

Abstract
Bacteria are protected by a polymer of peptidoglycan that serves as an exoskeleton1. In Staphylococcus aureus, the peptidoglycan assembly enzymes relocate during the cell cycle from the periphery, where they are active during growth, to the division site where they build the partition between daughter cells2–4. But how peptidoglycan synthesis is regulated throughout the cell cycle is poorly understood5,6. Here we used a transposon screen to identify a membrane protein complex that spatially regulates S. aureus peptidoglycan synthesis. This complex consists of an amidase that removes stem peptides from uncrosslinked peptidoglycan and a partner protein that controls its activity. Amidases typically hydrolyze crosslinked peptidoglycan between daughter cells so they can separate7. However, this amidase controls cell growth. In its absence, peptidoglycan synthesis becomes spatially dysregulated, causing cells to grow so large that cell division is defective. We show that the cell growth and division defects due to loss of this amidase can be mitigated by attenuating the polymerase activity of the major S. aureus peptidoglycan synthase. Our findings lead to a model wherein the amidase complex regulates the density of peptidoglycan assembly sites to control peptidoglycan synthase activity at a given cellular location. Removal of stem peptides from peptidoglycan at the cell periphery promotes peptidoglycan synthase relocation to midcell during cell division. This mechanism ensures that cell expansion is properly coordinated with cell division.
The bacterial cell wall, which is largely composed of peptidoglycan, is essential for survival. The chemical steps in cell wall biosynthesis are conserved, but how the activities of peptidoglycan biosynthetic enzymes are coordinated with growth and division is unclear5,6. To make peptidoglycan, glycan chains are first polymerized from a disaccharide-peptide precursor, Lipid II, and then crosslinked to the existing matrix via their attached peptides (Fig. 1a). Crosslinking is carried out by the transpeptidase domain of penicillin-binding proteins (PBPs) using a mechanism that involves formation of a covalent adduct between the enzymes and the stem peptides on nascent peptidoglycan8. There are two modes of peptidoglycan synthesis during the cell cycle. One mode occurs at the cell periphery and leads to an expansion in cell size; the other occurs during cell division when a cross wall, or septum, is formed between two daughter cells2–4. The first protein that assembles at the division site is the highly conserved tubulin homolog FtsZ, which forms a ring-shaped structure called the Z-ring that serves as a scaffold for the ordered recruitment of the rest of the divisome9. In S. aureus, proper placement of FtsZ depends on controlling cell size10–12, which in turn requires spatial control over peptidoglycan synthesis.
Figure 1: LytH is an amidase involved in cell growth and division.
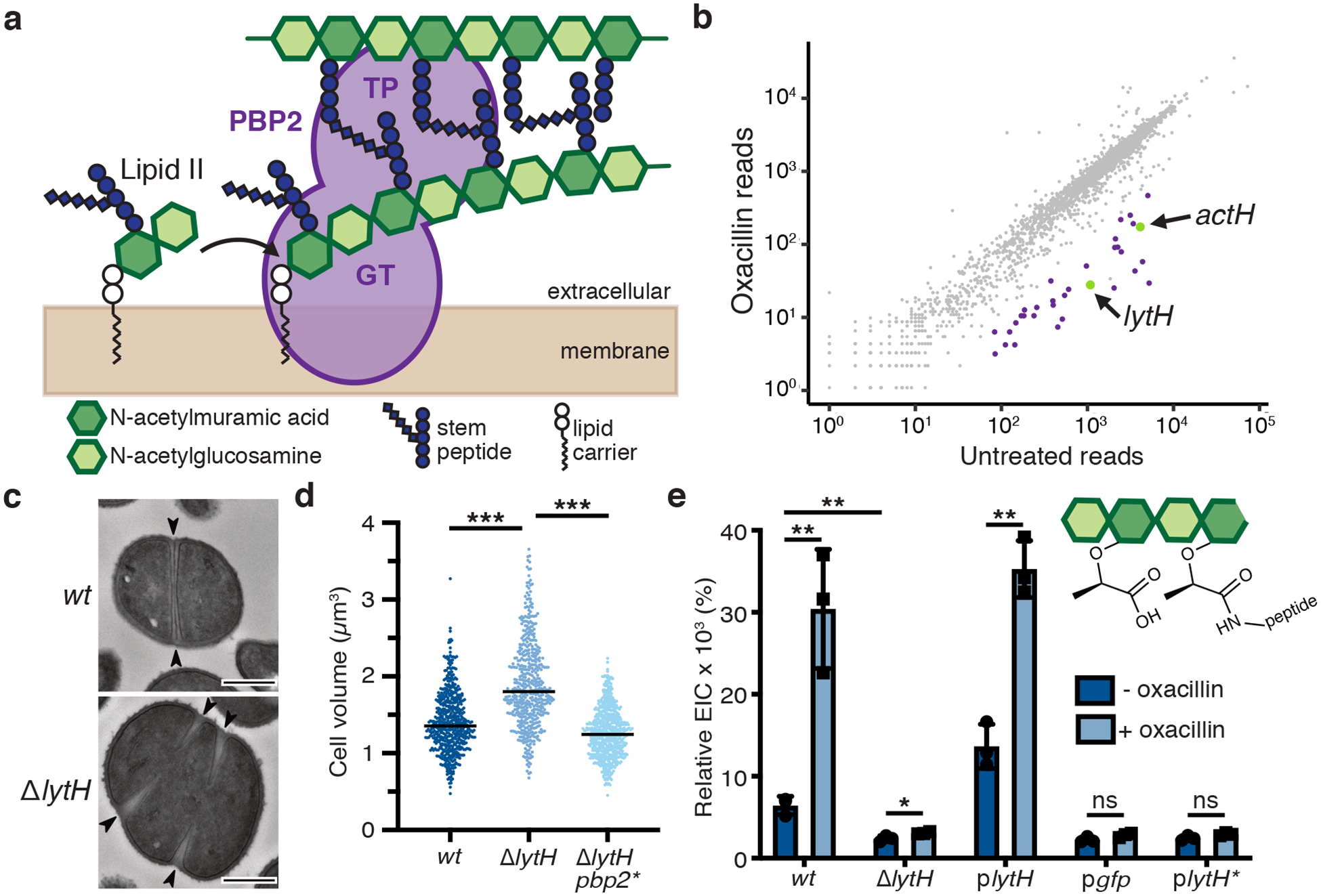
a, Peptidoglycan is assembled from Lipid II. PBP2 catalyzes the polymerization (GT, glycosyltransferase domain) of Lipid II and crosslinking (TP, transpeptidase domain) of glycan strands into the existing matrix. b, Transposon sequencing reads in lytH and actH (green) were depleted with oxacillin. The plot shows transposon reads for each gene (dot) of strain HG003 under the two conditions. Significantly depleted genes are denoted in green and purple, and are listed in Supplementary Table 1. c, Transmission electron micrographs: WT cells have a cross wall at midcell (arrows), but ΔlytH cells have multiple partial septa and are larger. Scale bars, 500 nm. d, Scatter plots showing cell size with the median of each distribution indicated by a black line. pbp2* denotes pbp2F158L. P-values were determined by two-sided Mann-Whitney U tests. From left to right: P = 4.58×10−50 and 8.10×10−86 (***P < 0.001); counted 566 (wt), 516 (ΔlytH), and 615 (ΔlytH pbp2*) phase 1 cells. e, Sacculi analysis: A tetrasaccharide-monopeptide was more abundant in cells expressing lytH and increased under oxacillin treatment. plytH, pgfp, and plytH* denote strains expressing lytHWT, gfp, or lytHD195A, respectively, from a plasmid in a ΔlytH background. Data represent mean ± SD of the relative extracted ion count (EIC). P-values were determined by unpaired, two-tailed t-tests (*P < 0.05; **P < 0.01; ns, not significant). From top left to right error bar: P = 0.0040, 0.0048, 0.0387, 0.0010, 0.0826, and 0.0788. Data are representative of two (c-d) and three (e) independent experiments.
To identify new regulators of peptidoglycan synthesis in S. aureus, we performed a transposon screen13 using sublethal oxacillin to partially inhibit transpeptidase activity. We hypothesized that genes whose absence was synthetically lethal with partial loss of transpeptidase activity might regulate peptidoglycan synthesis. Reads due to transposon insertions were depleted in a number of genes, but we focused on lytH, which encodes a putative amidase, because inactivation of a cell wall hydrolase typically protects, rather than sensitizes, cells towards a β-lactam14 (Fig. 1b and Supplementary Table 1). A spot-titer assay confirmed that lytH deletion sensitized cells to oxacillin (Extended Data Fig. 1a). Amidases are hydrolytic enzymes that cleave stem peptides from peptidoglycan to separate daughter cells following cell division7. The ΔlytH mutant exhibited division defects, but they were not the final cell separation defects previously observed for the S. aureus amidases Atl15 and Sle116. Instead, lytH deletion led to misplacement of nascent septa (Fig. 1c and Extended Data Fig. 1b), larger cell size (Fig. 1d), and reduced growth rate (Extended Data Fig. 1c). The nature of the defects implied that LytH affects cell division at an earlier stage than amidases that effect cell separation.
To assess LytH amidase activity, we compared the chemical composition of sacculi from wild-type (WT) and ΔlytH cells. Purified sacculi were digested with mutanolysin to generate muropeptides for LC-MS analysis (Extended Data Fig. 2a,b). We then searched for muropeptides consistent with amidase processing and identified a species having an exact mass that suggested a tetrasaccharide-monopeptide structure17. This species was more abundant in sacculi from cells expressing a catalytically-active18 copy of lytH than from ΔlytH cells (Fig. 1e). Using LC-MS/MS, we confirmed the identity of this species as a tetrasaccharide with only a single stem peptide attached (Extended Data Fig. 2c–e). We were able to detect this tetrasaccharide-monopeptide species because mutanolysin cannot cleave MurNAc-GlcNAc linkages unless the MurNAc residue carries a stem peptide19. The tetrasaccharide-monopeptide likely represents only a fraction of LytH products, as longer glycan chains extensively lacking stem peptides would not be detected with our analytical method.
Having evidence of LytH amidase activity in cells, we attempted to reconstitute its activity in vitro, but these efforts were unsuccessful (Extended Data Fig. 3a–d). We speculated that LytH requires a partner protein for activation. To identify potential activators, we performed co-immunoprecipitation and isolated a polytopic membrane protein containing seven predicted transmembrane helices and a C-terminal domain with three tetratricopeptide repeats (TPRs) (Fig. 2a and Extended Data Fig. 3e). This membrane protein was previously designated Rbd because it resembles a rhomboid protease12 (Extended Data Fig. 3f and Supplementary Table 2). A mutant lacking this gene was sensitive to oxacillin (Fig. 1b and Extended Data Fig. 4a,b) and displayed cell division defects similar to the ΔlytH mutant (Fig. 2b), supporting a role for the uncharacterized protein in activating LytH.
Figure 2: The LytH-ActH complex cleaves stem peptides from uncrosslinked peptidoglycan.
