

Abstract
Neutrophil trafficking is essential for a strong and productive immune response to infection and injury. During acute inflammation, signals from resident immune cells, fibroblasts, and the endothelium help to prime, attract, and activate circulating neutrophils at sites of inflammation. Due to current limitations with in vitro and animal models, our understanding of these events is incomplete. In this paper, we describe a microfluidic technology and incorporates a lumen-based vascular component with a high degree of spatiotemporal control to facilitate the study of neutrophil trafficking using primary human cells. The improved spatiotemporal control allows functional selection of neutrophils based on their migratory capacity. We use this technology to investigate neutrophil-endothelial interactions and find that these interactions are necessary for robust neutrophil chemotaxis to interleukin-8 (IL-8) and priming of the neutrophils. In agreement with previous studies, we observed that transendothelial migration (TEM) is required for neutrophils to enter a primed phenotypic state. TEM neutrophils not only produce a significantly higher amount of reactive oxygen species (ROS) when treated with PMA, but also upregulate genes involved in ROS production (CYBB, NCF1, NFKB1, NFKBIA), cell adhesion (CEACAM-8, ITGAM), and chemokine receptors (CXCR2, TNFRSF1A). These results suggest that neutrophil-endothelial interactions are crucial to neutrophil chemotaxis and ROS generation.
1. Introduction
Neutrophils are a type of white blood cell, or leukocyte, that circulate throughout the body and act as first responders to infection and cell damage. Neutrophils possess multiple different mechanisms with which they react to infection/injury namely phagocytosis, release of toxic granules, production of reactive oxygen species (ROS), and the formation of neutrophil extracellular traps (NETs). Trafficking of neutrophils to sites of infection and injury is essential for optimal functioning of the immune system. Endothelial vessels help traffic neutrophils to sites of inflammation and facilitate their activation and entry into the interstitium.1 Neutrophils first enter the interstitium in response to inflammatory cytokines, haptotactic and chemotactic gradients, and upregulation of cell surface adhesion proteins on endothelial cells in a process called transendothelial migration (TEM), and then continue to migrate to sites of infection or injury (Figure 1).2,3,4,5 This neutrophil-endothelial interaction serves to not only aid neutrophil entry into the interstitium, but also induces phenotypic changes that cause the neutrophil to enter an enhanced state of responsiveness, termed priming.6 Priming is known to occur after exposure to pro-inflammatory cytokines, pathogen-based products, and adhesion, and this process alters a neutrophils ability to phagocytose, release granules, form NETs, and generate ROS. Neutrophil TEM, chemotaxis, and priming have been extensively studied in 2D cell culture and animal models such as zebrafish and mice.7,8 While these studies have provided valuable insights into neutrophil biology, it remains unclear to what extent these findings translate to human biology.9
Fig 1: Mechanisms of neutrophil trafficking.
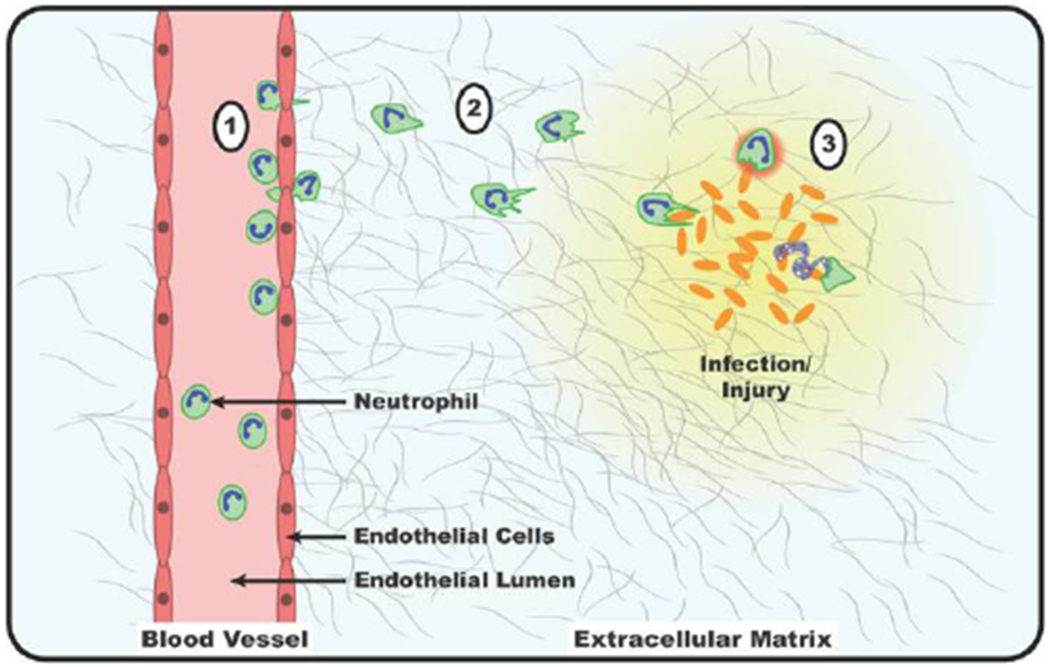
1) Circulating neutrophils encounter inflammatory signals which cause them to interact with and arrest on the wall of the blood vessel, adhere to endothelial cells, and undergo TEM. 2) Neutrophils migrate down gradients of chemokines in a process called chemotaxis. 3) Once at the site of infection/injury, neutrophils phagocytose pathogens or cell debris, release toxic granules, produce ROS, or undergo NETosis.
Transwells and 2D cell culture plates are the predominant tool used for assessing neutrophil TEM, chemotaxis, and priming in vitro. While transwells and culture plates allow for the incorporation of blood-vessel components including endothelial cells, and extracellular matrices (ECM), their spatial configuration, and working volumes lack relevance to in vivo biology. Additionally, using these approaches makes it difficult to separate and functionally characterize different populations of neutrophils. There is need for a better in vitro system that can provide this spatiotemporal configuration.
