

Abstract
We report a novel strategy for bridging information transfer between electronics and biological systems within microdevices. This strategy relies on our “electrobiofabrication” toolbox that uses electrode-induced signals to assemble biopolymer films at spatially defined sites and then electrochemically “activates” the films for signal processing capabilities. Compared to conventional electrode surface modification approaches, our signal-guided assembly and activation strategy provides on-demand electrode functionalization, and greatly simplifies microfluidic sensor design and fabrication. Specifically, a chitosan film is selectively localized in a microdevice and is covalently modified with phenolic species. The redox active properties of the phenolic species enable the film to transduce molecular to electronic signals (i.e., “molectronic”). The resulting “molectronic” sensors are shown to facilitate the electrochemical analysis in real time of biomolecules, including small molecules and enzymes, to cell-based measurements such as cytotoxicity. We believe this strategy provides an alternative, simple, and promising avenue for connecting electronics to biological systems within microfluidic platforms, and eventually will enrich our abilities to study biology in a variety of contexts.
Introduction
Integrating electronics with microfluidics for biochemical analysis has drawn significant attention.1–6 However, electronics are not inherently favorable for acquiring information from biological systems due to their poor molecular and biological specificity.7 Several approaches have been developed to improve the connection between electronics and biological systems. The most common approach relies on using biorecognition components such as nucleic acids8, enzymes3,9, and antibodies6,10. Yet, functionalizing electrodes to include biomolecular species with spatial selectivity within microdevices remains a challenge. Extra cumbersome steps (e.g., photolithography) are usually required during device fabrication,1,10–12 which raises the complexity and cost in device manufacturing. Additionally, biorecognition components that are enclosed within microdevices are often not assembled in the best configuration, they undergo degradation and denaturing, and these lead to short device shelf-life (typically less than 7 days8). Both factors limit biodevice dissemination and use. Here, we propose a novel concept to bridge information transfer between electronics and biological systems inside microdevices. This concept is based on our “electrobiofabrication” toolbox.13–17 Such an electrobiofabrication toolbox simplifies the design and fabrication of biosensors by uncoupling electrode functionalization from device manufacturing. That is, biosensors can be stored as “inactivated” versions (i.e., plain electronics) that are “activated” (i.e., functionalized) immediately before use. “Inactivated” devices can be stored for long periods of time without concern for the decay of biological components. Electrobiofabrication makes use of the stimuli-responsive and self-assembling properties of several biological polymers. These biological polymers are natural or engineered materials that recognize and respond to electrode-imposed signals in order to undergo hierarchical assembly. Previous studies have focused on using self-assembly to localize biological components (e.g., enzymes, cells) that could be used for recognition. Here we are extending electrobiofabrication from physical self-assembly mechanisms to electrode-induced covalent modification of the films for the purpose of creating functionality for signal transduction.
To demonstrate this concept, we functionalize microelectronics with a bio-based redox capacitor (BBRC) film within microchannels.18,19 This functionalization involves two electrochemical steps: electrodepositing a thin film of aminopolysaccharide, chitosan, and then grafting phenol molecules into the chitosan film.20,21 That is, at low pH, chitosan is a soluble cationic polyelectrolyte, as its primary amines are protonated. At even slightly basic pH, the amines get deprotonated and chitosan becomes insoluble. Instead of precipitating as insoluble particles, when in proximity to an electrode-generated pH gradient, chitosan can form a three-dimensional hydrogel film.22 Scheme 1A depicts this electrodeposition mechanism onto a standard gold electrode. By controlling the intensity and the time of the applied electric signal, chitosan films of different thickness can be established.23 It is interesting that the nucleophilic property of the deposited chitosan film also allows the film to be “activated”. For instance, phenols can readily diffuse through the chitosan film and be anodically oxidized at the underlying electrode. The oxidized products undergo rapid grafting to the nucleophilic primary amines of the chitosan (Scheme 1B).24,25 The resulting film is referred to as a BBRC film (Scheme 1C) as it is prepared from biological compounds and possesses redox-capacitor properties (endowed by the addition of the phenol, catechol), including accepting, storing and donating electrons in a controllable fashion.26 Importantly, BBRC films can be removed with a strong acid wash and the underlying electrode is reusable.27,28 BBRC films are non-conducting (i.e., unable to exchange electrons directly with the underlying electrodes) but redox-active and can repeatedly exchange electrons with soluble redox-active species. That is, the presence of catechol in the chitosan films enables redox cycling, a process by which the initial transfer of electrons can be cycled repeatedly under an applied voltage in the presence of a redox mediator, providing amplified signal from the initial electron transfer. Scheme 1D & 1E illustrate the mechanisms of reductive- and oxidative-redox cycling of the film that serve to amplify an initial signal from a biological redox-active molecule and an electrochemical mediator, 1,1’-ferrocenedimethanol (Fc), respectively. During reduction, biological reductants can diffuse from the bulk solution into the BBRC film and quickly donate electrons to the phenolic moieties grafted in the film and convert quinone (Q) to catechol (QH2). During oxidation, a reducing mediator, Fc, diffuses through the film and is electrochemically converted to its oxidized state, Fc+, at the electrode. The oxidized Fc+ can diffuse into the BBRC film to accept electrons from the film and convert QH2 to Q moieties. Scheme 1F illustrates the thermodynamics that control electron transfer to/from the film. Through these redox-cycling mechanisms, BBRC films can be reversibly switched between two redox states, during which electrons can be stored or extracted. Based on the direction of electron flow, reductive- and oxidative-redox cycling are also referred to as “charging” and “discharging” the film, respectively. Importantly, the discharging process can be performed after film charging to extract (or “titrate”) the charged state of BBRC films. This mechanism provides a unique means to measure the level of biological reductants during film charging.
