

Abstract
Melanoma is the deadliest type of skin cancer with one of the fastest increasing incidence rates among solid tumors. The use of checkpoint inhibitors (e.g. αPD-1 antibody) has recently emerged as a viable alternative to conventional modes of therapy. However, increasing evidence points towards the need for a tumor priming step to improve intratumoral immune cell infiltration. IL-12 is an immune-activating cytokine with such potential and was explored in earlier clinical trials as a highly concentrated systemic infusion. This unfortunately led to severe adverse effects. From this perspective, the localization and gradual release of such a potent immunotherapeutic agent in the tumor microenvironment is desired. This manuscript reports the use of a heparin-based complex coacervate to deliver IL-12, in which heparin-binding motifs on IL-12 allow for its effective encapsulation. IL-12-encapsulated complex coacervates significantly improved the bioactivity of IL-12 and provided protection from proteolytic cleavage in-vitro. Indeed, a single injection of IL-12 coacervate significantly inhibits the in-vivo growth of treated and untreated, contralateral tumor growth in a syngeneic B16F10 mouse melanoma model. Furthermore, tumors in mice receiving IL-12 complex coacervate treatment displayed increased infiltration by natural killer (NK) cells and CD8α+ T cells, and a decreased presence of CD4+Foxp3+ regulatory T cells. This study provides proof-of-concept data supporting the use of complex coacervates for sustained delivery of immunostimulatory proteins as an effective therapeutic strategy against disseminated tumors.
Keywords: IL-12, Heparin, Complex Coacervate, Immunotherapy, Melanoma
Introduction
Cancer continues to be a leading cause of death according to the World Health Organization1 and now surpasses cardiovascular-related diseases as the leading cause of death in areas with high development2. In particular, melanoma is a highly-aggressive and deadly form of skin cancer and its incidence continues to rise in stark contrast to the observed decline of other cancers such as prostate, breast, and lung carcinomas3. Surgical resection continues to remain the clinical mainstay, particularly for early-stage disease. Conventional modes of chemotherapy such as the use of dacarbazine yield low response rates with 5 year survival rates between 2–6%4. This general ineffectiveness stems from the inherent refractory nature of melanoma cells to apoptotic cell death5.
Most recently, immunotherapies have emerged as viable alternatives, rapidly becoming first-line treatment options in melanoma patients with advance-stage disease. In particular, seminal advances have been made in the field of T cell immunobiology6, 7, 8, leading to the discovery, development, and clinical translation of immune checkpoint inhibitors that have dramatically altered the landscape of patient management. CTLA-4 and PD-1/PD-L1 are checkpoint molecules most commonly targeted to promote anti-cancer T cell activity in association with improved objective clinical response rates of 15.2%9 and up to 40%10, respectively. However, these early achievements have been tempered by the limited number of patients that receive durable clinical benefits when these agents are applied as monotherapies, as well as, concerns for severe immune-related adverse events (irAEs)11, 12, 13. Nevertheless, these studies corroborate the importance of tumor-infiltrating T cells and other immune cells to the therapeutic efficacy of these agents, suggesting that improved combination approaches may be developed by defining pre-treatment conditioning regimens that potentiate improved inflammatory immune infiltrates selectively within the tumor microenvironment (TME). In this context, initial treatment of tumor lesions with immune-activating cytokines may provide the foundation for further development of more effective combination immunotherapies for cancer patients. One such immune-activating cytokine is interleukin (IL)-2 that is FDA-approved for high-dose intravenous delivery14,15 to mitigate immunosuppressive tumor mechanisms and to coordinately stimulate innate NK cell responses and adaptive T cell responses. Unfortunately, such high systemic concentrations of rhIL-2 elicit a sepsis-like “cytokine storm” associated with severe irAEs (≥ grade 3)14. Furthermore, numerous studies investigating therapeutic IL-2 in-vivo have demonstrated biologic redundancy with other (less toxic) immune-activating cytokines such as IL-12p70 or IL-15 that can compensate under conditions of IL-2 deficiency15, 16.
Like IL-2, IL-12 also coordinately activates the innate and adaptive immune systems and elicits robust IFN-γ production from NK and T cells as a basis for its anti-tumor activity. Unlike IL-2, IL-12 has a longer half-life of 12 hours17 vs. minutes for IL-218. In addition, while IL-2 has been shown to induce the expansion of cancer-promoting CD4+CD25+Foxp3+ Tregs19, 20, IL-12 antagonizes Treg-mediated immunosuppression21. Despite these advantages, initial clinical trials utilizing IL-12 were stopped due to severe irAEs (i.e. GI bleeding) in a minority of patients, subsequently leading to the emergence of gene-based delivery approaches for IL-1222. Advances in IL-12 gene therapy have been counterbalanced by poor transfection efficiencies that continue to limit its clinical potential22. Given that clinical trials using recombinant IL-12 were stopped due to systemic toxicity, local controlled delivery of sufficient levels of IL-12 in the TME appears necessary for its safe and effective therapeutic application23.
Given these perspectives, complex coacervation represents an attractive strategy to overcome the drawbacks of systemic infusion of IL-12. Complex coacervates are characterized by short-range24 and long-range electrostatic interactions25 that lead to a soluble polyelectrolyte-rich phase that is amenable to injection delivery. Our lab has previously developed a heparin-based complex coacervation platform26, which utilizes the natural affinity of heparin for a large number of proteins. The combination of protein-heparin complexes with cationic poly(ethylene argininylaspartate diglyceride) (PEAD) results in the instantaneous formation of a polymer-rich soluble phase from which proteins are delivered. The use of heparin confers excellent biocompatibility and biomimetic properties. Indeed, IL-12 bound to heparin27 has been shown to increase its bioactivity by more than 6-fold compared to unbound naked IL-1228. This work is the first to examine the concept of complex coacervation-based delivery of recombinant cytokines for use as anti-cancer agents. Herein, we provide evidence for the safe and effective use of complex coacervates for localized and sustained delivery of therapeutic IL-12 into the TME.