Recent advances in 3D in vitro microscale models of human neutrophil-vasculature interactions have resulted in models with increased physiological relevance when compared to classical approaches such as 2D microfluidic devices. While 2D models of human neutrophil trafficking have been useful for characterizing responses to chemokines, and measuring NETosis and ROS generation in a heterogeneous population, their usefulness for answering questions involving how co-cultures, or 3D ECM components affect neutrophil biology is limited.10,11 Current efforts to model blood vessel-leukocyte interactions in vitro include strategies such as using self-organized capillaries in a 3D extracellular matrix,12 3D bio-printed blood vessels,13 and micromolded channels seeded with endothelial cells.14 While these methods incorporate improved spatial configurations of cells and ECMs, they ultimately lack manipulability which, again, limits their usefulness. Here we add the capability to isolate different neutrophil populations based on their chemotactic capabilities to a 3D organotypic endothelial model and use it to investigate how neutrophil-endothelial interactions affect neutrophil TEM, chemotaxis, and priming. Further use of this technology could provide future insight into understanding neutrophil heterogeneity, and inflammation resolution.15
In this paper, we combine two existing microfluidic platforms (LumeNext and Stacks) to create an improved platform for studying neutrophil trafficking. LumeNext is a recently established microfluidic technology by which luminal structures can be created from multiple cell types in a user-defined ECM.16,17 To create organotypic blood vessels, lumens are mold-casted in a hydrogel that is polymerized within a microfluidic device, and the resulting lumen is seeded with endothelial cells to form an endothelial microvessel. Stacks is a modular, open-microfluidic technology consisting of segmented polystyrene (PS) tube sections which can be filled with different ECM and cellular components, and then stacked on top of one another in the desired configuration.18 This platform enables the study and isolation of cells in a 3D ECM and is amenable to most molecular biology assays. LENS (LumENext-Stacks) was designed to facilitate the study of neutrophil-endothelial interactions using human primary cells. It is suggested that human neutrophil heterogeneity contributes to their differential priming by the endothelium and inflammatory microenvironment.15,19,20 Using LENS, we show that neutrophil-endothelial interactions are essential for robust neutrophil TEM/chemotaxis, and priming, in agreement with previous findings.21 We then isolate neutrophils by their migratory capacity and further quantify their ability to generate ROS, and transcriptional changes that occur during neutrophil extravasation in migratory and non-migratory phenotypes. These findings reveal new biological insights into human immunology. We find that human neutrophil-endothelial interactions stimulate the upregulation of genes involved with cellular adhesion, chemokine reception, and ROS production in migratory neutrophils, priming them to possess increased migratory and ROS producing capabilities. Additionally, we find that these interactions have little to no effect on neutrophil phenotype for a subset of non-migratory neutrophils, likely indicating that pre-existing neutrophil heterogeneity results in varying neutrophil primed states.
2. Materials and Methods
2.1. Cell Culture
HUVECs (Lonza, #C2519A) were maintained in endothelial basal media-2 (EBM-2)(Lonza, #CC-3121) supplemented with the EGM-2 Bullet Kit (human EGF (hEGF), hydrocortisone, gentamicin, amphotericin-B, VEGF, hFGF-B, insulin-like growth factor-1 (R3-IGF-1, ascorbic acid, heparin, and 2% fetal bovine serum)(Lonza, #CC-3162). HUVECs were passaged with 0.25% trypsin-EDTA (Thermo Fisher, #25200056) prior to confluency and used from passage 3-10.
2.2. Human Primary Neutrophil Purification
Neutrophils were purified from whole blood using the Miltenyi Biotec MACSxpress Neutrophil Isolation Kit per the manufacturer’s instructions (Miltenyi Biotec, #130-104-434) and residual red blood cells were lysed using MACSxpress Erythrocyte Depletion Kit (Miltenyi Biotec, #130-098-196). All donors were healthy and informed consent was obtained at the time of the blood draw according to the requirements of the institutional review board (IRB) per the declaration of Helsinki. Prior to loading, the purified neutrophils were stained with calcein AM at 10nM (Thermo Fisher, #C3100MP) according to the manufacturer’s instructions.
2.3. LENS Device Fabrication
The LENS PDMS base was fabricated as previously described by Jiménez-Torres et al. Briefly, LumeNext devices consist of two components: an open chamber fabricated in PDMS (Dow Corning, Sylgard 184, 10:1 curing agent ratio) from an SU-8 (MicroChem, SU8-100) master via traditional soft lithography22, and a PDMS rod that is form-casted from 25 gauge hypodermic needles. The rod is inserted into the central open chamber and the device is oxygen plasma bonded to a glass coverslip using a Diener Electronic Femto Plasma Surface System.
The LENS Stacks layers are fabricated using plastic micromilling methods.23 Briefly, the Stacks layers are machined out of 1.2 mm thick polystyrene sheets (Good Fellow, # 640–597-67) on a CNC mill (Tormach, PCNC 770 mill). The layers are then deburred and soaked in DI water for 24 hours to remove leftover coolant from the machining process. The Stacks layers have alignment posts that fit into both the PDMS base and adjoining Stacks layers. Stacks layers are placed on top of the LumeNext base where the alignment posts act to keep both components aligned during ECM polymerization and experimentation.
2.4. Device and ECM preparation
Prior to loading ECM solution and cells, devices were UV sterilized for 20 minutes. All subsequent steps were performed under sterile conditions in a biosafety hood. To enable ECM attachment, the PDMS chamber was functionalized with 1% polyethylenimine (Sigma-Aldrich, #408727) in DI water followed by 0.1% Glutaraldehyde (Sigma-Aldrich, #G6257) in DI water. Devices were then washed three times with DI water to titrate out residual glutaraldehyde. A collagen-I/fibronectin solution was prepared on ice by neutralizing high concentration rat tail collagen I (Corning, #354249) and fibronectin (Sigma-Aldrich, #F1141) to a pH of 7.2. The final collagen-I and fibronectin concentrations were 4 mg/mL and 10 μg/mL, respectively. The unpolymerized ECM was pipetted into the side ports of the device and allowed to crosslink at room temperature for 30 minutes before the devices were moved to a 37C cell incubator for an additional 30 minutes.
2.5. Cell Loading
Drops of cell culture media were added to the outlet ports (larger port) of each device and tweezers were used to remove the PDMS rods from the inlet ports (smaller port), resulting in a lumen filled with media. HUVECs were added to each lumen at 15,000 cells/μL. The devices were placed in an incubator and flipped upside-down every 20 minutes for a total time of 1 hour and 20 minutes to allow the HUVECs to adhere to all sides of the lumen. The lumens were then rinsed 3 times with media to wash out nonadherent cells, and the devices were placed in the incubator overnight to allow the cells to more firmly adhere and spread out.
2.6. Image Acquisition
Bright-field and fluorescent images were obtained using a Nikon TI Eclipse inverted microscope. Images were processed using Nikon Elements.
2.7. Image Analysis
Image analysis was done using the open-source software ImageJ. Neutrophils in the stacks layers were quantified by first z-projecting the z-stack taken of the device into a single plane. The number of cells in each stacks layer were then counted.