Scheme 1:
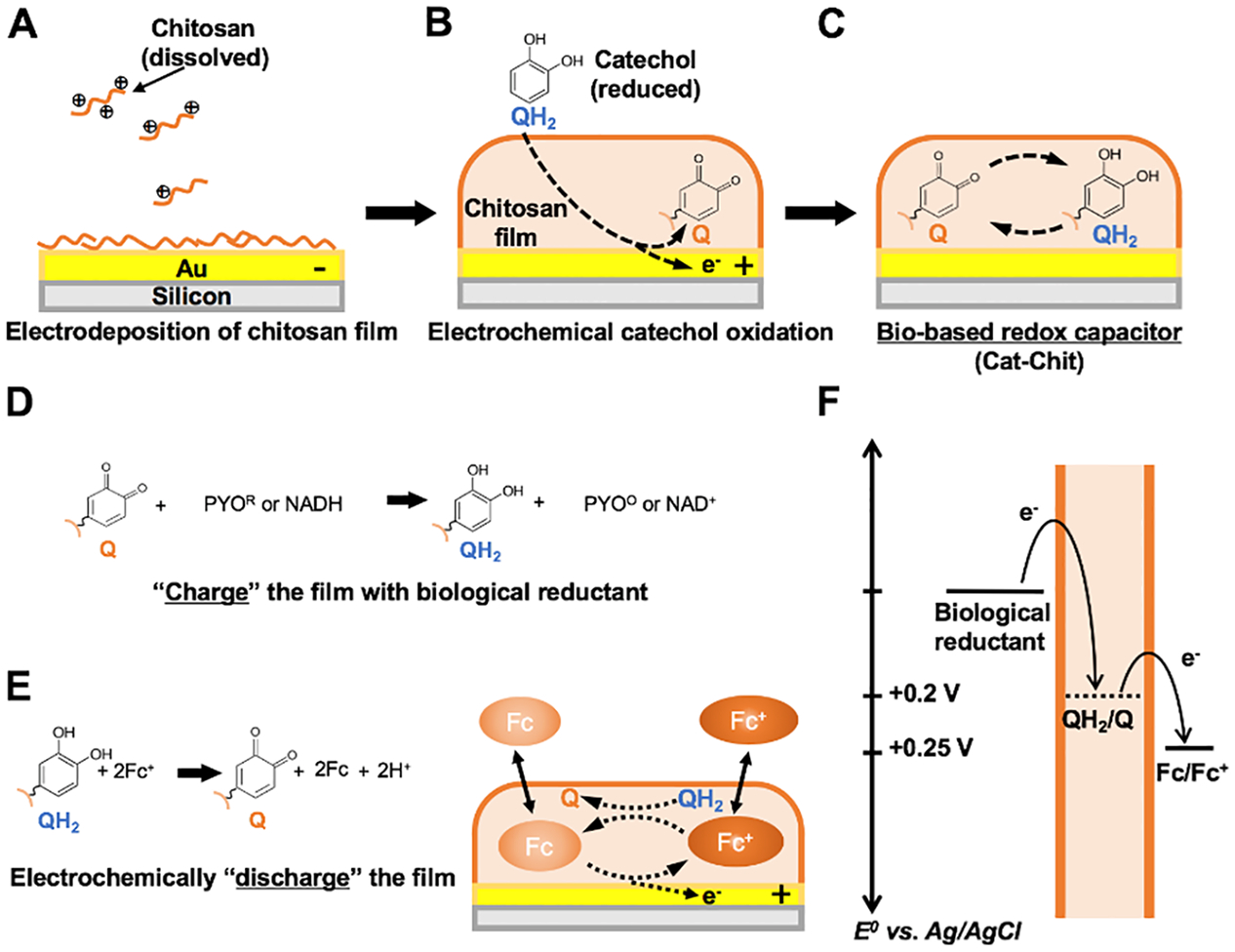
(A) Electrodeposition of chitosan film on a gold electrode. Dissolved chitosan responds to the pH change induced by a cathode initiating the formation of a three-dimensional hydrogel film on its surface. (B) Catechol grafting to the chitosan film is enabled by an anodic voltage. (C) A catechol molecule conjugated to the nucleophilic amino group of the chitosan can undergo oxidation and reduction reactions in the assembled bio-based redox capacitor (BBRC). (D) Reductive redox-cycling between biological reductants and the BBRC film. (E) Oxidative redox-recycling between Fc+/Fc and the BBRC film. (F) Thermodynamics of electron transfer. Q: quinone; QH2: catechol.
In this paper, we integrate BBRC films into an enclosed microchannel to build a “molecular to electronic” (“molectronic”) sensor for measuring analytes in biological samples ranging from small molecules (e.g., pyocyanin, a bacteria secreted toxin29–31), to enzymatic activity (e.g., lactate dehydrogenase (LDH), a biomarker for mammalian cell viability32–34). We note that electrochemical measurement of LDH activity, to our knowledge, is realized here for the first time within a microdevice. We further extend the application to assay the cytotoxicity of a drug mimic (e.g., Triton X-100) on cultures of mammalian cells.
Material and methods
Chemicals and materials
The following materials were purchased from Sigma-Aldrich (St. Louis, MO): chitosan (85% deacetylated), pyrocatechol, sodium L-lactate, β-nicotinamide adenine dinucleotide sodium salt (NAD+), pyocyanin (PYO) and hexaammineruthenium chloride (Ru3+). 1,1’Ferrocenedimethonal (Fc) was purchased from Acros Organics (New Jersey, NJ). All mammalian cell culture media, LIVE-DEAD viability/cytotoxicity kit and Pierce LDH cytotoxicity assay kits were obtained from Thermo Fisher Scientific (Waltham, MA).
Device fabrication
The molectronic sensor consists of an electrode layer and a microchannel layer (Figure 1C), both layers were made in the Maryland Micro and Nano Fabrication Center. The pattern of three-electrode system was custom designed on AutoCAD (Autodesk, Inc., Mill Valley, CA), converted, exposed, and developed onto a stainless-steel shadow mask. The pattern was transferred from the mask to circular glass coverslips by depositing 5 μm chromium and 50 μm gold sequentially using Metra Thermal Evaporator (Telemark, Battle Ground, WA). The pattern of the microchannel was designed on AutoCAD, converted, exposed, and developed onto a mylar mask. The pattern was transferred from the mask to a 4-inch silicon wafer covered with SU-8 3050 photoresist (MicroChem, Westborough, MA) by exposing the wafer to UV light (405 nm wavelength) at 23.4 mW cm−2 using an EVG 620 mask aligner (Electronic Visions Inc., Phoenix, AZ). The wafer was subsequently developed for 8 min in an SU-8 developer (MicroChem, Westborough, MA). The resulting SU-8 master can be reused almost indefinitely. The microchannel layer was made from polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning Co., Midland, MI) cast on the SU-8 master. Both electrode layer and microchannel layers were treated with oxygen plasma (IPC 4000 series plasma system) (Branson, Philadelphia, PA) and bonded with proper alignment. The device was equipped with a holder and rod electrodes for simple and stable connection to the external electrical source. The holder was 3D printed using a Connex 3 Object500 printer and MED610 ink (Stratasys, Eden Prairie, MN).
