

Abstract
2,3-Benzodiazepine compounds are an important family of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) antagonists that act in a noncompetitive manner. Due to the critical role of AMPARs in the synapse and various neurological diseases, significant scientific interest in elucidating the molecular basis of the function of the receptors has spiked. The analogues were synthesized to assess the functional consequence of removing the amine group of the phenyl ring, the potency and efficacy of inhibition by substituting a halogen group at the meta vs ortho position of the phenyl ring, and layout the prediction of potential drug candidates for AMPAR hyperactivation. Using the whole-cell patch-clamp technique, we assessed the effect of the derivative on the amplitude of various AMPA-type glutamate receptors and calculated the desensitization and deactivation rates before and after treatment of HEK293 cells. We noticed that the amino group is not necessary for inhibition as long as an electron-withdrawing group is placed on the meta position of the phenyl ring of BDZ. Furthermore, compound 4a significantly inhibited and affected the desensitization rate of the tested AMPARs but showed no effect on the deactivation rate. The current study paves the way to a better understanding of AMPARs and provides possible drug candidates of 2,3-BDZ different from the conventional derivatives.
Introduction
2,3-Benzodiazepine (2,3-BDZ) derivatives, also known as GYKI, are a group of synthetic drug candidates that noncompetitively inhibit α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs). In various acute neurological disorders such as cerebral ischemia and epilepsy as well as in chronic neurodegenerative pathologies such as Parkinson’s disease, Alzheimer’s disease (AD), Huntington’s chorea, and amyotrophic lateral sclerosis (ALS), excessive stimulation of AMPARs has been implicated.1−3 Consequently, chemotherapeutic applications provided strong motivation for the synthesis of 2,3-BDZ analogues due to their anticonvulsant and neuroprotective properties. Moreover, they have demonstrated higher potency and selectivity toward AMPA receptors than other compounds in animal and in vitro studies.4 The prototypic compound of the 2,3-BDZ family, 7,8-methylenedioxy-5H-2,3-benzodiazepine (GYKI 52466; Figure 1) was first introduced in the 1980s and has been used as a template and standard in the synthesis and activity evaluations of new GYKI compounds.1 While the 2,3-BDZs’ structures (Figure 1) have different pharmacological activity besides their effect on the central nervous system, they also possess anti-inflammatory,5 antimicrobial,6 vasopressin antagonist,7 endothelia antagonist,8 cholecystokinin antagonist,9 antithrombotic,10 anti-HIV,11 and antiproliferative activities.12 Hence, there is a keen interest in 2,3-BDZ for applications in numerous fields besides neurology.
Figure 1.

2,3-BDZ prototype and GYKI 52466 structure.
The crystal structure of AMPA-subtype ionotropic glutamate receptors shows that antiepileptic drugs bind to an allosteric site, located in the ion channel’s extracellular part. Noncompetitive inhibitors prevent channel openings by triggering an interaction network that results in a conformational change on the channel gate.13,14 Acting in a noncompetitive manner, 2,3-BDZ depresses the maximum of the sigmoid concentration–response curve. In other words, AMPA receptors cannot be maximally activated regardless of agonist concentration, hence preventing glutamate-induced neuronal death. On the contrary, at high agonist concentrations, the protective effect of competitive AMPA antagonists was absent.3,14 Moreover, a competitive AMPAR antagonist, 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo (F) quinoxaline (NBQX), and its analogues have been shown to increase gamma-aminobutyric acid (GABA) transmission in the cerebellum by non-AMPA-dependent mechanisms, as well as depolarize hippocampally and act at the KA (kainate) receptors, suggesting a loss of selectivity.4 These findings pivoted research toward noncompetitive antagonists for AMPARs, such as 2,3-BDZ derivatives. Previous work has identified three noncompetitive sites on the GluA2Qflip from different 2,3-BDZ analogues: (i) the “M” site, i.e., the methyl group in position 4 of the heptatonic ring is substituted with the methylenedioxy moiety in positions 7 and 8 of the aromatic ring, and numerous structure–activity relationship (SAR) studies on this show a chiral stereoselectivity of the R configuration for the methyl group.1,15 Moreover, it has been demonstrated that upon N-3 acylation the biological activity of the compound increases, and like the “E” site, a greater preferential in the closed channel state is observed. (ii) The E site, where the methylenedioxy is substituted with an ethylenedioxy group at the 7 and 8 positions of the aromatic ring and, unlike the M site, is not chiral or as potent. Finally, (iii) the “O” site, where the C-4 methyl group is replaced by a carbonyl moiety, prefers the open-channel state, and its N-3 acylation decreases the potency as shown by the Niu et al. group.2,16
The fundamental principle behind structure–activity relationships (SARs) is that molecular activity is a function of structure; as a result, molecules of similar structures have similar functions.4,17 By constructing a set of similar chemical structures, via a single molecule substitution, a mechanistic characterization can be deduced from the mode of action caused by these refinements.4 Providing more information from SAR studies enables a better understanding of and predictability for designing efficacious regulatory agents, such as inhibitors and can optimize their pharmacological profile through a higher potency and selectivity toward a specific protein or receptor.13 For this reason, we investigate the functional consequences of adding an electron-withdrawing group (i.e., chlorine atom) at the C-3 position vs C-2 position of the 2,3-benzodiazepine phenyl ring.
Previously reported SAR studies, specifically on the M site, demonstrated the importance of the 4-aminophenyl group for the antiepileptic effect of this class of compounds.18 Moreover, a lack of the amino group in the para position or its acetylation can tremendously decrease potency. However, the biological activity was increased upon a methyl addition at the ortho to the amino group position, which shared similar results to the addition of chlorine instead of a methyl group.1,19 These results suggested that the aminophenyl ring is a potential function moiety to be modified to improve potency. However, how the elimination of the amino group of the phenyl ring affects the O site potency has never been determined. Unlike the M or E site, the potency of the O site is reduced by the addition of alkyl or aryl-alkyl group on N-3 as it does not account for the steric hindrance in its side pocket. Thus, we hypothesize that by removing the amino group of the phenyl ring we can further enhance its potency by accommodating its small pocket in comparison to the E or M site.16
The current study synthesized two 2,3-BDZ analogues, (i) 5-(3-chlorophenyl)-7,9-dihydro-8H[1,3]dioxolo[4,5:4,5]benzo[1,2-d][1,2]diazepin-8-one, (ii) 5-(2-chlorophenyl)-7,9-dihydro-8H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-d][1,2]diazepin-8-one, as shown in Figure 1, on the assumption that (a) both compounds bind to the O site, (b) inhibition remains despite the amino group removal from the para position of the phenyl group, and (c) the chemical composition does not change when binding to different AMPAR subunits. The mechanisms of action of compounds were characterized to answer the following questions: what is the functional consequence of adding an electron-withdrawing group (i.e., chlorine atom) at C-3 position vs C-2 position of the aminophenyl ring? Which is better in terms of potency and efficacy? Would a methyl group have the same effect as a halogen? What is the significant role of the amino group in the phenyl ring? Consequently, we can provide a quantitative understanding of the functional consequences of these structural changes, based on the original structure of GYKI 52466.