2.8. ROS Analysis
Neutrophil ROS production was measured using 10μM dihydrorhodamine 123 (Thermo Fisher, D23806) after stimulation with or without 1μM Phorbol 12-myristate 13-acetate (Sigma-Aldrich, P8139). Mean fluorescence intensity per cell was measured using ImageJ.
2.9. RT-qPCR Analysis
Neutrophils were recovered from disassembled LENS devices by using a manual pipetman to collect non-migratory neutrophils from the lumen and Accutase (STEMCELL Technologies #07920), a cell detachment solution of proteolytic and collagenolytic enzymes, to collect migratory neutrophils from the Stacks layers. Their mRNA was then isolated in 15μL 10mM Tris buffer using Dynabeads mRNA DIRECT Purification Kit (Invitrogen, #61011) per the manufacturer’s instructions. Immediately preceding mRNA isolation, a reverse transcription reaction was run using iScript cDNA Synthesis kit (Bio-Rad, #170-8891) and the resultant cDNA was pre-amplified with SsoAdvanced PreAmp Supermix (Bio-Rad, #172-5160) and primers from Integrated DNA Technologies (Coralville, IA)(Supplementary Figure 3). Finally, qPCR reactions were run using iTaq Universal SYBR Green Supermix (Bio-Rad, #172-5121) in Roche’s Lightcycler 480 II (Roche Molecular Systems, Indianapolis, IN), and a ΔΔCt analysis was run.
2.10. Statistical Analysis
Data were analyzed (Prism 7.0; GraphPad Software) using one-way ANOVA. Tukey’s multiple comparison test with a 95% confidence interval was used when comparing different conditions.
3. Results
3.1. LENS Design
In order to create an organotypic model system to study neutrophil-endothelial interactions in real-time, with spatial control, we designed the LENS platform. The LENS technology consists of two basic components: a PDMS chamber that houses the endothelial lumen (LumeNext), and a stackable series of polystyrene (PS) tube sections (Stacks)(Figure 2). The PDMS chamber consists of two micromolded PDMS halves that fit together to form a space across which a PDMS rod can be threaded (Figure 2A). The rod is supported by struts on each end of the chamber to keep the rod from contacting surfaces within the chamber. Cut into the top of the chamber are five access ports; two gel-loading ports on either side of the rod, one access port directly above the middle of the rod which the stackable polystyrene tubes fit over, and two cell/media loading ports on either end of the rod (Figure 2B). The device is assembled by first aligning the two PDMS halves together, then threading the rod through the middle of the two halves, and lastly, the stackable tubes are placed on top of the access port (Figure 2C). Unpolymerized ECM is pipetted into the chamber and connected stacks layers through one of the gel loading ports and is allowed to set. This process creates a continuous ECM throughout the PDMS chamber and up into the Stacks layers (Figure 2D). The lumen structure is formed by pulling the PDMS rod from the device through the cell/media loading ports (Figure 2E). Cells and media can then be added to the lumen through one of the cell/media loading ports and allowed to adhere to the lumen-wall. Once assembled, soluble and/or cellular components can be added to the system using additional Stacks layers placed on top of the existing stacks layers (Figure 2F).
Fig 2: Schematic of LENS technology.
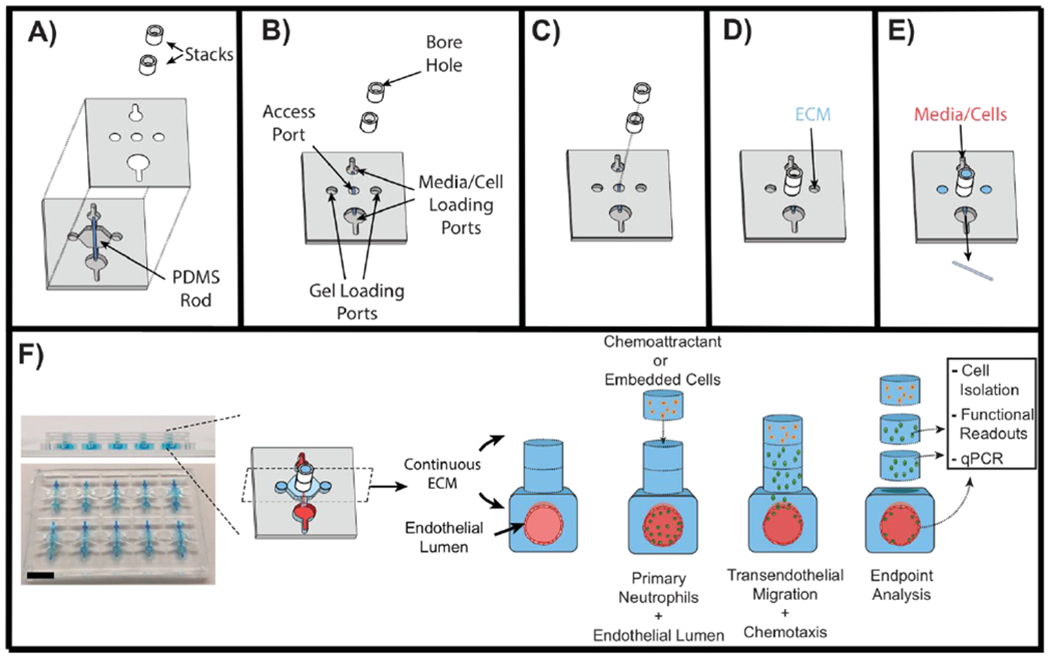
A – Exploded view diagram of all the components of LENS. The LumeNext base is composed of two PDMS halves with a PDMS rod inserted in the middle. The Stacks components consist of polystyrene tube sections. B – Once assembled, the LumeNext base creates a chamber with five ports. C – The Stacks layers stack on top of each other directly over the central access port. D – Unpolymerized ECM can be added to one of two gel loading ports. This creates a continuous ECM throughout the entire device. E – Once the ECM polymerizes, the PDMS rod can be pulled out of the device and media and/or cells can be added to the loading ports. F – Image of the LENS platform (Scale bar = 5mm) along with a schematic of a neutrophil migration assay. Primary neutrophils are loaded into an endothelial vessel and a chemoattractant (IL-8) is added to the top of the device. Neutrophils are allowed to migrate, after which the device is disassembled and the captured neutrophils can be further analyzed.