Figure 1:
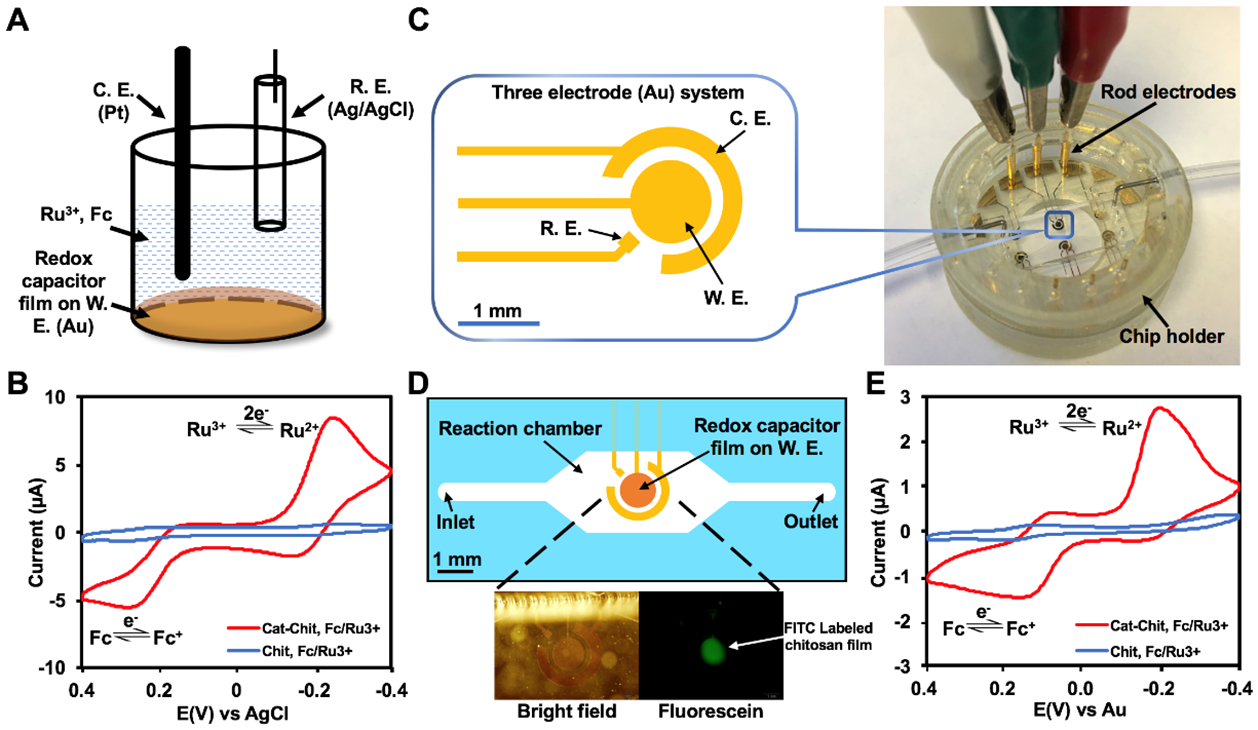
(A) Setup of a standard three-electrode electrochemical cell. The BBRC film is deposited on the working electrode. The system is characterized in a solution of Ru3+ and Fc. (B) Cyclic voltammogram (CV) comparing standard gold electrode modified with BBRC film (Cat-Chit) and chitosan film (Chit) using chamber in (A). (C) A miniaturized three-electrode electrochemical system designed and built inside a microdevice that is enclosed in a 3D printed chip holder. (D) Schematics showing the configuration of the molectronic sensor. The three-electrode system is contained within a reaction chamber. Lower panels depict bright field (BF) and fluorescent microscopic images of the functionalized electronics. (E) CV comparing the molectronic biosensor (Cat-Chit) and the control device (Chit) using electrode system in (D). W. E.: working electrode, C. E.: counter electrode, R. E.: reference electrode.
Selective functionalization of microelectrodes in microchannels with bio-based redox capacitor (BBRC) films
The working electrode in a three-electrode system was selectively functionalized with BBRC films in two steps.7,19,35 First, the target working electrode was connected to an external electrical power supply (2400 Sourcemeter, Keithley, Cleveland, OH) and set as the cathode. A nearby electrode was set as the anode. The microchannel (3 mm × 3 mm × 100 μm) was filled with 1% chitosan solution (dissolved in water and adjusted pH to 5.6 using 1.0 M HCl) to immerse both electrodes. Chitosan film was electrodeposited on the cathode by applying a constant current of 3 A/m2 for 30 s. Films deposited using these parameters are estimated to be 50 μm thick23, which is about half of the channel height (100 μm). Second, the device was connected to a potentiostat for catechol grafting. The channel was filled with catechol solution (5 mM in 0.1 M phosphate buffer (PB), pH 7.0), and a constant anodic potential of +0.6 V (vs Au) was applied for 3 min to oxidize the catechol. The oxidized catechol (i.e., o-quinone) undergoes grafting reactions to the chitosan film.18,26,36 All BBRC films were freshly prepared before each experiment. Fluorescein (FITC) labeled chitosan was used to assist film observation. In order to confirm the signal amplification capabilities of the film, the channel was filled with 50 μM Fc and 50 μM Ru3+ in PB (pH 7.0). Both the device and the Fc/Ru3+ solution was deoxidized with N2 gas for 30 minutes. Cyclic voltammetry (CV, from −0.4 V to 0.4 V) was applied for analysis. All experiments were conducted in a custom designed anaerobic chamber with constant N2 renewal. Results were compared between devices with the BBRC films and chitosan films only.
On-chip pyocyanin (PYO) measurement of conditioned medium (CM)
Pseudomonas aeruginosa (PAO1) and Salmonella typhimurium were obtained from American Type Culture Collection (Manassas, VA); Escherichia coli (W3110) was obtained from the Genetic Stock Center, Yale University (New Haven, CT). All were cultured in Luria broth (LB) medium at 37 °C with shaking (250 rpm). The conditioned media (CM) from overnight cultures were used for on-chip analysis of pyocyanin (PYO), a signal molecule secreted from PAO1. A standard curve for PYO measurement was generated using solutions with known PYO concentrations. The PYO samples of 0 μM, 5 μM, 10 μM, 15 μM and 20 μM were prepared in PB containing 50 μM Fc. Before each experiment, samples were deoxidized with N2 gas for 30 min. Samples were introduced into the device inlet via syringe. CV was performed on each sample for analysis. The device was washed with PB between each test. The supernatants (CM) of overnight bacterial cultures were collected and deoxidized with N2 gas. The deoxidized samples were introduced into the device for CV analysis. All experiments were conducted in a custom designed anaerobic chamber with constant N2 replacement. Results were compared between devices with BBRC films and with chitosan films only.
On-chip lactate dehydrogenase (LDH) measurement
Before experiments, the devices were filled with 50 μM Fc and BBRC films were discharged by applying anodic voltage of 0.4 V for 120 s. This discharging process was repeated until the film was fully discharged (i.e., no further change in chronocoulometry (CC) data, the film’s catechols were all in oxidized =O form). LDH stock solution (2750 unit/L, 1 unit of LDH produces 1 μM of NADH in 1 minute) was prepared in PB containing 50 μM Fc. LDH substrate solution was prepared with 20 mM L-lactate and 10 mM NAD+ in PB containing 50 μM Fc. For standard curve preparations, LDH stock solutions were spiked into the substrate solution to make samples with LDH concentrations of 0 unit/L, 15 unit/L, 30 unit/L, 45 unit/L, 60 unit/L, 90 unit/L and 120 unit/L. Time was set as t0 when the sample was mixed. Each sample was then immediately introduced into the device via syringe. At t = 10 min, CC was performed. Results were compared between the devices with BBRC films and with chitosan films (negative controls).
On-chip cytotoxicity - Effects of Triton X-100 on Caco-2 cell viability
Caco-2 cells were obtained from American Type Culture Collection (Manassas, VA). Cells were maintained using Dulbecco’s Modified Eagle Media (DMEM) supplied with 10% fetal bovine serum (FBS) in T75 flasks under 37 °C and 5% CO2 level and passaged every three days. At passage number 18, cells (~ 1.5×105 cell/cm2) were transferred to 35 mm petri dishes. All petri dishes were pre-sterilized with 70% ethanol and UV (40 min), and treated with 50 μg/ml type I collagen (Corning, Corning, NY) for 2 hours. Cells were cultured under 37 °C and 5% CO2 overlay. Caco-2 cell cultures at confluency were treated with Triton X-100 at levels of 0%, 0.001%, 0.01%, 0.015%, 0.02% and 0.1% (positive control) in DMEM for 2 hours. Then, the viability of each culture was assayed and visualized with a LIVE-DEAD viability/cytotoxicity kit. In parallel, the viability was quantified using the molectronic sensor and the Pierce LDH cytotoxicity assay kit. The supernatant of each culture was collected and diluted 10 times in the LDH substrate solution (this optimized dilution factor ensured that all measurements fell within the linear range of the calibration curve37,38). Time was set as t0 when the sample and the substrate were mixed. The mixture was introduced into the device via syringe. For all experiments, BBRC films were fully discharged before use as aforementioned. At t = 10 min, CC was applied. Based on our in vitro assays, the samples treated with 0.1% of Triton X-100 were set as the positive controls (i.e., 100% cytotoxicity) and the samples treated with DMEM served as negative controls. The cytotoxicity of each sample was calculated using equation (1):
| (1) |
Where [LDH] referred to the LDH level measured in each sample and [LDH]Max referred the LDH level in the positive control.