Results and Discussion
Identification of Receptor–Antagonist Interactions Using Molecular Modeling
Amino acid sequences and three-dimensional (3D) orientations of the allosteric binding sites are conserved between human and rat AMPA-subtype ionotropic glutamate receptors of both GluA1 and GluA2. The crystal structure of GluA2 (PDB code 5L1G(26)) was found with a 2,3-benzodiazepine containing a molecule, for the given reason: the docking studies were conducted on the GluA2 crystal structure to analyze the interaction network within the active site. Our molecular modeling studies showed that compounds 4a and 4b share a similar interaction network to the crystal ligand, a brominated analogue of a GYKI series inhibitor, GYKI-Br, which has been previously reported to interact at the O site.
4-Amino group of 3-bromophen-1-yl part of GYKI-Br makes a hydrogen bond with SER615, phenyl group of the same part makes van der Waals interactions with LEU620 and ASN791, and H atoms of the same aromatic ring make a weak hydrogen bond with the carbonyl group of SER516. The carboxamide group forms a hydrogen bond with ASN791. Finally, the dioxolobenzodiazepine ring forms π–π interaction with PHE623 amino acid. The predicted interactions of 4a showed higher similarity to the binding mode of GYKI-Br present in the crystal structure. This is related to the steric suitability of the binding pocket for the halogen atoms at the meta position of the phenyl ring.
Similarly, the dioxolobenzodiazepin ring of 4a forms a hydrogen bond with ASN791 and π–π interaction with PHE623, the 3-chlorobenzene ring of the same molecule forms van der Waals interactions with PRO520, LEU620, and ASN791, and H atoms of the same ring make a hydrogen bond with the carbonyl group of SER516. The predicted binding orientation of 4b shows a hydrogen bond with the 2,3-benzodiazepine core of the molecule and ASN791 amino acid. Additionally, a 2-chlorophenyl derivative makes van der Waals interactions with ASP519, LEU620, LEU624, LEU787, and ASN791 and the H atoms of the 2-chlorophenyl group make a weak hydrogen bond with SER516. The main reason for decreasing potency is thought to be related to the missing π–π interaction with PHE623 amino acid (Figure 2).
Figure 2.

Binding mode analysis of GYKI-Br obtained from crystal structures and docking results of AMPA-subtype ionotropic receptor antagonists 4a and 4b against GluA2 visualized with the main interacting residues of the allosteric binding site and summarized with a legend of protein–ligand interactions. There is a commonly shared interaction pattern with GYKI-Br, 4a, and 4b against the GluA2 receptor, including hydrogen bonds, π–π interactions, and hydrophobic interactions. The π–π interaction is lost between the PHE623 residue and 4b, which might be the reason for losing the activity against both GluA1 and GluA2. Additionally, keeping the hydrogen bond with SER615 might support the activity for both 4a and 4b.
Level of Inhibition from 4a vs 4b 2,3-BDZ Derivatives on Various AMPA-Type Receptors
Confirming the interaction of these compounds at the O site, we needed to analyze their activity for SAR comparisons and predictions. Thus, it was essential to quantify the inhibitory activity of these compounds and evaluate their possible drug potential for glutamate toxicity. Using whole-cell recordings of AMPAR-transfected HEK293 cells, we compared the current induced from glutamate alone (A) vs glutamate plus antagonist amplitude (AI) and ran a one-way analysis of variance (ANOVA) test to identify the significant inhibition resulted by these two compounds, as shown in Figure 3. Previously reported, O site docking structures favor an open-state channel; to ensure such a state, we used 10 mM ligand concentration, at which 95% corresponds to the open-channel form.
Figure 3.

Inhibition of 2,3-BDZ derivatives on various AMPA-Type receptors. The effect of 14 μM 4a and 4b 2,3-BDZ derivatives on the amplitude generated by different AMPAR receptors. (A) Whole-cell recording of amplitude after applying the cell with 10 mM glutamate alone (black), glutamate plus 4a (white), and glutamate plus 4b (gray), obtained from HEK239-expressing AMPAR cells. (B) Current traces from glutamate, glutamate plus 4a, and glutamate plus 4b pulses after a 20 ms washout treatment between every response. (C) Inhibition assays of different derivatives on GluA1, GluA2, GluA1/2, and GluA2/3. The whole-cell current recording was conducted at −60 mV, pH 7.4, and 22 °C. Graphs summarize weighted time constants for activation. Data shown are mean ± SEM; n = 6 (number of patch cells in the whole-cell configuration). Significance (one-way ANOVA): *p < 0.05; ** p < 0.01; *** p < 0.001; ns, not significant.
The above-described data elaborates on the difference in activity between the derivatives in terms of current induction. Before any antagonist treatment, the cells were supplied with 500 ms intervals of 10 mM glutamate to average the obtained amplitudes of GluA1, GluA2, GluA1/2, and GluA2/3 that were recorded at 967 ± 32, 1290 ± 62, 480 ± 31, and 395 ± 39 pA, respectively. However, upon treating the cells with the meta position 2,3-BDZ derivatives (4a), the recordings drastically drop to 144 ± 21, 208 ± 28, 87 ± 15, and 67 ± 24 pA, following the order of glutamate-induced recordings. The ratio, representing the significant inhibition A/AI of the derivative on GluA1, GluA2, GluA1/2, and GluA2/3, is the following: 6.7 ± 0.1, 6.2 ± 0.1, 5.5 ± 0.2, and 5.9 ± 0.3, respectively. Surprisingly, similar amplitudes were conducted between the glutamate-induced and ortho-induced treatment of the cells. Thus, the glutamate recordings were 715 ± 27, 847 ± 35, 481 ± 22, and 348 ± 26 pA, and comparatively, the current after the derivative treatment remained at 715 ± 27, 847 ± 35, 481 ± 22, and 348 ± 26 pA for GluA1, GluA2, GluA1/2, and GluA2/3, respectively.
