

Abstract
We describe a successful strategy to substantially enhance cell nucleation efficiency in polymer foams by using designer nanoparticles as nucleating agents. Bare and poly(dimethylsilane) (PDMS)-grafted raspberry-like silica nanoparticles with diameters ranging from ∼80 nm to ∼200 nm were synthesized and utilized as highly efficient cell nucleators in CO2-blown nanocellular polymethyl methacrylate (PMMA) foams. The successful synthesis of core–shell nanoparticles was confirmed by Fourier transform infrared spectroscopy, thermogravimetric analysis, Brunauer–Emmett–Teller measurements, and transmission electron microscopy. The cell size and cell density of the obtained PMMA micro- and nanocellular foams were determined by scanning electron microscopy. The results show that increased surface roughness enhances the nucleation efficiency of the designer silica particles. This effect is ascribed to a decreased nucleation free energy for foam cell nucleation in the nanocavities at the melt–nucleator interface. For PDMS grafted raspberry-like silica nanoparticles with diameters of 155 and 200 nm, multiple cell nucleation events were observed. These hybrid particles had nucleation efficiencies of 3.7 and 6.2, respectively. The surprising increase in nucleation efficiency to above unity is ascribed to the significant increase in CO2 absorption and capillary condensation in the corresponding PMMA during saturation. This increase results in the presence of large amounts of the physical blowing agent close to energetically favorable nucleation points. Additionally, it is shown that as a consequence of cell coalescence, the increased number of foam cells is rapidly reduced during the first seconds of foaming. Hence, the design of highly efficient nucleating particles, as well as careful selection of foam matrix materials, seems to be of pivotal importance for obtaining polymer cellular materials with cell dimensions at the nanoscale. These findings contribute to the fabrication of polymer foams with high thermal insulation capacity and have relevance in general to the area of cellular materials.
Keywords: nanocellular materials, polymer foam, nanocavity, CO2 capillary condensation, core−shell nanoparticles
Nanocellular polymer foams are receiving considerable attention from the scientific and industrial communities.1−6 This interest is ascribed to the unexpected and sometimes exciting foam properties when their cells have diameters on the order of a few tens to hundreds of nanometers. For instance, the thermal conductivity of nanocellular foams is significantly reduced as a result of the Knudsen effect provided that the foam density is sufficiently low.2,7 In addition, compared to that of microcellular foams with similar densities, the mechanical performance of these foams in terms of, for example, the Young’s modulus, impact properties and Shore hardness, is significantly improved.8−10 Thus, nanocellular polymer foams with low thermal conductivity can offer great alternative options when considering new classes of materials, for instance, to realize reductions in fuel consumption and CO2 emissions in transportation as well as to manage the thermal energy in buildings.
Nanocellular polymer foam structures can be obtained by various strategies, for example, selective extraction/decomposition,11,12 templating/imprinting,13−15 chemical/physical foaming,8,16−20etc. Among these methods, CO2-based batch foaming is of particular interest, which is ascribed to good control over the foaming conditions as well as to the low cost and environmental friendliness of CO2 as a physical blowing agent.9 In batch foaming, the polymer is first saturated with CO2 at a few to several hundreds of bars for a given time (on the order of hours to days). Subsequently, foaming is induced by a quick depressurization step that is typically followed by an increase in temperature. This results in an oversaturation of the dissolved CO2 in the polymer gas mixture leading to cell nucleation followed by rapid cell expansion.21 The disadvantage of batch foaming compared to other methods is that it has a relatively small batch size (i.e., on the scale of liters), which limits the industrial relevance of this process. On the other hand, nanocellular foam extrusion, that is, a continuous process, remains challenging, which is ascribed to the difficult control over key physical foaming parameters, for example, CO2 solubility, CO2 diffusivity, and foaming temperature.22−24
In addition to the production challenges, nanocellular foams with high porosities (>85%) and cell densities (>1014 cells cm–3) have been rarely reported.4,25−27 This is ascribed to the fact that it is difficult to nucleate such high cell densities in the early stages of foaming. In addition, on the time scale of foaming, cell coalesce is hard to avoid. Frequently used strategies to increase the nucleated cell density rely on increasing the physical blowing agent saturation pressure and/or increasing the pressure release rate.3,25,28
The introduction of nanostructured heterogeneous phases to polymers as heterogeneous nucleation sites prior to foaming has also been considered a promising approach. Examples include the use of phase-separated block copolymer domains29−32 and inorganic nanofillers.18,33−38
For instance, Rodríguez-Pérez and co-workers29 reported on CO2-assisted batch foaming of polymethyl methacrylate (PMMA) and poly(methyl methacrylate-block-butyl acrylate-block methyl methacrylate) (MAM) blends. The authors demonstrated that foam cells produced from these PMMA/MAM blends were nucleated in the nanostructured MAM micellar phase, which is ascribed to the high CO2-philicity of the poly(butyl acrylate) phase compared to the PMMA matrix. McClurg and co-workers31 demonstrated that the incorporation of A-B diblock copolymers containing poly(dimethylsiloxane) as the core block into polystyrene prior to foaming increases the cell density of microcellular polystyrene foams. The nanostructured poly(dimethylsilane) (PDMS) domains were considered efficient nucleation sites because of their high CO2-philicity as well as the low surface energy of PDMS.
Compared to block copolymer micelles, silica nanoparticles are of particular interest for use as heterogeneous nucleation agents in polymer foaming because of their low cost and easy preparation and the simplicity of employing various surface decoration strategies.39,40 For instance, Zhai and co-workers37 demonstrated that the incorporation of silica nanoparticles into polycarbonate prior to foaming resulted in a higher cell density and more uniform cell size distribution than those in the polycarbonate foams containing no silica nanoparticles as nucleating agents. Yang and co-workers41 reported that the decoration of silica nanoparticles with CO2-philic poly(ionic liquid) significantly enhances cell nucleation in CO2-assisted polystyrene foaming compared to polystyrene foams prepared with bare silica nanoparticles as nucleating agents. Despite these promising results, we note that the nanoparticles utilized so far for nucleation have suffered from poor nucleation efficiencies; that is, the number of foam cells obtained per number of nanoparticles per cm3 added to the polymer matrix prior to foaming was small. The nucleation efficiencies for these inorganic particle-based nanofillers are typically far below 0.01.18,41−43 This means that hundreds of added particles result in the formation of only one foam cell during foaming. In these calculations, it is assumed that homogeneous nucleation and cell coalescence can be neglected.3 We have reported the successful utilization of PDMS-grafted silica nanoparticles with a silica core diameter of 80 nm with a nucleation efficiency of approximately 0.5 for the preparation of CO2-blown batch PMMA foams.33 The good efficiency of these particles is ascribed to (i) the low surface energy of the PDMS shell, which reduces the nucleation energy barrier and (ii) the higher local CO2 concentration in the PDMS shell (∼75 wt %)44 than in the PMMA matrix (∼18 wt %).45 Furthermore, we demonstrated that the contribution of line tension significantly reduces the nucleation efficiency of PDMS-grafted nanoparticles with a silica core diameter below 40 nm under the used foaming conditions (i.e., CO2 saturation pressure of 55 bar and foaming at 40 °C).33 Thus, the use of smaller particles, for example, particles 10 nm in diameter, is considered unfavorable, although it would in principle allow for the addition of the required high number of potential nucleation sites (>1015 cm–3) at relatively low weight percentages of particle loading. In fact, increasing the weight percentage of particle loading for particles with sufficiently large diameters (>60 nm) to incorporate the required number of potential nucleation sites renders the resulting particle-filled polymer matrix barely foamable. Thus, we conclude that enhanced particle designs are needed to increase the nucleation efficiency to unity and beyond.