Instrumentation
All electrochemical measurements were performed using a CHI 420a electrochemical analyzer (CH instruments, Austin, TX). Optical detection of LDH was performed with a SpectraMax® M2e plate reader (Molecular Devices, San Jose, CA). Caco-2 cell cultures were imaged using a fluorescence microscopy (BX 60 microscope; Olympus) and a digital camera (Olympus DP72).
Statistical analysis
All assays were performed in triplicate. Results were expressed as mean ± standard error. One-way ANOVA was used to determine significant differences between groups. The level of significance was set at α = 0.05. The Pearson product-moment correlation coefficient was applied to measure the strength of linear correlation between groups. The level of significant correlation was set at r = 0.95.
Results and discussion
Selectively functionalizing microelectronics in microchannels for amplified electrochemical signals
Functionalizing electronics with BBRC films has previously been reported using standard electrochemical cells (Fig. 1A).7,19,35 That is, standard electrochemical cells containing a gold working electrode (gold/chromium deposited on silicon wafer), a platinum counter electrode and a Ag/AgCl reference electrode have been routinely deployed.18,39,40 We customized our electrochemical cell so that the gold working electrode can be properly placed and connected to an external power source. All three electrodes were connected to the same potentiostat and their working parts were immersed in the same solution. The signal amplification capability of the functionalized electrode was characterized using standard redox mediators, Ru3+ and Fc dissolved in 0.1 M PB. During CV, owing to the catechol in the BBRC, the electrochemical reduction of Ru3+ initiates reductive redox-cycling of the film through a similar mechanism described in Scheme 1D and the electrochemical oxidation of Fc initiates oxidative redox-cycling of the film as described in Scheme 1E. Specifically, redox cycling yields amplified Ru3+ reduction currents and Fc oxidation currents (Fig. 1B). Electrodes coated with unmodified chitosan serve as our negative control because chitosan by itself is non-conducting.26 It should also be noted that the BBRC films reversibly exchange electrons with these mediators although the film’s redox-capacity is very large, but finite.
Here, we extended this technique to a microfluidic platform for the first time. Microfluidics enables precise and automated fluid control41 and is envisioned to potentially enlarge the application scope of the molectronic sensor. Instead of inserting Ag/AgCl2 reference electrode through channel inlets42, we embedded a miniaturized three-electrode system in a simply designed microdevice (Fig. 1C). The device was stabilized in a 3D printed chip holder, through which the external power supply was connected. The three-electrode system was covered by a PDMS layer containing a reaction chamber. Through “electrobiofabrication”, we selectively functionalized the circular working electrode in situ with BBRC films, that is, via electroassembled chitosan and then grafted catechol, forming the BBRC. The entire functionalization procedure took 5.5 minutes to complete (30 seconds of chitosan deposition and 5 minutes of catechol grafting). Once functionalized, sensors were used within 24 hours. In Fig. 1D, fluorescein labeled chitosan was used enabling visualization of the BBRC film on the electrode. Then, functionalized electronics (i.e., the “molectronic” sensors) were tested to amplify electrochemical signals with standard redox mediators, Fc and Ru3+(Fig. 1E). CVs taken of the microsensing system showed that peak currents of the molectronic sensors were about 10 times higher than those of the control group. Since the CVs of Fig. 1B (immersed macroscale electrodes) and the Fig. 1E (the molectronic sensor) were nearly identical (only slight peak shifts owing largely to varied film thicknesses and different reference electrodes), our results indicate that BBRC films for the first time, were able to reveal redox activities from miniature samples (i.e., 2 μL) within a microdevice.
On-chip pyocyanin (PYO) measurement and bacterial sample analysis
The molectronic sensor was first leveraged in analyzing microbiological cell culture by magnifying the electrochemical signal of pyocyanin (PYO), a redox-active small molecule toxin produced by P. aeruginosa (Fig. 2A).29–31 Figure 2B&C illustrate the mechanisms of redox-recycling involved in PYO measurement. When secreted by P. aeruginosa, PYO is in its oxidative state (PYOO). Acting like an oxidizing mediator (e.g., Ru3+), PYOO can accept electrons from the cathode and be electrochemically measured. With the presence of the BBRC film, the reduced PYO (PYOR) can “charge” the redox capacitor film and be returned to its oxidative state (PYOO) (Fig. 2B). The reductive redox cycling reaction amplifies the reduction current of PYOO. However, as noted, the “charged” film (QH2) can return to its oxidative state (Q) via the oxidative redox-cycling with Fc/Fc+ in Figure 2C. The thermodynamics of electron transfer are depicted in Fig. 2D. Using CV, we found the reduction peak current of PYOO corresponded to −0.3V and its magnitude was linearly proportional to PYOO concentration (Fig. 2E and 2F). It should also be noted that increased PYOO proportionally “charged” the BBRC film, which in turn, raised the oxidation peak current of Fc.18