Concentration-Dependent Inhibition of the Derivatives on AMPA-Type Receptors
The levels of inhibition of 4a in comparison to 4b intrigued our interest to determine the potency of the drug in a concentration-dependent manner. Recording inhibition of different antagonist concentrations showcased the maximized potency as well as the plateau (14 μM), as seen in Figure 4. As previously demonstrated, 4b had no effect on inhibition, which is also evident here as the increase of concentration of the compound is indifferent to AMPAR activity. On the contrary, 4a drastically inhibits AMPARs even at a low concentration and maintains to inhibit as the concentration increases until it reached a plateau (14–18 μM). As a result, the antagonist concentration was fixed at 14 μM due to maximizing the level of inhibition concentration for all AMPARs. The concentration–inhibition curves were constructed by applying glutamate alone (A) at a concentration of 10 mM, followed by glutamate plus 2,3-BDZ derivatives (AI) at different concentrations (μM) for every recording point. Afterward, the cells were normalized back by applying glutamate alone. Each point used the 500 ms protocol followed by the 10 ms wash solution, and the resulting currents were normalized to the steady-state current obtained with agonist alone. Hence, the first point was recorded without any antagonist concentration, whereby the concentration increment for the recordings was 2 μM until a plateau was reached, as shown in Figure 4. Importantly, the cell was subjected to a new concentration after several washout intervals to ensure cell recovery. Finally, we determined the IC50 value on GluA1 for both compounds to ensure that the effectiveness of 4a is significant. From the dose–response curve of 4a, the IC50 on GluA1 was determined at 2.203 μM, as shown in Figure S3. As for 4b, the IC50 on GluA1 was determined at 8.476 μM (Figure S4), which is extremely higher than 4a and presents no potency potential. GYKI analogues are highly selective toward AMPARs. Here, we report a higher effect and potency toward homomeric forms of AMPARs than heteromeric types; nonetheless, the potency of 4a remains significant on all tested AMPARs.
Figure 4.
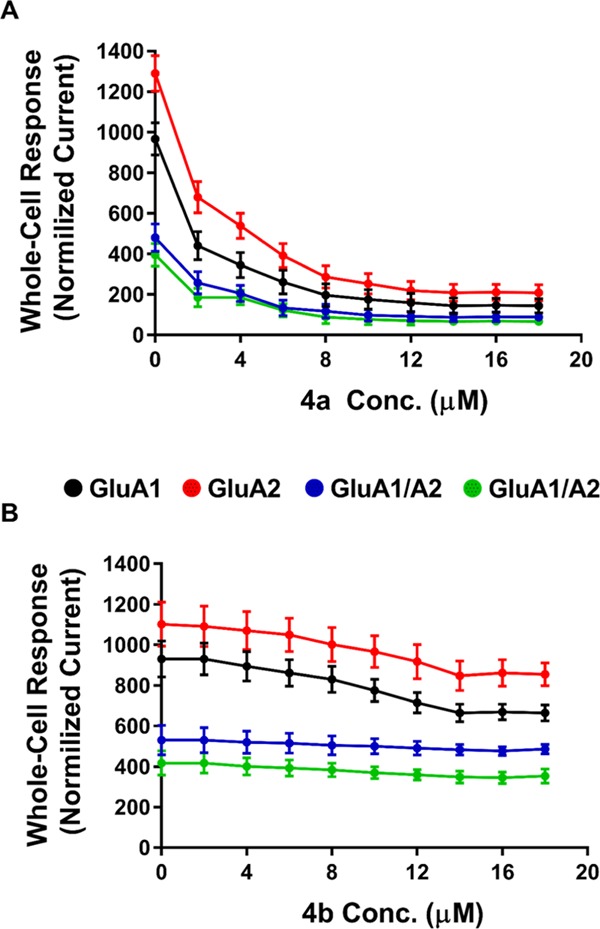
Concentration-dependent inhibition of the derivatives on AMPA-type receptors. To measure the inhibition of 4a (A) and 4b (B) on AMPARs, the cell was supplied with Glu (alone), then Glu plus 2,3-BDZ derivatives in μM concentration, and finally Glu alone using the 500 ms to ensure the healthiness of the cell. Between every concentration point, the cell was subjected to a wash solution for 10 ms to recover fully. After several washout intervals, the rest of the recording points were obtained using the same procedure with an increment of 2 μM between each concentration point. Data points (n = 6 cells) were normalized to the response of different 4a and 4b concentrations.
Effect of 4a and 4b 2,3-BDZ Derivatives on AMPAR Desensitization and Deactivation
The importance of the biophysical gating properties of AMPARs is needed to be assessed to confirm the functional consequence of substituting the halogen group of the phenyl ring at different positions. AMPAR-current deactivation and desensitization were fitted with two exponentials, and the weighted tau (τw) was calculated as τw = (τf × af) + (τs × as), where af and as are the relative amplitudes of the fast (τf) and slow (τs) exponential components. While 4b did not affect desensitization or deactivation rates, similar to its inhibiting activity, 4a seemed only to affect desensitization, as shown in Figure 5. The desensitization values before 4a application for GluA1, GluA2, GuA1/2, and GluA2/3 were 2.6 ± 0.1, 2.8 ± 0.1, 4.9 ± 0.1, and 2.5 ± 0.1 ms, respectively. After treatment with 4a, the desensitization values increased to 4.2 ± 0.2, 4.6 ± 0.2, 6.3 ± 0.2, and 3.8 ± 0.2 ms, following the previously mentioned order, which account for 0.24, 0.22, 0.16, and 0.26, respectively, when converted to the rate of desensitization. The most dramatic drop in rate was toward GluA1/2, which was unlike the inhibiting effect that had the highest potency on the GluA1 homomer. On the contrary, no effect on deactivation was observed by 4a, at a speed of 20 ms application of control vs 4a, the deactivation changed from 2.1 ± 0.1 to 2.3 ± 0.2 ms for GluA1 AMPAR-subtype, from 2.3 ± 0.1 to 2.5 ± 0.1 ms for GluA2 homomer, from 2.6 ± 0.2 to 2.5 ± 0.2 ms for GluA1/2, and from 2.7 ± 0.2 to 2.6 ± 0.2 ms GluA2/3 AMPAR heteromeric subunits, as shown in Figure 6. Insignificantly, 4b did not affect desensitization or deactivation; the whole-cell recording from this compound is provided in the Supporting Information.