We aimed to create a device that had a continuous ECM in the Stacks layers that could be easily disassembled and reassembled, thus requiring robust ECM retention within each layer. The Stacks layers needed to have both inner-bore dimensions that maximized intra-layer ECM retention during disassembly, and spatial dimensions small enough as to remain relevant to neutrophil migratory distances in vivo. Additionally, since the Stacks layers needed to be made of a stiff material, such as polystyrene, fabrication methods also played a role in limiting the device’s dimensions. To determine the optimum bore size for the Stacks layers, LENS devices consisting of a collagen I ECM and two stackable PS (Stacks) layers with a range of diameters (0.5mm, 0.75mm, 1mm) and heights (0.25mm, 0.5mm, 0.75mm), were assayed for collagen adherence to the side walls within each Stacks layer upon disassembly (Supplementary Figure 1). Stacks layers with the largest bore diameter and smallest height had the lowest collagen retention rate (16.67% +/− 15.2) whereas the Stacks layers with the smallest bore diameter and largest height had the best collagen retention rate (83.33% +/− 5.8). Due to micromachining fabrication considerations, the final bore diameter for the Stacks layers was kept at 0.5mm. Although 0.75mm is a relatively large distance in vivo, the high collagen retention rate warranted the use of this height for these validation experiments. In the future, the platform could be modified to recapitulate smaller in vitro distances.
3.2. Characterization of LENS Capabilities
The LENS technology was designed to facilitate the introduction and isolation of cells from the microdevice without significantly perturbing the microenvironment (Figure 2F). In order to validate these capabilities for the capture of neutrophils, neutrophil migration experiments were conducted in LENS devices to assay for the cell capture capabilities of the devices (Figure 2F). Leukocyte trafficking occurs in response to a chemokine gradient. We characterized the gradient within the device using a diffusion model in COMSOL to estimate gradient characteristics in LENS (Supplementary Figure 2). Using a porosity and density similar to a 4mg/mL collagen-I hydrogel and a diffusion coefficient of 8.974 x 10-7 cm2 s−1, consistent with 10kDa FITC-dextran (similar in size with known neutrophil chemokines), we computed diffusion profiles over time between 0 min to 90 min (Supplementary Figure 2A).24,25 Based on the COMSOL model, the gradient would begin to form at around 30 min and would persist for greater than 90 min (Supplementary Figure 2B).
Next, we investigated 10 kDa FITC-dextran diffusion in our 4 mg/mL collagen-I + 10 μg/mL fibronectin matrix, conditions shown to facilitate endothelial lumen formation and neutrophil migration.21 A single Stacks layer, containing collagen-I/fibronectin and 10 kDa FITC-dextran was placed on top of two other Stacks layers containing only collagen-I/fibronectin, which permitted the dextran to diffuse (Figure 3A). Multiple devices were assembled and diffusion was allowed to take place for 30, 60, and 90 minutes, after which the devices were disassembled and the fluorescence was quantified for each layer (Figure 3B). From time 0 min to 30 min there is a steep decline in the dextran gradient, however, from time 30 min to 90 min the deterioration of the gradient decreases and remains stable. Using the diffusion data in figure 3B we extrapolated a diffusion coefficient of 9.698 x 10-7 cm2 s-1 for the 10 kDa FITC-dextran which consistent with our COMSOL model and demonstrates that a chemokine gradient can be formed in the LENS platform.
Fig 3: Diffusion and neutrophil migration in the stacks layers.
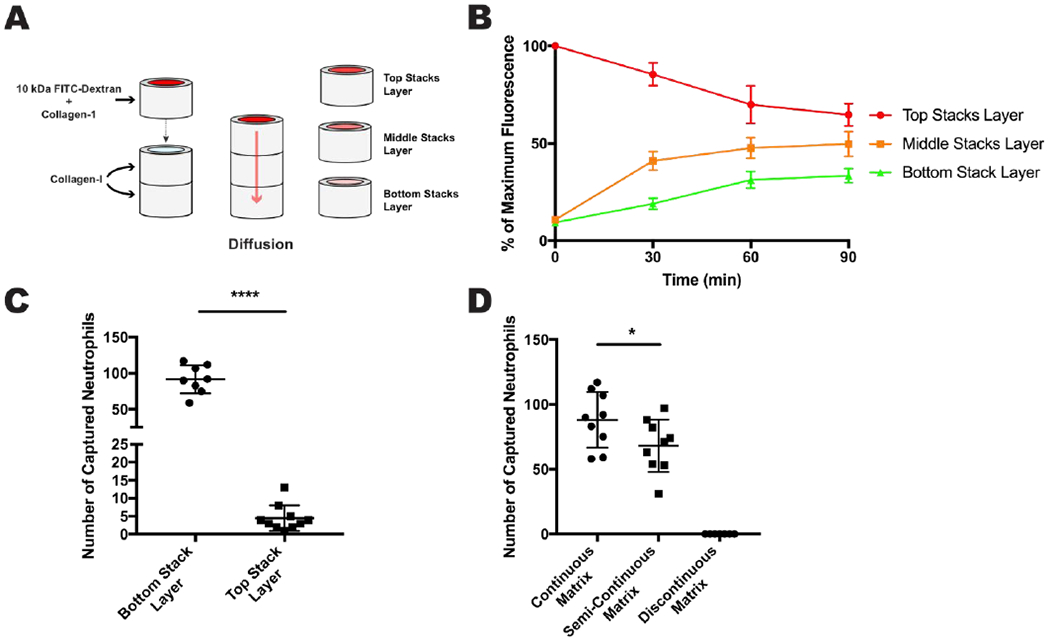
A - A single stacks layer containing collagen-1 and 10kDa FITC-Dextran was placed on top of two other stacks layers, both containing collagen-I only. The FITC-dextran was allowed to diffuse into the bottom two layers for a certain amount of time. B - Results of the diffusion experiment showing average maximum fluorescence for each stacks layer over 90 minutes. C - Results of the double Stacks layer migration experiment showing number of captured neutrophils per layer. D - Results for the ECM continuity experiments showing number of captured neutrophils per ECM condition. All data were quantified from neutrophils collected from 9 stacks layers across 3 independent experiments. Results are consistent across multiple donors. All bars represent mean plus SD. Asterisks represent significance between conditions. * = p < 0.05, **** = p < 0.0001
Being able to isolate migratory and non-migratory neutrophil phenotypes from LENS would allow for a range of cellular and molecular analyses and could offer valuable insight into human neutrophil biology. To test the cell isolation capabilities of LENS we ran a series of neutrophil migration experiments. A 4 mg/mL collagen-I and 10 μg/mL fibronectin continuous ECM was established inside two-Stacks layered devices along with a HUVEC vessel, like the setup in Figure 2F. Primary human, calcein-stained neutrophils were added to the endothelial lumen, and an IL-8 gradient, a known neutrophil chemoattractant which is secreted during inflammation, was established across the device. The neutrophils were allowed to migrate for 4 hours at 37C after which the device was disassembled, and the neutrophils in each Stacks layer were quantified (Figure 3C). While most migratory neutrophils reached the first stacks layer, ~10% of neutrophils migrated > 0.75 mm and were found within the second Stacks layer. These results confirm that neutrophil migration assays are compatible with LENS and that it has the resolution to differentiate between “fast” and “slow” migrating cells.