Figure 2:
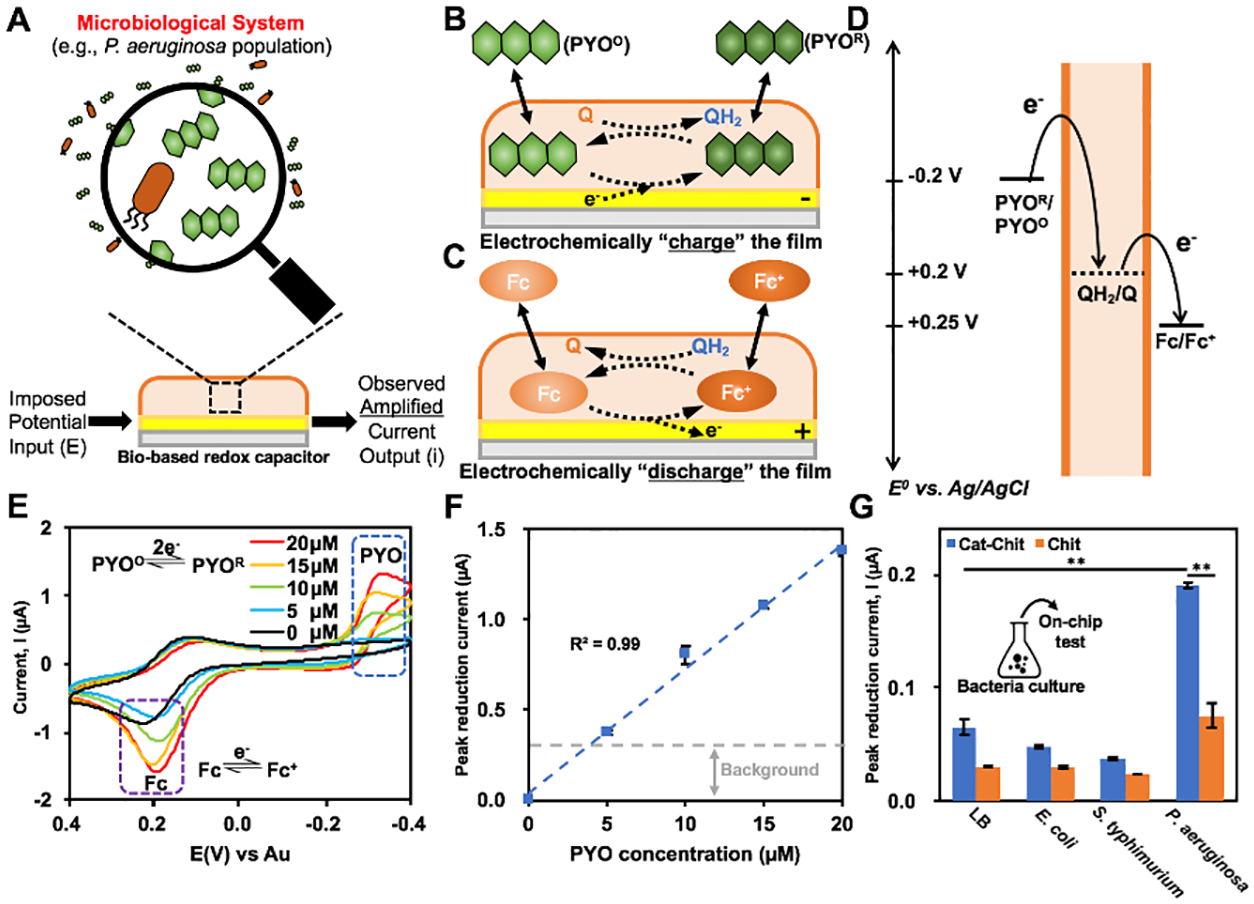
(A) Schematic illustrating the microbiological analysis using BBRC films made possible by amplifying electrochemical signals in the bacterial secretome. (B) Reductive redox-cycling between PYOO/PYOR and the BBRC film. (C) Oxidative redox-recycling between Fc+/Fc and the BBRC film. (D) Thermodynamics of electron transfer. (E) Cyclic voltammogram (CV) of PYOO in PB with concentrations from 0 μM to 20 μM. (F) Calibration curve of PYO measurement. (G) On-chip PYO measurements within LB and conditioned media (CM) from E. coli, S. Typhimurium and P. aeruginosa overnight cultures (**, p < 0.01). Results are compared between the molectronic sensor (Cat-Chit) and the control device (Chit). Q: quinone; QH2: catechol
To test the selectivity of the molectronic sensor towards bacterial identification, CM from several overnight cultures were collected and analyzed. The peak currents were measured at −0.3 V; CM from P. aeruginosa in the chitosan-only sensor control increased ~2-fold relative to uncultured LB media. When the molectronic sensor was used, the peak current increased nearly 8-fold for the case of P. aeruginosa (p < 0.01) (Figure 2G). This result confirmed the signal amplification capability of BBRC films (i.e., catechol modified chitosan films). Neither E. coli nor S. Typhimurium had secreted redox active metabolites during the preceding cultures. Interestingly, the absolute values of the reduction peak current in all cases reported in Figure 2G were lower than those in Figure 2F. This was presumably due to the complex components in LB media and bacterial CM that alter the electron flow between electrodes. In the case of LB alone, a non-zero current was obtained and in all cases the potential for redox cycling due to the presence of catechol was found to increase current. In sum, our results indicate that the molectronic sensor can selectively distinguish the presence of P. aeruginosa among other bacteria owing to the pyocyanin that it secretes during growth.
On-chip lactate dehydrogenase (LDH) measurement
In addition to small molecule analysis noted above, we evaluated the molectronic sensor’s ability to measure enzymatic activity, specifically, LDH. LDH is an enzyme abundant in the cytosol of mammalian cells. When cell membranes are damaged, LDH is often released into the surrounding media (Fig. 3A), consequently, measurement of LDH enzymatic activity in cell culture media is a standard method for evaluating cytotoxicity and is widely utilized in drug screening.32–34 An often used LDH-catalyzed reaction involves a coenzyme (NAD/NADH) which acts as an electron-shuttle mediator.43 During electrochemical measurements, this mediator functions to shuttle the electrons between the enzyme and electrode.44 Current LDH activity measurements rely on oxidants that oxidize NADH (e.g., Formazan45) and accordingly, undergo colorimetric changes.
Figure 3:
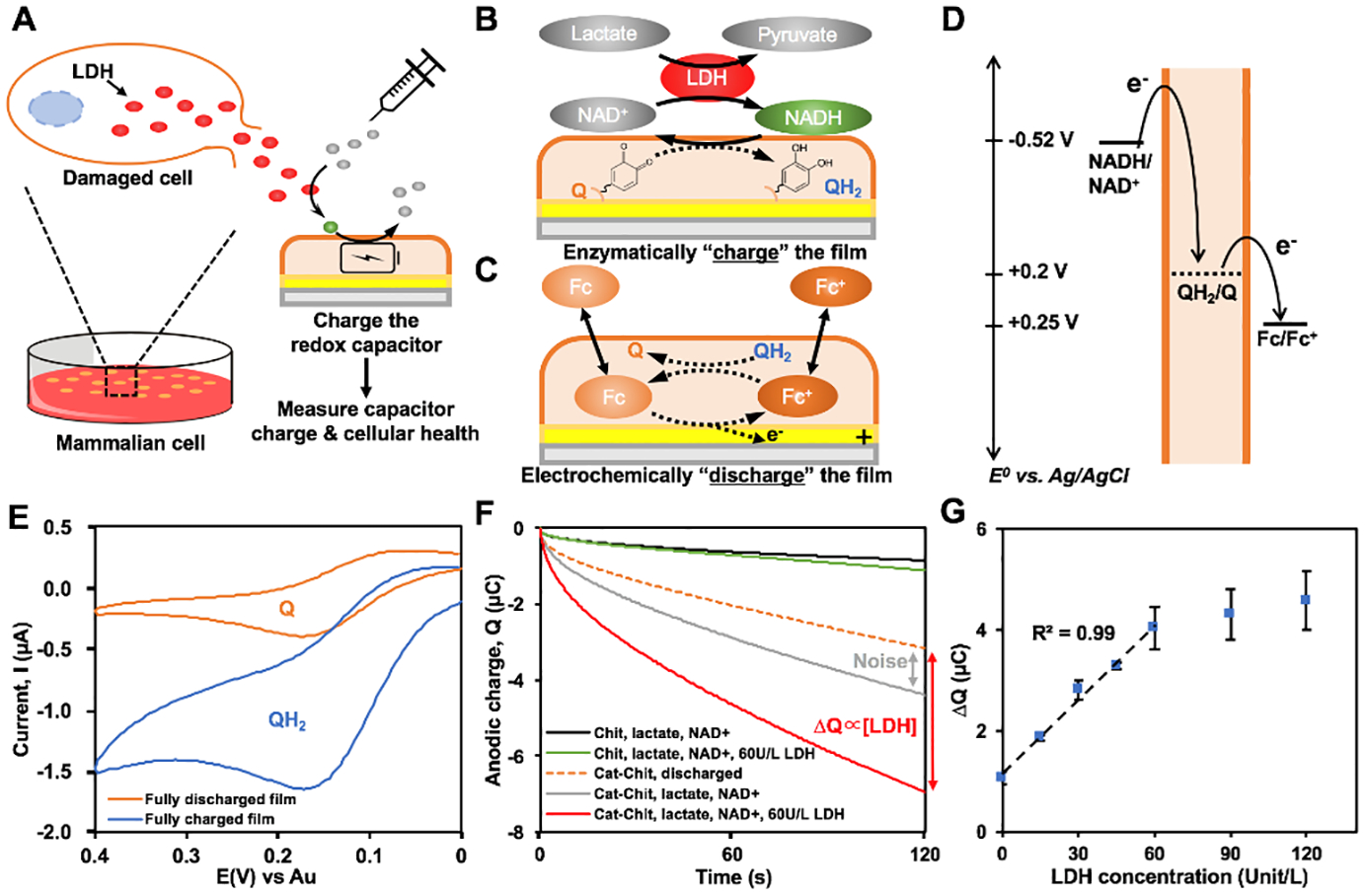
(A) Schematics showing that the membrane disruption of mammalian cells releases cytosolic lactate dehydrogenase (LDH) into surrounding environment. The released LDH can catalyze the added substrates (lactate & NAD+) and in turn charge the molectronic sensor in an analogous manner to battery. The final charge of the molectronic sensor can be measured to reflect the health state of tested cell culture. (B) Reaction catalyzed by LDH. Reductive redox-cycling between NAD+/NADH and the BBRC film. (C) Oxidative redox-recycling between Fc+/Fc and the BBRC film. (D) Thermodynamics of electron transfer. (E) Cyclic voltammogram (CV) of FC oxidation at the surface of electrodes modified with fully discharged (Q) and fully charged (QH2) BBRC films. (F) Chronocoulometry (CC) results comparing the molectronic sensor (Cat-Chit) and the control device (Chit). (G) Calibration curve of the molectronic sensor on LDH activity measurement. Q: quinone; QH2: catechol