Figure 5.
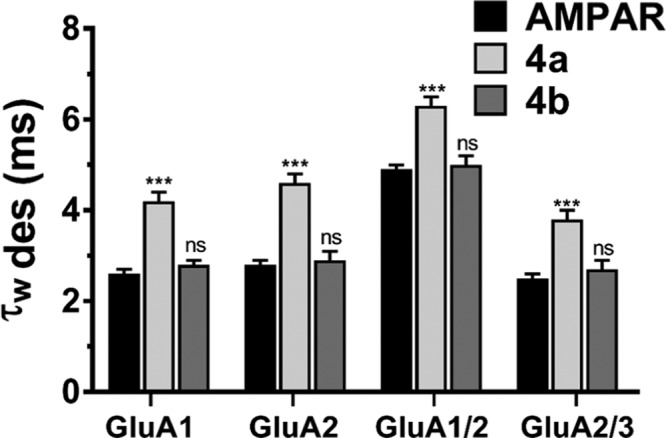
Effect of 4a and 4b on desensitization of recombinant AMPA receptors. The effects of glutamate alone (10 mM, black), Glu plus 4a (14 μM, white), and Glu plus 4b (14 μM, gray) on the desensitization of different AMPA-type receptors. Pulses on the response of AMPARs were set at 500 ms in whole-cell patched excised HEK293-expressing AMPAR cells- weighted time constants for desensitization (τW des). Data are shown as mean ± SEM; n = 6 (number of patch cells in the whole-cell configuration). Significance (one-way ANOVA): * p < 0.05; ** p < 0.01; *** p < 0.001; ns, not significant.
Figure 6.
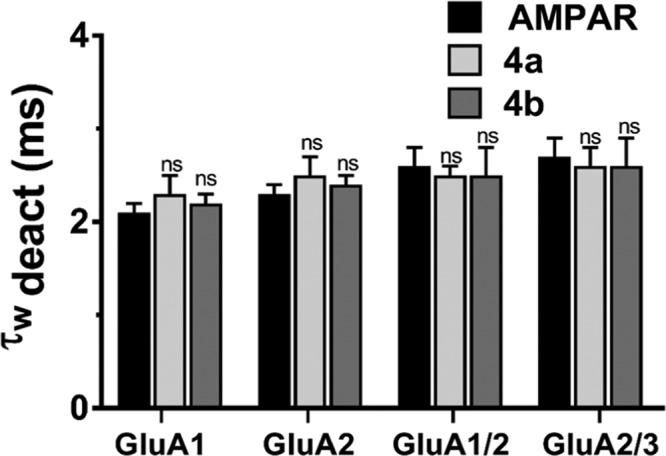
Effect of 4a and 4b on deactivation of different AMPA-type receptors. (A) Effects of glutamate alone (10 mM, black), Glu plus 4a (14 μM, white), and Glu plus 4b (14 μM, gray) on the deactivation of different AMPA-type receptors. Pulses on the response of AMPA receptors were set at 20 ms in whole-cell patched excised HEK293-expressing AMPAR cells. (B) Deactivation time in milliseconds (ms) from HEK293 cells expressing homomeric GluA2 alone or in combination with derivatives, representing the trace of the open tip junction currents that indicate the solution exchange in each experiment. The whole-cell current recording was conducted at −60 mV, pH 7.4, and 22 °C. Data shown are mean ± SEM; n = 6 (number of patch cells in the whole-cell configuration). Significance (one-way ANOVA): * p < 0.05; ** p < 0.01; *** p < 0.001; ns, not significant.
Conclusions
AMPA receptors are indispensable to the brain due to their critical role in learning and memory formation. However, excessive activation or overexpression of AMPARs has been linked to neurotoxicity and associated with numerous neurodegenerative diseases, such as ALS, Alzheimer’s disease, Parkinson’s disease, and epilepsy. This study has shown a structure-based activity relationship with novel 2,3-BDZ derivatives against various AMPA-type receptors. Predicted binding modes of the antagonists show slight changes in the interactions, which might be responsible for the differences in the observed selectivity against AMPA-subtype ionotropic receptors. Our electrophysiological investigations showed that the mechanism of inhibition of the meta chlorine position has higher biological activity than that of the ortho, which can be, in part, due to the reduced steric hindrance. In addition, we can conclude that the amino group of the phenyl ring is not necessary for inhibition; instead, an electron-withdrawing group can replace it. Moreover, both compounds seem to quite favorably inhibit the GluA2 homotetrameric AMPA receptors expressed in HEK293 cells. Surprisingly, the deactivation was not affected, while the desensitization rate was reduced from the 4a compound. Furthermore, the inhibition of the compound occurred in a noncompetitive manner, as the increase in glutamate concentration showed no correlation with the level of inhibition. Finally, in this study, we optimized the pharmacological profile of 2,3-BDZ by increasing its potency and selectivity and provided a better understanding of the AMPAR function and mechanism of inhibition. As part of our ongoing investigation, we aim to examine these compounds in vivo, i.e., AMPA toxicity in rat embryonic telencephalon cell culture.
Materials and Methods
Chemicals and Reagents
All of the reagents used were purchased from reliable resources: the following reagents were used in the research project: 3,4-(methylenedioxy)phenyl acetic acid, acetic acid, oxalyl chloride, sodium bicarbonate, ortho-chlorobenzoic acid, meta-chlorobenzoic acid, phosphorus pentoxide, magnesium sulfate, hydrazine hydrate, methanol, ethanol, n-hexane, ethyl acetate, and dichloromethane (DCM). Melting points were determined with an SMP-II digital melting point apparatus and were uncorrected. Infrared (IR) spectra were obtained using a PerkinElmer Spectrum 400 Fourier transform infrared (FTIR)/Fourier transform near-infrared (FTNIR) spectrometer. 1H NMR spectra were recorded in dimethyl sulfoxide (DMSO-d6) and performed using a Bruker Avance III 500 MHz FT-NMR high-performance digital spectrometer, and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 using a Bruker Avance III 400 MHz FT-NMR high-performance digital spectrometer, at the Faculty of Science, Department of Chemistry, The University of Jordan, Jordan. Tetramethylsilane was used as an internal standard. All chemical shifts were recorded as d (ppm). High-resolution mass spectra (HRMS) data were collected using a Shimadzu LCMS-IT-TOF by an electrospray ionization (ESI) (+) method, at the Doping and Narcotics Analysis Laboratory, Faculty of Pharmacy, Anadolu University, Turkey. Compounds were purified by chromatography and checked for purity using high-performance liquid chromatography (HPLC) before they were tested in biological assays (purity was 98–99%, as shown in Figures S1 and S2).