The effect of surface roughness (e.g., nanocavities) on the free energy of heterogeneous foam cell nucleation has not been included in the particle design process until now. Yang and co-workers reported46 a thermodynamic model and an analytical solution for the free energy of cell nucleation at the nanometer length scale in nanocavities. They showed that cells nucleated from a concave surface have a lower nucleation free energy than cells nucleated from flat convex surfaces (e.g., spherical particles).47,48 Inspired by these results, we considered surface roughness as an important and ignored design parameter for hybrid nucleating particle designs in enhanced nanocellular foaming. We chose a strategy to prepare silica nanoparticles with a superimposed roughened surface morphology. As the particles resemble a raspberry in appearance, we call them raspberry-like silica nanoparticles.
Since the preparation of raspberry-like silica nanoparticles is fairly easy and well described in the literature,49−53 we considered these surface roughened nanoparticles as ideal candidates to explore the effect of nanocavities on cell nucleation of nanocellular foams. For example, He and co-workers51 reported on a method to produce raspberry-like nanoparticles by a self-templated etching route. This method limits the particle diameter to 60 nm or larger. In fact, for the purpose of this work, this size is acceptable since it allowed us to study the effect of particle surface roughness on foam cell nucleation without considering other effects, such as particle curvature.43,54
Here, we show that raspberry-like silica nanoparticles are efficient foam cell nucleators with nucleation efficiencies that are comparable to their PDMS grafted nearly spherical counterparts. Upon decorating the raspberry-like silica nanoparticles with a few nanometer-thick PDMS shell, multiple foam cell nucleation events were observed from a single designer nanoparticle, and the highest nucleation efficiency obtained was 6.2 for particles with an overall core diameter of ∼200 nm. This nucleation efficiency is nearly 40 times higher than that of a pristine nearly spherical silica nanoparticle with a similar diameter. We qualitatively show that the high nucleation efficiency obtained for PDMS-grafted raspberry-like silica nanoparticles is ascribed to a significantly increased absorption of CO2 during saturation. This process results in an increased CO2 concentration close to the PDMS-decorated, energetically favorable nanocavities at the nucleating surface. The utilization of surface cavities combined with optimized surface chemistry for efficient heterogeneous nucleation at the macromolecular length scale is of interest for the production of nanocellular foams with high cell densities and thus superior thermal insulation power.
Results and Discussion
Core-Shell Raspberry-like Nanoparticle Synthesis and Characterization
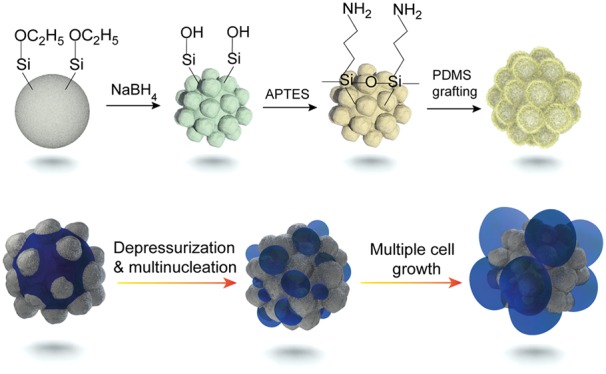
Raspberry-like silica nanoparticles (SiO2-R) were synthesized by the reaction of Stöber silica nanoparticles with NaBH4, followed by the surface grafting of PDMS to yield rough silica core PDMS-shell hybrid nanoparticles (SiO2-RP). The reaction scheme is depicted in Figure 1a. (Hereinafter we abbreviate “nanoparticle” with NP.) During the reaction of nearly smooth Stöber SiO2 (SiO2-S) NPs with NaBH4, the high basicity of the dissolved NaBH4 results in the dissociation of Si–O bonds and subsequently in the formation of soluble mono- and polysilicate species. Eventually the silicate species dissolved in the solution become supersaturated which leads to the formation of small SiO2 domains on the surface of the remaining silica NPs (Figure 1a, step 1).51,55,56
Figure 1.

(a) A schematic of the SiO2-RP NP preparation process. In (b) and (c) SEM images of SiO2-S and SiO2-R NPs with a silica particle diameter of ∼155 nm are shown, respectively. (d) Single reflection ATR-FTIR absorbance spectra of 155SiO2-S, 155SiO2-R, 155SiO2-R-NH2, 155SiO2-RP, and 155SiO2-SP NPs. The black arrows in the FTIR spectra indicate characteristic FTIR absorbance values of the (modified) silica NPs. (e) Non-isothermal TGA thermograms of the 155SiO2-S-NH2, 155SiO2-R-NH2, 155SiO2-SP, and 155SiO2-RP NPs.
Scanning electron microscopy (SEM) images of SiO2-S and SiO2-R NPs with a diameter of approximately 155 nm are shown in Figure 1b,c, respectively. The diameter of the SiO2-R NPs is defined as the diameter of a circle just large enough to enclose the SiO2-R NP outer surface. Particles with diameters between 80 and 200 nm were prepared (see Materials and Methods section). Hereinafter, the numbers preceding “SiO2” refer to the apparent particle diameter. For instance, 155SiO2-S refers to an NP with an average diameter of 155 nm.
From Figure 1c, it is obvious that small silica domains (with diameters of ∼20–30 nm) are present at the surface of the NPs after treatment with NaBH4, which is in agreement with the deposition of silicate species on the silica particle surface during the reaction.51 Following the successful synthesis of SiO2-R NPs with diameters of ∼80 nm, ∼120 nm, ∼155 nm, and ∼200 nm, a PDMS shell was grafted to the surface to yield the corresponding hybrid (SiO2-RP) NPs (Figure 1a). In brief, SiO2-R was derivatized with (3-aminopropyl)-triethoxysilane (APTES), resulting in the formation of amine-functionalized NPs (SiO2-R-NH2) (step 2). Subsequently, PDMS-grafted core–shell NPs were prepared by the “grafting to” approach with monoglycidyl ether-terminated PDMS (step 3). We note here that prolonged sonication or even melt blending did not destroy the surface roughness of the (hybrid) raspberry-like NPs; that is, the NPs are stable enough for utilization as nucleating particles in nanocellular foaming.
Figure 1d shows Fourier transform infrared (FTIR) absorbance spectra of SiO2, SiO2-R, SiO2-R-NH2, SiO2-RP, and nearly smooth PDMS-grafted NPs (SiO2-SP) with a (silica core) diameter of ∼155 nm. The ethoxy groups remaining after the Stöber reaction with tetraethyl orthosilicate (TEOS) are clearly observed in the FTIR spectra of the 155SiO2-S particles; that is, the CH2/CH3 bending absorbance band at 1452 cm–1 and the CH2/CH3 absorbance band at 2980 cm–1.57 After NaBH4 etching to yield surface-roughened NPs, these absorbance bands disappear, which indicates quantitative hydrolysis of the remaining ethoxy groups of the 155SiO2-R NPs.51 The bands at 2980 cm–1, 1450 cm–1, and 1380 cm–1 reappearing in the FTIR spectrum of 155SiO2-R-NH2 are assigned to the CH2 groups of the propyl spacer of surface tethered APTES. The presence of absorption bands ascribed to CH3 stretching at 2967 cm–1 and to C–H bending at 1263 cm–1 in the FTIR spectrum of 155SiO2-RP NPs confirms the successful grafting of PDMS.58 The same absorption bands are observed in the FTIR spectrum of SiO2-SP NPs; their synthesis is described in the Materials and Methods section.
To determine the amount of PDMS grafted to the silica NPs, thermogravimetric analysis (TGA) was employed. Figure 1e shows the weight loss versus temperature curves for non-isothermal TGA measurements of nearly spherical and raspberry-like, amino-functionalized and PDMS-grafted NPs with silica (core) diameters of approximately 155 nm. The results clearly show that the amount of PDMS covalently bound to the SiO2 NPs with a rough surface is significantly higher, that is, ∼10.3 wt %, than the amount covalently bound to the spherical NPs, that is, ∼3.0 wt %. This nearly 3.4-fold increase in grafted PDMS weight is ascribed to the increased specific surface area of the raspberry-like NPs compared to the spherical NPs, as shown in Figure S1. For the NPs with a diameter of 155 nm, the surface area increased from 32.4 m2 g–1 for the spherical NPs to 116.0 m2 g–1 for the raspberry-like NPs (see Figure S1). The nearly 3.6-fold increase in surface area is in good agreement with the 3.4-fold increase in amount of grafted PDMS. For the raspberry-like silica NPs with diameters of 200 and 120 nm, the increase in PDMS grafting wt % was in good agreement with the corresponding increase in surface area.