Materials and Methods
Materials
Ethylene glycol diglycidyl ether (EGDE) (Pfaltz & Bauer, Waterbury, CT), N -Boc-L-aspartic acid (Boc-Asp-OH), and N-Boc-L-arginine (Boc-Arg-OH) (Bachem Americas Inc.), tetra-«- butyl-ammonium bromide (TBAB, 98%+), trifluoroacetic acid (TFA, ≥99.5%), anhydrous dimethylformamide (DMF, ≥99.9%), N,N’-dicyclohexylcabodiimide (DCC, ≥99%), and 4- (dimethylamino)pyridine (DMAP, ≥99%) (Alfa Aesar, Ward Hill, MA), #-hydroxysuccinimide (NHS, ≥98.0%) (Acros Organics, Geel, Belgium), ethyl ether and dichloromethane (Pharmco-Aaper), heparin sodium USP (Scientific Protein Labs, Waunakee, WI), DMEM-high glucose supplemented with GlutaMAX, RPMI 1640 supplemented with L-glutamine and phenol red, 2.5% typsin with no phenol red and EDTA, 0.25% trypsin-EDTA, protease and phosphatase inhibitor mini tablets-EDTA free, Nunc MaxiSorp flat-bottom plates, goat anti-rat-Alexa Fluor 488, goat anti-rabbit-Alexa Fluor 647, BSA-Alexa Fluor 647, and Cytoseal 60 (Thermo Fisher Scientific, Waltham, MA), 96-well round bottom plates (Corning, Corning, NY), Eppendorf LoBind tubes (Eppendorf North America, Hauppauge, NY), B16F10 melanoma cells (ATCC, Manassas, VA), penicillin-streptomycin (Lonza, Basel, Switzerland), recombinant murine IL-12p70 (Peprotech, Rocky Hill, NJ), mouse IL-12 p70 antibody, mouse IL-12 biotinylated antibody, streptavidin- HRP, and substrate reagent pack (R&D Systems, Minneapolis, MN), mouse IFN-γ OptEIA ELISA kit, rat anti- mouse CD8α (clone 53–6.7), rat anti-mouse CD4, and rat anti-mouse NKp46 (clone 29A1.4) (BD, San Jose, CA), bacteroides heparinase II (New England Biolabs, Rowley, MA), FoxP3 rabbit mAb (Cell Signaling Technology, Danvers, MA), and rabbit aPD-L1 (Abcam, Cambridge, MA) were all used as received.
Synthesis of PEAD
PEAD was synthesized as previously described with minor modifications 29. Briefly, EGDE (1000 mg), Boc-Asp-OH (1338.8 mg), and TBAB (5 mg) were dissolved in 0.6 mL DMF. The mixture was reacted for 20 minutes at 120°C in a microwave reactor (Biotage, Uppsala, Sweden). The resulting intermediate polymer, poly(ethylene boc-aspartate diglyceride) was precipitated in ethyl ether overnight. The solvent was removed via rotary evaporation and dissolved in 20 mL DCM. Boc was removed by adding 5 mL of TFA dropwise ([TFA] = 2.5mM) and incubating for 2 hours at room temperature under stirring conditions. The solvent was subsequently removed with rotary evaporation and precipitated into ethyl ether. Multiple precipitation steps in ethyl ether were carried out to remove excess reagents, DMF, and TFA. The deprotected (PED) was then washed overnight in ethyl ether and dried under vacuum until further use. PEAD was prepared by combining PED (1000 mg), Boc-Arg-OH (1071.2 mg), NHS (449.4 mg), DCC (1047.4 mg), and DMAP (23.85 mg) in DMF and stirring at room temperature under N2 for 24 hours. The resulting insoluble dicyclohexylurea by-product was removed using centrifugation and filtration (0.22 μm). PEAD-boc was then precipitated into ethyl ether overnight. To remove the boc group, the solvent was removed via rotary evaporation, the resulting polymer was dissolved in 11 mL DCM, and 3 mL of TFA was added dropwise. The solution was stirred at room temperature for 2 hours. The solvent was subsequently removed via rotary evaporation and the deprotected final product was dissolved in 5 mL of methanol. At least three precipitation steps were carried out in ethyl ether, followed by two washes in ethanol. The final product was dissolved in deionized water and lyophilized for at least 72 hours before use.
Preparation of IL-12 Coacervates
10 mg/mL PEAD and heparin solutions were prepared in 0.9% saline and sterile filtered (0.22 μm). 5:1 mass ratios between PEAD:heparin were utilized based on their isoelectric charge as assessed via dynamic light scattering (Supplementary Figure 1). For stability and in-vitro release studies, 200 μL of complex coacervate was prepared, in which 1 μL of IL-12 was mixed with 33.3 μL of heparin. 166.7 μL of PEAD was then added to the IL-12-heparin complex to induce coacervation. For in-vivo studies, each mouse received 50 μL of treatment consisting of 25 μL protein and 25 μL coacervate, which were prepared in the same manner as in-vitro studies. IL-12 coacervate samples were prepared immediately before injection and any aggregated coacervate phase was gently resuspended by flicking the tube.
Cryo-SEM
Cryo-SEM images were taken using an FEI Strata 400 STEM FIB.
Stability of IL-12 in Trypsin
Complex coacervates were loaded with 100 ng of murine IL-12p70 for each sample (n = 4). Samples were centrifuged after which the supernatant was collected for subsequent ELISA analysis. 100 μL of 500 ng/mL trypsin was then added to the IL-12 coacervates or to 100 ng of IL-12 suspended in PBS and incubated for 0, 1, or 10 hours. At each time point, 100 μL of 1X protease inhibitor solution was added and incubated for 5 minutes at room temperature. To break apart the complex coacervate, 50 μL of heparinase cocktail solution (1 μL of 4000 units/mL Bacteroides Heparinase II, 25 μL Bacteroides Heparinase reaction buffer, and 24 μL of 20X PBS) was added to each sample and further incubated for 1 hour at 37°C. Samples were subsequently frozen at −20°C until analysis via ELISA.
