

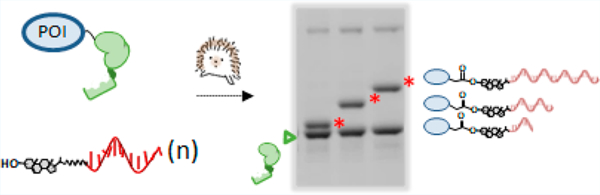
Abstract
Hedgehog (Hh) precursor proteins contain an autoprocessing domain called HhC whose native function is protein cleavage and C-terminal glycine sterylation. The transformation catalyzed by HhC occurs in cis from a precursor protein and exhibits wide tolerance toward both sterol and protein substrates. Here, we repurpose HhC as a 1:1 protein−nucleic acid ligase, with the sterol serving as a molecular linker. A procedure is described for preparing HhC-active sterylated DNA, called steramers, using aqueous compatible chemistry and commercial reagents. Steramers have KM values of 7−11 μM and reaction t1/2 values of ∼10 min. Modularity of the HhC/steramer method is demonstrated using four different proteins along with structured and unstructured sterylated nucleic acids. The resulting protein−DNA conjugates retain the native solution properties and biochemical function. Unlike self-tagging domains, HhC does not remain fused to the conjugate; rather, enzymatic activity is mechanistically coupled to conjugate release. That unique feature of HhC, coupled with efficient kinetics and substrate tolerance, may ease access and open new applications for these suprabiological chimeras.
Graphical Abstract
General methods to prepare chimeric protein nucleic acids (ProNAcs) have been sought for various applications, from probing immune system function to editing genetic information.1–5 However, without a broadly tolerant enzyme for this ligation, no general technique has emerged. Early protocols to join the two entities made use of spontaneous coupling chemistry involving side chain functional groups of cysteine and lysine residues. Disulfide bonding, for example, linked antibody to oligonucleotide for ultrasensitive bioanalytical detection.6 This and other pioneering studies brought attention to the potential for synergistic function with protein and nucleic acid in the conjugate state.7 However, inefficiencies of spontaneous coupling and limited regiospecificity presented obstacles to downstream applications. Chemical approaches have continued to evolve,8–10 and at the same time interest has grown in alternatives like biocatalysis.1,11–21
Here we report a new approach for protein−DNA conjugation that takes advantage of a promiscuous autoprocessing domain HhC/Hog, hereafter referred to as HhC, found in the hedgehog family of proteins. The native activity of HhC (25 kDa) is protein C-terminal glycine sterylation.22 As depicted in Figure 1, the reaction occurs in cis, starting with HhC, and its protein substrate, the hedgehog ligand (HhN), joined in a single polypeptide. Sterylation takes place at the protein substrate/HhC junction through an acyl relay analogous to self-splicing inteins. The reaction is complete when HhC is displaced by a sterol molecule.23 Thus, substrate sterylation and cleavage from HhC are mechanistically coupled.
Figure 1.
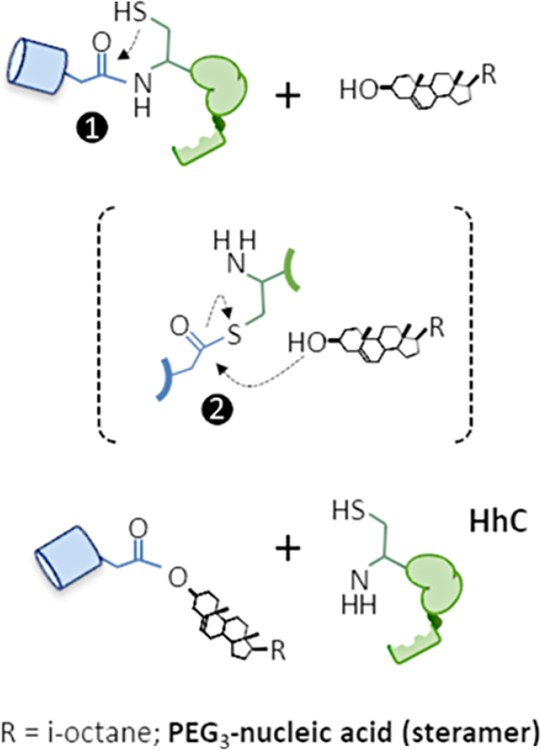
Autoprocessing activity of native and repurposed hedgehog domain, HhC. Precursor polypeptide is composed of substrate protein (blue) and HhC (green). Following peptide rearrangement at a conserved Gly-Cys to form an internal thioester intermediate, step 1, the two fragments split apart in step 2 through displacement by cholesterol in the native transformation, or by sterylated nucleic acid described herein. All catalytic activity resides in HhC.