Based on the grafted PDMS mass obtained by TGA measurements, the molar mass of the grafted PDMS chains (i.e., 1000 g mol–1), and the Brunauer–Emmett–Teller (BET) surface area of the NPs, the PDMS grafting densities were calculated to be ∼0.6 chains nm–2 for the 155SiO2-SP and 155-SiO2-RP NPs. This number is lower than the earlier reported value of 0.9 for SiO2-SP NPs with diameters from 12 to 120 nm.59 This difference is ascribed to the known overestimation of the surface area of Stöber silica NPs determined by BET measurements60−62 compared to the NP surface area calculated by simple arithmetic estimation based on the particle diameter. When using this simple arithmetic method for the determination of the grafting density of the SiO2-SP NPs with a particle diameter from 80 to 200 nm, grafting density values close to 0.9 chains nm–2 were obtained. This result is in good agreement with values for SiO2-SP NPs reported in our previous work.59 Based on these results, we note that BET analysis is used here only to qualitatively show the significant differences in the surface area of the particles before and after roughening their surface.
Finally, transmission electron microscopy (TEM) was used to confirm the core–shell structure of the PDMS-grafted NPs. Figure 2a–d shows TEM images of 155 nm bare and PDMS-grafted spherical and raspberry-like NPs. From these TEM images, it is clear that there is a thin PDMS shell around the 155SiO2-SP (Figure 2b) and 155SiO2-RP (Figure 2d) NPs. The thickness of the PDMS shell on the NPs was estimated to be approximately 6.0 ± 1.5 nm, which agrees well with previous results reported for SiO2-SP NPs with silica-core diameters from 12 to 120 nm.59
Figure 2.

TEM images of (a) 155SiO2-S, (b) 155SiO2-SP, (c) 155SiO2-R, and (d) 155SiO2-RP NPs.
Following the successful synthesis and characterization of the NPs, we employed them as nucleating agents in CO2-assisted batch foaming of PMMA as presented and discussed in the next sections.
Nanocomposite Foaming and Multinucleation
Prior to foaming, the designer NPs were melt-blended in a PMMA matrix and pressed into films with a thickness of approximately 200 μm. For comparison of the nucleation efficiency, NPs with the same volume number density (i.e., 1.07 × 1013 particles cm–3) were used for each nanocomposite film. Thus, assuming that the particles are well dispersed in the PMMA films and that each particle can nucleate the same number of foam cells, all nanocomposite films had the same volume number density of potential nucleation sites prior to foaming. The particles were well dispersed in PMMA prior to foaming, as observed from SEM images of cross-sectioned PMMA nanocomposite films (see Figure S3, Supporting Information).
The CO2 saturation pressure used for foaming was 55 bar, and following quick depressurization (i.e., within 2 s), foams were prepared at a foaming temperature of 40 °C for 1 s. This relatively short foaming time was chosen since we were primarily interested in the nucleation efficiency of the prepared particles and not in the well-developed foam morphology at the end stages of foaming (i.e., after several tens of seconds to min). Figure 3a–d shows SEM images of cross-sectioned PMMA foams nucleated by 155SiO2-S, 155SiO2-SP, 155SiO2-R, and 155SiO2-RP NPs. It is clear that for the 1 s foaming time, the PMMA foams have thick cell walls and struts. Furthermore, the 155SiO2-S, 155SiO2-SP, and 155SiO2-R NPs are present at the polymer foam cell wall surface. The 155SiO2-S NPs are more embedded in the PMMA cell wall than the 155SiO2-SP NPs, which can be attributed to the low surface energy and high CO2-philicity of the decorated PDMS shell compared with the bare particles, resulting in the different interfacial interactions between the nanoparticles and the polymer gas mixture.63 This is in good agreement with the reported results for the nucleation behavior of pristine SiO2 and corresponding PDMS-grafted core–shell NPs with (core) diameters up to 120 nm.33,59
Figure 3.

SEM images of cross-sectioned PMMA foams nucleated by (a) 155SiO2-S, (b) 155SiO2SP, (c) 155SiO2-R, (d) 155SiO2-RP, (e) 80SiO2-RP, and (f) 200SiO2-RP NPs. The white arrows in (d) and (f) point toward the particles showing multiple cell nucleation events per particle. The insets show higher magnification images of nucleating NPs in cross-sectioned PMMA foam cells. The saturation pressure, foaming temperature, and foaming time were 55 bar, 40 °C, and 1 s, respectively.
Interestingly, when examining the 155SiO2-RP NPs embedded in the PMMA foams, we observed that most of these particles are surrounded by a few primary polymer foam cells (see Figure 3d). Of particular interest is that the cell walls in between these primary foam cells are much thinner than the non-nucleated/expanded PMMA matrix surrounding them and that the size of the cells around the 155SiO2-RP particles is fairly uniform. This observation points toward simultaneous multiple heterogeneous nucleation events per added particle. In addition, the thin wall between some foam cells appears to be ruptured or partly disappeared. This result indicates that cell coalescence occurs during the initial stages of nucleation. An explanation for this behavior is provided later.
The occurrence of multiple nucleation events per 155SiO2-RP particles is promising for developing nanocellular foams with increased cell densities. To successfully exploit this possibility, it is of pivotal importance to obtain a deeper understanding of cell nucleation as a function of the surface roughening feature sizes. Therefore, we prepared SiO2-R and SiO2-RP NPs with diameters of ∼80 nm, ∼120 nm, and ∼200 nm to explore this idea. Figure S2 shows SEM images of 80SiO2-R, 120SiO2-R, and 200SiO2-R NPs. It is clear that the feature sizes of the roughened surfaces are different among the particles with different diameters, that is, 80SiO2-R has only a few small protrusions at its surface, while 200SiO2-R reveals numerous larger protruding features (Figure S2). It turned out that for the etching method used the initial Stöber silica NP size has an impact on the particle roughness.51
The SEM images of the cross-sectioned PMMA foams nucleated by 80SiO2-RP (Figure 3e) and 200SiO2-RP (Figure 3f) reveal distinct differences in the foam morphology as well. For instance, the images of the foam nucleated by 200SiO2-RP NPs clearly show the presence of multiple cells around single nucleating particles; however, no multiple cell nucleation events are observed around single 80SiO2-RP NPs.
Below, we present how the nucleation efficiency of SiO2-RP NPs depends on the particle surface roughness for a PMMA foam prepared with a foaming time of 1 s. Figure 4 shows the cell size (4a), cell density (4b), and nucleation efficiency (4c) values for foams nucleated by SiO2-S, SiO2-SP, SiO2-R, and SiO2-RP NPs as a function of the silica particle diameter.
Figure 4.

(a) Foam cell size, (b) cell density, and (c) cell nucleation efficiency for PMMA foams nucleated by SiO2-S, SiO2-SP, SiO2-R, and SiO2-RP NPs with diameters of ∼80 nm, ∼120 nm, ∼155 nm, and ∼200 nm. The saturation pressure, foaming temperature, and foaming time were 55 bar, 40 °C, and 1 s, respectively. The insets in (c) show SEM images of cross-sectioned PMMA foam cells showing the presence of multiple cells around single 155SiO2-RP and 200SiO2-RP NPs.