In-vitro Release
IL-12 loaded complex coacervates were prepared as described above. Each sample was loaded with 500 ng of murine IL-12p70 (n = 3). For the group loaded with both IL-12 and BSA, 0.1% BSA in saline was used to dilute IL-12. 0.9% saline without BSA was used for all other preparations. After initial formation of the complex coacervates, samples were immediately centrifuged to induce their coalescence. The supernatant was collected and analyzed via ELISA to indirectly assess loading efficiency. The coacervate pellet was gently resuspended in 0.1% BSA in saline for subsequent time points. At each time point, samples were spun down and the supernatant was collected and frozen at −20°C until analysis via IL-12 ELISA. For IL-12, 100 μL of 2 μg/mL mouse IL-12 p70 antibody was added to 96 well Nunc MaxiSorp flat-bottom plates and incubated at 4°C overnight. After washing with wash buffer (PBS supplemented with 0.05% Tween 20) 4 times, wells were blocked with PBS supplemented with 1% BSA for 2 hours at room temperature. Wells were then washed 4 times and 100 μL of sample was added to each well. After 2 hours incubation at room temperature, samples were washed with wash buffer 4 times, followed by incubation with 100 μL of 0.2 μg/mL biotinylated mouse IL-12 p70 antibody. After incubation at room temperature for 2 hours, samples were washed again with wash buffer 4 times. 100 μL of streptavidin-HRP (1:200 v/v dilution) was added to each well and incubated at room temperature for 20 minutes. After 4 washes with wash buffer, samples were incubated with 100 μL of substrate solution and incubated for 20 minutes in the dark. 50 μL stop solution was added to stop the reaction, after which samples were read at 450 nm and 540 nm.
Bioactivity Assay for IL-12 Releasates
Splenocytes (5 × 105 cells/well) from naive wild-type C57BL/6J mice were incubated with 10fold dilutions (1000 pg/ml to 0.1 pg/ml) of either IL-12 releasate, IL-12 standard, or blank releasate in a 96-well round bottom plate in 200 μL total volume of complete medium (RPMI supplemented with 10% FBS, 1X penicillin-streptomycin, 2 mM L-glutamine, and 50 |jM 2- mercaptoethanol). Cells were incubated for 72 hours at 37°C in a 5% CO2 atmosphere. Culture supernatants were harvested and a mouse IFN-γ ELISA was performed according to the manufacturer’s protocol to determine IL-12-induced IFN-γ production.
In-vivo Studies
6–8 week old female C57BL/6J mice (strain 000664) were purchased from Jackson Laboratory. After at least three days of acclimation, mice were shaved and inoculated with bilateral injections of 100,000–200,000 B16F10 cells per site. B16F10 cells were maintained and propagated in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. After 7–10 days post-inoculation, or when visible tumors reached a size of 25–50 mm2, mice were randomized into groups, ear-punched, and subcutaneously injected with 50 μL of the appropriate treatment. Subcutaneous peritumoral (p.t.) treatment injections were only given to one primary site and include the following: IL-12-loaded complex coacervates (n=11), IL-12 in saline (n=6), and blank complex coacervates (n=11). Tumor dimensions for both the primary and contralateral sites were measured every 2–3 days using a digital caliper. On days 5 and 12 post-treatment, mice were sacrificed to analyze tumor explants via immunofluorescence. Mice were euthanized when tumors reached a size larger than 400 mm2. Other causes for euthanasia included ulceration and bleeding of the tumor, lethargy, and cachexia. Data was compiled from two independent experiments.
Immunofluorescent Sectioning and Imaging
Tumor explants were immediately fixed in 4% (w/v) PFA for 3 hours at 4 °C followed by an overnight incubation step in 30% (w/v) sucrose. Samples were then embedded in OCT (Tissue-Tek), frozen on dry ice, sectioned using a cryomicrotome at 10 μm thickness, and stored at −20°C until staining. Slides were air-dried at room temperature for 20 minutes and incubated in PBS to remove OCT. Samples requiring permeabilization were incubated with 0.2% Triton X-100 in PBS for 10 minutes at room temperature and subsequently washed in PBS. Samples were blocked with 10% goat serum and 0.1% Triton X-100 and incubated at room temperature for 1 hour, after which primary antibody diluted in staining buffer (2% goat serum and 0.1% Triton X-100) was added. After an overnight incubation step at 4°C, samples were washed in staining buffer, and incubated with secondary antibody diluted in staining buffer. Samples were incubated at room temperature in the dark for 1 hour and washed in staining buffer. DAPI counterstaining was performed, washed in PBS and deionized water, and mounted in Cytoseal 60. Immunofluorescence images were captured on an inverted microscope (Eclipse Ti, Nikon). Fluorescent images were quantified using ImageJ (NIH), in which the total fluorescent intensity was divided by the average intensity from 20 individual cells; at least 4 separate fields of view were analyzed during this step.
Statistical Analysis
All statistical analysis was performed using Prism Version 8.1.1 (Graphpad Software, San Diego, CA). Detailed methods are provided for each experiment in figure captions.
Results
Heparin-Binding Proteins are Encapsulated in Soluble Complex Coacervates
The complex coacervate platform used in this study takes advantage of the natural binding affinity between IL-12 and heparin. IL-12-heparin complexes form further polymer- and protein- rich phases upon the addition of PEAD (Figure 1A). When initially clear solutions of heparin and PEAD are combined, a turbid solution forms instantaneously, indicating the formation of coacervates (Figure 1B). The complex coacervates are formulated in mass ratios of 5:1 = PEAD:heparin to yield isoelectrically-charged droplets (Supplementary Figure 1A). To visualize the loading capacity of our heparin-based complex coacervate platform, Alexa Fluor 647-conjugated BSA (BSA-647; blue) was encapsulated as a model protein. After centrifugation to accelerate coacervate sedimentation, BSA-647 is visualized together with the coacervate pellet (Figure 1B), indicating its successful encapsulation. In the absence of centrifugation, coacervate droplets initially have an average hydrodynamic diameter of 204.9 ± 7.7 nm (Supplementary Figure 1B). Over time, such droplets coalesce over time to form larger agglomerates; smaller coacervate droplets (yellow arrows) are observed surrounding larger droplets (yellow asterisks) as assessed by cryo-SEM.