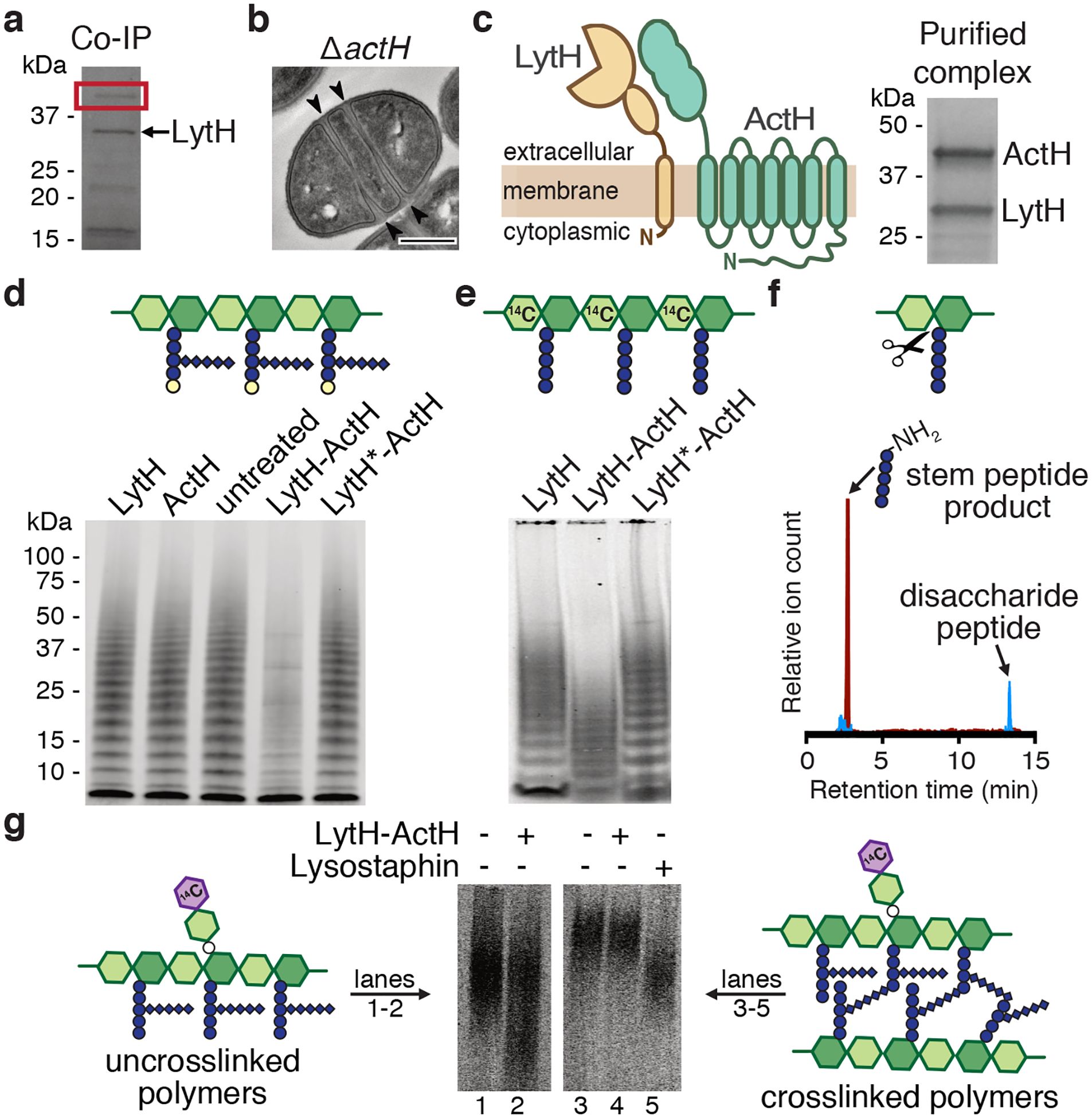
a, A pull-down identified ActH (red box) as a LytH-interaction partner. b, An actH deletion mutant exhibited septal defects similar to ΔlytH division defects. Scale bar, 500 nm. c, LytH and ActH purified as a complex after co-expression in E. coli. A schematic of the predicted topology is shown. d, LytH-ActH removed stem peptides from uncrosslinked glycan strands synthesized in vitro. Glycan strands were labeled with biotin-D-Lys (BDL, yellow spheres), separated by SDS-PAGE, and immunodetected with streptavidin. LytH* denotes the catalytic mutant LytHD195A. e, Uncrosslinked glycan strands containing a [14C]-radiolabel in the glycan backbone undergo a downward mass shift upon LytH-ActH removal of stem peptides. [14C]-glycan strands were detected by autoradiography. f, LC-MS analysis confirmed that LytH-ActH releases stem peptides from uncrosslinked polymers. g, LytH-ActH did not detectably cleave peptidoglycan crosslinks. Uncrosslinked and crosslinked polymers labeled with a [14C]-disaccharide branch on the polymer backbone were treated with the complex; a mass shift indicating cleavage was only observed for uncrosslinked polymers. The lysostaphin control cleaved crosslinks. [14C]-labeled polymers were detected by autoradiography. Data are representative of two (a-b, d) and three (c, e, g) independent experiments.
We co-expressed LytH and its putative protein partner and purified a stable complex (Fig. 2c and Extended Data Fig. 4c). We first tested activity of the complex on peptidoglycan polymers prepared from native S. aureus Lipid II20 using a mutant polymerase21 that can form short glycan strands but cannot crosslink them (Extended Data. Fig. 3c). To visualize the polymers, we labeled the stem peptides with biotin-D-Lys (BDL) for immunoblotting22. No changes in the polymers were observed with LytH treatment alone, but in the presence of the complex, we saw a reduction in the intensity of BDL-labeled glycan strands (Fig. 2d and Extended Data Fig. 4d,e). When a radiolabel was incorporated into the glycan backbone23, we observed a reduction in polymer size for all the different radiolabeled glycan backbones tested (Fig. 2e and Extended Data Fig. 5a,b). These changes, which were only observed for complexes containing catalytically-active LytH, were consistent with removal of stem peptides from the polymers. We were able to detect the tetrasaccharide-monopeptide by LC-MS analysis (Extended Data Fig. 6). We were also able to detect the stem peptide released from synthetic peptidoglycan polymers24 (Fig. 2f). Taken together, these experiments confirmed that the LytH complex removes stem peptides from peptidoglycan. We have renamed the membrane protein partner ActH (activator of LytH).
Many amidases efficiently degrade sacculi because they can act on crosslinked peptidoglycan. We tested the LytH-ActH complex with purified sacculi but did not detect hydrolytic activity, suggesting the complex prefers uncrosslinked substrates (Extended Data Fig. 5c). To directly assess substrate preference, we needed matched substrates that only differed in whether they were crosslinked. We made uncrosslinked glycan strands from native S. aureus Lipid II, introduced a radiolabel on the glycan backbone, and subjected a portion of the labeled material to crosslinking25 (Extended Data Fig. 5d). Treatment with the LytH complex showed that the uncrosslinked material reacted to produce lower molecular weight material, but the crosslinked material did not detectably react in the presence of the complex (Fig. 2g and Extended Data Fig. 5e). We concluded that LytH is unlike previously characterized amidases involved in cell separation because it strongly prefers uncrosslinked peptidoglycan as a substrate. Consistent with the preference observed in vitro using synthetic peptidoglycan, we observed a substantial increase in abundance of the tetrasaccharide-monopeptide species in digests of sacculi from lytHWT cells that were treated with sublethal oxacillin to reduce crosslinking (Fig. 1e). Hence, the in vitro and cellular evidence indicates that LytH removes stem peptides from membrane-proximal peptidoglycan that is not, or at least not extensively, crosslinked.
We next investigated how LytH could regulate cell division. The division defects resulting from lytH deletion implied LytH is important for FtsZ assembly. We found using an FtsZ-sGFP fusion3 that FtsZ was frequently mislocalized in ΔlytH cells of different sizes (Fig. 3a). Therefore, LytH is needed for the correct positioning of FtsZ, and the divisome, at midcell. Because FtsZ assembly defects have been observed in other mutants that grow to aberrantly large size10–12, we speculated that the defects in divisome placement were due to uncontrolled cell growth. We exploited the observation that the ΔlytH mutant is temperature-sensitive to select for suppressors that might provide more information about LytH function (Fig. 3b). We identified multiple suppressors with distinct mutations in the polymerase domain of PBP2, a bifunctional enzyme that is the major peptidoglycan synthase26 in S. aureus (Supplementary Table 3). Overexpression of pbp2WT, but not a pbp2 suppressor allele, was toxic in a ΔlytH background and resulted in exacerbated division defects, suggesting the suppressor mutations reduced PBP2 activity (Fig. 3c and Extended Data Fig. 7a,b). We purified one of the PBP2 suppressors, PBP2F158L, and confirmed that it produced fewer and shorter glycan strands than PBP2WT (Fig. 3d and Extended Data Fig. 7c,d). The pbp2 suppressor also restored ΔlytH cells to WT size (Fig. 1d and Extended Data Fig. 8) and corrected FtsZ mislocalization defects (Fig. 3a). These results showed that reducing PBP2 polymerase activity compensates for loss of LytH. If so, one would predict that moenomycin treatment21, which selectively inhibits PBP2-mediated peptidoglycan polymerization, would also rescue growth of the ΔlytH mutant at elevated temperature; indeed, this was the case (Fig. 3e). Taken together, these findings suggested that LytH controls cell growth by regulating PBP2-mediated synthesis of new peptidoglycan.
Figure 3: Slowing peptidoglycan synthesis compensates for loss of LytH.