As can be clearly seen from Figure 4a, the average cell size nucleated with SiO2-RP NPs is smaller than that nucleated with SiO2-S, SiO2-SP and SiO2-R NPs with the same diameter. In addition, upon increasing the particle diameter from 80 to 200 nm, the average cell size decreases, and the cell size distribution becomes narrower. SEM images of cross-sectioned PMMA foams nucleated by 80SiO2-RP particles did not reveal any multiple cell nucleation events per particle (see Figure 3e), while for 120SiO2-RP, this phenomenon was observed for some particles (Figure S4). The decrease in the number of multiple nucleation events is ascribed to the lower surface roughness of the 80-SiO2-RP and 120SiO2-RP NPs, that is, resulting in a reduced number of effective nanocavities. However, for the PMMA foams nucleated by 155SiO2-RP and 200SiO2-RP particles, multiple cell nucleation events were observed, resulting in a higher CO2 consumption during cell growth than that during the growth of the foams without multiple cell nucleation events. This finding explains the reduction in cell size to 160 ± 50 nm and 110 ± 60 nm for the foams nucleated with 155SiO2-RP and 200SiO2-RP particles, respectively.
The occurrence of multiple cell nucleation events for SiO2-RP NPs with various roughnesses has, as expected, a significant effect on the cell density and nucleation efficiency values (Figure 4b,c). Compared to the other particle types, the 200SiO2-RP NPs cause, similar to the 155SiO2-RP NPs, a significant increase in cell density and cell nucleation efficiency. In fact, for the 200SiO2-RP NPs, the nucleation efficiency was ∼6.2, which is the highest nucleation efficiency reported thus far.
In addition, 120SiO2-RP particles have a higher nucleation efficiency than 120SiO2-S, 120SiO2-R, and 120SiO2-SP NPs, as well (see Figure 4c). This result is again qualitatively explained by the presence of the nanocavities. However, 80SiO2-RP NPs show a comparable nucleation efficiency with 80SiO2-SP NPs, which is ascribed to the presence of mainly convex surface domains at its particle surface. Compared to concave surface roughness, convex structures have a higher cell nucleation free energy.47,48
From Figure 4, it is obvious that for the particle sizes evaluated, the SiO2-R NPs have nucleation efficiencies comparable to those of the SiO2-SP NPs, while the SiO2-S particles are the least effective nucleating agents. Overall, it is shown that NPs designed with a concave surface roughness are more efficient for use as nucleation agents than the nearly spherical NPs and that a combination of roughness with a CO2-philic low surface energy PDMS shell has a very large synergetic effect on PMMA foam cell nucleation.
Elucidation of the Multiple Foam Cell Nucleation Mechanism
The synergetic effect of a PDMS shell with the presence of cavities on the nucleation efficiency of SiO2-RP NPs was much higher than expected. In fact, we reasoned that for the higher number of cells to grow, more CO2 as a blowing agent must be available. Thus, the CO2 absorption of PMMA films containing SiO2-S, SiO2-SP, SiO2-R, and SiO2-RP particles with diameters of ∼80 nm, ∼120 nm, ∼155 nm, and ∼200 nm were determined to further elucidate the cell nucleation efficiency as a function of the particle type and size. The results are shown in Figure 5a. Clearly, PMMA films containing SiO2-S and SiO2-SP NPs have no significant differences in CO2 absorption from PMMA films without NPs regardless of the particle diameter. The same trend is observed for 80SiO2-R and 80SiO2-RP NPs. Hence, for these particles, the overall CO2 concentration is not altered, while locally in the PMMA films, the CO2 concentration may vary due to the presence of the PDMS shell. Upon increasing the surface roughness of the particles, for example, for the 120SiO2-R, 155SiO2-R, and 200SiO2-R NPs, the amount of CO2 absorbed is expected to significantly increase. The presence of a PDMS shell increases the amount absorbed even more for these NPs. For instance, for the 200SiO2-R and 200SiO2-RP NPs, the increase in the amount of CO2 absorbed was ∼4.7 wt % and ∼8.8 wt %, respectively, compared to the PMMA film containing no particles (∼18.2 wt %).
Figure 5.

(a) CO2 absorption by PMMA films containing SiO2-S, SiO2-SP, SiO2-R, and SiO2-RP particles with diameters between 80 and 200 nm. The saturation pressure and time were 55 bar and 4 h, respectively. The dashed line shows the CO2 absorption by PMMA containing no NPs. (b) Schematic representation of the nucleation of multiple cells from PDMS-grafted raspberry-like silica NPs. SEM images of cross-sectioned PMMA foams nucleated by 155SiO2-RP NPs for (c) 10 s, (d) 5 s, (e) 1 s, and (f) 0.3 s. In the insets, the foam cell wall around a single nanoparticle is shown. The saturation pressure and foaming temperature were 55 bar and 40 °C, respectively.
This increase in CO2 absorption for the rough particles is ascribed to capillary condensation of CO2 in the nanocavities.64−66 The even larger increase in CO2 absorption for the rough particles with a PDMS shell with diameters of ∼120 nm, ∼155 nm, and ∼200 nm compared to their bare counterparts is attributed to the presence of CO2-philic molecules attached to the nanocavity surfaces, which is known to potentially enhance the absorption of CO2.67−69
The higher physical blowing agent concentration close to the particle surface is favorable for increasing the nucleation rate, and combined with the presence of a low surface energy PDMS shell in the nanocavities, this property leads to a significant enhancement in the foam cell nucleation efficiency of these particles. Figure 5b shows a schematic representation of the assumed mechanism leading to multiple cell nucleation events for raspberry-like PDMS-grafted silica NPs. Upon saturation of PMMA films containing SiO2-RP NPs, CO2 capillary condensation in the cavities results in the presence of significantly increased amounts of CO2 in close proximity to the nucleating particle surface. The nanocavities are energetically favorable nucleating sites and, together with the increased amount of available CO2, result in more than one nucleating event per particle (step A) and subsequent cell growth (step B) and cell coalescence (step C). This mechanism is further experimentally confirmed by the development of the foam cell morphology as a function of the foaming time for PMMA foams nucleated by 155SiO2-RP NPs, as shown in Figure 5c–f. Upon increasing the foaming time, the cell walls between these primary foam cells nucleated from the same single 155SiO2-RP appear to be ruptured or partly disappeared, leading to cell coalescence. In addition, the cell size and cell nucleation efficiency for PMMA foams, nucleated with 155SiO2-S, 155SiO2-SP, 155SiO2-R, and 155SiO2-RP NPs, were also determined as a function of the foaming time (see Figure S5).
Overall, we experimentally demonstrated that multiple cell nucleation events on a single NP can be obtained through the use of PDMS-decorated core–shell raspberry-like NPs as nucleation agents. These designer NPs exhibit significantly higher cell nucleation efficiency in PMMA nanocellular foaming than their bare and nearly spherical counterparts. We also utilized 155SiO2-RP NPs in the CO2-assisted batch foaming of polystyrene, and the corresponding SEM images also revealed the occurrence of multiple cell nucleation events (see Figure S6). Hence, these hybrid core–shell NPs are very promising as highly efficient cell nucleation agents in polymer nanocellular foaming in general.
To fully exploit the high nucleation efficiency of SiO2-RP particles, it is of pivotal importance to find foaming strategies and foam matrixes that both allow the introduction of a high number of cell nuclei and ensure early cell stabilization during the foaming process to prevent cell coalescence. For instance, without optimization of any of the foaming parameters or the composition of the PMMA nanocomposite matrix, we obtained a high cell density of ∼6.6 × 1013 cells cm–3, which is already very close to the order of magnitude of interest (i.e., 1014 cells cm–3). Future work must be directed toward developing new strategies for sufficiently increasing the surface roughness of NPs with a particle diameter <100 nm. This objective is essential since at the same weight percentage of particle loading, a higher number of effective nucleation agents is available when smaller particles are used.
Conclusion
In summary, bare and PDMS-decorated nearly spherical and raspberry-like silica NPs were synthesized and utilized as efficient nucleation agents in CO2-blown PMMA nanocellular foams. Raspberry-like nanoparticles with a thin PDMS shell exhibit a higher nucleation efficiency than their nearly spherical counterparts. Multiple nucleating events per particle were observed for SiO2-RP NPs with core diameters of 155 and 200 nm. The highest nucleation efficiency obtained was 6.2 for 200SiO2-RP NPs. The significantly higher nucleation efficiency (well above unity) of the core–shell raspberry-like nanoparticles is attributed to the synergistic effect between the thin PDMS shell and the presence of cavities on the nanoparticles surface as well as the higher CO2 absorption. This results in an increased physical blowing agent concentration available for nucleation close to the energetically favorable nanocavities serving as nucleating sites. Hence, PDMS-decorated core–shell raspberry-like nanoparticles are very promising for use as an interesting class of highly efficient nucleation agents. To improve our understanding of multiple cell nucleation events, future work should be directed toward the quantification of the relationship between nucleation energy barrier and surface cavity structure dimensions as well optimization of foaming conditions to stabilize the multiple nucleated cells during the early stages of foaming.