Figure 1. Heparin-based Complex Coacervate Behavior.

A. Schematic detailing the complexation of IL-12 and heparin with PEAD. B. 10 mg/mL heparin and PEAD solutions in 0.9% saline instantaneously form a turbid solution upon mixing. BSA-647 (blue) is localized together with the empty coacervate. C. Cryo-SEM images show both small (yellow arrows) and large (yellow asterisk) complex coacervates (left scale bar: 10 μm; right scale bar: 5 μm).
Complex Coacervates Protect and Enhance Bioactivity of IL-12
The TME presents a challenging milieu for cytokines due to its hypoxic and acidic nature. The effect of IL-12 encapsulation was assessed on its bioactivity by incubating free IL-12 and IL-12 loaded into complex coacervates in the presence of trypsin, a broad-spectrum protease (Figure 2A). At set time points, coacervates were disrupted using a combination of 2X PBS and heparinase II; heparinase II degrades heparin in a time-dependent manner and shows complete degradation after 1 hour (Supplementary Figure 2), allowing the complex coacervate to disassemble. At both 1h and 10h post-incubation at 37°C, a significantly improved protective effect was observed when IL-12 was encapsulated in coacervates. Specifically, 83.9 ± 10.5% and 46.0 ± 5.2% of initial IL-12 remained after 1h of incubation with trypsin. By 10h, only 2.1 ± 0.6% of the IL-12 remained when unprotected vs. 70.1 ± 18.7% when encapsulated. The release behavior of IL-12 from the complex coacervates was then assessed for two different formulations (Figure 2B). The first (black squares) included both BSA and IL-12 during the coacervate complexation step while the second (white squares) did not include BSA. Both formulations showed a near complete encapsulation efficiency of >99.99% as assessed by ELISA. A near 100% cumulative release was observed over 14 days when IL-12 is co-encapsulated with BSA, presumably due to the increased competition for heparin by both BSA30 and IL-12. On the other hand, when only IL-12 is encapsulated, it is released in a slower manner. Given the general quicker degradation rates invivo, we opted to advance studies using the second formulation without BSA in all subsequent experiments. Finally, the bioactivity of the IL-12 released from the complex coacervates was examined using a splenocyte stimulation assay (Figure 2C). A range of IL-12 concentrations were prepared from IL-12 releasate (black squares) or stock IL-12 (white squares) and incubated with freshly-isolated C57BL/6J splenocytes and assessed for their ability to secrete IFN-γ. A dose-responsive effect was observed for both experimental groups, in which lower concentrations of IL-12 yielded less IFN-γ secretion from responder splenocytes. Remarkably, the bioactivity of IL-12 releasate was 65.6% and 762.7% higher than that of stock IL-12 at 10 pg/mL and 1 pg/mL, respectively. This increase in bioactivity indicates that IL-12 is most likely released as a complex with heparin, as heparin-bound IL-12 has been previously shown to increase IL-12 bioactivity28.
Figure 2. Coacervate Protects and Enhances IL-12 Bioactivity.

A. 100 ng of unprotected or encapsulated IL-12 was incubated with 50 ng of trypsin at 37°C. IL-12 levels were quantified using a cytokine-specific ELISA (n = 4, ****p<0.0001). B. IL-12 was encapsulated in the presence or absence of BSA and assessed for its release in 0.9% saline containing 0.1% BSA (n = 3). C. C57BL/6J mouse splenocytes were cultured with known concentrations of IL-12 releasate (day 2 samples) or stock IL-12. Three days later, supernatants were assessed for IFN-γ levels by ELISA (n = 3, ****p<0.0001).
A Single p.t. Injection of IL-12 Coacervate Inhibits Disseminated Tumor Growth
The utility of the IL-12 complex coacervates to serve as an effective therapy was determined in translational mouse models using syngeneic B16F10 melanoma cells. Age-matched C57BL/6J mice were inoculated s.c. bilaterally, with p.t. treatment of a single tumor lesion beginning when tumors reach a size of 25–50 mm2 (approximately 7–10 days post-inoculation). A pilot dose escalation study was carried out using 1, 10, or 30 μg of IL-12 per complex coacervate treatment injection. Given that there was no difference in tumor growth between mice receiving blank coacervates and PBS (Supplementary Figure 3), mice treated with blank coacervates were taken as negative controls. Individual tumor growth curves for the treated and untreated (contralateral) tumor sites indicate that higher amounts of p.t.-delivered IL-12 resulted in superior tumor growth inhibition without evidence of systemic toxicities (Figure 3A), with variation in tumor size among mice also decreasing with increasing IL-12 dose. Mice receiving the lowest dose of 1 μg exhibit lower survival rates, with euthanasia required due to tumor size. Mice treated with blank coacervates (white circles) show typical growth kinetics of B16–10 cells in a mouse. While increasing doses of IL-12 appeared to improve the inhibition of tumor growth (Figure 3B), there was no statistically significant difference in treated tumor sizes across the different IL-12 dose cohorts. Interestingly, a similar trend was observed for tumor growth on the untreated, contralateral flank, suggesting treatment-associated systemic immune-mediated benefits (Supplementary Figure 4). In terms of overall survival, there was no significant difference observed between the IL-12 treatment groups (Figure 3C). It should be noted, however, that a log-rank test for trend indicates a significant trend for increased therapy benefit with increased dose of delivered IL-12 (**p=0.003). Given these results, we opted to use 10 μg of IL-12 per complex coacervate injection.
Figure 3. Dose Escalation Study of IL-12 Coacervates in mice bearing established B16-F10 melanomas.