Synthesis of 2,3-BDZ Derivatives
Benzodiazepine Derivatives 4a and 4b
Benzodiazepine derivatives were synthesized in three steps, as outlined in Scheme 1. To collect high yield of ester (as shown in step 2), two methods were used: the first one by employing hydrochloric acid in methanol for 30 min, and20 the second method, which gave higher yield, used thionyl chloride or oxalyl chloride that had been added dropwise to methanol solvent and stirred for 30 min in an ice bath.21 The IR spectra of ester (step 2) showed the disappearance of the broad peak that belonged to the acetic acid group of step 1. Ketoesters 3a and 3b were synthesized by dissolving the ester (step 2) in dichloromethane with benzoic acid derivatives in the presence of an excess of phosphorus pentoxide, followed by stirring at room temperature for approximately 16 h. The subsequent treatment of methyl 2-[6-(3-chlorobenzoyl)-2H-1,3-benzodioxol-5-yl]acetate (3a) and methyl 2-[6-(2-chlorobenzoyl)-2H-1,3-benzodioxol-5-yl]acetate (3b) with hydrazine hydrate in ethanol with acetic acid afforded benzodiazepine derivatives 4a and 4b, respectively.20,22−25
Scheme 1. (A) MeOH, Oxalyl Chloride, rt, 30 min; (B) P2O5, (CH2Cl2), rt, 16 h; and (C) NH2NH2, Acetic Acid, Ethanol, Reflux, 24 h.

Experimental Section
Synthesis of Methyl 2-(2H-1,3-benzodioxol-5-yl)acetate
Dissolved the 3,4-(methylenedioxy)-phenyl acetic acid (1) (2 g, 11.10 mmol) in methanol, then cooled in an ice bath at 0 °C, and added oxalyl chloride (1 mL, 11.70 mmol) dropwise, and stirred the reaction mixture for 30 min. The reaction mixture was evaporated to dryness, then diluted with ethyl acetate, and then washed with saturated sodium bicarbonate (NaHCO3) and distilled water, respectively. The organic layer was dried with magnesium sulfate drying agent, filtered, evaporated again to concentrate it, and then purified by silica gel column chromatography using the n-hexane/ethyl acetate solvent system (50:50) to afford a yellow oily compound (step 2) in 95% yield.
General Procedure for Ketoester (3a and 3b) Synthesis
To a stirred solution of dichloromethane (50 mL) and methyl 2-(2H-1,3-benzodioxol-5-yl)acetate compound (step 2) (500 mg, 2.57 mmol), ortho- or meta-chlorobenzoic acid (523 mg, 3.34 mmol) and phosphorus pentoxide (3 g) were added (Scheme 1). Afterward, the mixture was further stirred at room temperature for 16–20 h, then distilled water (50 mL) was cautiously added, and the mixture extracted with ethyl acetate (2 × 50 mL). The organic layer was separated and then treated with 1M NaOH (50 mL), saturated sodium chloride (50 mL), and distilled water (2 × 50 mL). The organic layer was dried with a drying agent magnesium sulfate, filtered and evaporated under reduced pressure, and then purified by silica gel column chromatography.
Methyl 2-[6-(3-chlorobenzoyl)-2H-1,3-benzodioxol-5-yl]acetate (3a)
Purified by silica gel column chromatography using the n-hexane/ethyl acetate solvent system (80:20%). Crude green semisolid, yield 66%; IR (FTIR/FTNIR-ATR):1737 cm–1 ester carbonyl (C=O), 1661 cm–1 keton carbonyl (C=O). 1H NMR (DMSO-d6, 500 MHz) δ ppm: 3.49 (s, 3H, COOCH3), 3.78 (s, 2H, −CH2–C=O), 6.15 (s, 2H, −OCH2O), 6.95 (s, 1H, Ar), 7.07 (s, 1H, Ar), 7.55–7.61 (m, 2H, Ar), 7.63 (t, 1H, J = 2 Hz, Ar), 7.74 (d, 1H, J = 7.5 Hz, Ar) 13C NMR (DMSO-d, 400 MHz) δ ppm: 194.65, 171.04, 149.46, 145.63, 139.64, 133.23, 132.50, 130.43, 129.80, 128.95, 128.34, 127.83, 112.19, 109.89, 102.01, 51.46, 37.81. HRMS (m/z): [M + H]+ calcd. for C17H13O5Cl 333.0524, found 333.0517.
Methyl 2-[6-(2-chlorobenzoyl)-2H-1,3-benzodioxol-5-yl]acetate (3b)
Purified by silica gel column chromatography using the n-hexane/ethyl acetate solvent system (75:25%). Crude yellow solid powder, m.p. 80–82, yield 70%; IR (FTIR/FTNIR-ATR): 1727 cm–1 ester carbonyl (C=O), 1663 cm–1 keton carbonyl (C=O). 1H NMR (DMSO-d6, 500 MHz) δ ppm: 3.59 (s, 3H, COOCH3), 3.94 (s, 2H, −CH2–C=O), 6.14 (s, 2H, −OCH2O), 6.72 (s, 1H, Ar), 7.11 (s, 1H, Ar), 7.40 (d, 1H, J = 7.5 Hz, Ar), 7.47 (t, 1H, J = 7.5 Hz, Ar), 7.54–7.59 (m, 2H, Ar). 13C NMR (DMSO-d, 400 MHz) δ ppm: 194.28, 170.92, 150.68, 146.06, 138.85, 132.30, 131.65, 129.85, 129.22, 127.20, 112.94, 111.22, 102.34, 51.38, 38.83. HRMS (m/z): [M + H]+ calcd. for C17H13O5Cl 333.0524, found 333.0523.