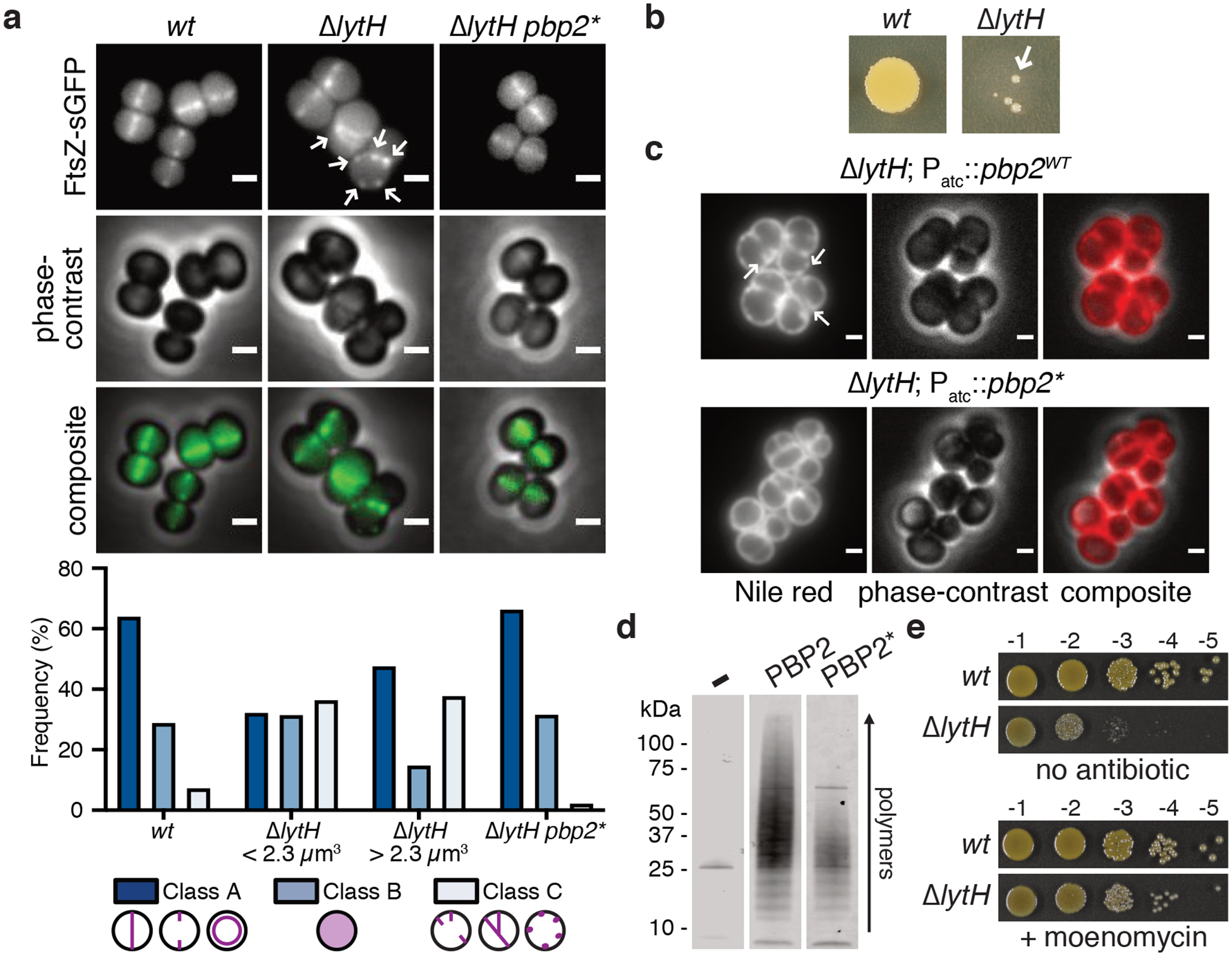
a, FtsZ was frequently mislocalized in the absence of LytH (arrows). Quantitation of FtsZ-sGFP localization either at midcell where a nascent or complete septum was formed (Class A), as diffuse fluorescence (Class B), or as peripheral puncta/multiple septa (Class C). ΔlytH cells were divided into two categories – cells that are smaller or larger than the median cell volume of 2.3 μm14 – which were separately analyzed. The suppressor allele pbp2F158L (pbp2*) corrected ΔlytH defects. From left to right, n = 965, 550, 549, and 1963 cells. Scale bars, 1 μm. b, High-temperature (42°C) sensitivity of lytH deletion was suppressed (arrow) by reducing PBP2 activity. c, Overexpression of pbp2WT from a plasmid in a lytH deletion background exacerbated cell division defects. Overexpression of a suppressor allele, pbp2N220→KDLN (pbp2*), was not toxic. In these strains, the endogenous copy of pbp2WT was still intact. Cells were stained with the membrane dye Nile red. Scale bars, 1 μm. d, Purified PBP2 and a representative variant, PBP2F158L (PBP2*), were incubated with Lipid II to assess polymerization activity. e, Reducing peptidoglycan synthesis activity with a compound suppressed ΔlytH high-temperature sensitivity. Moenomycin inhibits PBP2. Data are representative of two (a-e) independent experiments.
We used fluorescence microscopy to examine whether peptidoglycan synthesis was dysregulated in the absence of LytH. To monitor sites of peptidoglycan synthesis, we first labeled cells with fluorescein-D-lysine (FDL)27, which is exchanged into stem peptides by transpeptidases. Fluorescent bands at midcell reflect septal peptidoglycan synthesis. Compared with WT, the ΔlytH mutant showed fewer cells with signal at midcell and more cells with signal at misplaced septa or peripheral puncta (Extended Data Fig. 9a). By measuring the ratio of fluorescence intensity at the septum vs. periphery in cells that have a single complete septum (Extended Data Fig. 9b), we found a change in the spatial distribution of FDL signal (Fig. 4a). This change was independent of cell size as ΔlytH cells across a range of cell volumes showed reduced septal to peripheral fluorescence compared to WT cells (Extended Data Fig. 9c,d). A reduction in fluorescence ratio could result from decreased septal fluorescence and/or increased peripheral fluorescence. To directly compare the septal or peripheral signal between WT and the ΔlytH mutant, we labeled a mixed population of WT and ΔlytH cells with FDL and imaged the two strains simultaneously in the same frame. One strain was selectively labeled with the nuclear dye DAPI so that we could distinguish WT from ΔlytH cells in the mixed population. We found that FDL-labeled stem peptides were depleted at the septum in the absence of LytH (Fig. 4b and Extended Data Fig. 9e,f). To determine whether the transpeptidase activity that was observed is due to reaction at sites of new peptidoglycan synthesis, we used a pulse-chase approach28 to selectively detect nascent peptidoglycan with fluorescent vancomycin, which binds stem peptides having terminal D-Ala-D-Ala residues. We found that newly synthesized peptidoglycan was depleted at the septum relative to the periphery in ΔlytH cells (Fig. 4c and Extended Data Fig. 9g). Together, these experiments showed that the change in transpeptidase activity, as assessed by FDL incorporation, occurs because production of new peptidoglycan is spatially altered in ΔlytH cells.
Figure 4: LytH spatially regulates peptidoglycan synthesis.
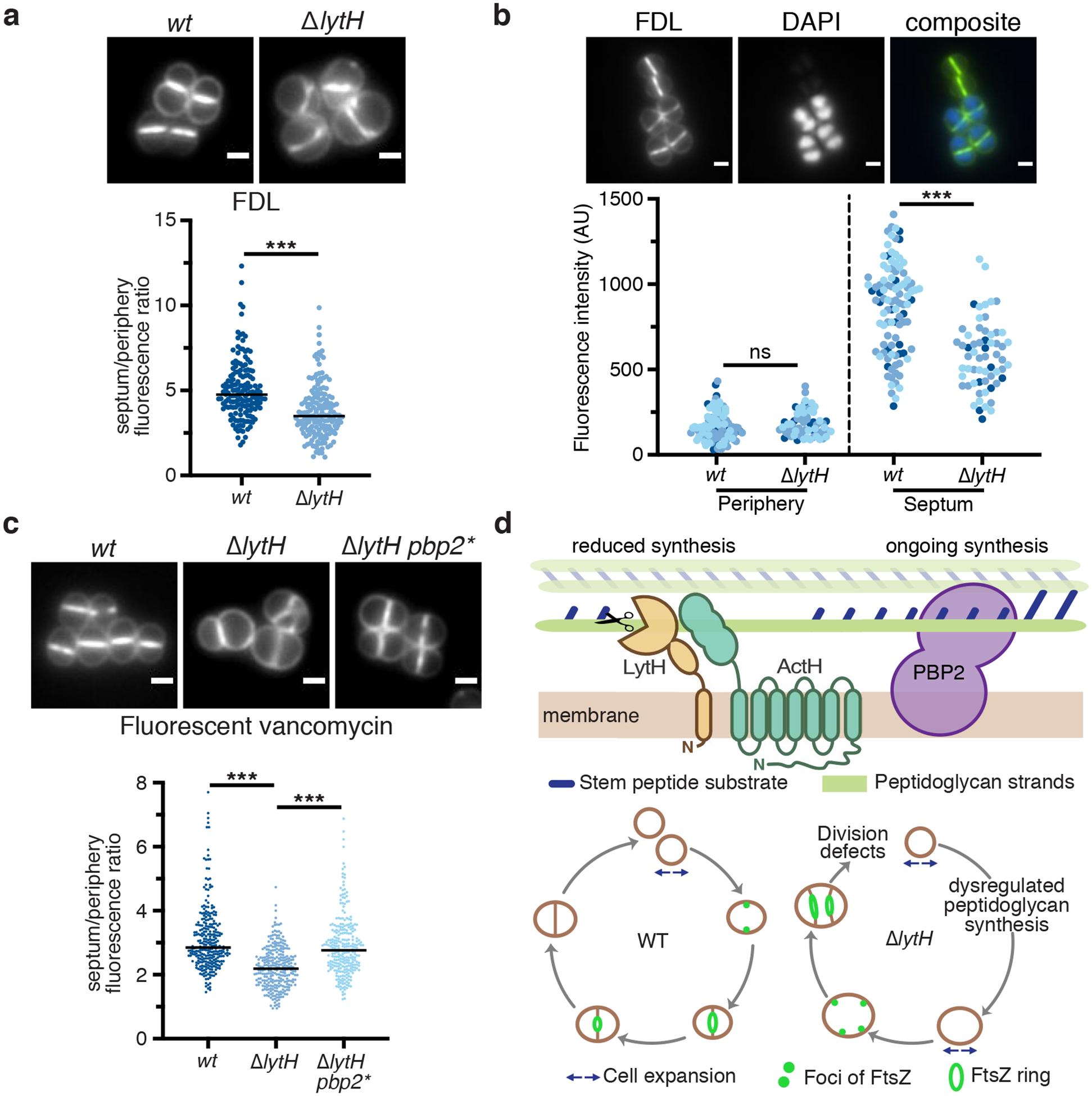
a, Cells were labeled with fluorescein-D-lysine (FDL) to mark sites of transpeptidase activity; ΔlytH cells showed decreased septal to peripheral signal. Fluorescence ratios were calculated using only cells with a single complete septum. FDL incorporation was catalyzed by the transpeptidases PBP1, PBP2, and PBP3; pbp4 was deleted to reduce background47. In all plots, the median is marked by a black line, each dot represents a single cell, and p-values were determined by two-sided Mann-Whitney U tests. In this plot: n = 159 (wt) and 167 (ΔlytH) cells; P = 1.34×10−12 (***P < 0.001). Scale bars, 1 μm. b, A mixed population of WT and ΔlytH cells was labeled with FDL and imaged in the same frame to directly compare the signal at the septum and periphery; ΔlytH cells showed decreased septal activity. In these images, ΔlytH cells were stained with DAPI to distinguish them from WT cells. Fluorescence intensity measurements correspond to the median intensity at the periphery or septum. Different colored dots represent cells analyzed from different fields of view: n = 100 (wt) and 61 (ΔlytH) cells. From left to right: P = 0.0864 and 4.49×10−10 (***P < 0.001; ns, not significant). Scale bars, 1 μm. c, Newly synthesized peptidoglycan was labeled with fluorescent vancomycin; a decrease in septal to peripheral signal was observed for ΔlytH cells expressing pbp2WT. pbp2* represents the suppressor allele pbp2F158L. From left to right: P = 3.87×10−32 and 1.51×10−19 (***P < 0.001); counted 283 (wt), 287 (ΔlytH), and 278 (ΔlytH pbp2*) cells. Scale bars, 1 μm. d, Model for LytH function. Control of cell size is required for FtsZ localization at midcell to properly initiate division. Free stem peptides limit rapid diffusion of PBP2 to midcell during division, leading to increased cell size due to dysregulated peptidoglycan synthesis. LytH regulates stem peptide abundance on membrane-proximal peptidoglycan: in its absence, cells enlarge, FtsZ becomes mislocalized, and division initiates at aberrant sites. Data are representative of two (a-c) independent experiments.