A. 6–8 week old female C57BL/6J mice were inoculated s.c. with 100,000–200,000 B16F10 cells in each flank and allowed to establish tumors for 7 days. Tumors on the right flank were treated i.t. with IL-12 coacervates to deliver varying doses of IL-12 into the TME. Tumor sizes were monitored every 3–4 days, until time of required euthanasia or death. B. Average size of treated tumors was monitored over time, with no significant difference observed between the various IL-12 treatment cohorts (n = 5 mice/group). C. Kaplan-Meier survival curves of treated mice indicates a significance in trend as assessed by logrank test for trend (n = 5, **p<0.003).
We next assessed the effect of p.t. injections of recombinant IL-12, either encapsulated in our complex coacervate platform or suspended in 0.9% saline (IL/12/saline). Average tumor growth curves at the primary site show a significant inhibition of growth when mice are treated with IL-12 coacervate as opposed to IL-12/saline (Figure 4A). Not surprisingly, treatment with IL-12/saline showed efficacy over blank complex coacervate treatment. Interestingly, this trend in significance was replicated at the contralateral site (Figure 3B), which suggests development of systemic therapeutic immune responses. Individual growth plots of mice further corroborated the significance observed for differences between treatment cohorts (Supplementary Figure 5).
Figure 4. IL-12 Delivery Significantly Inhibits Systemic Tumor Growth.

A. C57BL/6 mice were treated with a single injection of one of the following: 10 μg IL-12 encapsulated in complex coacervates, 10 μg of IL-12 in saline (IL-12/saline), or blank coacervates. Tumor size was monitored over time on treatment. B. Average tumor growth curves were also determined for untreated contralateral site tumors (n = 11 for IL-12 coacervate group, n = 6 for IL-12/saline group, n = 11 for the blank coacervate group, **p<0.01, ***p<0.001 as assessed using the Holm- Sidak method).
Significant Lymphocyte Accumulation Observed in Tumor Microenvironment
To better understand the mechanism of inhibition of tumor growth, we probed the TME for immune cell infiltrates at different time points. The early immune response was assessed by examining NKp46+ cell infiltration into the tumor at a relatively early timepoint of 5 days post-treatment (Figure 5A). Mice receiving IL-12 coacervate treatment displayed a significant increase in NK cell infiltration (yellow) compared to those receiving IL-12/saline or blank coacervate. Quantification of the immunofluorescence images further corroborated this trend (Figure 5C). Given the role of IL-12 in enhancing T cell survival and proliferation31, we then assessed the TME for presence of CD8α+ T cells, which are capable of mediating antigen- specific killing of the tumor cells. At 12 days post-treatment, we observed a massive CD8α+ T cell infiltrate into tumors selectively in mice receiving treatment consisting of IL-12 coacervates vs. mice receiving IL-12/saline or blank coacervates (Figure 5B top row and Figure 5D). Given that IL-12 has also been shown to mediate the reversal of Treg immunosuppression by decreasing Treg frequencies21, we probed the TME for CD4+ FoxP3+ Tregs (Figure 5B bottom row and Supplementary Figure 6). Remarkably, we did not observe Treg cells in the TME of mice receiving IL-12 coacervate-based treatment. On the other hand, large numbers of Treg were detected in the TME of mice receiving treatment with blank coacervates. Collectively, the intratumoral increase in CD8α+ T cells and decrease in Tregs in mice receiving IL-12 coacervates yielded a profound increase in the CD8:Treg ratio within the TME in association with inhibited tumor growth.
Figure 5. Treatment with IL-12 Coacervates Improves Innate and Adaptive Immune Cell Infiltration in the TME.
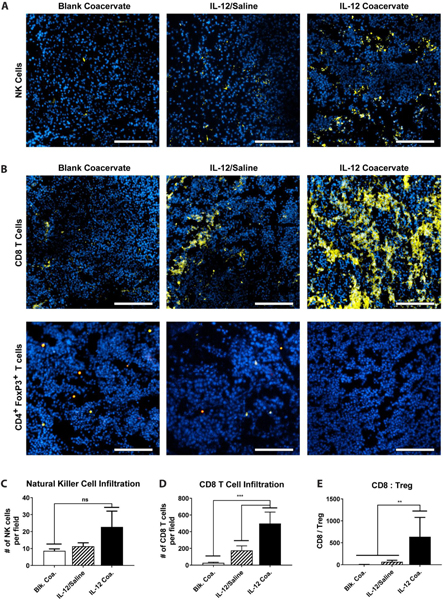
A. Mice receiving various treatments were euthanized 5 days post-treatment, with resected tumors assessed for the presence of NKp46+ cells in the tumor (blue: nucleus, yellow: NKp46, scale bar: 200 μm). B. TME presence of CD8α+ cells (top) and CD4+FoxP3+ cells (bottom) were assessed 12 days post-treatment (blue: nucleus, yellow: CD8α, red: CD4, teal: FoxP3, scale bar: 200 μm). The fluorescence signal was quantified from at least five different fields to yield average numbers of C. NKp46+ cells and D. CD8α+ cells (*p<0.05, ***p<0.001, ****p<0.0001) per unit tissue area. E. CD8α+:CD4+FoxP3+ was quantified for the different treatment groups (**p<0.01).