The transformation can be reconstituted in vitro using a simple buffer/detergent system without accessory proteins or energy source; all catalytic activity is encoded in HhC.23,24
We and others have found that HhC maintains activity toward analogs of cholesterol25–28 and toward proteins unrelated to its natural substrate as long as the substrate protein’s C-terminal residue is glycine.24,29 We sought to harness that promiscuity with sterylated oligonucleotides, which we call steramers. In steramers, the aliphatic side chain of cholesterol is replaced by PEGylated single-stranded DNA. Substantial differences in polarity, molecular weight, and solubility notwithstanding, HhC accepts these hybrid molecules as efficient substrates, generating site-specific, 1:1, ProNAcs. Because HhC is an enzymatic agent, the overall transformation is nearly traceless.
RESULTS AND DISCUSSION
In our ongoing screening of sterols as alternative HhC substrates,27,28,30 we were intrigued by the robust activity and the potential utility of pregnenolone, a naturally occurring 21 carbon sterol with a ketone group at carbon 20 (Figure 2A). Steady state kinetic analysis indicated that the maximum rate of reaction of pregnenolone is within measurement error of the cholesterol rate, and exhibits reasonable affinity (KM) of 11 μM, compared with cholesterol’s KM of 1 μM. In addition, we noted chemical bifunctionality in the pregnenolone structure: the 3-β hydroxyl group and steroid skeleton provide critical elements for HhC recognition, while the ketone group offers a site for chemical elaboration. Ketone groups react with alkoxy amines to form oximes through aqueous compatible chemistry used widely in bioconjugation.31–33 From these considerations, we hypothesized that pregnenolone might be exploited as a molecular linker for synthetic HhC catalyzed conjugations. With a specific view toward ProNAc biocatalysis, we pursued the idea of using pregnenolone to create sterol-nucleic acids. These so-called steramers were designed with a bis-alkoxyamine PEG3 moiety as a spacer to join the ketone group of pregnenolone to aldehyde-modified oligonucleotide. All reagents were available commercially, including bis-alkoxyamine PEG3, and adding a single aldehyde moiety to oligonucleotides is considered a standard modification.34
Figure 2.
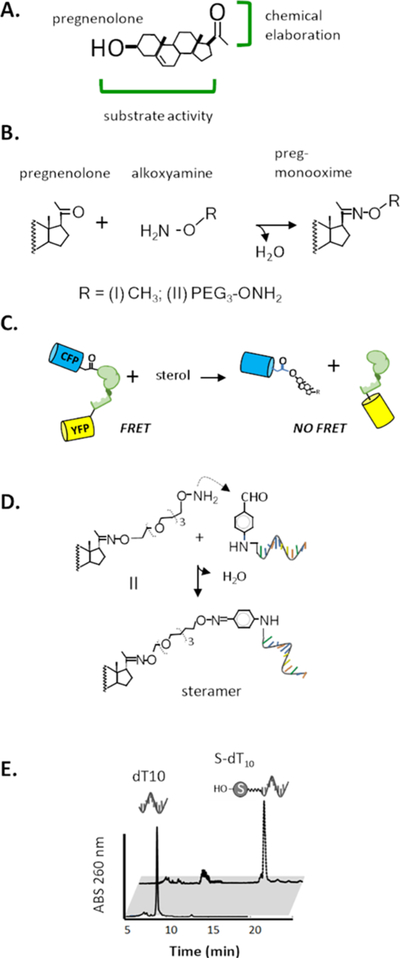
Components for protein nucleic acid conjugation using HhC biocatalysis. (A) Pregnenolone, a bifunctional sterol. (B) Oxime formation: condensation of pregnenolone and alkoxyamines. (C) FRET assay for HhC activity: optical reporter monitors HhC sterylation activity by loss of FRET from precursor, C−H−Y. (D) Steramers: sterylation of benzaldehyde modified nucleic acid using oxime bond chemistry. (E) Separation of steramer from starting oligonucleotide by RP-HPLC.