Cell migration is sensitive to many ECM properties including composition, porosity, stiffness, and especially continuity.26 A method for adding cells to the ECM in LENS would be useful for studying cell-sourced chemoattractants and juxtacrine signalling, though the current method of placing a Stacks layer containing embedded cells on top of a LENS device would result in a discontinuous matrix and possibly exclude neutrophils from trafficking properly. To overcome this ECM continuity issue, we used unpolymerized collagen-I to connect the additional Stacks layer to the LENS device, bridging the two ECMS, and assayed for neutrophil migration into the added layer (Figure 3D). In these experiments, there was a significant difference between the number of neutrophils captured in the continuous matrix condition and the semi-continuous condition with the latter capturing 23% fewer neutrophils, there were no neutrophils captured in the discontinuous matrix. This result indicates that while neutrophils prefer a continuous matrix, the semi-continuous collagen-I matrix is a viable alternative to a completely discontinuous matrix, and proves that cells can be added to the LENS matrix with minimal effects on neutrophil trafficking.
3.3. LENS as a tool to Study Neutrophil Trafficking
During the characterization of LENS, we have shown that this technology can facilitate the addition and retrieval of cells to a microscale device without disturbing the microenvironment significantly. To demonstrate the utility of these innovations, we used LENS to study the effect that endothelial cells have on neutrophil TEM/chemotaxis, and priming.
Neutrophil-endothelial interactions are critical for effective immune system function, however, quantifying this behavior and isolating cells for downstream analysis is difficult to do in current organotypic models.27 We first investigated the downstream effects of endothelial contact on neutrophil TEM/chemotaxis (Figure 4). Neutrophils were added to the lumen of LENS devices which was either an empty lumen with no endothelial cells or a HUVEC microvessel, and an IL-8 gradient was established vertically through the stacks layers. Neutrophils were allowed to migrate for four hours, after which the devices were disassembled, and the number of captured neutrophils were quantified. In empty (no endothelium) lumen conditions, an average of 1.17 neutrophils were captured in the stacks layer in the absence of a chemokine gradient, and 2.88 neutrophils were captured when exposed to an IL-8 gradient. Alternatively, in the endothelial lumen conditions, an average of 10.5 neutrophils were captured in the absence of a chemokine gradient, and 137.63 neutrophils were captured in the IL-8 condition. These results demonstrate LENS’s ability to isolate populations of migratory neutrophils and suggests that it can be used to facilitate analysis of these cells further. Additionally, these results confirm that the endothelium significantly enhances a neutrophil’s ability to sense and respond to chemokine gradients. While outside the scope of this, IL-8/glycosaminoglycan (GAG) dimerization and presentation on the luminal surface of the endothelium,28 activation of neutrophil Integrin receptors,29 and secretion of cytokines21 could potentially contribute to these observations. While these factors contribute to enhancing neutrophil TEM and chemotaxis, the effect neutrophil-endothelial interactions have on neutrophil behavioral phenotype and downstream signaling is less clear.