IL-12 Coacervate Treatment Increases Tumor PD-L1 Expression
Finally, we examined the TME for treatment-associated changes in expression of the immune checkpoint molecule PD-L1, a ligand for PD-1 that is normally increased in the presence of IFN-γ (produced by Type-1 infiltrating NK cells and T cells). We observed that tumors in mice receiving the IL-12 complex coacervate but not control treatment demonstrated a major, significant increase in PD-L1 expression post-therapy (Figure 6A), thereby providing indirect evidence that IL-12 complex coacervates recruit and activate NK and/or CD8 T cells to locally secrete IFN-γ in the therapeutic TME. Quantification of PD-L1 expression indicates a nearly 30-fold increase in PD-L1 expression in mice treated with IL-12 coacervate over those treated with IL-12/saline or blank coacervates (Figure 6B). Given the immune-suppressive role of lymphocyte PD-1 binding to PD-L1 on tumor cells, the large increase in PD-L1 expression in mice receiving the IL-12 coacervate preparation may limit the therapeutic activity mediated by the large increase in intratumoral NKp46+ and CD8α+ cells. Therefore, despite the significant differences observed in tumor size when mice were treated with IL-12 coacervates, tumor regression was not noted and significant differences in overall survival were minimal for IL-12 based therapies (Figure 6C). Hence, although there was a significant survival benefit for tumor-bearing mice treated with IL-12 coacervates compared to those receiving blank coacervates (p=0.0037, log-rank test) and the difference between mice receiving IL-12/saline vs. blank coacervates trended towards significance (p=0.1358, log-rank test), there was no significant difference in survival for mice receiving IL-12 coacervate vs. IL-12/saline (p=0.2392, log-rank test).
Figure 6. Treatment with IL-12 Coacervates Increases PD-L1 Expression in the TME.

A. Intratumoral expression of PD-L1 was analyzed via immunofluorescence 12 days post-treatment (blue: nucleus, yellow: PD-L1, scale bar: 200 μm). B. PD-L1+ fluorescence signal is quantified to represent the average number of PD-L1+ cells ± SD (****p<0.0001). C. Kaplan-Meier survival curves of mice indicates a non-significant difference in mice receiving IL-12 coacervate vs. IL-12/saline. However, a significant difference was observed between mice receiving IL-12 coacervates vs. blank coacervates (n = 11 for IL-12 coacervate group, n = 6 for IL-12/saline group, n = 11 for blank coacervate group). D. Schematic illustration supporting the need to coordinately inhibit PD-/PD-L1 checkpoint interactions to optimize the therapeutic benefits associated with IL-12 coacervate-based treatment of melanoma.
Discussion
Skin cancer constitutes nearly 80% of all cancer diagnoses; approximately 5.5 million out of 7.1 million cancers diagnosed in 2017 were found to be of the skin3. While the bulk of these skin cancers are relatively benign forms of basal cell or squamous cell carcinomas that are generally easily cured, melanoma in stark contrast is an aggressive cancer with high-morbidity. Melanomas are currently most effectively treated with immunotherapies. IL-2 represents one of the earliest immunotherapeutic agents used to treat melanoma and other forms of cancer. While effective in a small minority (< 20%) of melanoma patients, the significant toxicity of high-dose regimens has tempered enthusiasm for IL-2’s use since its FDA approval in 199832, 33. Other pro- inflammatory cytokines including IL-12 have demonstrated promising results in pre-clinical studies as a cancer therapeutic agent. However, early clinical trials using systemically-delivered recombinant IL-12 were halted due to severe hematologic and organ toxicities in most of the enrolled patients, including two deaths22. Subsequent studies determined that a priming/desensitizing dose of IL-12 was required before the initiation of daily IL-12 injections to mitigate a severe burst of IFN-γ production, akin to that observed under sepsis-like conditions23. Unfortunately, while this IL-12 condition regimen attenuates systemic toxicity, it also blunts IFN-γ-dependent therapeutic responses, thereby limiting patient clinical benefits. As a consequence, IL-12 based interventional approaches have been skewed to more tightly-regulated applications such as vaccines and gene therapies. While significant advances have been made in these areas, subpar transfection efficiencies and the potential for undesired off-target genetic changes have further limited such regimens. Furthermore, the advent of checkpoint inhibitors has led to an explosion of related studies. Unfortunately, checkpoint inhibitors are not without their issues as well, some of which include systemic toxicities and the limited forms of disease that are responsive to such interventions. Importantly, the effective use of checkpoint inhibitors requires priming/conditioning of the TME for infiltration by pro-inflammatory innate and/or adaptive immune cells, such as NK cells and T cells11, 12, 13, 34.
From this perspective, tumor pre-conditioning with immunomodulatory compounds that increase inflammatory infiltrates within the TME is crucial to provide improved therapeutic responsiveness to combination immunotherapy interventions (i.e. conditioning + checkpoint blockade) (Figure 6D). Our study examines a previously unexplored avenue of using complex coacervation to more effectively and safely deliver potent biologic modifiers, including (systemically-toxic) recombinant cytokines such as IL-12. Notable roles for IL-12 include, but are not limited to the following: 1) activation of NK cell activity, in which NK cells activated by IL-12 display an enhanced level of cytolytic activity against tumor/target cells35, 36, 2) enhanced activation of cytotoxic CD8+ T cells37, 3) dendritic cell (DC)-mediated increases in T helper 1 (Th1) responses38, 4) upregulation of MHC I antigen presentation on tumor cells39, and 5) anti-angiogenic activity via upregulated local production of interferon gamma-induced protein 10 (IP- 10; aka CXCL10)40.
Our heparin-based complex coacervation platform represents a departure from standard forms of controlled delivery, in which the following advantages are proposed: 1) the low interfacial energy between the coacervate and non-coacervate phase25 results in an emulsion (Figures 1B and 1C) unlikely to be taken up by cells, 2) the utilization of anionic heparin acts to stabilize and enhance the bioactivity of therapeutic proteins41 (Figure 2C), 3) the affinity between heparin and proteins promotes highly-efficient encapsulation (> 99.99% for IL-12 in our system), and 4) the degradation of cationic poly(ethylene argininylaspartate diglyceride) does not generate a pro-tumor acidic environment. Surprisingly, the use of complex coacervation has not yet been examined for its utility in delivering therapeutic proteins/cytokines in anti-cancer applications. Given the dynamic nature of complex coacervation, it is important to consider that other proteins may compete with rIL-12 for heparin binding sites, allowing for pro-tumor proteins (i.e. FGF-2, VEGF) to replace IL-12 molecules within the coacervate formulation. Our data indicates that encapsulation of IL-12 in the absence of BSA yields slower release of IL-12 when compared with coacervate preparations generated with BSA (Figure 2B). Given that both formulations were released in sink conditions of saline containing excess BSA, the lack of expedited release in the BSA-free formulation suggests that IL-12 is not easily replaced. It should be noted, however, that other proteins with higher binding affinities do exist and their impact on the integrity of our platform have not yet been examined. Furthermore, while we utilized trypsin as a broad- spectrum protease to demonstrate coacervate-mediated protection of IL-12, trypsin does not fully reflect the complexity of proteases in the TME.