Materials and Methods
Materials
PMMA was purchased from Arkema (VM100, i.e., a PMMA-co-EA polymer, ρ = 1.18 g cm–3) (La Garenne-Colombes, France). Absolute ethanol for analysis was purchased from Merck (Darmstad, Germany). Absolute tetrahydrofuran (THF), 2-propanol (99.8%), and chloroform (99.9%) were purchased from Biosolve (Valkenswaard, The Netherlands). Monoglycidyl ether-terminated poly(dimethylsilane) (PDMS-G) (Mw = 1000 g mol–1), ammonium hydroxide solution (28–30%), (3-aminopropyl)triethoxysilane (APTES, ≥98%), sodium borohydride purum (NaBH4, p.a., ≥96%, gas-volumetric), polyvinylpyrrolidone with a molar mass of 40,000 g mol–1 (PVP40), and tetraethyl orthosilicate (TEOS, ≥99.0%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Milli-Q water was produced by a Millipore Synergy system (Billerica, MA, USA). Unless otherwise mentioned, all other chemicals were used as received.
Stöber Silica Nanoparticle (SiO2) Synthesis
SiO2 particles with diameters of ∼80 nm, ∼120 nm, ∼155 nm, ∼180 nm, ∼200 nm, and ∼310 nm were prepared by the Stöber method. To prepare NPs with a diameter of ∼155 nm, 100 mL of ethanol was mixed with 8 mL of Milli-Q water and 10 mL of TEOS in the presence of 5 mL of ammonium hydroxide while stirring at 500 rpm at 50 °C. After 4.5 h, the SiO2 dispersion obtained was centrifuged at 10,000 rpm for 30 min. Subsequently, the collected SiO2 was redispersed in ethanol and centrifuged again. This washing step was repeated 2 more times, followed by vacuum-drying the collected SiO2 nanoparticles at room temperature for 12 h. To prepare NPs with a diameter of ∼80 nm, 168 mL of ethanol was mixed with 28 mL of Milli-Q water and 30 mL of TEOS in the presence of 2 mL of ammonium hydroxide while stirring for 1.5 h at 500 rpm at room temperature. To synthesize the ∼120 nm particles, 100 mL of ethanol was mixed with 8 mL of Milli-Q water and 5 mL of TEOS in a round-bottom flask stirring at 500 rpm, and subsequently 5 mL of ammonium hydroxide was added and reacted for 3 h at 50 °C. To synthesize the ∼180 nm particles, 100 mL of ethanol was mixed with 8 mL of Milli-Q water and 10 mL of TEOS in the presence of 5.3 mL of ammonium hydroxide in a 250 mL round-bottom flask while stirring at 500 rpm. The reaction was conducted for 4.5 h at 50 °C. To synthesize the ∼200 nm particles, 100 mL of ethanol was mixed with 8 mL of Milli-Q water and 10 mL of TEOS in the presence of 5.6 mL of ammonium hydroxide in a 250 mL round-bottom flask while stirring at 500 rpm. The reaction was conducted for 4.5 h at 50 °C. To synthesize the ∼310 nm particles, 100 mL of ethanol was mixed with 8 mL of Milli-Q water and 10 mL of TEOS in a round-bottom flask stirring at 500 rpm, and subsequently, 7.0 mL of ammonium hydroxide was added and reacted for 4.5 h at 50 °C. The collection, washing, and drying steps of these nanoparticles were the same as those described for the ∼155 nm nanoparticles.
Nanoparticles Hydrolysis
To introduce silanol groups on the surface of the prepared SiO2 nanoparticles, the particles were redispersed in Milli-Q water by sonication (BRANSON 2510, Canada) for 1 h. Subsequently, hydrochloric acid was added to the dispersion while stirring at 500 rpm until the pH of the solution reached a value of approximately 1. After 4 h, the dispersion was centrifuged at 10,000 rpm for 30 min. The collected nanoparticles were redispersed in Milli-Q water and centrifuged again. This washing step was repeated 2 more times, followed by drying the silanol-functionalized nanoparticles (SiO2-OH) under vacuum at room temperature for 12 h.
APTES Modification of Silica Nanoparticles
First, 1.5 g of SiO2-OH nanoparticles were redispersed in 50 mL of ethanol, followed by the addition of 7.5 mL of APTES. The dispersion was left stirring at 500 rpm at room temperature for 17 h. The APTES-functionalized nanoparticles (SiO2-NH2) were collected by centrifugation at 10,000 rpm for 30 min, redispersed in ethanol, and centrifuged again. This washing step was repeated 2 more times, followed by drying the collected SiO2-NH2 nanoparticles under vacuum at room temperature for 12 h.
PDMS-G Grafting to Silica Nanoparticles
First, 1.0 g of SiO2-NH2 NPs was redispersed in 20.5 mL of THF and 15 g of PDMS-G while stirring at 500 rpm for 1 h, followed by sonication for 1 h. Subsequently, THF was removed by rotary evaporation, and the resulting silica nanoparticle dispersion in PDMS-G was immersed in an oil bath thermostated at 80 °C for 17 h. After cooling to room temperature, the reaction mixture was washed with THF and centrifuged at 10,000 rpm for 30 min. This washing step was repeated 2 more times, followed by vacuum-drying the PDMS-G-grafted silica nanoparticles (SiO2-SP) at room temperature for 12 h.
Synthesis and Modification of Raspberry-like Nanoparticles
To synthesize NPs with enhanced surface roughness of ∼80 nm, 0.3 g of Stöber silica NPs of ∼80 nm and 0.25 g of PVP were redispersed in 10 mL of Milli-Q water by sonication for 30 min. Subsequently, 0.6 g of NaBH4 was added to the dispersion while stirring at 500 rpm at room temperature for another 30 min. The resulting mixture was reacted at 50 °C for 1 h and 15 min. Stöber silica nanoparticles with diameters of ∼155 nm, ∼180 nm, and ∼310 nm were used to synthesize surface-roughened nanoparticles of ∼120 nm, ∼155 nm, and ∼200 nm, respectively, and the reactions were conducted for 3 h and 20 min, 3.5 h, and 6 h, respectively. The other steps for the synthesis of these surface-roughened NPs (with diameters from 120 to 200 nm) were the same as described for the synthesis of rough surface NPs of ∼80 nm. Subsequently, the obtained NPs featuring rough surfaces were collected by centrifugation at 10,000 rpm for 30 min, redispersed in Milli-Q water, and centrifuged again. This washing step was repeated 2 more times, followed by drying the collected nanoparticles under vacuum at room temperature for 12 h. The processes of amino functionalization of and PDMS-G grafting to the surface-roughened nanoparticles were the same as those described for the Stöber silica nanoparticles.
Nanocomposite Preparation
Nanocomposites were prepared by dispersing the same amount of (functionalized) silica nanoparticles (1.07 × 1013 cm–3) in PMMA with a mini-extruder (DSM Xplore, The Netherlands). In a typical procedure, a dry blend of nanoparticles and PMMA was fed to the extruder followed by internal mixing for 3 min. The barrel temperature was set to 155 °C, and the screw speed was 100 rpm. Subsequently, the PMMA nanocomposite was collected and left to cool to room temperature.
Film Preparation
A hot press (Fontijne, The Netherlands) was used to press ∼0.2 mm-thick nanocomposite films in a mold (4 × 3 cm). The press temperature, applied load, and press time were 180 °C, 250 kN, and 10 min, respectively.