Our selection of pbp2 suppressors that reduced peptidoglycan synthesis activity and corrected cell size suggested PBP2 contributes to the dysregulated synthesis of peptidoglycan. Reducing PBP2 activity restored the normal spatial distribution of peptidoglycan synthesis (Fig. 4c). We examined PBP2 localization using a sGFP-PBP2 fusion29 and found that the ΔlytH mutant showed reduced PBP2 signal at the septum relative to the periphery (Extended Data Fig. 10a). Delocalization of PBP2 was independent of cell size (Extended Data Fig. 10b). It also did not appear to be a consequence solely of FtsZ mislocalization because we observed PBP2 signal at peripheral regions where FtsZ was not found. Moreover, quantitative assessment showed reduced colocalization of FtsZ and PBP2 in the ΔlytH mutant (Supplementary Fig. 19c). Taken together, our findings indicate that LytH spatially regulates peptidoglycan synthesis to control cell size and allow for proper assembly of the divisome.
The two key findings in this paper are the discovery of the first direct regulator for an amidase in a Gram-positive organism and the demonstration that this amidase controls cell growth. Amidases are a class of cell wall hydrolases previously shown to be important for separating daughter cells after cell division by cleaving crosslinked peptidoglycan7. We have shown that the amidase studied here, LytH, instead cleaves stem peptides from uncrosslinked peptidoglycan. We have also shown that LytH regulates where peptidoglycan synthesis occurs to control cell size and cell division.
A key question is how this amidase, which acts on membrane-proximal peptidoglycan, can spatially regulate peptidoglycan synthesis activity. We propose the following model (Fig. 4d). Previous studies found that S. aureus PBP2 is recruited to midcell following Z-ring assembly3 and recruitment of MurJ30,31, the flippase that exports the peptidoglycan precursor, Lipid II, to the cell surface3. It was shown that PBP2 is recruited to midcell by substrates present there with which it covalently reacts32. Our findings suggest that it is also necessary to control the rate of cell expansion by altering the density of peptidoglycan synthesis sites, which ensures that the division machinery properly localizes to midcell at the appropriate time. We have found that PBP2 can react with stem peptides in preassembled glycan strands even in the absence of ongoing polymerization by the polymerase domain (Supplementary Table 4). This means that excess free stem peptides in nascent or partially crosslinked peptidoglycan can transiently trap PBP2 so that it cannot relocate freely to another site in the cell. Because Lipid II can diffuse freely, peripheral peptidoglycan synthesis will continue unless binding sites on peptidoglycan polymer that capture the synthase at the periphery are sufficiently reduced. In the absence of LytH, there is less synthesis at the septum; peripheral synthesis increases but the signal is diluted across a larger cell volume, so the local intensity appears similar as in WT. Trimming of excess stem peptides slows peptidoglycan synthesis to keep cell expansion in check. According to this model, the more uncrosslinked stem peptides there are, the more important LytH is for regulating peptidoglycan synthesis to control cell size. LytH deletion mutants have substantial cell size, growth rate, and division defects even when grown at 30°C, and the morphological aberrations become more pronounced with increased temperature until LytH becomes essential for viability. LytH thus plays a key role in controlling the spatial distribution of peptidoglycan synthesis to keep cell expansion coordinated with cell division.
We note that actH (rbd) forms a synthetic lethal pair with noc12, which encodes the nucleoid occlusion protein that coordinates DNA replication with division and prevents FtsZ assembly over the chromosomes (lytH barely missed the cutoff for synthetic lethality with noc). In the absence of Noc, cells grow larger than normal, resulting in division defects. Therefore, Noc has been described as a spatial regulator of cell division10,12. Our findings that the LytH-ActH complex regulates cell growth and division explain the synthetic lethal relationship with Noc.
The need to regulate cell wall hydrolases to control their activity in cells has long been appreciated. The peptidoglycan cell wall is essential for bacterial survival and perturbations that compromise cell wall integrity reduce cell viability. At the same time, cell wall hydrolases that cleave bonds within peptidoglycan are important for cell wall remodeling and cell separation7. Hydrolytic activity must be regulated to ensure that hydrolases act when and where they should in coordination with peptidoglycan synthesis to ensure cell wall integrity5. The first cell wall hydrolase regulators, NlpD and EnvC33, were only discovered recently; both are lipoproteins that activate the cell-separation amidases in Gram-negative bacteria. In the Gram-positive organisms Streptococcus pneumoniae and Bacillus subtilis, the ABC transporter-like FtsEX complex has been implicated in regulating the hydrolases PcsB34–36 and CwlO37,38, but a direct activation mechanism has not yet been demonstrated.
Here we have shown that ActH directly interacts with LytH and activates its amidase activity. A topology prediction of ActH revealed three parts to the protein: a large intracellular region, a polytopic rhomboid fold, and an extracellular TPR domain (Extended Data Fig. 3e). Rhomboid proteins are a ubiquitous family of intramembrane proteases present across all kingdoms of life, but their physiological function in bacteria is largely unknown39. Our work establishes a role for rhomboid proteins in bacterial cell wall biogenesis and we suggest that ActH orthologs may play a similar role in amidase activation in some other bacteria. How ActH activates LytH is under investigation, although we note that ActH does not activate LytH by proteolytic processing because the amidase remains intact both in cells and in vitro (Supplementary Table 5). Moreover, the catalytic serine40 of ActH is dispensable for activation of LytH (Extended Data Fig. 4e). A better understanding of the domain requirements for ActH activation of LytH will help focus the search for additional regulators of cell wall hydrolases.
Methods
Bacterial growth conditions.
Strains are listed in Supplementary Table 6. All S. aureus strains were grown in tryptic soy broth (TSB), unless otherwise indicated, containing appropriate antibiotics at temperatures ranging from 30–42°C with aeration. All E. coli strains were grown in Lysogeny broth (LB), unless otherwise indicated, containing appropriate antibiotics at temperatures ranging from 30–37°C with aeration. Plasmids were cloned using E. coli XL1-Blue, Stellar, and NovaBlue(DE3) for general purposes and E. coli NEB 10-beta for pTarKO constructs. Plasmids isolated from E. coli DC10B were directly electroporated into S. aureus HG003 strains without first passaging through S. aureus RN4220. The E. coli C43(DE3) strain was used for overexpression of LytH-His6, His6-ActH, LytH-1x FLAG, His6-ActHS269A, His6-ActH-LytHWT-1x FLAG, and His6-ActH-LytHD195A-1x FLAG. The E. coli BL21(DE3) strain was used for overexpression of LytH-His6 truncations and PBP2-His8 constructs. Antibiotics were used at the following concentrations for S. aureus strains: kanamycin (50 μg/mL), neomycin (50 μg/mL), tetracycline (3 μg/mL), chloramphenicol (10 μg/mL), and erythromycin (10 μg/mL). Antibiotics were used at the following concentrations for E. coli strains: carbenicillin (100 μg/mL) and kanamycin (50 μg/mL).
Plasmid construction.
Plasmids and oligonucleotides are listed in Supplementary Tables 7 and 8, respectively. DNA sequencing of plasmids was conducted through Eton Bioscience and the Dana-Farber/Harvard Cancer Center DNA Sequencing Facility. Genomic DNA prepared from S. aureus strain RN4220 was used as the template in PCR reactions to amplify S. aureus genes.
pTP63 expression plasmids.
To construct pTP6312 plasmids containing anhydrotetracycline (atc)-inducible constructs, the gene sequence with its native ribosome-binding site (−25) was amplified by PCR. When needed, a C-terminal 1x FLAG tag encoded in a primer was appended to the gene sequence. For plasmid pTD10, primers oTD38 and oTD39 were used. For plasmid pTD11, primers oTD40 and oTD41 were used. For plasmid pTD18, primers oTD54 and oTD58 were used. For plasmid pTD19, primers oTD56 and oTD57 were used. The gene fragments were cloned between the KpnI and BlpI restriction sites of the pTP63 vector.
pLOW expression plasmids.
To construct pLOW41 plasmids containing IPTG-inducible constructs, the gene sequence with the native rpoB ribosome-binding site (-25) was amplified by PCR. When needed, a C-terminal 1x FLAG tag encoded in a primer was appended to the gene sequence. For plasmid pTD30, primers oTD86 and oTD85 were used. For plasmid pTD31, primers oTD87 and oTD85 were used. For plasmid pTD34, a gBlock gene fragment was synthesized by IDT. For plasmid pTD39, primers oTD86, oTD111, oTD104, and oTD106 were used; in this case, overlap PCR was performed to stitch together the two fragments. The gene fragments were cloned between the SalI and BamHI restriction sites of the pLOW vector.
pTD2 and pTD3.
To construct plasmid pTD2 for His6-LytHWT [E41-A291] overexpression and purification, the gene sequence for lytH was amplified by PCR using primers oTD15 and oTD17. To construct plasmid pTD3 for His6-LytHWT [T102-A291] overexpression and purification, the gene sequence for lytH was amplified by PCR using primers oTD16 and oTD17. The gene fragments were cloned between the NdeI and BamHI restriction sites of the pET15b vector. LytHWT [E41-A291] encodes a LytH truncation missing the predicted transmembrane domain. LytHWT [T102-A291] encodes a LytH truncation missing the predicted transmembrane and SH3 domains.
pTD6.
The kanR marker (primers oTD3 and oTD4) and the 1-kb sequences upstream (primers oTD24 and oTD25) and downstream (primers oTD7 and oTD8) of the lytH open reading frame were amplified by PCR. Overlap PCR was performed to stitch together the three fragments, and the resulting fragment was cloned between the BamHI and SalI restriction sites of pKFC42.
pTD42.
This construct is for LytHWT [M1-A291]-His6 overexpression and purification. The gene sequence for lytH was amplified by PCR using primers oTD117 and oTD116. The gene fragment was cloned between the NcoI and XhoI restriction sites of the pET28b vector.
pTD47.
This plasmid is used to make a tetR-marked deletion of actH. The tetR marker (primers oTD145 and oTD146) and the 1-kb sequences upstream (primers oTD141 and oTD142) and downstream (primers oTD143 and oTD144) of the actH open reading frame were amplified by PCR. Overlap PCR was performed to stitch together the three fragments, and the resulting fragment was cloned between the BamHI and SalI restriction sites of the pTarKO vector43.
pTD48.
This construct is for PBP2F158L [K60-S716]-His8 overexpression and purification. The pET42a(+)-pbp2 plasmid20 template was linearized by PCR using primers oTD148 and oTD149. The linearized DNA was phosphorylated at the 5’ ends using T4 polynucleotide kinase, and the ends were ligated to re-produce the circular DNA. Plasmid pET42a(+)-pbp2E114Q was constructed using QuikChange site-directed mutagenesis (StrataGene) with primers YQ1 and YQ2 and plasmid template pET42a(+)-pbp2.
pTD51.
This construct is for the dual expression of His6-actH [N2-K487]-lytH [M1-A291]-1x FLAG. First, the gene sequence for lytH was amplified by PCR using primers oTD166 and oTD167, and the gene fragment was cloned between the NdeI and KpnI restriction sites of the pETDuet-1 vector. Next, the gene sequence for actH was amplified by PCR using primers oTD160 and oTD161, and the gene fragment was cloned between the EcoRI and HindIII restriction sites of the previously made pETDuet-1-lytH vector.
pTD52.
This construct is for His6-actH [N2-K487] overexpression and purification. First, plasmid pTD51 was digested with NcoI and HindIII restriction enzymes. The excised NcoI-His6-actH [N2-K487]-HindIII fragment was then cloned between the NcoI and HindIII restriction sites of the pET28b vector.
pTD54.
This construct is for the dual expression of His6-actH [N2-K487]-lytHD195A [M1-A291]-1x FLAG. pTD51 plasmid template was linearized by PCR using primers oTD103 and oTD104. The linearized DNA was phosphorylated at the 5’ ends using T4 polynucleotide kinase, and the ends were ligated to re-produce the circular DNA.
pTD71.