Interestingly, NKp46+ cell infiltrate in the IL-12/saline treatment group was unexpectedly low (Figure 5A). In this regard, it remains possible that 5 days post-treatment may not be optimal (i.e. too late) for examining NK cell infiltration into the TME. Of course, it is also possible that treatment with IL-12 complex coacervates led to the sustained delivery of IL-12 and consequently extended local NK cell activation and proliferation. Furthermore, NKp46 is known to be expressed on a small subset of NKT cells42; hence it is formally possible that some of the NKp46+ cells in the TME of mice treated with IL-12 coacervate may be NKT cells. Similarly, CD8α may also be expressed on DC subsets that play a role in antigen cross-presentation43, 44, which could activate CD8α+ T cells within the TME and/or tumor-draining lymph nodes. These phenotypic subtleties will be further evaluated in extended future studies. Finally, given the important role of IL-12 in driving Type-1 CD8+ T cell proliferation and survival, the presence of robust populations of CD8α+ T cells in tumors of mice treated with IL-12 coacervate (Figure 5B) strongly supports the capacity of our platform to enhance/prolong IL-12 bioactivity in the TME. While our findings confirm previous reports that IL-12 (delivered in a coacervate formulation) decreases Treg frequencies and Foxp3 levels in Tregs21, which would circumvent one level of regulatory (pro-tumor) immunity, we also observed that coacervate-delivered IL-12 leads to large increases in PD-L1 expression in the TME (Figure 6A). We therefore hypothesize that delivery of both IL-12 and blocking anti-PD1/anti-PD-L1 antibodies or combined treatment with coacervate IL-12 + systemic delivery of checkpoint blockade antibodies will enhance therapeutic benefits leading to tumor regression and extended overall survival benefits in treated mice.
Conclusions
This study demonstrates heparin-based complex coacervates are an effective means to deliver and extend the therapeutic action of IL-12 in pre-clinical murine melanoma models. This study is the first to examine the utility of recombinant protein delivery via complex coacervates in anti-cancer applications. The coacervate protects IL-12 within the proteolytic/inactivating TME, leading to enhanced local IL-12 bioactivity in the absence of systemic toxicity. Mice treated with IL-12 coacervates exhibited significantly reduced rates of tumor (both directly treated and distal untreated) growth compared to therapy control cohorts. Notably, treatment benefits were associated with increased levels of NKp46+ and CD8α+ cells, reduced levels of CD4+FoxP3+Treg cells, and enhanced expression of PD-L1 in the TME. Future studies combining coacervate delivery of recombinant proteins such as IL-12 along with immune checkpoint inhibitors are clearly warranted and may define novel and effective treatment options for translation into the clinic.
Supplementary Material
Highlights.
First to demonstrate complex coacervate-mediated protein delivery for cancer
Complex coacervates protect/enhance bioactivity of immunostimulatory protein IL-12
Single injection of IL-12 coacervate systemically inhibits tumor growth in-vivo
Acknowledgements
We would like to thank Scientific Protein Laboratories, LLC for their generous donation of clinical-grade heparin. This work was supported by Cornell University startup funds to YW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016. World Health Organization; (2018). [Google Scholar]
- 2.Cao B, Bray F, Beltrán-Sánchez H, Ginsburg O, Soneji S, Soerjomataram I. Benchmarking Life Expectancy and Cancer Mortality: Global Comparison with Cardiovascular Disease 1981–2010. BMJ 357, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Facts & Figures 2017. In: American Cancer Society (2017). [Google Scholar]
- 4.Domingues B, Lopes JM, Soares P, Pópulo H. Melanoma Treatment in Review. Immunotargets Ther 7, 35–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soengas MS, Lowe SW. Apoptosis and Melanoma Chemoresistance. Oncogene 22, 3138–3151 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Brunner MC, Chambers CA, Chan FK-M, Hanke J, Winoto A, Allison JP. Ctla-4-Mediated Inhibition of Early Events of T Cell Proliferation. The Journal of Immunology 162, 5813–5820 (1999). [PubMed] [Google Scholar]
- 7.Freeman GJ, et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. The Journal of Experimental Medicine 192, 1027 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-Like Autoimmune Diseases by Disruption of the Pd-1 Gene Encoding an Itim Motif-Carrying Immunoreceptor. Immunity 11, 141–151 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Seidel JA, Otsuka A, Kabashima K . Anti-Pd-1 and Anti-Ctla-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol 8, 86–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid O, et al. Safety and Tumor Responses with Lambrolizumab (Anti-Pd-1) in Melanoma. New England Journal of Medicine 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haanen J. Converting Cold into Hot Tumors by Combining Immunotherapies. Cell 170, 1055–1056 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Tang H, Qiao J, Fu YX. Immunotherapy and Tumor Microenvironment. Cancer letters 370, 85–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu G, et al. Low-Dose Decitabine Enhances the Effect of Pd-1 Blockade in Colorectal Cancer with Microsatellite Stability by Re-Modulating the Tumor Microenvironment. Cellular & Molecular Immunology 16, 401–409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acquavella N, et al. Toxicity and Activity of a Twice Daily High-Dose Bolus Interleukin 2 Regimen in Patients with Metastatic Melanoma and Metastatic Renal Cell Cancer. Journal of Immunotherapy 31, 569–576 (2008). [DOI] [PubMed] [Google Scholar]
- 15.D’Souza WN, Lefrancois L. Il-2 Is Not Required for the Initiation ofCd8 T Cell Cycling but Sustains Expansion. Journal of Immunology 171, 5727–5735 (2003). [DOI] [PubMed] [Google Scholar]