We first established substrate activity of pregnenolone mono-oximes by generating adducts of pregnenolone with methoxyamine and bis-alkoxyamine PEG3 (Figure 2B). Compounds I and II, respectively, were prepared in one step and >80% yield in 9/1 methanol/triethylammonium acetate (TEAA), pH 7, at room temperature. NMR studies confirmed their hydrolytic stability, with t1/2 of ∼40 h for oxime hydrolysis at 23 °C, pH 7 (Supporting Information).32 Steady state substrate kinetics of HhC toward I and II were determined by a continuous assay using a FRET-active construct, C−H−Y, where Cyan and Yellow fluorescent proteins flank HhC.24 Catalytic turnover separates H−Y from the C-sterol so that initial rates of FRET loss report HhC activity (Figure 2C). Pregnenolone oximes I and II were found to react at rates approximating those of unmodified pregnenolone (Table 1). Observed KM values were in the low micromolar range, and under saturating sterol concentration (50 μM), the t1/2 for conjugations was ∼10 min. To corroborate those results, we carried out HhC reactions using a precursor protein whose activity was monitored by SDS-PAGE.35 In this construct of SHhN-DHhC, DHhC is the HhC from Drosophila hedgehog, the same fragment used in C−H−Y, while SHhN is the Sonic Hh signaling protein from human. SHhN-DHhC (45 kDa) is autoprocessed into DHhC (25 kDa) and the respective SHhN−sterol conjugate (≥20 kDa). As judged by SDS-PAGE, reactions with SHhN-DHhC proceeded to near-completion with natural and synthetic sterols in accord with the FRET assay (vide infra and Supporting Information).
Table 1.
Kinetic Parameters of Sterol and Steramer Substrates for Protein−DNA Conjugation Catalyzed by HhC
| substrate | kmax(s−1) (×10−3) | t1/2(min) | Km (μM) | relative proficiency |
|---|---|---|---|---|
| cholesterol | 1.6 | 7.4 | 0.70 | |
| pregnenolone | 1.6 | 7.3 | 11 | 1 |
| preg-methoxime (I) | 2.8 | 4.2 | 5.3 | 3.6 |
| II | 1.2 | 9.3 | 3.0 | 2.8 |
| S-dT10 | 1.8 | 6.4 | 9.2 | 1.3 |
| S-dT30 | 2.0 | 5.8 | 7.1 | 1.9 |
| S-dT39 | 1.1 | 11 | 11 | 0.7 |
We next assembled prototype steramers by oxime coupling II to commercial, 5′-benzaldehyde modified single stranded, deoxythymidine oligonucleotides of 10, 20, and 30 nucleotides (Figure 2D). Oxime formation was carried out as illustrated above, at room temperature in 9:1, methanol/TEAA buffer. Following 16 h incubation, oligonucleotide sterylation was assessed with RP-HPLC by comparing elution peaks of unsterylated and sterylated oligonucleotide. Sterylation extended the oligonucleotide retention time by >10 min. Results consistently showed >90% conversion of oligonucleotide to steramer (Figure 2E). Excess II was then removed by n-butanol extraction.36 After lyophilization to remove residual n-butanol, steramers readily dissolved in Tris-EDTA (TE) buffer.
Manifesting promiscuity, HhC readily accepted prototype steramers, S-dT10, S-dT20, and S-dT30 as alternative substrates for ProNAc biocatalysis. We assessed substrate activity of the steramers first by using SDS-PAGE which allowed direct detection of precursor consumption and conjugate formation. In addition to SHhN-DHhC used above, we explored reaction scope with a second precursor, T4Lysozyme-DHhC, comprising substrate protein unrelated to Hh. In the SDS-PAGE of Figure 3A, following control reactions (lanes 1−3), samples in the final three lanes show precursor protein conjugations with S-dT10, S-dT20, and S-dT30 (bands highlighted with red stars). Comparing the results from two different precursor proteins, we noted the following: first, for both native and heterologous HhC precursors, the extents of conjugation with substrate cholesterol, II, and the three steramers were similar, suggesting substrate modularity and compatibility with the steramer’s unnatural polyanionic tail of appended oligonucleotide; second, the apparent molecular weight of the conjugates is consistent with 1:1, stoichiometric ProNAcs; last, the ProNAc oxime linkages appeared stable as indicated by their integrity during SDS-PAGE, which included heat denaturation (95 °C, 2 min).