Synthesis of 7,8-Methylenedioxy-1-(3-chlorophenyl)-3,5-dihydro-2,3-benzodiazepin-4(4H)-one (4a)
Methyl 2-[6-(3-chlorobenzoyl)-2H-1,3-benzodioxol-5-yl]acetate (3a) (400 mg, 1.2 mmol) was dissolved in ethanol (25 mL), then hydrazine hydrate (214 μL, 3.44 mmol) and acetic acid (100 μL) were added and refluxed for 24 h, then the solvent was removed under vacuum pressure, and the resulting residue was purified by silica gel column chromatography using the n-hexane/ethyl acetate solvent system (50:50%). Crude semibrown solid powder, m.p. 214–125.5 °C, purity 98%, yield 72%; IR (FTIR/FTNIR-ATR): 1661 cm–1 carbonyl (C=O). 1H NMR (DMSO-d6, 500 MHz) δ ppm: 3.41 (s, 2H, −CH2–C=O), 6.11 (s, 2H, −OCH2O−), 6.64 (s, 1H, ArH), 7.10 (s, 1H, ArH), 7.44–7.50 (m, 2H, ArH), 7.54–7.55 (m, 2H, ArH), 11.03 (s, 1H, NH). 13C NMR (CDCl3, 400 MHz) δ ppm: 175.05, 163.21, 155.85, 151.46, 145.63, 138.46, 137.07, 135.65, 134.76, 133.64, 133.08, 129.25, 113.15, 107.40, 46.55. HRMS (m/z): [M + H]+ calcd. for C16H11ClN2O3 315.0531, found. 315.0527.
Synthesis of 7,8-Methylenedioxy-1-(2-chlorophenyl)-3,5-dihydro-2,3-benzodiazepin-4(4H)-one (4b)
Methyl 2-[6-(2-chlorobenzoyl)–2H-1,3-benzodioxol-5-yl]acetate (3b) (400 mg, 1.2 mmol) was dissolved in ethanol (25 mL), then hydrazine hydrate (214 μL, 3.44 mmol) and acetic acid (100 μL) were added and reflux for 24 h, then the solvent was removed under vacuum pressure, and the resulting residue was purified by silica gel column chromatography using the n-hexane/ethyl acetate solvent system (50:50%). Crude yellow solid powder, m.p. 189.5–191.5 °C, purity 99%, yield 69%; IR (FTIR/FTNIR-ATR): 1658 cm–1 carbonyl (C=O). 1H NMR (DMSO-d6, 500 MHz) δ ppm: 3.47 (s, 2H, −CH2-C=O), 6.08 (s, 2H, −OCH2O−), 6.30 (s, 1H, ArH), 7.10 (s, 1H, ArH), 7.50–7.52 (m, 3H, ArH), 7.61–7.62 (m, 1H, ArH), 11.13 (s, 1H, NH), 13C NMR (CDCl3, 400 MHz) δ ppm: 174.63, 162.85, 155.51, 151.74, 142.90, 136.58, 136.25, 135.85, 135.08, 132.78, 133.08, 113.15, 111.63, 107.42, 46.61. HRMS (m/z): [M + H]+ calcd. for C16H11ClN2O3 315.0531, found. 315.0533.
Molecular Docking Studies
The molecules bind to an allosteric binding site, which is conserved between human and rat AMPA-subtype ionotropic glutamate receptors GluA1 and GluA2. The crystal structure of AMPA GluA2 was complexed with a similar heterocyclic ring containing a noncompetitive inhibitor molecule GYKI-Br. Potential protein–ligand interactions of the molecules were revealed by conducting docking studies with the crystal structure (PDB code 5L1G,26 resolution 4.5 Å) using Glide 8.4.27 The ligands were drawn using Maestro 12.1 graphical user interface28 and prepared by LigPrep29 (pH 7.0 ± 2.0) to assign the atom types and the protonation states with OPLS3e force field. The same force field was used for proteins, and predicted positions of the missing side chains were added with the Protein Preparation Wizard.30 The grids were generated, and docking simulations were done in single-precision mode (GlideScore SP). The docking results of 2,3- benzodiazepine derivatives were visualized at GluA1 and GluA2 active sites, the resulting binding modes were evaluated with PLIP 1.4.4,30 and the resulting poses were visualized with PyMOL 2.3.31
Whole-Cell Recordings
DNA preparation, cDNA transient transfection, and cell culturing of human embryonic kidney cells (HEK293) expressing the flip isoform of AMPAR subunits have been previously described.32−35 Electrophysiological recordings took place 36–48 h after the chemical mediated transfection took place, whereby the cells were replated on coverslips coated with laminin. The highly fluorescent cells displaying the cotransfected enhanced green fluorescent protein were selected for a giga-seal for the current recording. Borosilicate glass was used to fabricate the patch electrodes with a resistance of 2–4 MΩ. At a temperature of 22 °C, a membrane potential of −60 mV, a sampling frequency of 10 kHz, and a low-pass filter set at 2 kHz, an integrated patch amplifier (IPA) was used to execute the whole-cell patch-clamp technique (IPA, Sutter Instruments, Novato, CA) using their SutterPatch Software v. 1.1.1 to digitize membrane currents for a short period. The extracellular solution contained (values are in mM): 150 NaCl, 2.8 KCl, 0.5 MgCl2, 2 CaCl2, and 10 HEPES, adjusted to pH 7.4 with NaOH. The pipette solution contains (values are in mM): 110 CsF, 30 CsCl, 4 NaCl, 0.5 CaCl2, 10 trypsin–EDTA solution B (0.25%), EDTA (0.05%), and 10 HEPES, adjusted to pH 7.2 with CsOH. Double-barrel glass (theta tube) was used for constant washing out of the cell from one barrel while the other supplying the cell with the compound of interest using a high-speed piezo solution switcher (Automate Scientific, Berkeley, CA). A 10–90% solution exchange occurred typically at 500 ms, represented by the open tip potential that was recorded during the application of solutions of different ionic strengths that resulted from the expulsion of the patch from the electrode to estimate the speed of solution exchange. Six viable cells were considered for the sample size to obtain the average inhibition by the derivative of interest. To ensure the safety of the cell and validate the results of the antagonist, the cell was then resupplied with the control (glutamate alone) after compound washout, which, to consider viable, should be almost identical to the glutamate-induced current before the application of the antagonist. The exact data analysis of this process is provided in the Supporting Information. Data acquired were analyzed using Igor Pro7 (Wave Metrics, Inc). Receptor desensitization (τdes) and deactivation rates were estimated by a single exponential fitting of the current decay curve, starting from 95% of the peak to the baseline current. The currents were evoked by the application of 10 mM glutamate for desensitization (500 ms) and 20 ms of the same concentrated glutamate for deactivation.
Acknowledgments
The authors would like to thank An-Najah National University for funding this study (grant number ANNU-1920-Sc011), the Dean of Scientific Research, and Anadolu University for the support in chemical analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04000.