This is a cadmium chloride (CdCl2)-inducible S. aureus expression vector for an FtsZ-sGFP sandwich fusion3. Plasmid pCN5144 was digested with ApaI and XhoI restriction enzymes. The excised ermR resistance cassette was then cloned between the ApaI and XhoI restriction sites of the pCN-ftsZ55–56sGFP vector.
pTD72.
This is an anhydrotetracycline-inducible S. aureus expression vector for a C-terminal FtsZ [M1-R390]-mCherry fluorescent fusion with a 5-amino acid spacer sequence. This fusion protein was designed using a published sequence for an FtsZ-mCherry fluorescent fusion3. The gene sequence for ftsZ was amplified by PCR using primers oTD248 and oTD249. The sequence for mCherry45 was amplified by PCR from a gBlock gene fragment synthesized by IDT using primers oTD250 and oTD227. Overlap PCR was performed to stitch together the two fragments using primers oTD248 and oTD227, and the resulting fragment was cloned between the KpnI and BlpI restriction sites of the pTP63 vector.
pTD78.
This construct is for LytHWT [M1-A291]-1x FLAG overexpression and purification. The gene sequence for lytH was amplified by PCR using primers oTD117 and oTD258. The gene fragment was cloned between the NcoI and HindIII restriction sites of the pET28b vector.
pTD79.
This construct is for His6-ActHS269A [N2-K487] overexpression and purification. pTD52 plasmid template was linearized by PCR using primers oTD259 and oTD260. The linearized DNA was phosphorylated at the 5’ ends using T4 polynucleotide kinase, and the ends were ligated to re-produce the circular DNA.
Strain construction.
To construct the ΔlytH::kanR marked deletion mutant, a previously published method was used42. Briefly, plasmid pTD6 was electroporated into S. aureus strain RN4220 and transformants were selected on 10 μg/mL chloramphenicol at 30°C. Transformants were cultured at 42°C to promote chromosomal integration of the plasmid; integrants were selected on 5 μg/mL chloramphenicol at 42°C. Confirmed integrants were passaged in TSB without antibiotics at 30°C for several days to promote plasmid excision from the chromosome. Deletion mutants were selected on 50 μg/mL kanamycin and 50 μg/mL neomycin at 30°C. The ΔlytH::kanR deletion was confirmed by colony PCR and sequencing, and the marked deletion was transduced into other strain backgrounds to construct strains TD024, TD240, TD300, and PBF002.
To construct strains containing pTP63 constructs12, the plasmids were first electroporated into S. aureus strain TD011 and transformants were selected on 10 μg/mL chloramphenicol at 30°C. The pTP63 constructs were transduced from these transformants into strain TD024 to construct strains TD036, TD037, TD074, and TD075.
To construct strains containing pLOW constructs41, the plasmids were first electroporated into S. aureus strain RN4220 and transformants were selected on 10 μg/mL erythromycin at 30°C. The pLOW constructs were transduced from these transformants into strain TD024 to construct strains TD134, TD135, TD157, and TD164.
To construct the ΔactH::tetR marked deletion mutant, a previously published method43 was used. Briefly, plasmid pTD47 was UV-irradiated and electroporated into S. aureus strain TD140, and double-crossover mutants were selected on 3 μg/mL tetracycline and 10 μg/mL targocil. The ΔactH::tetR deletion was confirmed by colony PCR, and after expressing pKFC-tarO, the marked deletion was transduced into strains HG003, TD024, and TD134 to construct strains TD177, TD178, and TD179, respectively.
To construct strains expressing a CdCl2-inducible fluorescent FtsZ-sGFP sandwich fusion3, plasmid pTD71 was first electroporated into S. aureus strain RN4220 and transformants were selected on 10 μg/mL erythromycin. From the transformants, plasmid pTD71 was transduced into strains HG003, TD024, and TD104 to construct strains TD268, TD269, and TD282, respectively.
To construct strains expressing a sGFP-PBP2 fusion29 at the native pbp2 locus, the plasmid pMAD-sGFPpbp2 was first electroporated into S. aureus strain RN4220 and then transduced into S. aureus strains HG003 and NCTC8325–4; transformants were selected on 50 μg/mL kanamycin and 50 μg/mL neomycin. Chromosomal integration and excision of pMAD-sGFPpbp2 to construct a clean replacement of the native pbp2 allele with sgfp-pbp2 was performed as previously described29. Briefly, cells were grown at 43°C in the presence of 50 μg/mL kanamycin and 50 μg/mL neomycin to promote the integration of pMAD-sGFPpbp2 into the chromosome by homologous recombination. After growing cells at 30°C without antibiotic, a second homologous recombination event promotes excision of the plasmid. PCR was used to confirm the genetic replacement in strains TD299 and PBF001.
To construct the TD305 ezrA deletion strain, a ΔezrA::kanR marked deletion was transduced from strain TD27143 into strain TD215. To construct the PBF003 ezrA deletion strain, the plasmid pBCBAJ00311 was transduced into strain PBF001 using phage 80α46. Chromosomal integration and excision of pBCBAJ003 to construct a clean knockout of the ezrA coding sequence on the chromosome was performed as previously described11. Briefly, to promote the integration of pBCBAJ003 at the ezrA locus via homologous recombination, PBF001 harboring the plasmid was grown at 43°C in the presence of 10 μg/mL erythromycin. Plasmid excision occurred after a second homologous recombination event, in cells grown at 30°C in the absence of antibiotic, to yield strain PBF003. Gene deletion was confirmed by PCR.
To construct dual fluorescent strains that simultaneously express sGFP-PBP229 and FtsZ-mCherry, plasmid pTD72 was first electroporated into S. aureus strain TD011 and transformants were selected on 10 μg/mL chloramphenicol at 30°C. The pTD72 construct was then transduced from the transformant into strains TD299 and TD300 to construct strains TD303 and TD304, respectively.
Phage transduction was performed using a previously published protocol, unless otherwise indicated43. Briefly, S. aureus donor cultures were mixed with staphylococcal phage ϕ85 and 10 mM CaCl2. After 30 min of incubation at room temperature, top agar (0.7% agar in TSB media) containing 7 mM CaCl2 was added, and the mixture was poured on tryptic soy agar (TSA) plates. The plates were incubated overnight at 30°C, and TSB was added to the plates the next day. After overnight incubation, the phage lysate was harvested from the plate and filtered. The phage lysate was used to transduce S. aureus recipient cultures by mixing recipient cells, phage lysate, LB media, and CaCl2. Transductants were selected on TSA plates containing the appropriate antibiotics and 0.05% sodium citrate. Colonies that appeared on the plates were passaged twice on sodium citrate, and transductants were confirmed using colony PCR.
Materials.
All reagents and chemicals were purchased from Sigma-Aldrich, unless otherwise indicated. Biotin-D-Lys (BDL) was prepared from Fmoc-biotin-D-lysine as previously described47. PCR primers were purchased from Integrated DNA Technologies. Phusion high-fidelity PCR master mix, restriction endonucleases, and T4 polynucleotide kinase were purchased from New England Biolabs, and KOD DNA polymerase was obtained from EMD Millipore. The In-fusion HD Cloning Plus kit was purchased from Takara Bio USA. Recombinant lysostaphin was purchased from AMBI Products. Mutanolysin from Streptomyces globisporus ATCC 21553 was purchased from Sigma-Aldrich. Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega). Synthetic Lipid I was prepared as previously reported48,49. The radiolabeled WTA precursor, [14C]-LIIAWTA, was prepared as previously reported50. Native Lipid II was extracted from S. aureus RN4220 culture using a published protocol20. Fluorescein-D-lysine (FDL) was synthesized as previously reported27. BODIPY FL vancomycin was purchased from ThermoFisher Scientific. D-serine was purchased from Alfa Aesar. DAPI (diamidino-2-phenylindole) solution was purchased from ThermoFisher Scientific.
Overexpression and purification of SgtB: E. coli BL21(DE3) was transformed with plasmid pET24b(+)-sgtBY181D encoding S. aureus SgtBY181D-His6 or plasmid pMgt1 encoding S. aureus SgtBWT-His6. These proteins were expressed and purified as reported previously21.
Overexpression and purification of S. aureus LcpB: E. coli BL21(DE3) was transformed with plasmid pLcpB encoding S. aureus His8-LcpB [S31-N405]. This protein was expressed and purified as previously reported23.
Overexpression and purification of PBPX: E. coli BL21(DE3) was transformed with plasmid pMW1010 encoding E. faecalis His6-PBPX [T36-P429]. This protein was expressed and purified as reported previously22.
Transposon sequencing.
A high-density transposon library of S. aureus HG003 was prepared51, treated with 37 μg/mL oxacillin, and sequenced as previously reported13. The oxacillin and control sequencing data can be found in the National Center for Biotechnology Information BioSample database using accession numbers SAMN08025141 and SAMN08025168, respectively. The data was processed as reported previously13, and genes significantly enriched and depleted under oxacillin treatment were identified using a two-sided Mann-Whitney U test corrected for multiple hypothesis testing via the Benjamini-Hochberg method. All scripts used for this analysis can be found at https://github.com/SuzanneWalkerLab/TnSeqMOAPrediction.
Transmission electron microscopy.
S. aureus strains were inoculated in TSB and the cultures were grown at 30°C overnight with aeration. The overnight cultures were diluted 1:100 into fresh TSB and grown to mid-log phase at temperatures ranging from 30–42°C. Cells were collected, added to an equal volume of fixative (1.25% formaldehyde, 2.5% glutaraldehyde, 0.03% picric acid in 0.1 M sodium cacodylate buffer, pH 7.4), and pelleted for the fixation process. TEM samples were prepared by the Electron Microscopy Facility at Harvard Medical School (Department of Cell Biology), and images were captured on the JEOL 1200EX instrument.
Spot dilution assay.
S. aureus strains were inoculated in TSB with antibiotics and the cultures were grown at 30°C overnight with aeration. Overnight cultures were diluted 1:100 into fresh TSB without antibiotics and grown to mid-log phase. The cultures were normalized, ten-fold dilutions were prepared in TSB, and 5 μL of each dilution was spotted onto TSA plates containing the appropriate antibiotics (0.1–0.125 μg/mL oxacillin or 0.01 μg/mL moenomycin A) and inducer. Plates were incubated overnight at temperatures ranging from 30–42°C. A Nikon D3400 DSLR camera fitted with an AF Micro-Nikkor 60 mm f/2.8D lens was used to take pictures of the plates.
Growth rate analysis.
S. aureus strains were inoculated in TSB and the cultures were grown at 30°C overnight with aeration. Overnight cultures were diluted to an initial OD600 of 0.01 into fresh TSB. The cultures were grown at 37°C with aeration. Every hour for the first 2 hours, and subsequently every 30 min for an additional 5 hours, the optical density (OD600) of the cultures was measured. At these time points, 10−1 to 10−8 dilutions of the cultures were prepared and plated on TSA; these plates were incubated overnight at 30°C and colonies were counted the next day. OD600 readings and colony-forming units (CFUs) were tracked over time.
Muropeptide analysis.