Here, we leveraged the capacitor feature of the molectronic sensor to electrochemically measure LDH activity in a microdevice (Fig. 3A). Figure 3B&C illustrates the mechanisms involved. Briefly, LDH catalyzes reversible reactions between lactate and pyruvate with NAD+/NADH as the coenzyme. When the reaction proceeds from lactate to pyruvate, generated NADH serves to “charge” the BBRC film (i.e., reduce quinone to catechol) and is then returned to the oxidized (NAD+) state. This cycling between NADH and the BBRC film thus facilitates signal processing by amplifying the signal output. It should be noted that the enzymatic film charging takes place without the need of electrical input. Importantly, the electrons stored in the charged film can be extracted (or “titrated”) by the oxidative-redox cycling reaction of Fc (i.e., discharging) in Figure 3C. The thermodynamics of electron transfer are depicted in Figure 3D. To test this possibility, we prepared films for the fully charged state, where the film’s catechols were all in reduced QH2 form (positive control) and the fully discharged state, where the film’s catechols were all in oxidized Q form (negative control) by the electrochemical redox-cycling reactions. Figure 3E shows that more electrons can be titrated from the fully charged films, compared with fully discharged films, as expected. That is, the capability of amplifying electrochemical signals between two control films is attributed to the charging state of film (Fig. 3E) and this difference is utilized to reflect the LDH activity in samples.
In order to accurately titrate the electrons in enzymatically charged films, chronocoulometry (CC) was employed. In Figure 3F, the fully discharged films were incubated in various solutions with no applied voltage and then each resulting film was titrated by the Fc-redox cycling reaction.7 The red line in Figure 3F shows a large oxidative charge curve of the film that has been incubated with LDH and substrate (lactate and NAD+). However, when the film was incubated in solutions lacking LDH, the gray line in Figure 3F resulted. This shows a smaller oxidative charge transfer, and was more similar to the negative control that was fully discharged (dotted line). The results of Figure 3F indicate that the addition of LDH and the substrate enzymatically charged the BBRC film. Then, the charged film was titrated by measuring the amplified oxidative charge during Fc oxidation. The magnitude of this amplification is then dependent on the number of reduced catechols in the film, which, in turn, were found to be linearly proportional to the LDH activity (i.e., concentration) (Fig. 3G). That is, we found the response was linear up to 60 unit/L, where the signal was seen to saturate. The signal saturation was presumably due to the limited contact efficiency (confined space for diffusion) between NADH in the bulk solution and the film within microchannel.43
On-chip cytotoxicity - Effects of Triton X-100 on Caco-2 cell viability
Since LDH activity can be used for determining cytotoxicity, we cultivated mammalian cells and challenged them with cytotoxic surfactant, Triton X-100, which permeabilizes cell membranes.46 That is, a human epithelial colorectal adenocarcinoma cell line, Caco-2, was used as a model cell culture. Caco-2 cell is commonly used to resemble the enterocytes lining the human small intestine, which is pivotal in drug absorption.47,48 Caco-2 cell cultures were exposed to Triton X-100 at various concentrations for 120 min, followed by on-chip viability measurements and LDH activity assays (Fig. 4A). The viability of Caco-2 cells was examined using a commercial LIVE/DEAD viability kit (see Methods), while the LDH activity was measured using both the molectronic sensor and a commercialized cytotoxicity kit in parallel. The LIVE/DEAD viability kit stains live cells with green fluorescence (catalyzed by cytosolic esterase and retained intracellularly) while dead or permeabilized cells fluoresce red (generated by nucleic acid binding stain that only enters cells with damaged membrane). Microscopic images in Fig. 4B reveal that cell viability was highest at the lowest Triton X-100 concentrations. In addition, it was evident in the bright field (BF) and green fluorescence images that as the Triton X-100 concentration increased, the Caco-2 cells started to shrink and lyse. While the number of live cells decreased, the number of dead cells increased dramatically (sample treated with 0.02% Triton X-100 showed decreased cell number due to cell detachment from substrate). The qualitative results depicted in Fig. 4B were quantified in Fig. 4C using both the molectronic sensor and the commercial cytotoxicity kit, which measures LDH activity level via colorimetric means. All results were normalized to the positive control (cells treated with 0.1% Triton X-100, not shown). Both methods demonstrated similar values and patterns (r = 0.99, p = 0.94) that also corresponded to the microscopic results. In sum, these data demonstrate concordance between an electrobiofabricated molectronic sensor that can be assembled in situ within fluidic devices versus the commercialized kits that required pooled samples. Perhaps more importantly, the in situ measurement shown here is quantitative and augments visible fluorescence microscopy measurements that are difficult to quantify particularly at high cell densities.