- 16.D’Souza WN, Schluns KS, Masopust D, Lefrancois L. Essential Role for Il-2 in the Regulation of Antiviral Extralymphoid Cd8 T Cell Responses. Journal of Immunology 168, 5566–5572 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Bajetta E, et al. Pilot Study of Subcutaneous Recombinant Human Interleukin 12 in Metastatic Melanoma. Clinical Cancer Research 4, 75–85 (1998). [PubMed] [Google Scholar]
- 18.Donohue JH, Rosenberg SA. The Fate of Interleukin-2 after in Vivo Administration. Journal of Immunology 130, 2203–2208 (1983). [PubMed] [Google Scholar]
- 19.Ahmadzadeh M, Rosenberg SA. Il-2 Administration Increases Cd4(+)Cd25(Hi) Foxp3(+) Regulatory T Cells in Cancer Patients. Blood 107, 2409–2414 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Cruz LM, Klein L. Development and Function of Agonist-Induced Cd25+Foxp3+ Regulatory T Cells in the Absence of Interleukin 2 Signaling. Nature immunology 6, 1152–1159 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Zhao J, Zhao J, Perlman S. Differential Effects of Il-12 on Tregs and Non-Treg T Cells: Roles of f-r, Il-2 and Il-2r. PLoS ONE 7, e46241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: Still a Promising Candidate for Tumor Immunotherapy? Cancer Immunology, Immunotherapy 63, 419–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonard JP, et al. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12-Associated Toxicity and Interferon-r Production. Blood 90, 2541–2548 (1997). [PubMed] [Google Scholar]
- 24.Kim S, et al. Complexation and Coacervation of Like-Charged Polyelectrolytes Inspired by Mussels. Proceedings of the National Academy of Sciences 113, E847–E853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin J, et al. Interfacial Tension of Polyelectrolyte Complex Coacervate Phases. ACS Macro Letters 3, 565–568 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Chu H, Johnson NR, Mason NS, Wang Y. A [Polycation:Heparin] Complex Releases Growth Factors with Enhanced Bioactivity. Journal of Controlled Release 150, 157–163 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Hasan M, Najjam S, Gordon MY, Gibbs RV, Rider CC. Il-12 Is a Heparin-Binding Cytokine. The Journal of Immunology 162, 1064–1070 (1999). [PubMed] [Google Scholar]
- 28.Jayanthi S, et al. Modulation of Interleukin-12 Activity in the Presence of Heparin. Scientific reports 7, 5360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zern BJ, Chu H, Osunkoya AO, Gao J, Wang Y. A Biocompatible Arginine-Based Polycation. Advanced Functional Materials 21, 434–440 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hattori T, Kimura K, Seyrek E, Dubin PL. Binding of Bovine Serum Albumin to Heparin Determined by Turbidimetric Titration and Frontal Analysis Continuous Capillary Electrophoresis. Analytical biochemistry 295, 158–167 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Starbeck-Miller GR, Xue H-H, Harty JT. Il-12 and Type I Interferon Prolong the Division of Activated Cd8 T Cells by Maintaining High-Affinity Il-2 Signaling in Vivo. The Journal of Experimental Medicine 211, 105–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkins MB, et al. High-Dose Recombinant Interleukin 2 Therapy for Patients with Metastatic Melanoma: Analysis of 270 Patients Treated between 1985 and 1993. Journal of Clinical Oncology 17, 2105–2116 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Bright R, Coventry BJ, Eardley-Harris N, Briggs N. Clinical Response Rates from Interleukin-2 Therapy for Metastatic Melanoma over 30 Years’ Experience: A Meta-Analysis of 3312 Patients. Journal of Immunotherapy 40, 21–30 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Mahoney KM, Rennert PD, Freeman GJ. Combination Cancer Immunotherapy and New Immunomodulatory Targets. Nat Reviews Drug Discovery 14, 561–584 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Chehimi J, et al. Natural Killer (Nk) Cell Stimulatory Factor Increases the Cytotoxic Activity of Nk Cells from Both Healthy Donors and Human Immunodeficiency Virus-Infected Patients. The Journal of Experimental Medicine 175, 789–796 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Zhang J, Niu J, Zhou Z, Zhang J, Tian Z. Interleukin-12 Improves Cytotoxicity of Natural Killer Cells Via Upregulated Expression of Nkg2d. Human immunology 69, 490–500 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM. I1–12 Produced by Dendritic Cells Augments Cd8+ T Cell Activation through the Production of the Chemokines Ccll and Ccl17. Journal of Immunology 181, 8576–8584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heufer C, et al. Interleukin-12 Is Produced by Dendritic Cells and Mediates T Helper 1 Development as Well as Interferon-Gamma Production by T Helper 1 Cells. European journal of immunology 26, 659–668 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Suzuki S, et al. Exogenous Recombinant Human Il-12 Augments Mhc Class I Antigen Expression on Human Cancer Cells in Vitro. Tohoku Journal of Experimental Medicine 185, 223–226 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Angiolillo AL, Sgadari C, Tosato G. A Role for the Interferon-Inducible Protein 10 in Inhibition of Angiogenesis by Interleukin-12. Annals of the New York Academy of Sciences 795, 158–167 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Capila I, Linhardt RJ. Heparin-Protein Interactions. Angewandte Chemie International Edition 41, 390–412 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Yu J, et al. Nkp46 Identifies an Nkt Cell Subset Susceptible to Leukemic Transformation in Mouse and Human. J Clin Invest 121, 1456–1470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.den Haan JM, Lehar SM, Bevan MJ. Cd8(+) but Not Cd8(−) Dendritic Cells Cross-Prime Cytotoxic T Cells in Vivo. The Journal of Experimental Medicine 192, 1685–1696 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preynat-Seauve O, Schuler P, Contassot E, Beermann F, Huard B, French LE. Tumor- Infiltrating Dendritic Cells Are Potent Antigen-Presenting Cells Able to Activate T Cells and Mediate Tumor Rejection. The Journal of Immunology 176, 61–67 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.