Fig 4: Neutrophil migration to IL-8 with and without and endothelial lumen.
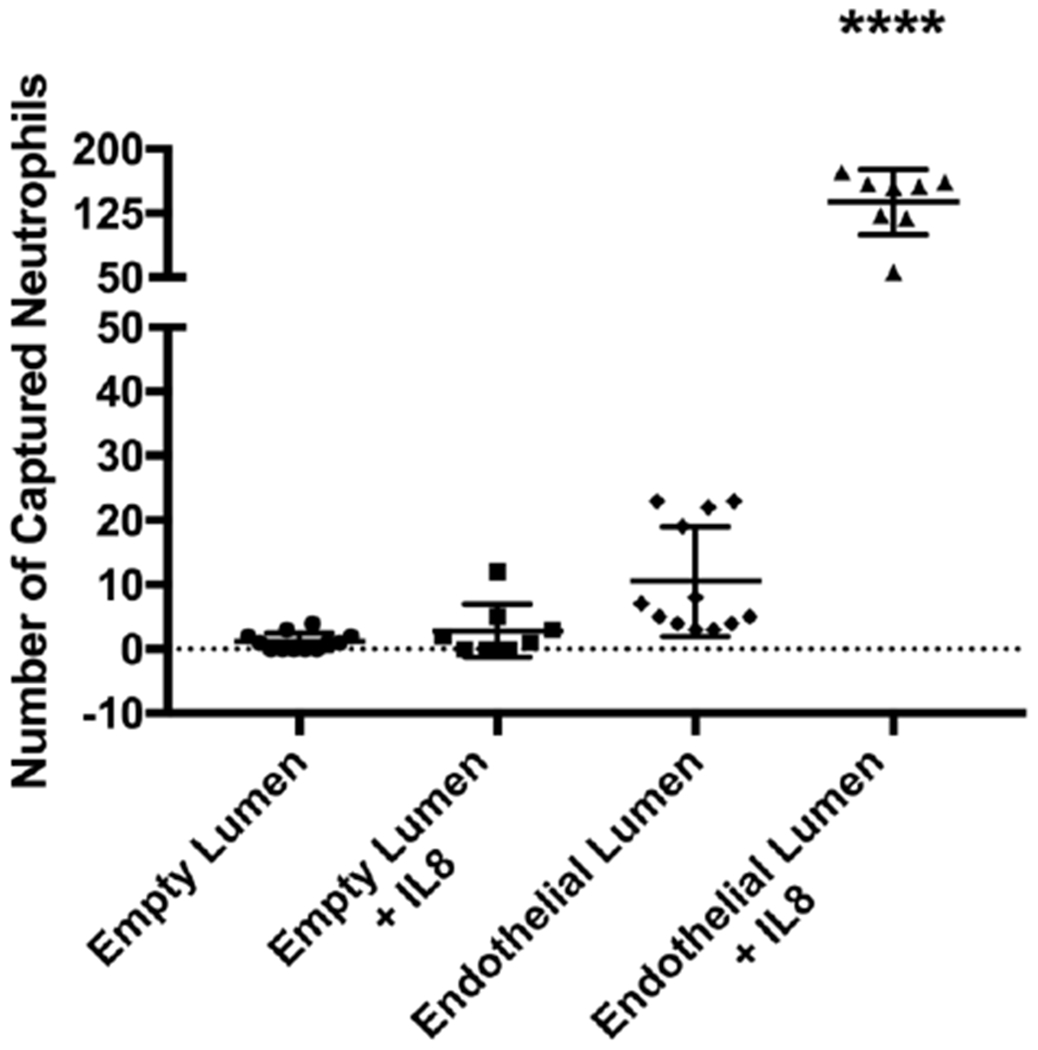
Results showing neutrophil migration in different microenvironmental conditions. All bars represent mean plus SD. All data were quantified from neutrophils collected from 12 stacks layers for no IL-8 conditions, and 8 stacks layers for +IL-8 conditions across 3 independent experiments. Results are consistent across multiple donors. Asterisks represent significance between conditions. **** = p < 0.0001
It has previously been shown in mice that extravasation of neutrophils across the endothelium into the interstitium alters their phenotype and induces changes in transcription.30,31 To test if the same is true for human cells, we first looked at how TEM affects a neutrophil’s ability to produce ROS. Using single stacks-layer LENS devices containing HUVEC lumens, we ran a series of IL-8 migration experiments. After four hours of migration, devices were disassembled, both the lumen and stacks components were treated with either dihydrorhodamine 123 (DHR), or DHR and Phorbol 12-myristate 13-acetate (PMA), and then the mean fluorescence intensity per cell (MFIPC) was measured over four additional hours (Figure 5). In the no-PMA control, neutrophil ROS production remained relatively low, peaking at 240 after three hours. There was a significant increase in ROS production during hours one and two for the migratory/HUVEC condition compared to the other PMA conditions. There were no significant differences within the other PMA treated samples. Differences in ROS generation between migratory and nonmigratory neutrophils in the same condition suggests an existing heterogeneity within the isolated neutrophil population. Furthermore, these results show that neutrophils that have undergone TEM, and not just chemotaxis, are primed to elicit a more robust ROS response. Whether this difference in functional phenotypes is pre-existing or acquired within the system remains unresolved.
Fig 5: Neutrophil ROS –
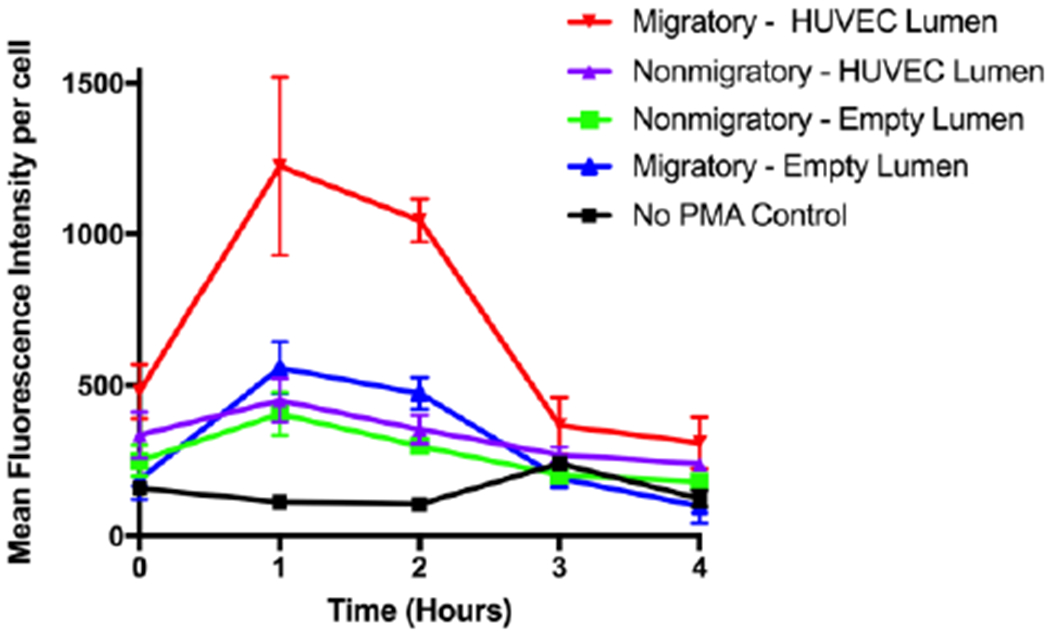
Average fluorescence measurements over four hours from migratory and nonmigratory neutrophils stained with DHR 123 and treated or not treated with PMA. Measurements were consistent across multiple donors.
Next we looked at how TEM altered gene transcription. To test this, migratory neutrophils (i.e. neutrophils that were captured in a stack layer), and non-migratory neutrophils (i.e. neutrophils that remained inside the lumen), were isolated from LENS devices containing endothelial and empty lumens, and an RT-qPCR analysis was conducted (Figure 6). Our genes of interest focused on both ROS production, and those previously shown to be altered during neutrophil priming, such as genes involved with chemotaxis, cell adhesion, and receptor signalling.6,32,33,34
Fig 6: Gene expression is different in motile and non-motile neutrophils.