Figure 3.

Protein nucleic acid (ProNAc) conjugation catalyzed by HhC using substrate steramers. (A) Gel based validation of 1:1 ProNAc synthesis. Reactions of native-like hedgehog precursor, SHhN-DHhC, and heterologous precursor, T4Lysozyme-DHhC, at 2 μM following incubation in buffer control (NS, no sterol), and substrates: cholesterol (chol, C), II, steramers, present at initial concentration of 50 μM. Molecular weights of the conjugates (red asterisks): SHhN:S-dT10, 23.3 kDa; SHhN:S-dT20, 26.7 kDa SHhN:S-dT30, 29.4 kDa; T4Lysozyme:S-dT10, 22.8 kDa; T4Lysozyme:S-dT20, 26.1 kDa; T4Lysozyme:S-dT30, 29.4 kDa. Product DHhC is marked with a green triangle. Percent precursor consumed, %PC. (B) Conjugation kinetics from FRET assay using C−H−Y precursor. Michaelis−Menten plot (upper) and representative kinetic trace (lower). Lower plot shows reaction using 50 μM substrate and 0.1 μM C−H−Y.
With substrate activity and conjugate stability established, we determined catalytic constants using the FRET assay. Steramers S-dT10 and S-dT30 were added to C−H−Y at 10 different concentrations, ranging from 58 to 0.11 μM. Initial rates of reaction measured by FRET signal loss were plotted as a function of steramer concentration and showed saturation behavior, allowing determination of KM value (Figure 3B, upper). At the highest steramer concentration, the progress curves conformed to a first order exponential decay, where the derived rate constant is kmax (Figure 3B, lower, and Table 1). Kinetic values for the steramers were in agreement with those of pregnenolone, with nearly identical kmax values and KM values (Table 1). Together, these results demonstrated catalytic efficiency with composite sterol-ssDNAs as alternative substrates for HhC biocatalysis and encouraged further exploration.
Steramers containing DNA with secondary structure also proved to be suitable substrates for HhC biocatalysis. Our prototype steramers, with varying tracks of deoxythymidine, are expected to have simple, elongated structures. Oligonucleotides with higher order structure, i.e., Watson−Crick hydrogen bonding interactions, offer a broader array of functionality. We therefore investigated the compatibility of a hairpin forming (Tm = 60 °C), 39-mer DNA oligonucleotide.37 As above, we purchased this ssDNA oligonucleotide with a 5′-benzaldehyde modification, then through oxime formation, coupled it to II, yielding steramer, S-dHP39. We tested S-dHP39 as a substrate for ProNAc generation using the FRET-active precursor, C−H−Y. The added structural complexity of S-dHP39 showed only a modest effect on the maximum rate, slower by ∼2-fold, and slight weakening of ground state binding (<1.5-fold) in comparison with the deoxythymidine steramers (Table 1). Using an initial steramer concentration of 50 μM, the reaction appeared complete after 30 min (Figure 4A). Michaelis−Menten plot of initial rates versus steramer concentration also appeared normal (Figure 4B). We conclude that the oligonucleotide component of the steramer has minimal interaction with HhC during biocatalysis. As above, we also confirmed ProNAc generation by separating product mixtures by SDS-PAGE. Here, gels were stained first with GelRed, a DNA dye, then by Coomassie blue for total protein (Figure 4C, top and bottom). While Coomassie blue staining proved relatively weak, the ProNAc, CFP:S-dHP39, gave a clear signal with the DNA stain. The apparent molecular weight of CFP:S-dHP39 based on gel mobility was again consistent with 1:1 protein-to-DNA. This finding suggests that structured and unstructured DNA alike could serve almost interchangeably as substrates for HhC biocatalysis.
Figure 4.