Figure 4:
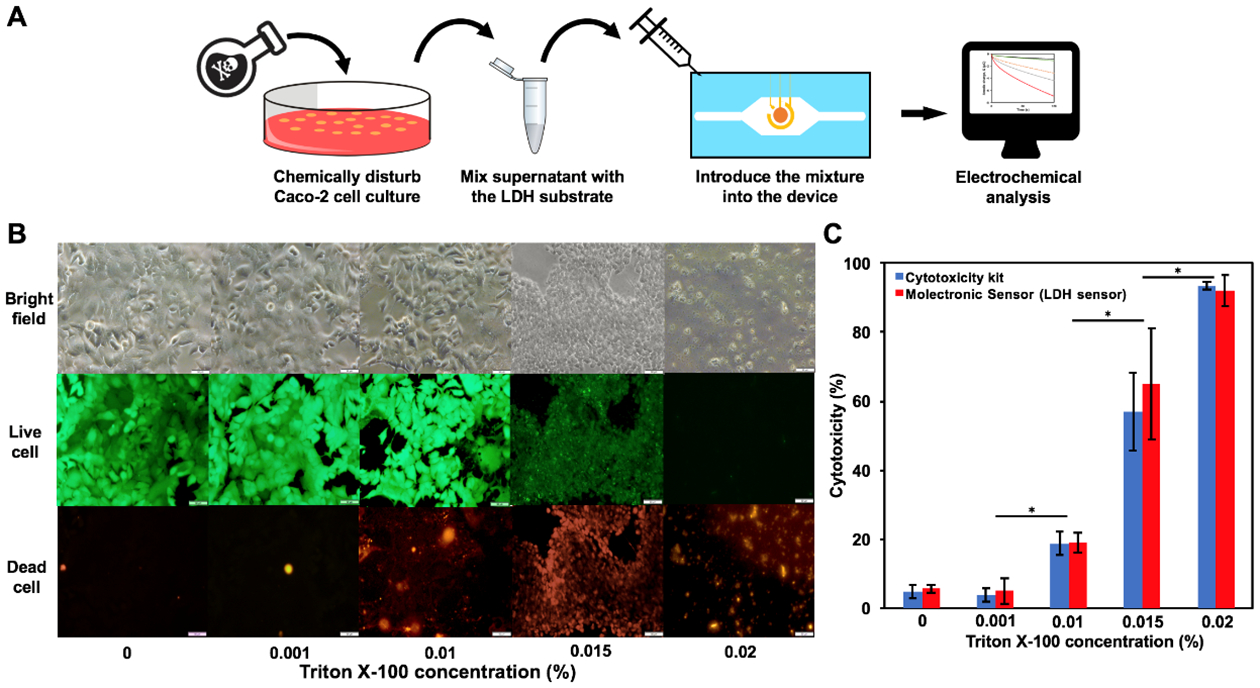
(A) Schematics demonstrating cytotoxicity assay steps using the molectronic sensor. (B) Microscopic results of Caco-2 cell cultures treated with various concentration of Triton X-100. Green and red fluorescent images depict live and dead cells after 120 min, respectively. Scale bar = 50 μm. BF: bright field. (C) Cytotoxicity measurements by the molectronic sensor and the commercialized cytotoxicity kit (*, p < 0.05). All results are normalized to data from the positive control, in which cell cultures are treated with 0.1% Triton X-100.
Conclusion
In this study, we proposed and developed a novel concept, based on our “electrobiofabrication” toolbox, to simplify the bridging between electronics and biological systems within microdevices. This concept permits biosensors to be assembled and functionalized on-demand and with minimal equipment – just the application of voltage in solutions for assembly of polysaccharide chitosan and then grafting of catechols to enable signal amplification via redox cycling. This process takes minutes and can be built into the experimental protocols instead of the chip design and manufacture processes. That is, this methodology avoids one of the most problematic issues of using biological components in biosensing: the decay of labile biorecognition components that occurs during manufacturing processes and long-term device storage. Specifically, we demonstrated three important advantages of the molectronic sensor. First, through “electrobiofabrication”, target electrodes fabricated inside completely packaged microdevices are functionalized with BBRC films with high spatial selectivity. Second, the molecular electronic properties of the BBRC films provide enhanced functional properties (e.g., signal amplification) that facilitate signal processing. Finally, the generic capacitor feature of BBRC film allows the molectronic sensor to accept electrons from biological reductants. We demonstrated the molectronic sensor in the analysis of several biological samples ranging from small molecules (e.g., PYO), to enzymatic activity (e.g., LDH), to monitoring the status of cellular health (e.g., cytotoxicity).
We believe such “electrobiofabrication” extends our means to develop microfluidic biosensors. We expect biosensors that are functionalized using this technique might be embedded in microfluidic platforms for broader applications. For instance, functionalized sensors with enhanced specificity and sensitivity can be incorporated in microfluidic cell culture systems (e.g., organ-on-a-chip49–52) for self-contained and automated drug screening and toxicity tests. The interrogated volumes using this system are on the order of nanoliters. Also, we have recently immobilized engineered bacteria onto BBRC films for expanded biological functions beyond biorecognition, such as signaling.53 While we envision this work provides an alternative, simple, and promising avenue for bridging electronics to biological systems assembled within microfluidic platforms, we can envision many additional applications perhaps not confined by fluidics but equally as effective – the sum of these approaches will enrich our abilities to study biology in a variety of contexts.
Acknowledgements
The authors thank Dr. David Quan for the help in mammalian cell culture. The authors thank Ms. Joanne Chan for the help in preparing the 3D printing file. This work was supported with funds from the National Science Foundation (DMREF #1435957), the National Institutes of Health (R21EB024102) and Defense Threat Reduction Agency (HDTRA1-13-0037).
Notes and references
- 1.Yan J, Pedrosa VA, Enomoto J and Simonian AL, Biomicrofluidics, 2011, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dumitrescu I, Yancey DF and Crooks RM, Lab Chip, 2012, 12, 986. [DOI] [PubMed] [Google Scholar]
- 3.Weltin A, Slotwinski K, Kieninger J, Moser I, Jobst G, Wego M and Urban GA, Lab Chip, 2014, 14, 138–146. [DOI] [PubMed] [Google Scholar]
- 4.Luka G, Ahmadi A, Najjaran H, Alocilja E, Derosa M, Wolthers K, Malki A, Aziz H, Althani A and Hoorfar M, Sensors (Switzerland), 2015, 15, 30011–30031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Q and Yuan YJ, Micromachines, , DOI: 10.3390/mi7060096. [DOI] [Google Scholar]
- 6.Lee GH, Lee JK, Kim JH, Choi HS, Kim J, Lee SH and Lee HY, Sci. Rep, 2017, 7, 1–8.28127051 [Google Scholar]
- 7.Kim E, Liu Y, Bentley WE and Payne GF, Adv. Funct. Mater, 2012, 22, 1409–1416. [Google Scholar]
- 8.Shin SR, Zhang YS, Kim DJ, Manbohi A, Avci H, Silvestri A, Aleman J, Hu N, Kilic T, Keung W, Righi M, Assawes P, Alhadrami HA, Li RA, Dokmeci MR and Khademhosseini A, Anal. Chem, 2016, 88, 10019–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bavli D, Prill S, Ezra E, Levy G, Cohen M, Vinken M and Vanfleteren J, PNAS, 2016, 113, E2231–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Mousavi Shaegh SA, Massa S, Riahi R, Chae S, Hu N, Avci H, Zhang W, Silvestri A, Sanati Nezhad A, Manbohi A, De Ferrari F, Polini A, Calzone G, Shaikh N, Alerasool P, Budina E, Kang J, Bhise N, Ribas J, Pourmand A, Skardal A, Shupe T, Bishop CE, Dokmeci MR, Atala A and Khademhosseini A, Proc. Natl. Acad. Sci, 2017, 114, E2293–E2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali MA, Srivastava S, Solanki PR, Reddy V, Agrawal VV, Kim C, John R and Malhotra BD, Sci. Rep, 2013, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esteves-Villanueva JO, Trzeciakiewicz H and Martic S, Analyst, 2014, 139, 2823–31. [DOI] [PubMed] [Google Scholar]