S. aureus strains were inoculated in TSB with antibiotics and the cultures were grown at 30°C overnight with aeration. Overnight cultures were diluted to OD600 = 0.02 in fresh TSB with the appropriate antibiotics and grown for 6–8 hours at 37°C. For the oxacillin treatment condition, cells were grown in TSB supplemented with 0.03125 μg/mL oxacillin. Cells were harvested (10,000 rpm for 5 min) from 2 mL of each culture. The cell pellets were processed for muropeptide analysis as previously described52, with the following modifications. For mutanolysin digestion, the pellet was resuspended in a 100 μL digestion buffer of 12.5 mM NaH2PO4, pH 5.5, and 10 μL of mutanolysin (4000 U/mL stock in H2O), and the mixture was incubated at 37°C for 16 hr on an orbital shaker (300 rpm). The lyophilized materials were resuspended in 100 μL of H2O and 15 μL was injected for LC-MS separation of muropeptides.
LC-MS was conducted using an Agilent Technologies 1200 series HPLC in line with an Agilent 6520 Q-TOF mass spectrometer and electrospray ionization (ESI) operating in positive ion mode and the low (1700 m/z) mass range. The Agilent Q-TOF was partially funded by the Taplin Funds for Discovery program.
The muropeptide fragments were separated on a Waters Symmetry Shield RP18 column (5 μm, 3.9 × 150 mm) with a matching column guard using previously published LC-MS conditions20: 0.5 mL/min solvent A (H2O, 0.1% formic acid) for 5 min followed by a linear gradient of 0 to 40% solvent B (acetonitrile, 0.1% formic acid) over 25 min.
For targeted LC-MS/MS identification of the tetrasaccharide-monopeptide species (866.3864 [M+2H]2+), 25 μL of muropeptide sample from oxacillin-treated wild-type cells was injected. The same method and column as above were used, with the following modifications: prec. m/z = 866.3881; Z = 2; retention time = 14.3 min; delta retention time = 2 min; iso. width = ~4 m/z.
LC-MS data were analyzed using the Agilent MassHunter Workstation Qualitative Analysis software version B.06.00. Theoretical isotope distributions were obtained using the Thermo Xcalibur software version 3.0.63. Muropeptide species were extracted using their theoretical m/z values with a mass error of ± 20 ppm. The observed and theoretical isotope distributions were compared to confirm the species. The relative ion count for a species of interest was calculated by dividing its extracted ion count (EIC) by the sum of the EICs for the following species within the same experiment and sample (we refer to this sum as the total ion count, TIC): monomer (1253.5856 [M+H]+), dimer (1209.0617 [M+2H]2+), trimer (1194.2204 [M+3H]3+), and tetramer (1186.7998 [M+4H]4+). These are the theoretical mass-to-charge ratios used to extract the ions.
Pairwise comparison of muropeptide abundance.
LC-MS total ion chromatograms were converted from Agilent Masshunter .d format to .mzdata format and uploaded to the XCMS platform (The Scripps Research Institute). Pairwise comparison of muropeptide abundance was performed with the HPLC/Q-TOF parameter. Pairwise data were sorted using the following parameters to distinguish relevant species from background signal: retention time (rtmed) between 12–18 min for this LC-MS method; fold enrichment greater than 5; maxint > 10000; and dataset1_mean > 10000.
LytH topology prediction and homology model.
LytH transmembrane topology was predicted using TMHMM Server version 2.0. LytH homology model was generated using the Phyre2 server and displayed in MacPyMol version 1.6. A sequence alignment of S. aureus LytH and E. coli AmiC was generated using Clustal Omega to identify the catalytic residues in LytH belonging to the amidase_3 family of proteins18.
Overexpression and purification of full-length LytH-His6.
E. coli C43(DE3) was transformed with plasmid pTD42 encoding full-length S. aureus LytH [M1-A291] with a C-terminal His6 tag. Colonies were inoculated into 50 mL Terrific broth supplemented with 50 μg/mL kanamycin, and the culture was grown overnight at 37°C with aeration. The overnight culture was diluted 1:100 into 1.5 L Terrific broth supplemented with 50 μg/mL kanamycin and grown at 37°C with aeration until mid-log phase, at which point the culture was shifted to 16°C. LytH expression was induced by the addition of 1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG), and the culture was grown for another 18–20 hr at 16°C with aeration. Cells were harvested (3600 rpm, 20 min, 4°C), resuspended in 30 mL Buffer A (25 mM HEPES, pH 6.8, 200 mM NaCl, 10% glycerol) supplemented with 1 mg/mL lysozyme and 250 μg/mL DNase, and lysed using an EmulsiFlex-C3 cell disruptor (Avestin). The cell lysate was centrifuged at low speed (10000×g, 5 min, 4°C) to collect and remove cell debris, and the supernatant was transferred for ultracentrifugation (35000 rpm, 45 min, 4°C). The membrane pellet was resuspended in 30 mL Buffer A supplemented with 1% n-Dodecyl β-D-maltoside (DDM, Avanti Polar Lipids), homogenized with a Dounce homogenizer, and the homogenate was tumbled at 4°C for 1 hr. A second ultracentrifugation step (35000 rpm, 45 min, 4°C) was performed to remove non-homogenized membranes, and the supernatant was applied to 2 mL of pre-washed Ni-NTA resin (Qiagen) at 4°C. The Ni-NTA resin was pre-equilibrated in Buffer B (25 mM HEPES, pH 7.5, 200 mM NaCl, 10% glycerol, 1% DDM, 10 mM imidazole). After collecting the flowthrough, the resin was washed with 3 times 10 CV of Buffer A containing: 1% DDM/10 mM imidazole, 0.25% DDM/50 mM imidazole, and 0.1% DDM/80 mM imidazole. The protein was eluted with 10 CV of Buffer A supplemented with 0.05% DDM and 400 mM imidazole. The eluted LytH protein was concentrated using a 50 kD MWCO Amicon Ultra Centrifugal Filter Unit (EMD Millipore). LytH was further purified using size-exclusion chromatography with a Superdex 200 Increase 10/300 GL column (GE Life Sciences) equilibrated in Buffer C (25 mM HEPES, pH 6.8, 200 mM NaCl, 10% glycerol, 0.05% DDM). The final protein concentration was measured via Nanodrop using the calculated extinction coefficient. Purified LytH protein was diluted to 20 μM in Buffer C, aliquoted, and stored at −80°C.
Biotin-D-lysine labeling of glycan strands.
A previously published method was adapted to label and detect glycan strands22. Briefly, 1X reaction buffer (50 mM HEPES, pH 7.5, 10 mM CaCl2, 60 μM Zn(OAc)2), BDL (3 mM), Lipid II (5 μM), and SgtBY181D (5 μM) were combined in a final volume of 10 μL (5% DMSO). The reactions were incubated at room temperature for 30 min and then quenched at 100°C for 5 min. LytH, ActH, LytHWT-ActH, LytHD195A-ActH, or LytH truncation mutants (1 μL of 20 μM stock) was added next. For the experiment described in Extended Data Fig. 4e, LytH and ActH were separately purified, a 40 μM stock of each protein was mixed 1:1 (v/v), and 1 μL of the pre-mixed proteins was used for the reactions. The reactions were incubated at room temperature for 16 hr and then quenched at 100°C for 5 min. To label glycan strands, E. faecalis PBPX (1 μL of 40 μM stock) was added and the reactions were incubated at room temperature for 1 hr. Finally, reactions were quenched by addition of an equal volume of 2X SDS loading buffer and analyzed using a previously published protocol22, with the following modifications. Five μL of each reaction was loaded into a 4–20% gradient polyacrylamide gel and run at 180V for 40 min. The products were transferred onto a 0.2 μm PVDF membrane (BioRad) and the membrane was blocked with SuperBlock TBS blocking buffer (ThermoFisher Scientific) for 1.5 hr at room temperature. Biotinylated products were probed using IRDye 800CW streptavidin (LI-COR) diluted 1:5000 in SuperBlock TBS buffer for 1 hr at room temperature. The blot was washed several times with 1X TBS, imaged using an Odyssey CLx imaging system (LI-COR), and analyzed with ImageJ.
Co-immunoprecipitation with 1x FLAG-tagged proteins.
S. aureus strains TD134, TD135, and TD157 carrying IPTG-inducible, FLAG-tagged full-length LytH, GFP, and truncated LytH, respectively, were grown in 25 mL TSB supplemented with the appropriate antibiotics at 30°C overnight with aeration. Overnight cultures were diluted 1:100 into 1 L TSB supplemented with 10 μg/mL erythromycin and 1 mM IPTG. Cultures were grown at 37°C for 6–8 hr with aeration to induce the expression of the FLAG-tagged proteins. Five hundred mL of cultures were harvested (3600 rpm, 15 min, 4°C) and the cell pellet was resuspended in 30 mL of lysis buffer (1X PBS, pH 7.4, 10 μg/mL DNase, 20 μg/mL RNase, 200 μg/mL lysostaphin, 5 mM MgCl2, 1 complete EDTA-free protease inhibitor tablet (Roche)). The resuspended cells were incubated at 37°C for 1 hr on a shaking platform to promote cell lysis, then cooled on ice, and further lysed using an EmulsiFlex-C3 cell disruptor. The lysate was spun down (10000×g, 15 min, 4°C) and the supernatant was transferred for ultracentrifugation (35000 rpm, 45 min, 4°C). The membrane pellet was resuspended in 3 mL of membrane extraction buffer (1X PBS, pH 7.4, 500 mM NaCl, 1% Triton X-100), homogenized with a Dounce homogenizer, and the homogenate was tumbled at 4°C for 1 hr. A second centrifugation step (10000×g, 15 min, 4°C) was performed to remove the remaining cell debris. The supernatant (~3 mL) was added to 3 mL of no salt wash buffer (1X PBS, pH 7.4, 1% Triton X-100) to dilute the salt. The diluted sample was batch incubated with 50 μL of pre-washed α-FLAG M2 magnetic beads (Sigma) at 4°C for 1 hr on a rocker. The α-FLAG M2 magnetic beads were pre-equilibrated in 1X PBS, pH 7.4. After incubation, the samples were transferred to a magnetic tube rack to separate the magnetic beads containing bound proteins from the supernatant. The beads were washed with 5x 500 μL of wash buffer (1X PBS, pH 7.4, 200 mM NaCl, 1% Triton X-100). To elute bound proteins, the magnetic beads were resuspended in 125 μL of elution buffer (1X PBS, pH 7.4, 200 mM NaCl, 1% Triton X-100, 150 μg/mL 3x FLAG peptide) and the samples were incubated for 15 min at room temperature to allow the proteins to elute. The tubes were then transferred to the magnetic rack and the supernatant containing eluted proteins was collected. A second elution step with 125 μL of elution buffer was performed. The eluate and wash fractions were heated at 37°C for 15 min in SDS loading buffer and then separated on a 4–20% gradient polyacrylamide gel. The gel was stained with InstantBlue Protein Stain (Expedeon) for 1 hr at room temperature, destained with H2O, and imaged using a FluorChem R system (ProteinSimple). Protein bands were excised and stored in deionized H2O prior to submission for mass spectrometry analysis at the Taplin Mass Spectrometry Facility.
N-terminal Edman sequencing of LytH.