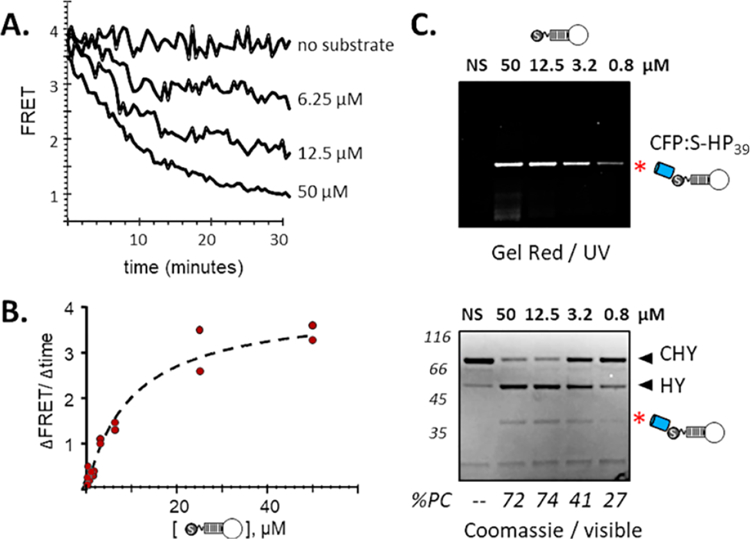
ProNAc conjugation catalyzed by HhC using hairpin-forming steramer, S-dHP39. (A) Kinetic analysis: activity of C−H−Y toward S-dHP39 at the indicated concentrations, monitored by loss of FRET. (B) Michaelis−Menten: initial rate of reaction using FRET assay plotted as a function of increasing S-dHP39. (C) Gel validation of ProNAc formation: reactions of C−H−Y in buffer only (NS), and decreasing concentrations of S-dHP39. ProNAcs were detected using the DNA stain, GelRed (upper); total protein was visualized with Coomassie blue dye (lower). Calculated molecular weight of conjugate, CFP:S-dHP39 is 38.8 kDa. Percent precursor consumed, %PC.
Last, we examined whether native function and solution properties of the ProNAc were retained post-conjugation. To test the possibility that steramer attachment could confer undesired effects, such as aggregate formation and compromised solubility,38,39 we prepared HhC precursor where the protein substrate is the monomeric enzyme, nanoluciferase (Nluc).35 C-terminally His6-tagged Nluc-DHhC precursor was overexpressed and purified from E. coli, and then ProNAcs were generated using S-dT10 and S-dT30 (Figure 5A). Conjugation was again successful and the resulting reaction mixtures with Nluc:S-dT10 and Nluc:S-dT30 were passed over a Ni-NTA column to remove unreacted His-tagged precursor and product His-tagged DHhC, and then separated by size exclusion chromatography (SEC). Sterylated oligonucleotide and corresponding nonsterylated oligonucleotide produced nearly identical chromatograms on SEC, differing only by 0.2 min,40 indicating that steramers were aggregation resistant. Accordingly, traces for separation of Nluc:S-dT10 were consistent with monomeric conjugate (Figure 5B, middle chromatogram) as were NLuc:S-dT30 (Figure 5B, bottom chromatogram). In these chromatograms, the only other peak present was excess steramer, which is not removed by Ni-NTA chromatography (Figure 5B, black triangles). Following SEC separation, enzymatic activity of unmodified nanoluciferase and steramer conjugated nanoluciferase were determined (Figure 5B, right). Luminescence readings were within 10%, indicating that enzymatic activity in the ProNAc state was unaltered. In summary, ProNAcs generated by HhC using steramers exhibited native solution properties and native function.
Figure 5.
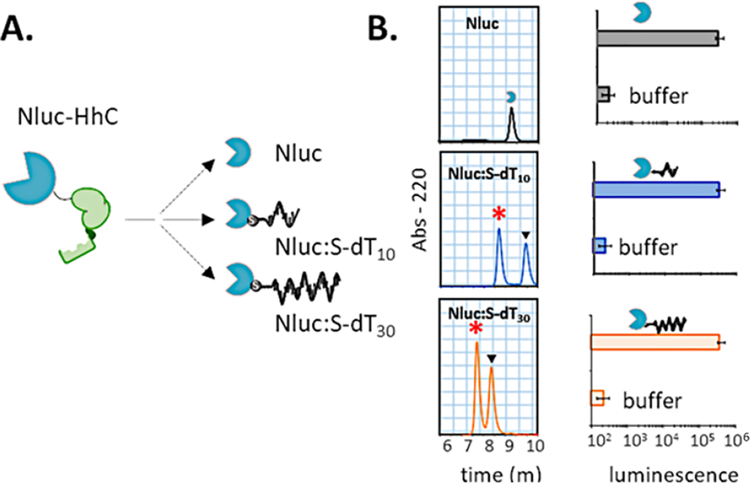
Retention of monodispersity and function following steramer conjugation. (A) Autoprocessing of nanoluciferase (Nluc) precursor, Nluc-DHhC. (B) Solution properties and enzymatic activity of native and steramer conjugated Nluc. Unmodified enzyme (top) and ProNAc conjugates of Nluc (asterisk) were separated by size exclusion chromatography (left), then analyzed for enzymatic activity (right). Black triangles in chromatogram indicate excess steramer.