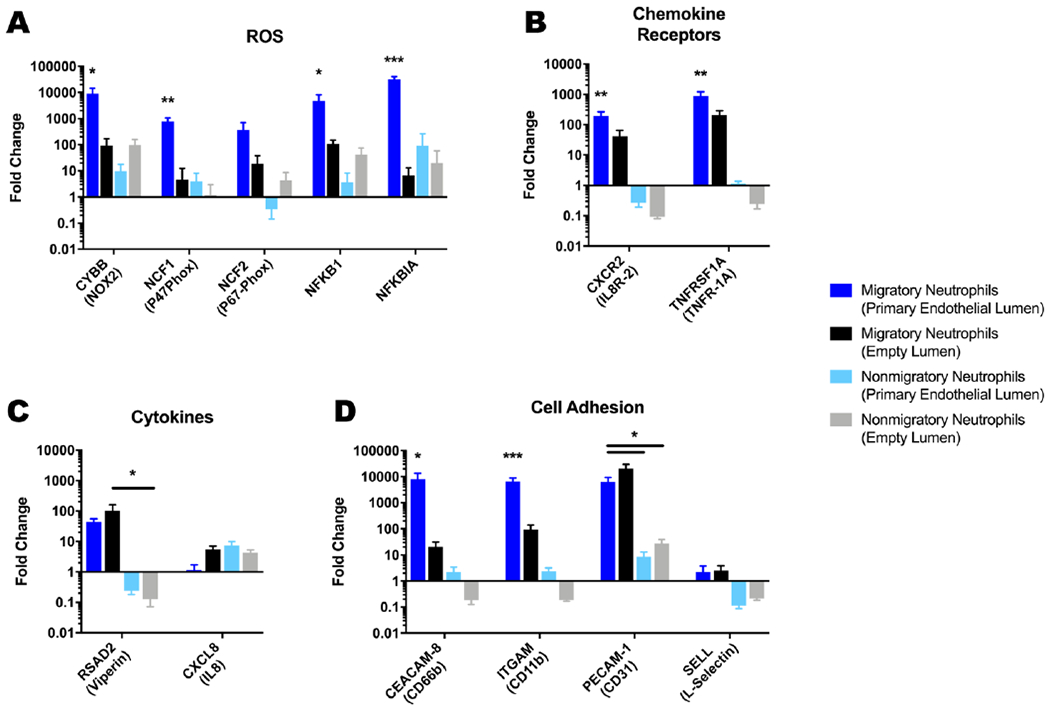
qPCR results comparing gene expression of migratory and non-migratory neutrophils in LENS devices containing an empty collagen-I lumen or a HUVEC lumen investigating genes involved with A – production of ROS in neutrophils. B – neutrophil chemokine pathways. C – secreted cytokines. D – neutrophil adhesion. All bars represent mean plus SD. All data were quantified from neutrophils collected from 9 stacks layers for migratory neutrophils in endothelial lumen conditions, 30 stacks layers for migratory neutrophils in empty lumen conditions, and 9 lumens for both non-migratory neutrophil conditions across 3 independent experiments. Results are consistent across multiple donors. Asterisks represent significance between conditions. * = p < 0.05, ** = p < 0.01, *** = p < 0.001
For the genes associated with ROS generation, we found that the expression for all but NCF2 were significantly more upregulated for the migratory neutrophils in the endothelial lumen condition (>1000-fold) compared to the other conditions (<100-fold), though the trend in expression was similar for NCF2(Figure 6A). Amongst the other conditions, i.e. migratory/empty lumen, non-migratory/empty lumen, and non-migratory/endothelial lumen, there was no significant differences in gene expression for these genes. These results correlate with our ROS measurements and strongly suggest that the migratory neutrophils are acquiring a primed phenotype during TEM.
Both genes associated with chemokine receptors, CXCR2, and TNFRSF-1A, were significantly more upregulated in migratory neutrophils in endothelial lumen conditions compared to the other conditions with fold increases of 190, and 882 respectively (Figure 6B). Migratory neutrophils in empty lumen conditions also displayed a significant increase in expression compared to both non-migratory conditions, although at a lower level with fold changes for CXCR2 and TNFRSF-1A were 40, and 209 respectively.
We also investigated chemokine genes involved in neutrophil migration (Figure 6C). RSAD2 was upregulated for both conditions involving migratory neutrophils, though only the neutrophils in the empty lumen condition displayed significance. Alternatively, both nonmigratory neutrophil conditions had decreased expression for RSAD2. CXCL8 was unchanged in migratory neutrophils in the endothelial condition, while it was slightly upregulated, between 4- and 7-fold, in the other conditions. While the endothelium has been associated with enhanced neutrophil priming, we did not expect all of our genes of interest would be altered.35,36
CEACAM-8, ITGAM, PECAM1, and SELL all encode for genes associated with cellular adhesion and have been shown to be upregulated during priming, TEM and chemotaxis.37,38,39 For all of these genes except SELL, migratory neutrophils in endothelial lumen conditions had >1000-fold increase in gene expression (Figure 6D). Expression of CEACAM-8 and ITGAM were significantly higher than all other conditions for the migratory neutrophils in an endothelial lumen. The difference in upregulation of CEACAM-8 and ITGAM between the two migratory neutrophil conditions, like the genes for ROS and chemokine reception, further illustrates the effect the endothelium has on neutrophil priming.
LENS allowed us to isolate phenotypically pure neutrophil populations. Due to this capability, we were able to directly correlate transcriptional changes in neutrophils to changes in functional phenotypes. We found that while there exists heterogeneity within isolated neutrophil populations, endothelial-neutrophil interactions are necessary for robust neutrophil chemotaxis and ROS generation. These changes correspond to the upregulation in transcription of genes associated with, but not limited to ROS production, chemokine reception, and cell adhesion.
4. Conclusion
In this study, we developed an in vitro microscale technology able to produce organotypic endothelial-lumens with a simple way to add or capture cells from a device without disturbing the microenvironment. This added spatiotemporal control allowed us to model neutrophil priming, TEM, and chemotaxis with a focus on studying endothelial-neutrophil interactions.
In this study, we were able to directly correlate transcriptional changes in neutrophils to changes in functional phenotypes. This correlation was made possible by the design of LENS, and would’ve been difficult to do using other conventional methods such as 2D microfluidic devices or transwells. We found that while there exists heterogeneity within isolated neutrophil populations, endothelial-neutrophil interactions enhance neutrophil chemotaxis and ROS generation. These changes are at least partly due to the upregulation in transcription of genes associated with, but not limited to ROS production, chemokine reception, and cell adhesion.
In summary, the LENS platform is a new microfluidic technology that enables the study and isolation of migratory cells in a highly relevant and highly controllable manner. We used LENS to look at neutrophil-endothelial interactions using human cells generating results consistent with those found in non-organotypic, 2D models of human neutrophil migration, implying LENS could be a valuable tool for further studies involving human neutrophil trafficking.
One area of interest that could adapt well to this technology is neutrophil reverse migration and inflammation resolution pathways.2,40 Neutrophils, during inflammation resolution, were assumed to all undergo apoptosis followed by macrophage clearance of the apoptotic bodies. However, recent evidence in mouse and zebrafish models, as well as in human studies suggest that some neutrophils can migrate away from sites of inflammation and re-enter circulation in a process termed neutrophil reverse migration. While modelling this process using 2D microfluidics is useful, incorporating ECM and cellular components could give us a more physiologically relevant picture of this event. Additionally, modelling reverse migration in a system with a high degree of spatiotemporal control like LENS could provide insight into why only certain neutrophils reverse migrate.
Supplementary Material
Acknowledgements
This work was supported in part by the NIH: National Institutes of Allergy and Infectious Diseases through R01 AI134749, and the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
Conflicts of interest
David J. Beebe holds equity in Bellbrook Labs, LLC, Tasso Inc., Salus Discovery LLC, LynxBiosciences Inc., Turba LLC, Stacks to the Future, LLC and Onexio Biosystems, LLC.