C-terminal FLAG-tagged LytH was expressed in strain TD134 and purified from detergent-solubilized membranes as described above. The eluate was separated on a 4–20% gradient polyacrylamide gel. Separated proteins were transferred to a 0.2 μm methanol-activated PVDF membrane. After the transfer, the membrane was washed with deionized H2O (5 washes, 5 min each). The membrane was then soaked in staining solution (0.02% Coomassie Brilliant Blue R-250 (ThermoFisher Scientific), 40% methanol, 5% acetic acid) for 30 sec at room temperature. The staining solution was removed and the membrane was immediately soaked in destaining solution (40% methanol, 5% acetic acid) for 1 min at room temperature. The destaining solution was removed and the membrane was washed with deionized H2O (6 washes, 5 min each). After the washes, the membrane was placed on a Whatman filter paper to air-dry completely. Protein bands were visible against a clear background. The LytH protein band was excised and submitted for Edman sequencing analysis at the Tufts University Core Facility.
ActH topology prediction and phylogeny analysis.
ActH transmembrane topology was predicted using TMHMM Server version 2.0. The domain structure of ActH was determined using the NCBI blastp (protein-protein BLAST) suite. Using NCBI CDART (Conserved Domain Architecture Retrieval Tool), ActH homologs with a similar domain structure as ActH, containing a rhomboid domain and tetratricopeptide repeats, were identified. Most homologs were found in the Firmicutes. Several homologs from each genus were then crosschecked using the TMHMM Server version 2.0 to confirm that their predicted membrane topologies were similar to that of ActH. The confirmed 228 CDART hits were loaded through the NCBI Batch Entrez database using their NCBI taxonomy IDs, and the 16S rRNA sequences from 196 species were extracted in FASTA format. These 16S rRNA sequences were aligned and phylogenic analysis was performed using Geneious version 9.1.5 as follows. First, a multiple sequence alignment of the 16S rRNA sequences was generated based on the ClustalW algorithm. The built-in Geneious Tree Builder function was used to generate a cladogram based on the Tamura-Nei genetic distance model and neighbor-joining method.
Overexpression and purification of LytH-ActH complexes, ActHWT, and ActHS269A.
E. coli C43(DE3) was transformed with plasmid pTD51 encoding S. aureus LytHWT-1x FLAG and His6-ActH, plasmid pTD54 encoding S. aureus LytHD195A-1x FLAG and His6-ActH, plasmid pTD52 encoding S. aureus His6-ActHWT, or plasmid pTD79 encoding S. aureus His6-ActHS269A. Colonies were inoculated into 50 mL Terrific broth supplemented with the appropriate antibiotic, and the culture was grown overnight at 37°C with aeration. The overnight culture was diluted 1:100 into 2 x 1 L Terrific broth supplemented with 100 μg/mL carbenicillin and grown at 37°C with aeration until mid-log phase, at which point the culture was shifted to 20°C. Dual protein expression was induced by the addition of 1 mM IPTG, and the culture was grown for another 18–20 hr at 20°C with aeration. Cells were harvested (3600 rpm, 20 min, 4°C) and resuspended in 30 mL Buffer A (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol) supplemented with 1 mg/mL lysozyme, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl, ThermoFisher Scientific), and 250 μg/mL DNase. The resuspended cells were stirred at 4°C for 30 min to disperse clumps and lysed using an EmulsiFlex-C3 cell disruptor. The cell lysate was centrifuged at low speed (10000 xg, 5 min, 4°C) to collect and remove cell debris, and the supernatant was transferred for ultracentrifugation (32000 rpm, 45 min, 4°C). The membrane pellet was resuspended in 30 mL of Buffer A supplemented with 1% DDM and 1 mM TCEP, and homogenized with a Dounce homogenizer. The homogenate was tumbled at 4°C for 1 hr. A second ultracentrifugation step (32000 rpm, 45 min, 4°C) was performed to remove non-homogenized membranes, and the supernatant was applied to 1 mL of pre-washed TALON metal affinity resin (Takara Clontech) at 4°C. The TALON resin was pre-equilibrated in Buffer A supplemented with 0.05% DDM and 1 mM imidazole. After collecting the flowthrough, the resin was washed with 40 CV of Buffer A containing: 1% DDM/2 mM imidazole, 0.2% DDM/4 mM imidazole, 0.1% DDM/6 mM imidazole, 0.05% DDM/8 mM imidazole, and 0.05% DDM/10 mM imidazole. The protein was eluted with 20 CV of Buffer A supplemented with 0.05% DDM and 150 mM imidazole. The eluted protein complex was concentrated using a 100 kD MWCO Amicon Ultra Centrifugal Filter Unit (EMD Millipore). The concentrated protein complex was further purified using size-exclusion chromatography with a Superose-6 Increase 10/300-GL (GE Life Sciences) column equilibrated in Buffer B (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol, 0.05% DDM). The final protein concentration was measured via Nanodrop using the calculated extinction coefficient. Purified proteins were diluted to 20 μM in Buffer B, aliquoted, and stored at −80°C.
Overexpression and purification of LytH-FLAG.
E. coli C43(DE3) was transformed with plasmid pTD78 encoding S. aureus LytHWT [M1-A291]-1x FLAG. Protein expression was induced as described for the LytH-ActH complexes. Cells were harvested and resuspended in 30 mL Buffer A (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol) supplemented with 1 mg/mL lysozyme, 250 μg/mL DNase, and 1 mM 4-benzenesulfonyl fluoride hydrochloride (AEBSF, ThermoFisher Scientific). The resuspended cells were lysed using an EmulsiFlex-C3 cell disruptor and the membrane fraction was isolated by ultracentrifugation. The membrane pellet was resuspended in 30 mL of Buffer B (50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol) supplemented with 1% DDM, and homogenized with a Dounce homogenizer. The homogenate was tumbled at 4°C for 1 hr, and a second ultracentrifugation step was performed to remove non-homogenized membranes. All subsequent purification steps were performed at 4°C. The supernatant was applied to 1 mL of anti-FLAG G1 affinity resin (Genscript) that was pre-equilibrated with Buffer B. The flowthrough was collected and reapplied onto the column 5 times to allow LytH to bind to the resin. After collecting the final flowthrough, the resin was washed with 20 CV of Buffer B containing a detergent gradient: 0.2% DDM, 0.1% DDM, and 0.05% DDM. The protein was eluted with 10 CV of Buffer A supplemented with 0.05% DDM and 0.2 mg/mL FLAG peptide. The eluted protein was concentrated using a 50 kD MWCO Amicon Ultra Centrifugal Filter Unit (EMD Millipore). The concentrated protein was further purified by size-exclusion chromatography on a Superdex 200 Increase 10/300 GL column (GE Life Sciences) column equilibrated in Buffer C (50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol, 0.05% DDM). The final protein concentration was measured via Nanodrop using the calculated extinction coefficient. Purified proteins were diluted to 40 μM in Buffer C, aliquoted, and stored at −80°C.
Overexpression and purification of truncated LytH.
E. coli BL21(DE3) was transformed with plasmid pTD2 encoding S. aureus His6-LytHWT [E41-A291] or plasmid pTD3 encoding S. aureus His6-LytHWT [T102-A291]. Colonies were inoculated into 25 mL LB supplemented with 100 μg/mL carbenicillin, and the culture was grown overnight at 37°C with aeration. The overnight culture was diluted 1:100 into 1.5 L LB supplemented with 100 μg/mL carbenicillin and grown at 37°C with aeration until mid-log phase, at which point the culture was shifted to 16°C. Protein expression was induced by the addition of 1 mM IPTG, and the culture was grown for another 18–20 hr at 16°C with aeration. Cells were harvested (3600 rpm, 20 min, 4°C) and resuspended in 30 mL Lysis Buffer (100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.6% 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS, Anatrace), 0.5% Triton X-100 reduced, 100 μg/mL lysozyme, 10 μg/mL DNase, and 1 mM phenylmethylsulfonyl fluoride (PMSF)). The resuspended cells were lysed using an EmulsiFlex-C3 cell disruptor. The cell lysate was centrifuged at low speed (10000 xg, 5 min, 4°C) to collect and remove cell debris, and the supernatant was transferred for ultracentrifugation (32000 rpm, 45 min, 4°C). After ultracentrifugation, the supernatant was applied to 2 mL of pre-washed Ni-NTA resin (Qiagen) at 4°C. The Ni-NTA resin was pre-equilibrated in Buffer A (100 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20% glycerol) supplemented with 2 mM imidazole. After collecting the flowthrough, the resin was washed with 20 CV of Buffer A containing: 10, 20, and 40 mM imidazole. The protein was eluted with 5 CV of Buffer A supplemented with 500 mM imidazole. The eluted protein was concentrated using a 10 kD MWCO Amicon Ultra Centrifugal Filter Unit (EMD Millipore). The concentrated protein was further purified using size-exclusion chromatography with a Superdex 75 10/300 GL column (GE Life Sciences) equilibrated in Buffer A. The final protein concentration was measured via Nanodrop using the calculated extinction coefficient. Purified proteins were diluted to 50 μM in Buffer A, aliquoted, and stored at −80°C.
Preparation of radiolabeled peptidoglycan polymers.
To prepare radiolabeled peptidoglycan oligomers, a [14C]-label was initially incorporated by incubating synthetic Lipid I with UDP-[14C]-GlcNAc (specific activity= 300 nCi/nmol) using MurG as previously published23. [14C]-Lipid II was incubated at room temperature with SgtBY181D (2 μM) in 1X TGase buffer (50 mM HEPES, pH 7.5, and 10 mM CaCl2) in 20% DMSO for 20 min.
To prepare WTA-modified oligomers, synthetic peptidoglycan oligomers and native peptidoglycan oligomers were first prepared cold. To prepare synthetic, cold peptidoglycan oligomers, a batch of synthetic Lipid II (20 μM) was incubated with SgtBY181D (2 μM) at room temperature in 1X TGase buffer (50 mM HEPES, pH 7.5, and 10 mM CaCl2) in 20% DMSO for 20 min. To prepare native peptidoglycan oligomers, a batch of native Lipid II (8 μM) was incubated with SgtBY181D (800 nM) at room temperature in 1X TGase buffer and 20% DMSO. Polymerization proceeded for 8 min before heat inactivation of SgtBY181D at 95°C for 10 min. After cooling to room temperature, the radiolabeled WTA-disaccharide precursor, [14C]-LIIAWTA (4.5 μM, specific activity = 300 nCi/nmol), and LcpB (2 μM) were added to each batch of peptidoglycan oligomers. Reactions proceeded for 2.5 hr at room temperature. LcpB was subsequently heat-inactivated at 95°C for 10 min.
PAGE analysis of amidase activity.
Radiolabeled peptidoglycan oligomer mixtures as prepared above were incubated with 2 μM LytH alone, LytHD195A-ActH, or LytHWT-ActH and 600 μM Zn(OAc)2. Reactions were incubated at room temperature overnight. Subsequently, reactions were quenched with methanol (equal volume; 10 μL) and dried to completion with a speed vacuum. Reactions were re-hydrated with 10 μL 2X SDS loading dye. The reactions were loaded on acrylamide gels, which have been described previously53. Tris/SDS gels (dimensions being 20 cm × 16 cm, H × W; 1.0 mM thickness) were prepared with 10 % acrylamide. The two running buffers were composed as follows: the anode buffer was 100 mM Tris (pH 8.8), and the cathode buffer consisted of 100 mM Tris (pH 8.25), 100 mM Tricine, and 0.1% SDS. The gels ran at constant voltage (30 mA) for 5 hr and were subsequently dried using a gel dryer (Labconco). Dried gels were incubated with a general phosphor screen for approximately 12–48 hr. The autoradiographs were imaged using a Typhoon 9400 imager (GE Healthcare) and analyzed using ImageJ software.