CONCLUSIONS
In this work, we introduced a biocatalytic approach to 1:1 protein nucleic acid conjugates (ProNAcs) that combines a promiscuous enzymatic agent, HhC, with hybrid substrates, called steramers. Modularity was verified using four unrelated substrate proteins—nanoluciferase, T4 lysozyme, cyan fluorescent protein, and the native hedgehog substrate protein—and steramers comprising unstructured and structured ssDNA, ranging in length from 10 to 39 nucleotides. Our preliminary experiments suggest that compatibility extends to steramers of structured RNA.41 The catalytic element, HhC, has a relatively strong affinity (low KM value) for sterol molecules, and our kinetic studies indicate that binding strength and reaction velocity were maintained toward steramers. Because HhC and its substrate are part of the same polypeptide (precursor), ProNAc conjugations are two component events that can be driven with steramer at 20−50 μM and nanomolar HhC precursor protein. Chemi-enzymatic techniques, like expressed protein ligation, require a “label” molecule concentration in the millimolar range. Unlike conventional self-tagging domains, HhC splits off from the ProNAc conjugate, and thus foreign tag elements are minimized to a single C-terminal glycine residue and sterol linker, the smallest “scar” that we know of for biocatalytic conjugations. Consistent with this feature, we observed similar biochemical properties of steramer-conjugated and steramer-free protein. Thus, the present approach offers an efficient means to functional, nearly tag-free ProNAc for applications to either fundamental studies or biotechnology. Future iterations could involve biorthogonal sterol linkers with complementary HhC variants for in vivo ProNAc synthesis in plant and animal cells.
Nonetheless, the ProNAc ligase activity of HhC carries limitations in its present form. Conjugation is restricted to the target protein’s C-terminus, and this could be inconsistent with function or ProNAc application in the absence of suitable means to reposition the N and C terminal residues, such as circular permutation. Second, the oxime bonds of the steramer are reversible; under dilute conditions, over extended incubation, these bonds will hydrolyze. In addition, the sterol linker of the ProNAc that tethers protein to nucleic acid is hydrophobic, a property that could conceivably interfere with activity by promoting unintended interactions, although we did not observe this pitfall here. An ideal ProNAc ligase brings about direct, regiospecific coupling of protein to nucleic acid, without an intervening self-tagging domain, foreign recognition sequence, unnatural amino acid, or engineered substrate element.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge generous support from the Harpur College Faculty Grant Program; SUNY Research Foundation; and National Cancer Institute (Grant R01 CA206592). We acknowledge Dr. Juergen Schulte for expert advice on NMR.
ABBREVIATIONS
- RP-HPLC
Reverse Phase High Performance Liquid Chromatography
- SDS-PAGE
Sodium Dodecyl Sulfate Polyacrylamide Electrophoresis
- SEC
Size Exclusion Chromatography
- FRET
Forster Resonance Energy Transfer
- Nluc
nanoluciferase
- PEG
polyethylene glycol
- ProNAc
protein nucleic acid
- HhC
Hedgehog C-terminal domain
- KM
concentration of substrate required to achieve 1/2 maximum initial reaction rate
- kmax
maximum first order rate of reaction
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.9b00550.
Protein preparation, kinetic methods, chemical synthesis, protein and oligonucleotide sequences (PDF)
The authors declare the following competing financial interest(s): Authors BPC and TSO are coinventors on a patent application pending at USPTO. This is mentioned in the manuscript NOTES.