Batch Foaming of Nanocomposite Films
The nanocomposite PMMA films were saturated with CO2 (55 bar) in an autoclave for 4 h at room temperature followed by rapid depressurization. Subsequently, the PMMA nanocomposite films were immersed in a water bath thermostated at 40 °C for different foaming times (0.3 s, 1 s, 5 s, 10 s, or 30 s), after which the samples were quenched in an ice bath for 30 min. The samples were left to dry in air for at least 12 h prior to further analysis.
Fourier Transform Infrared Spectroscopy
FTIR spectra were collected with a Bruker ALPHA single attenuated total reflection (ATR) FTIR spectrometer equipped with a single reflection ATR crystal (Bruker Optic GmbH, Ettlingen, Germany). The spectra were collected in the range of 400–4000 cm–1 (spectral resolution of 4 cm–1, 128 scans). Background spectra were recorded against air.
Thermogravimetric Analysis
The weight loss of the (modified) particles as a function of temperature was measured with a TGA400 (PerkinElmer, Inc., Waltham, MA, USA). A sample weighing ∼3–6 mg was loaded into a platinum pan, and the temperature was set to 50 °C to stabilize. Subsequently, the sample was heated to 900 °C at a heating rate of 20 °C min–1. The applied air flow was 20 mL min–1.
Transmission Electron Microscopy
TEM was performed with an FEI/Philips CM300 system (Eindhoven, The Netherlands). Diluted particle dispersions in THF were deposited on the carbon side of a carbon/copper grid (HC200-Cu) (EMS, Germany) prior to imaging. Images were obtained in the bright-field mode with a 300 kV acceleration voltage.
Scanning Electron Microscopy
For SEM, a high-resolution scanning electron microscope (JEOL Field Emission JSM-633OF, JEOL Benelux, Nieuw-Vennep, The Netherlands) was employed at an acceleration voltage of 5 keV. Prior to analysis, the nanocomposite foams were freeze fractured following cooling in liquid nitrogen for 5 min.
Surface Area Measurement for Nanoparticles
The specific surface area of the nanoparticles was measured from the nitrogen adsorption isotherm obtained at 300 °C using an ASAP 2010 sorptometer (Micromeritics). Approximately 100 mg of nanoparticle powder was used for each measurement.
CO2 Absorption Measurement
To measure the absorption of CO2, PMMA nanocomposite films containing the same amount of the respective nanoparticles were saturated in CO2 at 55 bar for 4 h. The weights of the PMMA (nanocomposite) films before and after CO2 saturation were measured.
Calculation of PDMS Grafting Density
The grafting density (D) of the PDMS-decorated NP surface was determined according to eq 1:
| 1 |
where K = 6.02 × 1023 mol–1, which is Avogadro’s number, and m is the weight of PDMS grafted per gram of NPs (determined from TGA), M = 1000 g·mol–1, which is the molar mass of the PDMS used, and S is the surface area per gram of NPs, which was determined by BET.
Calculation of Cell Density and Nucleation Efficiency
The cell size and cell density were obtained by analyzing the obtained cross-sectional SEM images. The cell density Nv of the foams was calculated according to Kumar’s theoretical approximation.70 No direct measurements of cell dimensions over the micrograph are required in this method; only the micrograph area (A) and the total number of cells (n) contained therein are measured. Together with the magnification factor of the micrograph (M), the values enabling Nv to be calculated according to eq 2:
| 2 |
By combining Nv with the volume expansion ratio (B) of nanocomposite films after foaming, the cell numbers per cm3 of unfoamed materials (N) can be calculated according to eq 3:
| 3 |
The cell densities mentioned in the main text are all refer to N. In addition, the nucleation efficiency (f) of nanoparticles during foaming can be calculated as
| 4 |
where C is the number of nanoparticles per cm3 (i.e., 1.07 × 1013) used for foaming.
Figure S7 shows how we calculated the number of cells in the occasion of multiple cell nucleating events occurring around a particle.
Acknowledgments
The authors would like to thank the MESA+ Institute for Nanotechnology of the University of Twente for financial support. S.L. acknowledges the China Scholarship Council for funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b06837.
BET results, SEM images for SiO2-R nanoparticles with different diameters, nanoparticles dispersion in PMMA, and cross-sectioned PMMA foams nucleated by 120SiO2-RP. Cell size and nucleation efficiency as a function of foaming time, and SEM images of cross-sectioned polystyrene foams nucleated by 150SiO2-RP. SEM images show the number of cells in the occasion of multiple cell nucleating events occurring around a particle (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Notario B.; Pinto J.; Rodriguez-Perez M. Nanoporous Polymeric Materials: ANew Class of Materials with Enhanced Properties. Prog. Mater. Sci. 2016, 78, 93–139. 10.1016/j.pmatsci.2016.02.002. [DOI] [Google Scholar]
- Forest C.; Chaumont P.; Cassagnau P.; Swoboda B.; Sonntag P. Polymer Nano-Foams for Insulating Applications Prepared from CO2 Foaming. Prog. Polym. Sci. 2015, 41, 122–145. 10.1016/j.progpolymsci.2014.07.001. [DOI] [Google Scholar]
- Liu S.; Duvigneau J.; Vancso G. J. Nanocellular Polymer Foams as Promising High Performance Thermal Insulation Materials. Eur. Polym. J. 2015, 65, 33–45. 10.1016/j.eurpolymj.2015.01.039. [DOI] [Google Scholar]
- Costeux S. CO2-Blown Nanocellular Foams. J. Appl. Polym. Sci. 2014, 131, 41293. 10.1002/app.41293. [DOI] [Google Scholar]
- Chen L.; Rende D.; Schadler L. S.; Ozisik R. Polymer Nanocomposite Foams. J. Mater. Chem. A 2013, 1, 3837–3850. 10.1039/c2ta00086e. [DOI] [Google Scholar]
- Eaves D.Handbook of Polymer Foams; Rapra Technology Ltd.: Shrewsbury, 2004; pp 1–6. [Google Scholar]
- Notario B.; Pinto J.; Solorzano E.; De Saja J. A.; Dumon M.; Rodríguez-Pérez M. A. Experimental Validation of the Knudsen Effect in Nanocellular Polymeric Foams. Polymer 2015, 56, 57–67. 10.1016/j.polymer.2014.10.006. [DOI] [Google Scholar]
- Pinto J.; Notario B.; Verdejo R.; Dumon M.; Costeux S.; Rodriguez-Perez M. A. Molecular Confinement of Solid and Gaseous Phases of Self-Standing Bulk Nanoporous Polymers Inducing Enhanced and Unexpected Physical Properties. Polymer 2017, 113, 27–33. 10.1016/j.polymer.2017.02.046. [DOI] [Google Scholar]
- Lee S.-T.Polymeric Foams: Technology and Developments in Regulation, Process, and Products; CRC Press: Boca Raton, 2009; pp 20–25. [Google Scholar]