HPLC results of compounds 4a and 4b; dose–response curves for compounds 4a and 4b; and whole-cell recordings for compounds 4a and 4b (pdf)
Author Contributions
M.Q., N.J., M.H., and A.O. conceived and designed the current study and analyzed the data obtained. This paper was written by M.Q., M.H., N.J., A.O., and N.E. and drafted by all authors. The authors read and approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Niu L. Mechanism-based design of 2, 3-benzodiazepine inhibitors for AMPA receptors. Acta Pharm. Sin. B 2015, 5, 500–505. 10.1016/j.apsb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Niu L. Mechanism of inhibition of the GluA2 AMPA receptor channel opening by talampanel and its enantiomer: the stereochemistry of the 4-methyl group on the diazepine ring of 2, 3-benzodiazepine derivatives. ACS Chem. Neurosci. 2013, 4, 635–644. 10.1021/cn3002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Sheng Z.; Niu L. Mechanism of inhibition of the GluA2 AMPA receptor channel opening: consequences of adding an N-3 methylcarbamoyl group to the diazepine ring of 2, 3-benzodiazepine derivatives. Biochemistry 2011, 50, 7284–7293. 10.1021/bi2007977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappala M.; Grasso S.; Micale N.; Polimeni S.; De Micheli C. Synthesis and structure-activity relationships of 2, 3-benzodiazepines as AMPA receptor antagonists. Mini-Rev. Med. Chem. 2001, 1, 243–253. 10.2174/1389557013406783. [DOI] [PubMed] [Google Scholar]
- Grossi G.; Di Braccio M.; Roma G.; Ballabeni V.; Tognolini M.; Calcina F.; Barocelli E. 1, 5-Benzodiazepines: Part XIII. Substituted 4H-[1, 2, 4] triazolo [4, 3-a][1, 5] benzodiazepin-5-amines and 4H-imidazo [1, 2-a][1, 5] benzodiazepin-5-amines as analgesic, anti-inflammatory and/or antipyretic agents with low acute toxicity. Eur. J. Med. Chem. 2002, 37, 933–944. 10.1016/S0223-5234(02)01400-9. [DOI] [PubMed] [Google Scholar]
- Chadha S.; Paul S.; Kapoor K. Synthesis and biological screening of 4-(5-alkyl-2-isoxazolin-3-yl)-2-aryl-2, 3-dihydro-1H-1, 5-benzodiazepines. J. Chem. Pharm. Res. 2011, 3, 331–340. [Google Scholar]
- Ohtake Y.; Naito A.; Hasegawa H.; Kawano K.; Morizono D.; Taniguchi M.; Tanaka Y.; Matsukawa H.; Naito K.; Oguma T.; et al. Novel vasopressin V2 receptor-selective antagonists, pyrrolo [2, 1-a] quinoxaline and pyrrolo [2, 1-c][1, 4] benzodiazepine derivatives. Bioorg. Med. Chem. 1999, 7, 1247–1254. 10.1016/S0968-0896(99)00049-8. [DOI] [PubMed] [Google Scholar]
- Xia J.; Li J.; Sun H. Insights into ET A subtype selectivity of benzodiazepine endothelin receptor antagonists by 3D-QSAR approaches. J. Mol. Model. 2012, 18, 1299–1311. 10.1007/s00894-011-1153-x. [DOI] [PubMed] [Google Scholar]
- Roberts K.; Ursini A.; Barnaby R.; Cassarà P. G.; Corsi M.; Curotto G.; Donati D.; Feriani A.; Finizia G.; Marchioro C.; et al. Synthesis and structure–activity relationship of new 1, 5-dialkyl-1, 5-benzodiazepines as cholecystokinin-2 receptor antagonists. Bioorg. Med. Chem. 2011, 19, 4257–4273. 10.1016/j.bmc.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Hsiao G.; Shen M.-Y.; Chou D.-S.; Chang Y.; Lee L.-W.; Lin C.-H.; Sheu J.-R. Mechanisms of antiplatelet and antithrombotic activity of midazolam in in vitro and in vivo studies. Eur. J. Pharmacol. 2004, 487, 159–166. 10.1016/j.ejphar.2004.01.026. [DOI] [PubMed] [Google Scholar]
- Sardana S.; Madan A. K. Predicting anti-HIV activity of TIBO derivatives: a computational approach using a novel topological descriptor. J. Mol. Model. 2002, 8, 258–265. 10.1007/s00894-002-0093-x. [DOI] [PubMed] [Google Scholar]
- Dourlat J.; Liu W.-Q.; Gresh N.; Garbay C. Novel 1, 4-benzodiazepine derivatives with antiproliferative properties on tumor cell lines. Bioorg. Med. Chem. Lett. 2007, 17, 2527–2530. 10.1016/j.bmcl.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Szénási G.; Vegh M.; Szabo G.; Kertesz S.; Kapus G.; Albert M.; Greff Z.; Ling I.; Barkoczy J.; Simig G.; et al. 2, 3-benzodiazepine-type AMPA receptor antagonists and their neuroprotective effects. Neurochem. Int. 2008, 52, 166–183. 10.1016/j.neuint.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Szénási G.; Hársing L. G. Jr Pharmacology and prospective therapeutic usefulness of negative allosteric modulators of AMPA receptors. Drug Discovery Today: Ther. Strategies 2004, 1, 69–76. 10.1016/j.ddstr.2004.08.018. [DOI] [Google Scholar]
- Marinelli S.; Gatta F.; Sagratella S. Effects of GYKI 52466 and some 2, 3-benzodiazepine derivatives on hippocampal in vitro basal neuronal excitability and 4-aminopyridine epileptic activity. Eur. J. Pharmacol. 2000, 391, 75–80. 10.1016/S0014-2999(00)00050-9. [DOI] [PubMed] [Google Scholar]
- Niu L. Mechanism-based design of 2, 3-benzodiazepine inhibitors for AMPA receptors. Acta Pharm. Sin. B 2015, 5, 500–505. 10.1016/j.apsb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micale N.; Colleoni S.; Postorino G.; Pellicanò A.; Zappalà M.; Lazzaro J.; Diana V.; Cagnotto A.; Mennini T.; Grasso S. Structure–activity study of 2, 3-benzodiazepin-4-ones noncompetitive AMPAR antagonists: Identification of the 1-(4-amino-3-methylphenyl)-3, 5-dihydro-7, 8-ethylenedioxy-4H-2, 3-benzodiazepin-4-one as neuroprotective agent. Bioorg. Med. Chem. 2008, 16, 2200–2211. 10.1016/j.bmc.2007.11.080. [DOI] [PubMed] [Google Scholar]