REFERENCES
- (1).Taylor MJ, Husain K, Gartner ZJ, Mayor S, and Vale RD (2017) A DNA-Based T Cell Receptor Reveals a Role for Receptor Clustering in Ligand Discrimination. Cell 169, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Savic N, Ringnalda FC, Berk C, Bargsten K, Hall J, Jinek M, and Schwank G (2019) In vitro Generation of CRISPR-Cas9 Complexes with Covalently Bound Repair Templates for Genome Editing in Mammalian Cells. Bio Protoc 9, e3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vogel P, Moschref M, Li Q, Merkle T, Selvasaravanan KD, Li JB, and Stafforst T (2018) Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gu L, Li C, Aach J, Hill DE, Vidal M, and Church GM (2014) Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature 515, 554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nielsen TB, Thomsen RP, Mortensen MR, Kjems J, Nielsen PF, Nielsen TE, Kodal ALB, Clo E, and Gothelf KV (2019) Peptide-Directed DNA-Templated Protein Labelling for The Assembly of a Pseudo-IgM. Angew. Chem., Int. Ed 58, 9068–9072. [DOI] [PubMed] [Google Scholar]
- (6).Sano T, Smith CL, and Cantor CR (1992) Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 258, 120–122. [DOI] [PubMed] [Google Scholar]
- (7).Niemeyer CM (2010) Semisynthetic DNA-protein conjugates for biosensing and nanofabrication. Angew. Chem., Int. Ed 49, 1200–1216. [DOI] [PubMed] [Google Scholar]
- (8).Tsai CT, Robinson PV, Spencer CA, and Bertozzi CR (2016) Ultrasensitive Antibody Detection by Agglutination-PCR (ADAP). ACS Cent. Sci 2, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liang SI, McFarland JM, Rabuka D, and Gartner ZJ (2014) A modular approach for assembling aldehyde-tagged proteins on DNA scaffolds. J. Am. Chem. Soc 136, 10850–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liszczak G, and Muir TW (2019) Nucleic Acid-Barcoding Technologies: Converting DNA Sequencing into a Broad-Spectrum Molecular Counter. Angew. Chem., Int. Ed 58, 4144–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Singh V, Wang S, Chan KM, Clark SA, and Kool ET (2013) Genetically encoded multispectral labeling of proteins with polyfluorophores on a DNA backbone. J. Am. Chem. Soc 135, 6184–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- (13).Gu GJ, Friedman M, Jost C, Johnsson K, Kamali-Moghaddam M, Pluckthun A, Landegren U, and Soderberg O (2013) Protein tag-mediated conjugation of oligonucleotides to recombinant affinity binders for proximity ligation. New Biotechnol 30, 144–152. [DOI] [PubMed] [Google Scholar]
- (14).Lovendahl KN, Hayward AN, and Gordon WR (2017) Sequence-Directed Covalent Protein-DNA Linkages in a Single Step Using HUH-Tags. J. Am. Chem. Soc 139, 7030–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Takahara M, Wakabayashi R, Minamihata K, Goto M, and Kamiya N (2017) Primary Amine-Clustered DNA Aptamer for DNA-Protein Conjugation Catalyzed by Microbial Transglutaminase. Bioconjugate Chem 28, 2954–2961. [DOI] [PubMed] [Google Scholar]
- (16).Sorensen RS, Okholm AH, Schaffert D, Kodal AL, Gothelf KV, and Kjems J (2013) Enzymatic ligation of large biomolecules to DNA. ACS Nano 7, 8098–8104. [DOI] [PubMed] [Google Scholar]
- (17).Duckworth BP, Chen Y, Wollack JW, Sham Y, Mueller JD, Taton TA, and Distefano MD (2007) A universal method for the preparation of covalent protein-DNA conjugates for use in creating protein nanostructures. Angew. Chem., Int. Ed 46, 8819–8822. [DOI] [PubMed] [Google Scholar]
- (18).Bernardinelli G, and Hogberg B (2017) Entirely enzymatic nanofabrication of DNA-protein conjugates. Nucleic Acids Res 45, No. e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Burgahn T, Garrecht R, Rabe KS, and Niemeyer CM (2019) Solid-Phase Synthesis and Purification of Protein-DNA Origami Nanostructures. Chem. - Eur. J 25, 3483–3488. [DOI] [PubMed] [Google Scholar]
- (20).Chu C, Wong OY, and Silverman SK (2014) A generalizable DNA-catalyzed approach to peptide-nucleic acid conjugation. ChemBioChem 15, 1905–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang Y, Park KY, Suazo KF, and Distefano MD (2018) Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Porter JA, Young KE, and Beachy PA (1996) Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259. [DOI] [PubMed] [Google Scholar]
- (23).Hall TM, Porter JA, Young KE, Koonin EV, Beachy PA, and Leahy DJ (1997) Crystal structure of a Hedgehog autoprocessing domain: homology between Hedgehog and self-splicing proteins. Cell 91, 85–97. [DOI] [PubMed] [Google Scholar]
