7, 40228 10.1038/srep40228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardi F.; Worth G. A.; Wade R. C. Local interactions of aromatic residues in short peptides in aqueous solution: a combined database and energetic analysis. Folding Des. 1997, 2, S62–S68. 10.1016/S1359-0278(97)00066-7. [DOI] [PubMed] [Google Scholar]
- Mohamed M. F.; Abdelkhalek A.; Seleem M. N. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707 10.1038/srep29707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krut O.; Sommer H.; Krönke M. Antibiotic-induced persistence of cytotoxic Staphylococcus aureus in non-phagocytic cells. J. Antimicrob. Chemother. 2004, 53, 167–173. 10.1093/jac/dkh076. [DOI] [PubMed] [Google Scholar]
- Bechara C.; Sagan S. Cell-penetrating peptides: 20 years later, where do we stand?. FEBS Lett. 2013, 587, 1693–1702. 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed M. F.; Hamed M. I.; Panitch A.; Seleem M. N. Targeting methicillin-resistant Staphylococcus aureus with short salt-resistant synthetic peptides. Antimicrob. Agents Chemother. 2014, 58, 4113–4122. 10.1128/AAC.02578-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Kandi S. K.; Manohar S.; Mukhopadhyay K.; Rawat D. S. Monocarbonyl curcuminoids with improved stability as antibacterial agents against Staphylococcus aureus and their mechanistic studies. ACS Omega 2019, 4, 675–687. 10.1021/acsomega.8b02625. [DOI] [Google Scholar]
- Marathe S. A.; Sen M.; Dasgupta I.; Chakravortty D. Differential modulation of intracellular survival of cytosolic and vacuolar pathogens by curcumin. Antimicrob. Agents Chemother. 2012, 56, 5555–5567. 10.1128/AAC.00496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]