References
- 1.Kolaczkowska E and Kubes P, Nat. Rev. Immunol, 2013, 13, 159–175. [DOI] [PubMed] [Google Scholar]
- 2.De Oliveira S, Rosowski EE and Huttenlocher A, Nat. Rev. Immunol, 2016, 16, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mai J, Virtue A, Shen J, Wang H and Yang XF, J. Hematol. Oncol, 2013, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson S, Martínez-Burgo B, Sepuru KM, Rajarathnam K, Kirby JA, Sheerin NS and Ali S, Int. J. Mol. Sci, 2017, 18, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joseph PRB, Sawant KV and Rajarathnam K, Open Biol., DOI: 10.1098/rsob.170168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miralda I, Uriarte SM and McLeish KR, Front. Cell. Infect. Microbiol, 2017, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harvie EA and Huttenlocher A, J. Leukoc. Biol, 2015, 98, 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao L and Reese TA, Trends Immunol, 2017, 38, 181–193. [DOI] [PubMed] [Google Scholar]
- 9.Mestas J and Hughes CCW, J. Immunol, 2004, 172, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 10.Hamza B and Irimia D, Lab Chip, 2015, 15, 2625–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moussavi-Harami SF, Mladinich KM, Sackmann EK, Shelef MA, Starnes TW, Guckenberger DJ, Huttenlocher A and Beebe DJ, Integr. Biol, 2016, 8, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kusuma S, Shen Y-I, Hanjaya-Putra D, Mali P, Cheng L and Gerecht S, Proc. Natl. Acad. Sci, 2013, 110, 12601–12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu W, Qu X, Zhu J, Ma X, Patel S, Liu J, Wang P, Lai CSE, Gou M, Xu Y, Zhang K and Chen S, Biomaterials, 2017, 124, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsvirkun D, Grichine A, Duperray A, Misbah C and Bureau L, Sci. Rep, 2017, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestre-Roig C, Hidalgo A and Soehnlein O, Blood, 2016, 127, 2173–2181. [DOI] [PubMed] [Google Scholar]
- 16.Jiménez-Torres JA, Peery SL, Sung KE and Beebe DJ, Adv. Healthc. Mater, 2016, 5, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A and Beebe DJ, Adv. Healthc. Mater, 2018, 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu J, Berthier E, Craig A, de Groot TE, Sparks S., Ingram P, Jarrard D, Huang W, Beebe D, Theberge AB, Nat. Biomed. Eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deniset JF and Kubes P, J. Leukoc. Biol, 2018, 1–10. [DOI] [PubMed] [Google Scholar]
- 20.Scapini P and Cassatella M, Blood, 2014, 124, 710–720. [DOI] [PubMed] [Google Scholar]
- 21.Hind LE, Ingram PN, Beebe DJ and Huttenlocher A, Blood, 2018, 132, blood-2018-05-848465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Y and Whitesides GM, Angew. Chemie Int. Ed, 1998, 37, 550–575. [DOI] [PubMed] [Google Scholar]
- 23.Guckenberger DJ, de Groot T, Wan AM-D, Beebe D Young E, Lab Chip, 2015, 15, 2364–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gribbon P and Hardingham TE, Biophys. J, 1998, 75, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hettiaratchi MH, Schudel A, Rouse T, García AJ, Thomas SN, Guldberg RE and McDevitt TC, APL Bioeng., 2018, 2, 026110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charras G and Sahai E, Nat. Rev. Mol. Cell Biol, 2014, 15, 813–824. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt S, Moser M and Sperandio M, Mol. Immunol, 2013, 55, 49–58. [DOI] [PubMed] [Google Scholar]
- 28.Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AEI, Wells TNC, Parks WC, Wight TN, Martin TR and Frevert CW, J. Immunol, 2010, 184, 2677–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pichert A, Schlorke D, Franz S and Arnhold J, Biomatter, 2012, 2, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao Y, Matsushima H, Ohtola JA, Geng S, Lu R and Takashima A, J. Immunol, 2014, 194, 1211–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakschevitz FS, Visser MB, Sun C and Glogauer M, Cell. Mol. Immunol, 2015, 12, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang K, Sun X, Chen Y, Shen Y and Jarvis JN, BMC Med. Genomics, 2015, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ecker S, Chen L, Pancaldi V, Bagger FO, Fernández JM, Carrillo de Santa Pau E, Juan D, Mann AL, Watt S, Casale FP, Sidiropoulos N, Rapin N, Merkel A, Stunnenberg HG, Stegle O, Frontini M, Downes K, Kuijpers TW, Kuijpers TW, Rico D, Valencia A, Beck S, Soranzo N, Paul DS, Albers CA, Amstislavskiy V, Ashford S, Bomba L, Bujold D, Burden F, Busche S, Caron M, Chen SH, Cheung WA, Clarke L, Colgiu I, Datta A, Delaneau O, Elding H, Farrow S, Garrido-Martín D, Ge B, Guigo R, Iotchkova V, Kundu K, Kwan T, Lambourne JJ, Lowy E, Mead D, Pourfarzad F, Redensek A, Rehnstrom K, Rendon A, Richardson D, Risch T, Rowlston S, Shao X, Simon MM, Sultan M, Walter K, Wilder SP, Yan Y, Antonarakis SE, Bourque G, Dermitzakis ET, Flicek P, Lehrach H, Martens JHA, Yaspo ML, Ouwehand WH, Genome Biol., 2017, 18, 1–17.28077169 [Google Scholar]
- 34.Galli SJ, Borregaard N and Wynn TA, Nat. Immunol, 2011, 12, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drost EM and MacNee W, Eur. J. Immunol, 2002, 32, 393–403. [DOI] [PubMed] [Google Scholar]
- 36.FAN J and MALIK AB, Nat. Med, 2003, 9, 315–321. [DOI] [PubMed] [Google Scholar]
- 37.Fortunati E, Kazemier KM, Grutters JC, Koenderman L and Van Den Bosch VJMM, Clin. Exp. Immunol, 2009, 155, 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dangerfield J, Larbi KY, Huang M-T, Dewar A and Nourshargh S, J. Exp. Med, 2002, 196, 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B and Nourshargh S, Blood, 2009, 113, 6246–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powell D, Tauzin S, Hind LE, Deng Q, Beebe DJ and Huttenlocher A, Cell Rep., 2017, 19, 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.