- Chimirri A.; De Sarro G.; De Sarro A.; Gitto R.; Grasso S.; Quartarone S.; Zappala M.; Giusti P.; Libri V.; Constanti A. 1-Aryl-3, 5-dihydro-4 H-2, 3-benzodiazepin-4-ones: Novel AMPA Receptor Antagonists. J. Med. Chem. 1997, 40, 1258–1269. 10.1021/jm960506l. [DOI] [PubMed] [Google Scholar]
- Ben-Yaacov A.; Gillor M.; Haham T.; Parsai A.; Qneibi M.; Stern-Bach Y. Molecular mechanism of AMPA receptor modulation by TARP/stargazin. Neuron 2017, 93, 1126.e4–1137. e4. 10.1016/j.neuron.2017.01.032. [DOI] [PubMed] [Google Scholar]
- Zappalà M.; Postorino G.; Micale N.; Caccamese S.; Parrinello N.; Grazioso G.; Roda G.; Menniti F. S.; De Sarro G.; Grassor S. Synthesis, Chiral Resolution, and Enantiopharmacology of a Potent 2,3-Benzodiazepine Derivative as Noncompetitive AMPA Receptor Antagonist. J. Med. Chem. 2006, 49, 575–581. 10.1021/jm050552y. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S.; Pal B. C.; Parasuraman J.; Roy S.; Chakrabotry J. B.; Mukherjee C.; Mahato K.; Konar A.; Rakshit S.; Mandal L.; Ganguly D.; Paul K.; Manna A.; Vinayagam J.; Pal C.. Inhibitors of phosphatidylinositol-3-kinase (PI3) and inducers of nitric oxide (NO). 2016.
- Menniti F. S.; Chenard B. L.; Collins M. B.; Ducat M. F.; Elliott M. L.; Ewing F. E.; Huang J. I.; Kelly K. A.; Lazzaro J. T.; Pagnozzi M. J. Characterization of the binding site for a novel class of noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonists. Mol. Pharmacol. 2000, 58, 1310–1317. 10.1124/mol.58.6.1310. [DOI] [PubMed] [Google Scholar]
- Zappalà M.; Postorino G.; Micale N.; Caccamese S.; Parrinello N.; Grazioso G.; Roda G.; Menniti F. S.; De Sarro G.; Grasso S. Synthesis, chiral resolution, and enantiopharmacology of a potent 2, 3-benzodiazepine derivative as noncompetitive AMPA receptor antagonist. J. Med. Chem. 2006, 49, 575–581. 10.1021/jm050552y. [DOI] [PubMed] [Google Scholar]
- Micale N.; Colleoni S.; Postorino G.; Pellicano A.; Zappala M.; Lazzaro J.; Diana V.; Cagnotto A.; Menninic T.; Grassoa S. Structure–activity study of 2,3-benzodiazepin-4-ones noncompetitive AMPAR antagonists: Identification of the 1-(4-amino-3-methylphenyl)-3,5-dihydro-7,8-ethylenedioxy- 4H-2,3-benzodiazepin-4-one as neuroprotective agent. Bioorg. Med. Chem. J. 2008, 16, 2200–2211. 10.1016/j.bmc.2007.11.080. [DOI] [PubMed] [Google Scholar]
- Zappala M.; Pellicano A.; Micale N.; Menniti F. S.; Ferreri G.; De Sarro G.; Grassoa S.; De Michelid C. New 7,8-ethylenedioxy-2,3-benzodiazepines as noncompetitive AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 167–170. 10.1016/j.bmcl.2005.09.029. [DOI] [PubMed] [Google Scholar]
- Yelshanskaya M. V.; Singh A. K.; Sampson J. M.; Narangoda C.; Kurnikova M.; Sobolevsky A. I. Structural bases of noncompetitive inhibition of AMPA-subtype ionotropic glutamate receptors by antiepileptic drugs. Neuron 2016, 91, 1305–1315. 10.1016/j.neuron.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnasamy K.; Saravanan M.; Poomani K. Investigation of binding mechanism and downregulation of elacestrant for wild and L536S mutant estrogen receptor-α through molecular dynamics simulation and binding free energy analysis. J. Comput. Chem. 2020, 41, 97–109. 10.1002/jcc.26076. [DOI] [PubMed] [Google Scholar]
- Release S.Maestro, Schrödinger, LLC; Schrödinger Inc: New York, 2017. [Google Scholar]
- Release S.LigPrep, Schrödinger, LLC; New York, 2017. [Google Scholar]
- Salentin S.; Schreiber S.; Haupt V. J.; Adasme M. F.; Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger L.The PyMOL molecular graphics system Version 2010, 1 (5), .
- Qneibi M.; Hamed O.; Fares O.; Jaradat N.; Natsheh A.-R.; AbuHasan Q.; Emwas N.; Al-Kerm R.; Al-Kerm R. The inhibitory role of curcumin derivatives on AMPA receptor subunits and their effect on the gating biophysical properties. Eur. J. Pharm. Sci. 2019, 136, 104951 10.1016/j.ejps.2019.06.005. [DOI] [PubMed] [Google Scholar]
- Qneibi M.; Jaradat N.; Emwas N. Effect of Geraniol and Citronellol Essential Oils on the Biophysical Gating Properties of AMPA Receptors. Appl. Sci. 2019, 9, 4693–4701. 10.3390/app9214693. [DOI] [Google Scholar]
- Qneibi M.; Hamed O.; Natsheh A.-R.; Fares O.; Jaradat N.; Emwas N.; AbuHasan Q.; Al-Kerm R.; Al-Kerm R. Inhibition and assessment of the biophysical gating properties of GluA2 and GluA2/A3 AMPA receptors using curcumin derivatives. PloS One 2019, 14, e0221132 10.1371/journal.pone.0221132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qneibi M.; Jaradat N.; Hawash M.; Zaid A. N.; Natsheh A.-R.; Yousef R.; AbuHasan Q. The Neuroprotective Role of Origanum syriacum L. and Lavandula dentata L. Essential Oils through Their Effects on AMPA Receptors. Biomed. Res. Int. 2019, 2019, 5640173 10.1155/2019/5640173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.