- (24).Owen TS, Ngoje G, Lageman TJ, Bordeau BM, Belfort M, and Callahan BP (2015) Forster resonance energy transfer-based cholesterolysis assay identifies a novel hedgehog inhibitor. Anal. Biochem 488, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Heal WP, Jovanovic B, Bessin S, Wright MH, Magee AI, and Tate EW (2011) Bioorthogonal chemical tagging of protein cholesterylation in living cells. Chem. Commun. (Cambridge, U. K.) 47, 4081–4083. [DOI] [PubMed] [Google Scholar]
- (26).Mann RK, and Beachy PA (2004) Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem 73, 891–923. [DOI] [PubMed] [Google Scholar]
- (27).Ciulla DA, Jorgensen MT, Giner JL, and Callahan BP (2018) Chemical Bypass of General Base Catalysis in Hedgehog Protein Cholesterolysis Using a Hyper-Nucleophilic Substrate. J. Am. Chem. Soc 140, 916–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bordeau BM, Ciulla DA, and Callahan BP (2016) Hedgehog Proteins Consume Steroidal CYP17A1 Antagonists: Potential Therapeutic Significance in Advanced Prostate Cancer. ChemMedChem 11, 1983–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, and Salic A (2012) Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep 2, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ciulla DA, Wagner AG, Liu X, Cooper CL, Jorgensen MT, Wang C, Goyal P, Banavali NK, Pezzullo JL, Giner JL, and Callahan BP (2019) Sterol A-ring plasticity in hedgehog protein cholesterolysis supports a primitive substrate selectivity mechanism. Chem. Commun. (Cambridge, U. K.) 55, 1829–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kalia J, and Raines RT (2008) Hydrolytic stability of hydrazones and oximes. Angew. Chem., Int. Ed 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kolmel DK, and Kool ET (2017) Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev 117, 10358–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gu H, Ghosh S, Staples RJ, and Bane SL (2019) β-Hydroxy-Stabilized Boron-Nitrogen Heterocycles Enable Rapid and Efficient C-Terminal Protein Modification. Bioconjugate Chem DOI: 10.1021/acs.bioconjchem.9b00534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Podyminogin MA, Lukhtanov EA, and Reed MW (2001) Attachment of benzaldehyde-modified oligodeoxynucleotide probes to semicarbazide-coated glass. Nucleic Acids Res 29, 5090–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, et al. (2012) Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol 7, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sawadogo M, and Van Dyke MW (1991) A rapid method for the purification of deprotected oligodeoxynucleotides. Nucleic Acids Res 19, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Du H, Disney MD, Miller BL, and Krauss TD (2003) Hybridization-based unquenching of DNA hairpins on au surfaces: prototypical ″molecular beacon″ biosensors. J. Am. Chem. Soc 125, 4012–4013. [DOI] [PubMed] [Google Scholar]
- (38).Mozhdehi D, Luginbuhl KM, Dzuricky M, Costa SA, Xiong S, Huang FC, Lewis MM, Zelenetz SR, Colby CD, and Chilkoti A (2019) Genetically Encoded Cholesterol-Modified Polypeptides. J. Am. Chem. Soc 141, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Peters C, Wolf A, Wagner M, Kuhlmann J, and Waldmann H (2004) The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc. Natl. Acad. Sci. U. S. A 101, 8531–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). Elution volumes on SEC for steramers and for unmodified oligonucleotides suggested exaggerated molecular weight values. Comparison of values for the DNA samples to elution volumes for protein calibration standards, gave apparent molecular weights for S-dT10 of 25 kDa and for S-dT30 of 68 kDa, while the calculated formula weights for the two steramers are 3 and 9 kDa, respectively. Their unsterylated counterparts eluted earlier, suggesting even larger molecular weight values. The low accuracy of those estimates, greater by eight times, is due to the nonglobular nature of long linear chain ssDNA. Those derived values were taken as the apparent contribution from S-dT10 and S-dT30 to the respective ProNAc. With that assumption, Nluc:S-dT10 and Nluc:S-dT30 elute from the SEC column at the positions expected for monomeric 1:1 conjugates.
- (41).Zhang X, and Callahan BP Unpublished.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.