- Notario B.; Pinto J.; Rodríguez-Pérez M. Towards a New Generation of Polymeric Foams: PMMA Nanocellular Foams with Enhanced Physical Properties. Polymer 2015, 63, 116–126. 10.1016/j.polymer.2015.03.003. [DOI] [Google Scholar]
- Li L.; Shen X.; Hong S. W.; Hayward R. C.; Russell T. P. Fabrication of Co-Continuous Nanostructured and Porous Polymer Membranes: Spinodal Decomposition of Homopolymer and Random Copolymer Blends. Angew. Chem., Int. Ed. 2012, 51, 4089–4094. 10.1002/anie.201107867. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Chang D.; Liu J.; Luo Y. Conducting Polymer Aerogels from Supercritical CO2 Drying PEDOT-PSS Hydrogels. J. Mater. Chem. 2010, 20, 5080–5085. 10.1039/c0jm00050g. [DOI] [Google Scholar]
- Hedrick J. L.; Miller R. D.; Hawker C. J.; Carter K. R.; Volksen W.; Yoon D. Y.; Trollsås M. Templating Nanoporosity in Thin-Film Dielectric Insulators. Adv. Mater. 1998, 10, 1049–1053. . [DOI] [Google Scholar]
- Park S. H.; Xia Y. Macroporous Membranes with Highly Ordered and Three-Dimensionally Interconnected Spherical Pores. Adv. Mater. 1998, 10, 1045–1048. . [DOI] [Google Scholar]
- Du Fresne Von Hohenesche C.; Schmidt D.; Schadler V. Nanoporous Melamine-Formaldehyde Gels by Microemulsion Templating. Chem. Mater. 2008, 20, 6124–6129. 10.1021/cm8015319. [DOI] [Google Scholar]
- Bledzki A. K.; Faruk O. Effects of the Chemical Foaming Agents, Injection Parameters, and Melt-Flow Index on the Microstructure and Mechanical Properties of Microcellular Injection-Molded Wood-Fiber/Polypropylene Composites. J. Appl. Polym. Sci. 2005, 97, 1090–1096. 10.1002/app.21685. [DOI] [Google Scholar]
- Klempner D.Handbook of Polymeric Foams and Foam Technology; Hanser: Munich, 1991; pp 6–13. [Google Scholar]
- Zeng C.; Han X.; Lee L. J.; Koelling K. W.; Tomasko D. L. Polymer-Clay Nanocomposite Foams Prepared Using Carbon Dioxide. Adv. Mater. 2003, 15, 1743–1747. 10.1002/adma.200305065. [DOI] [Google Scholar]
- Park C. B.; Behravesh A. H.; Venter R. D. Low Density Microcellular Foam Processing in Extrusion Using CO2. Polym. Eng. Sci. 1998, 38, 1812–1823. 10.1002/pen.10351. [DOI] [Google Scholar]
- Tomasko D. L.; Li H.; Liu D.; Han X.; Wingert M. J.; Lee L. J.; Koelling K. W. A Review of CO2 Applications in the Processing of Polymers. Ind. Eng. Chem. Res. 2003, 42, 6431–6456. 10.1021/ie030199z. [DOI] [Google Scholar]
- Jacobs L. J.; Kemmere M. F.; Keurentjes J. T. Sustainable Polymer Foaming Using High Pressure Carbon Dioxide: A Review on Fundamentals, Processes and Applications. Green Chem. 2008, 10, 731–738. 10.1039/b801895b. [DOI] [Google Scholar]
- Aram E.; Mehdipour-Ataei S. A Review on the Micro-and Nanoporous Polymeric Foams: Preparation and Properties. Int. J. Polym. Mater. 2016, 65, 358–375. 10.1080/00914037.2015.1129948. [DOI] [Google Scholar]
- Costeux S.; Lantz D. R.; Beaudoin D. A.; Barger M. A.. Continuous Process for Extruding Nanoporous Foam. U.S. Patent 9145478B2, 2015.
- Sandler J. K. W.; Francis T.; Lopes P. M. S.. Nanoporous Polymer Foams. U.S. Patent 8529808B2, 2013.
- Costeux S.; Zhu L. Low Density Thermoplastic Nanofoams Nucleated by Nanoparticles. Polymer 2013, 54, 2785–2795. 10.1016/j.polymer.2013.03.052. [DOI] [Google Scholar]
- Costeux S.; Bunker S. P.; Jeon H. K. Homogeneous Nanocellular Foams from Styrenic-Acrylic Polymer Blends. J. Mater. Res. 2013, 28, 2351–2365. 10.1557/jmr.2013.100. [DOI] [Google Scholar]
- Liao Z.-E.; Yeh S.-K.; Chu C.-C.; Tseng T.-W. Critical Parameters of Generating PMMA Nanocellular Foam. Annu. Technol. Conf.-Soc. Plast. Eng. 2016, 1773–1778. [Google Scholar]
- Tammaro D.; Astarita A.; Di Maio E.; Iannace S. Polystyrene Foaming at High Pressure Drop Rates. Ind. Eng. Chem. Res. 2016, 55, 5696–5701. 10.1021/acs.iecr.5b04911. [DOI] [Google Scholar]
- Pinto J.; Dumon M.; Pedros M.; Reglero J.; Rodriguez-Perez M. A. Nanocellular CO2 Foaming of PMMA Assisted by Block Copolymer Nanostructuration. Chem. Eng. J. 2014, 243, 428–435. 10.1016/j.cej.2014.01.021. [DOI] [Google Scholar]
- Li L.; Nemoto T.; Sugiyama K.; Yokoyama H. CO2 Foaming in Thin Films of Block Copolymer Containing Fluorinated Blocks. Macromolecules 2006, 39, 4746–4755. 10.1021/ma060325l. [DOI] [Google Scholar]
- Spitael P.; Macosko C. W.; McClurg R. B. Block Copolymer Micelles for Nucleation of Microcellular Thermoplastic Foams. Macromolecules 2004, 37, 6874–6882. 10.1021/ma049712q. [DOI] [Google Scholar]
- Yokoyama H.; Sugiyama K. Nanocellular Structures in Block Copolymers with CO2-Philic Blocks Using CO2 as a Blowing Agent: Crossover from Micro-to Nanocellular Structures with Depressurization Temperature. Macromolecules 2005, 38, 10516–10522. 10.1021/ma051757j. [DOI] [Google Scholar]
- Liu S.; Zoetebier B.; Hulsman L.; Zhang Y.; Duvigneau J.; Vancso G. J. Nanocellular Polymer Foams Nucleated by Core-Shell Nanoparticles. Polymer 2016, 104, 22–30. 10.1016/j.polymer.2016.09.016. [DOI] [Google Scholar]
- Lee L. J.; Zeng C.; Cao X.; Han X.; Shen J.; Xu G. Polymer Nanocomposite Foams. Compos. Sci. Technol. 2005, 65, 2344–2363. 10.1016/j.compscitech.2005.06.016. [DOI] [Google Scholar]
- Ji G.; Zhai W.; Lin D.; Ren Q.; Zheng W.; Jung D. W. Microcellular Foaming of Poly(Lactic Acid)/Silica Nanocomposites in Compressed CO2: Critical Influence of Crystallite Size on Cell Morphology and Foam Expansion. Ind. Eng. Chem. Res. 2013, 52, 6390–6398. 10.1021/ie302281c. [DOI] [Google Scholar]
- Rende D.; Schadler L. S.; Ozisik R. Controlling Foam Morphology of Poly(Methyl Methacrylate) via Surface Chemistry and Concentration of Silica Nanoparticles and Supercritical Carbon Dioxide Process Parameters. J. Chem. 2013, 2013, 1–13. 10.1155/2013/864926. [DOI] [Google Scholar]
- Zhai W.; Yu J.; Wu L.; Ma W.; He J. Heterogeneous Nucleation Uniformizing Cell Size Distribution in Microcellular Nanocomposites Foams. Polymer 2006, 47, 7580–7589. 10.1016/j.polymer.2006.08.034. [DOI] [Google Scholar]
- Yuan H.; Xiong Y.; Luo G.; Shen Q.; Zhang L. The Influence of In Situ Synthesized Nanoparticles on Microstructure and Compression Properties of Polymer Foams during Supercritical Carbon Dioxide Foaming. J. Appl. Polym. Sci. 2017, 134, 44629. 10.1002/app.44629. [DOI] [Google Scholar]
- Liberman A.; Mendez N.; Trogler W. C.; Kummel A. C. Synthesis and Surface Functionalization of Silica Nanoparticles for Nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. 10.1016/j.surfrep.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagwe R. P.; Hilliard L. R.; Tan W. Surface Modification of Silica Nanoparticles to Reduce Aggregation and Nonspecific Binding. Langmuir 2006, 22, 4357–4362. 10.1021/la052797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Sang Y.; Chen F.; Fei Z.; Zhong M. Synthesis of Silica Particles Grafted with Poly(Ionic Liquid) and Their Nucleation Effect on Microcellular Foaming of Polystyrene Using Supercritical Carbon Dioxide. J. Supercrit. Fluids 2012, 62, 197–203. 10.1016/j.supflu.2011.11.024. [DOI] [Google Scholar]