- 13.Wu LQ and Payne GF, Trends Biotechnol, 2004, 22, 593–599. [DOI] [PubMed] [Google Scholar]
- 14.Yi H, Wu L, Ghodssi R, Rubloff GW, Payne GF and Bentley WE, Langmuir, 2005, 21, 2104–2107. [DOI] [PubMed] [Google Scholar]
- 15.Park JJ, Luo X, Yi H, Ghodssi R and Rubloff GW, Lab Chip, 2006, 1, 1315–1321. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Kim E, Ghodssi R, Rubloff GW, Culver JN, Bentley WE and Payne GF, Biofabrication, 2010, 2, 022002. [DOI] [PubMed] [Google Scholar]
- 17.Koev ST, Dykstra PH, Luo X, Rubloff GW, Bentley WE, Payne GF and Ghodssi R, Lab Chip, 2010, 10, 3026–3042. [DOI] [PubMed] [Google Scholar]
- 18.Kim E, Gordonov T, Bentley WE and Payne GF, Anal. Chem, 2013, 85, 2102–2108. [DOI] [PubMed] [Google Scholar]
- 19.Kim E, Leverage WT, Liu Y, White IM, Bentley WE and Payne GF, Analyst, 2014, 139, 32–43. [DOI] [PubMed] [Google Scholar]
- 20.Wu LQ, Gadre AP, Yi H, Kastantin MJ, Rubloff GW, Bentley WE, Payne GF and Ghodssi R, Langmuir, 2002, 18, 8620–8625. [Google Scholar]
- 21.Redepenning J, Venkataraman G, Chen J and Stafford N, J. Biomed. Mater. Res, 2003, 66A, 411–416. [DOI] [PubMed] [Google Scholar]
- 22.Ladet S, David L and Domard A, Nature, 2008, 452, 76–79. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Luo X, Betz J, Buckhout-White S, Bekdash O, Payne GF, Bentley WE and Rubloff GW, Soft Matter, 2010, 6, 3177. [Google Scholar]
- 24.Wu LQ, Ghodssi R, Elabd YA and Payne GF, Adv. Funct. Mater, 2005, 15, 189–195. [Google Scholar]
- 25.Wu LQ, McDermott MK, Zhu C, Ghodssi R and Payne GF, Adv. Funct. Mater, 2006, 16, 1967–1974. [Google Scholar]
- 26.Kim E, Liu Y, Shi X, Yang X, Bentley WE and Payne GF, Adv. Funct. Mater, 2010, 20, 2683–2694. [Google Scholar]
- 27.Park JJ, Luo X, Yi H, Valentine TM, Payne GF, Bentley WE and Rubloff GW, Lab Chip, 2006, 6, 1315–1321. [DOI] [PubMed] [Google Scholar]
- 28.Luo X, Lewandowski AT, Yi H, Payne GF, Ghodssi R, Bentley WE and Rubloff GW, Lab Chip, 2008, 8, 420–430. [DOI] [PubMed] [Google Scholar]
- 29.Lau GW, Hassett DJ, Ran H and Kong F, Trends Mol. Med, 2004, 10, 599–606. [DOI] [PubMed] [Google Scholar]
- 30.Jacob C, Jamier V and Ba LA, Curr. Opin. Chem. Biol, 2011, 15, 149–155. [DOI] [PubMed] [Google Scholar]
- 31.Okegbe C, Sakhtah H, Sekedat MD, Price-Whelan A and Dietrich LEP, Antioxid. Redox Signal, 2012, 16, 658–667. [DOI] [PubMed] [Google Scholar]
- 32.Racher AJ, Looby D and Griffiths JB, Cytotechnology, 1990, 3, 301–307. [DOI] [PubMed] [Google Scholar]
- 33.Haslam G, Wyatt D and Kitos PA, Cytotechnology, 2000, 32, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolterbeek HT and van der Meer AJGM, Assay Drug Dev. Technol, 2005, 3, 675–682. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Kim E, White IM, Bentley WE and Payne GF, Bioelectrochemistry, 2014, 98, 94–102. [DOI] [PubMed] [Google Scholar]
- 36.Kim E, Leverage WT, Liu Y, White IM, Bentley WE and Payne GF, Analyst, 2014, 139, 32–43. [DOI] [PubMed] [Google Scholar]
- 37.Bosetti M, Santin M, Lloyd AW, Denyer SP, Sabbatini M and Cannas M, J. Mater. Sci. Mater. Med, 2007, 18, 611–617. [DOI] [PubMed] [Google Scholar]
- 38.Soares-Bezerra RJ, Ferreira NCDS, Alberto AVP, Bonavita AG, Fidalgo-Neto AA, Calheiros AS, Frutuoso VDS and Alves LA, PLoS One, , DOI: 10.1371/journal.pone.0123089. [DOI] [Google Scholar]
- 39.Liba BD, Kim E, Martin AN, Liu Y, Bentley WE and Payne GF, Biofabrication, 2013, 5, 015008. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, Liu Y, Kim E, Bentley WE and Payne GF, Anal. Chem, 2016, 88, 7213–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitesides GM, Nature, 2006, 442, 368–73. [DOI] [PubMed] [Google Scholar]
- 42.Gray KM, Liba BD, Wang Y, Cheng Y, Rubloff GW, Bentley WE, Montembault A, Royaud I, David L and Payne GF, Biomacromolecules, 2012, 13, 1181–1189. [DOI] [PubMed] [Google Scholar]
- 43.Kim E, Liu Y, Baker CJ, Owens R, Xiao S, Bentley WE and Payne GF, Biomacromolecules, 2011, 12, 880–888. [DOI] [PubMed] [Google Scholar]
- 44.Rathee K, Dhull V, Dhull R and Singh S, Biochem. Biophys. Reports, 2016, 5, 35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Decker T and Lohmann-Matthes M-L, J. Immunol. Methods, 1988, 115, 61–69. [DOI] [PubMed] [Google Scholar]
- 46.Borner MM, Schneider E, Pirnia F, Sartor O, Trepel JB and Myers CE, FEBS Lett, 1994, 353, 129–132. [DOI] [PubMed] [Google Scholar]
- 47.Meunier V, Bourrie M, Berger Y and Fabre G, Cell Biol. Toxicol, 1995, 11, 187–194. [DOI] [PubMed] [Google Scholar]
- 48.Borchardt RT, AAPS J., 2011, 13, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung JH, Esch MB, Prot J-M, Long CJ, Smith A, Hickman JJ and Shuler ML, Lab Chip, 2013, 13, 1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhatia SN and Ingber DE, Nat. Biotechnol, 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- 51.Oleaga C, Bernabini C, Smith AST, Srinivasan B, Jackson M, McLamb W, Platt V, Bridges R, Cai Y, Santhanam N, Berry B, Najjar S, Akanda N, Guo X, Martin C, Ekman G, Esch MB, Langer J, Ouedraogo G, Cotovio J, Breton L, Shuler ML and Hickman JJ, Sci. Rep, 2016, 6, 20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng F, Fu F, Cheng Y, Wang C, Zhao Y and Gu Z, Biomaterials, 2016, 12, 2253–2282. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Li J, Tschirhart T, Terrell JL, Kim E, Tsao C-Y, Kelly DL, Bentley WE and Payne GF, Adv. Healthc. Mater, 2017, 1700789. [DOI] [PubMed] [Google Scholar]