- Siripurapu S.; DeSimone J. M.; Khan S. A.; Spontak R. J. Low-Temperature, Surface-Mediated Foaming of Polymer Films. Adv. Mater. 2004, 16, 989–994. 10.1002/adma.200306068. [DOI] [Google Scholar]
- Goren K.; Chen L.; Schadler L. S.; Ozisik R. Influence of Nanoparticle Surface Chemistry and Size on Supercritical Carbon Dioxide Processed Nanocomposite Foam Morphology. J. Supercrit. Fluids 2010, 51, 420–427. 10.1016/j.supflu.2009.09.007. [DOI] [Google Scholar]
- Miller M. B.; Luebke D. R.; Enick R. M. CO2-Philic Oligomers as Novel Solvents for CO2 Absorption. Energy Fuels 2010, 24, 6214–6219. 10.1021/ef101123e. [DOI] [Google Scholar]
- Wang H.; Li W. Selective Ultrasonic Foaming of Polymer for Biomedical Applications. J. Manuf. Sci. Eng. 2008, 130, 021004. 10.1115/1.2823078. [DOI] [Google Scholar]
- Liu Q.; Zhu Y.; Yang G.; Yang Q. Nucleation Thermodynamics inside Micro/Nanocavity. J. Mater. Sci. Technol. 2008, 24, 183–186. [Google Scholar]
- Qian M.; Ma J. The Characteristics of Heterogeneous Nucleation on Concave Surfaces and Implications for Directed Nucleation or Surface Activity by Surface Nanopatterning. J. Cryst. Growth 2012, 355, 73–77. 10.1016/j.jcrysgro.2012.06.031. [DOI] [Google Scholar]
- Maksimov A. O.; Kaverin A. M.; Baidakov V. G. Heterogeneous Vapor Bubble Nucleation on a Rough Surface. Langmuir 2013, 29, 3924–3934. 10.1021/la400340y. [DOI] [PubMed] [Google Scholar]
- Du X.; Liu X.; Chen H.; He J. Facile Fabrication of Raspberry-Like Composite Nanoparticles and Their Application as Building Blocks for Constructing Superhydrophilic Coatings. J. Phys. Chem. C 2009, 113, 9063–9070. 10.1021/jp9016344. [DOI] [Google Scholar]
- Qian Z.; Zhang Z.; Song L.; Liu H. A Novel Approach to Raspberry-Like Particles for Superhydrophobic Materials. J. Mater. Chem. 2009, 19, 1297–1304. 10.1039/b810808k. [DOI] [Google Scholar]
- Du X.; He J. A Self-Templated Etching Route to Surface-Rough Silica Nanoparticles for Superhydrophobic Coatings. ACS Appl. Mater. Interfaces 2011, 3, 1269–1276. 10.1021/am200079w. [DOI] [PubMed] [Google Scholar]
- Li D.; Zhu Y.; Mao C. One-Pot Synthesis of Surface Roughness Controlled Hollow Silica Spheres with Enhanced Drug Loading and Release Profiles under Ambient Conditions in Aqueous Solutions. J. Mater. Chem. B 2013, 1, 5515–5520. 10.1039/c3tb20733a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H.-J.; Lee Y.-L. Facile Method to Fabricate Raspberry-Like Particulate Films for Superhydrophobic Surfaces. Langmuir 2007, 23, 12687–12692. 10.1021/la702521u. [DOI] [PubMed] [Google Scholar]
- Zakiyan S. E.; Famili M. H. N.; Ako M. Controlling Foam Morphology of Polystyrene via Surface Chemistry, Size and Concentration of Nanosilica Particles. J. Mater. Sci. 2014, 49, 6225–6239. 10.1007/s10853-014-8347-4. [DOI] [Google Scholar]
- Zhang T.; Ge J.; Hu Y.; Zhang Q.; Aloni S.; Yin Y. Formation of Hollow Silica Colloids through a Spontaneous Dissolution-Regrowth Process. Angew. Chem. 2008, 120, 5890–5895. 10.1002/ange.200800927. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Zhang Q.; Ge J.; Goebl J.; Sun M.; Yan Y.; Liu Y.-s.; Chang C.; Guo J.; Yin Y. A Self-Templated Route to Hollow Silica Microspheres. J. Phys. Chem. C 2009, 113, 3168–3175. 10.1021/jp810360a. [DOI] [Google Scholar]
- Musić S.; Filipović-Vinceković N.; Sekovanić L. Precipitation of Amorphous SiO2 Particles and Their Properties. Braz. J. Chem. Eng. 2011, 28, 89–94. 10.1590/S0104-66322011000100011. [DOI] [Google Scholar]
- Kim H.; Kim H.-G.; Kim S.; Kim S. S. PDMS-Silica Composite Membranes with Silane Coupling for Propylene Separation. J. Membr. Sci. 2009, 344, 211–218. 10.1016/j.memsci.2009.08.004. [DOI] [Google Scholar]
- Liu S.; Eijkelenkamp R.; Duvigneau J.; Vancso G. J. Silica-Assisted Nucleation of Polymer Foam Cells with Nanoscopic Dimensions: Impact of Particle Size, Line Tension, and Surface Functionality. ACS Appl. Mater. Interfaces 2017, 9, 37929–37940. 10.1021/acsami.7b11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommes M. Physical Adsorption Characterization of Nanoporous Materials. Chem. Ing. Tech. 2010, 82, 1059–1073. 10.1002/cite.201000064. [DOI] [Google Scholar]
- Rouquerol F.Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Kidlington, Oxford, 2014; pp 237–251. [Google Scholar]
- Thommes M.; Cychosz K. A. Physical Adsorption Characterization of Nanoporous Materials: Progress and Challenges. Adsorption 2014, 20, 233–250. 10.1007/s10450-014-9606-z. [DOI] [Google Scholar]
- Liu S.; Pandey A.; Duvigneau J.; Vancso J.; Snoeijer J. H. Size-Dependent Submerging of Nanoparticles in Polymer Melts: Effect of Line Tension. Macromolecules 2018, 51, 2411–2417. 10.1021/acs.macromol.7b02353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ally J.; Molla S.; Mostowfi F. Condensation in Nanoporous Packed Beds. Langmuir 2016, 32, 4494–4499. 10.1021/acs.langmuir.6b01056. [DOI] [PubMed] [Google Scholar]
- Ullah R.; Atilhan M.; Diab A.; Deniz E.; Aparicio S.; Yavuz C. T. Synthesis, Characterization and Evaluation of Porous Polybenzimidazole Materials for CO2 Adsorption at High Pressures. Adsorption 2016, 22, 247–260. 10.1007/s10450-016-9762-4. [DOI] [Google Scholar]
- Jones W.; Isaac P.; Phillips D. The Adsorption of Carbon Dioxide and Nitrogen at High Pressures by Porous Plugs of Lampblack. Trans. Faraday Soc. 1959, 55, 1953–1958. 10.1039/tf9595501953. [DOI] [Google Scholar]
- Fujiki J.; Yamada H.; Yogo K. Enhanced Adsorption of Carbon Dioxide on Surface-Modified Mesoporous Silica-Supported Tetraethylenepentamine: Role of Surface Chemical Structure. Microporous Mesoporous Mater. 2015, 215, 76–83. 10.1016/j.micromeso.2015.05.037. [DOI] [Google Scholar]
- Sanz-Pérez E.; Arencibia A.; Sanz R.; Calleja G. An Investigation of the Textural Properties of Mesostructured Silica-Based Adsorbents for Predicting CO2 Adsorption Capacity. RSC Adv. 2015, 5, 103147–103154. 10.1039/C5RA19105J. [DOI] [Google Scholar]
- Fujiki J.; Yogo K. The Increased CO2 Adsorption Performance of Chitosan-Derived Activated Carbons with Nitrogen-Doping. Chem. Commun. 2016, 52, 186–189. 10.1039/C5CC06934C. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Suh N. P. A Process for Making Microcellular Thermoplastic Parts. Polym. Eng. Sci. 1990, 30, 1323–1329. 10.1002/pen.760302010. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.