Preparation of crosslinked WTA-oligomers.
To prepare native peptidoglycan oligomers, a batch of native Lipid II (8 μM) was incubated with SgtBY181D (800 nM) at room temperature in 1X TGase buffer (50 mM HEPES, pH 7.5, and 10 mM CaCl2) and 20% DMSO. Polymerization proceeded for 8 min before heat inactivation of SgtBY181D at 95°C for 10 min. After cooling to room temperature, the radiolabeled WTA-disaccharide precursor, [14C]-LIIAWTA (4.5 μM, specific activity = 300 nCi/nmol), and LcpB (2 μM) were added to each batch of peptidoglycan oligomers. Reactions proceeded for 2.5 hr at room temperature. LcpB was subsequently heat-inactivated at 95°C for 10 min. Following precipitation and removal of both SgtBY181D and LcpB, PBP4 (2 μM) was added to each reaction and incubated at room temperature for 2 hr followed by inactivation at 95°C for 10 min. In addition to an untreated control, reactions were then subjected to either LytHWT-ActH (2 μM) or lysostaphin (1 mg/ml) treatment overnight at room temperature.
Sacculi hydrolysis assay.
Sacculi was prepared from stationary-phase cells of S. aureus strain HG003 using a previously published protocol52, with the following modifications. Volumes of solutions were scaled appropriately for the number of cells harvested. After 1 M HCl treatment, the pellets were washed with dH2O, flash-frozen, and lyophilized to yield purified sacculi.
A 0.5 mg/mL solution of sacculi was prepared in reaction buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.02% DDM, 60 μM Zn(OAc)2). The sacculi hydrolysis assay was set up in non-treated 96-well plates (Genesee Scientific). To each well, 150 μL of sacculi solution was added. Lysostaphin (dissolved in dH2O) or the purified LytH-ActH complex (stored in 50 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol, 0.05% DDM) was mixed with the sacculi substrate. Reactions were prepared in triplicate using 0–125 nM of purified protein per reaction. The plate was covered with a lid and incubated at 25°C with shaking. Absorbance (OD600) was recorded at 20-min intervals for 20 hr using a SpectraMax Plus 384 microplate spectrophotometer (Molecular Devices). Hydrolysis of sacculi was monitored over time as a decrease in absorbance.
LC-MS analysis to assess LytH activity.
A previously published method was adapted to detect the products of LytH enzymatic activity by LC-MS22. Briefly, 1X reaction buffer (50 mM HEPES, pH 7.5, 10 mM CaCl2, 60 μM Zn(OAc)2), Lipid II (40 μM), and SgtBWT (2 μM) were combined in a final volume of 10–30 μL (10% DMSO). S. aureus native Lipid II or synthetic Lipid II was used. The reactions were incubated at room temperature for 1 hr and then quenched at 100°C for 5 min. LytHWT-ActH or LytHD195A-ActH (2 μM) was added and the reactions were incubated at room temperature for 16 hr. The reactions were quenched at 100°C for 5 min and incubated with 4 U of mutanolysin for 1.5 hr at 37°C on an orbital shaker (300 rpm), followed by another 4 U of mutanolysin for 1.5 hr at 37°C. The muropeptides were reduced with NaBH4 (10–30 μL of 10 mg/mL solution in H2O) for 30 min at room temperature, and 20% phosphoric acid (~1.4–5 μL) was added to adjust the pH to ~4. The mixtures were lyophilized and redissolved in 20 μL of H2O, and 15 μL was injected for LC-MS separation using the same method as described above.
Isolation of lytH suppressor mutants and whole-genome sequencing.
Suppressor mutants of ΔlytH at 42°C on TSA spotting assay plates were independently isolated. These candidate suppressor colonies were picked and restreaked on TSA to confirm growth at 42°C. Overnight cultures of the suppressor mutants were inoculated, genomic DNA was prepared from these cultures, and the DNA concentration was measured with the Qubit dsDNA HS Assay Kit (ThermoFisher Scientific). The genomic DNA was prepared for whole-genome sequencing using a previously published method12, with the following modifications. Tagmented samples were pooled and analyzed by TapeStation and quantitative PCR at the Biopolymers Facility, Harvard Medical School. Sequencing was performed on the Illumina MiSeq platform using the MiSeq v3 150 cycle Reagent Kit (Illumina). Sequencing reads were analyzed using Geneious version 9.1.5. The suppressor mutants were compared to the parental S. aureus HG003 ΔlytH deletion strain to identify single nucleotide polymorphisms and deletion and insertion mutations that were unique to the suppressor mutants. The NCTC8325 genome was used as the reference genome.
Overexpression and purification of PBP2.
E. coli BL21(DE3) was transformed with plasmid pET42a(+)-pbp2 encoding S. aureus PBP2WT [K60-S716] with a C-terminal His8 tag or plasmid pTD48 encoding PBP2F158L [K60-S716]-His8. These proteins were expressed and purified as reported previously20. PBP2S398G and PBP2E114Q were prepared in the same way. To confirm that purified PBP2 was properly folded, a Bocillin-binding assay was used as reported previously22. Briefly, PBP2 (250 nM) was incubated with varying concentrations of penicillin G (1000, 100, 0 U/mL) in 1X buffer (20 mM potassium phosphate, pH 7.5, 140 mM NaCl) in a 9 μL reaction for 1 hr at 37°C on an orbital shaker (600 rpm). Bocillin-FL (1 μL of 100 μM stock) was then added and the reactions were incubated for 30 min at 37°C on an orbital shaker (600 rpm). Reactions were quenched by addition of an equal volume of 2X SDS loading buffer and analyzed by SDS-PAGE. The gel was imaged on a Typhoon FLA 9500 and analyzed using ImageJ.
BDL-labeling assay to assess PBP2 activity.
A previously published method20 was adapted to assess PBP2 activity. Briefly, 1X reaction buffer (50 mM HEPES, pH 7.5, 10 mM CaCl2), BDL (3 mM), Lipid II (10 μM), and PBP2 (1 μM) were combined in a final volume of 10 μL (10% DMSO). The reaction was incubated at room temperature for 5–15 min and then heat-quenched at 100°C for 1 min. As appropriate, lysostaphin was added to resolve highly-crosslinked peptidoglycan: 0.5 μL of lysostaphin (1 mg/mL stock) was added and the reactions were incubated at 37°C for 3 hr on an orbital shaker (300 rpm). Reactions were quenched by addition of an equal volume of 2X SDS loading buffer, and analyzed by SDS-PAGE and western blotting analysis with streptavidin as described above.
LC-MS analysis of crosslinked muropeptides by different polymerases.
Native S. aureus Lipid II (25 μM) was incubated with i. SgtB (1 μM), ii. the transpeptidase-inactive PBP2S398G (1 μM), or iii. PBP2WT (2 μM) in a 20 μl reaction containing 20% DMSO and 1X reaction buffer (50 mM HEPES, pH 6.5, 2.5 mM MgCl2). For i. and ii., the polymerase was heat-inactivated at 95°C for 5 min after a 2-hr incubation at room temperature; after cooling to room temperature, the glycosyltransferase-inactive PBP2E114Q (1 μM) was added and the reactions were incubated at room temperature for an additional 2 hr alongside reaction iii. All reactions were then heat-inactivated at 95°C for 5 min. After cooling to room temperature, mutanolysin (1 U) was added to each reaction for 1.5 hr at 37°C followed by another 1 U aliquot for 1.5 hr. The cleaved disaccharide fragments were then reduced with NaBH4 treatment (10 μL of 10 mg/mL solution in H2O) for 30 minutes. Phosphoric acid (20%, 1.4 μL) was added to each reaction to adjust the pH to 4, and then the mixture was lyophilized and re-dissolved in 20 μl H2O for LC/MS analysis, conducted with ESI-MS operating in positive mode on a Bruker qTOF mass spectrometer. The same column and method as for muropeptide analysis were used.
Nile red labeling to measure cell size.
A previously published method54 was adapted for Nile red labeling of S. aureus for fluorescence microscopy. Overnight cultures were diluted to a starting OD600 of 0.02 in 3 mL TSB, grown at 37°C with aeration to mid-log phase, and normalized to the same OD600. To 1 mL of cells, 5 μg/mL Nile red was added. The cells were labeled for 5 min at room temperature on an orbital shaker (500 rpm). The cells were centrifuged (4000 xg, 2 min, 25°C) and most of the supernatant was removed prior to resuspending the cells in the remaining (~50 μL) supernatant.
Nile red labeling of PBP2 overexpression strains.
Overnight cultures were diluted to a starting OD600 of 0.01 in 3 mL TSB. One set of culture was supplemented with 0.4 μM of the inducer anhydrotetracycline to overexpress PBP2. The second set of culture was supplemented with 95% ethanol, the solvent used to dissolve anhydrotetracycline, as controls. Cultures were grown at 37°C with aeration to mid-log phase, normalized to the same OD600, and labeled with Nile red as described above. Cells were manually inspected and sorted by morphology using the FIJI Cell Counter plugin.
Fluorescein-D-lysine labeling to visualize sites of transpeptidase activity.
A previously published method47 was adapted for fluorescein-D-lysine (FDL) labeling of S. aureus. Overnight cultures were diluted to a starting OD600 of 0.02 in 20 mL TSB, grown at 37°C with aeration to mid-log phase, and normalized to the same OD600. One mL of culture was centrifuged (4000 xg, 1 min, 25°C), and the cell pellet was resuspended in 500 μL of TSB. To the resuspended cells, 100 μM FDL was added and the cells were labeled for 10 min at 37°C on an orbital shaker (300 rpm). The cells were centrifuged (4000 xg, 1 min, 25°C), washed twice with 200 μL 1X PBS, pH 7.4, and resuspended in 50 μL of 1X PBS, pH 7.4.
Fluorescein-D-lysine and DAPI labeling of a mixed cell population.
Overnight cultures were diluted to a starting OD600 of 0.02 in 3 mL TSB, grown at 37°C with aeration to mid-log phase, and normalized to the same OD600. To 1 mL of strain A, 2 μg/mL of DAPI solution (ThermoFisher Scientific) was added. DAPI was not added to strain B. Both strains were incubated for 5 min at 37°C on an orbital shaker (500 rpm). Following DAPI labeling, the cells were centrifuged (4000 xg, 1 min, 25°C), washed once with 500 μL of TSB, and resuspended in 500 μL of TSB. To prepare a mixed population, 250 μL of DAPI-labeled strain A was mixed with 250 μL of unlabeled strain B. The mixed cells were then further labeled with 100 μM FDL for 10 min at 37°C on an orbital shaker (500 rpm). The cells were centrifuged (4000 xg, 1 min, 25°C), washed once with 500 μL 1X PBS, pH 7.4, and resuspended in 100 μL of 1X PBS, pH 7.4. The reciprocal experiment was performed by labeling strain B, and not strain A, with DAPI prior to mixing the two strains and labeling the mixture with FDL. Imaging strains A and B simultaneously in the same frame enables direct comparison of FDL signal at the septum and periphery between the two strains in the mixed population.
Labeling of nascent peptidoglycan.
A published method28 was used to label newly synthesized peptidoglycan in S. aureus. Strains were grown overnight in TSB containing excess D-serine (0.125 M). Overnight cultures were diluted to a starting OD600 of 0.02 in 3 mL of the same medium and grown at 37°C with aeration to mid-log phase. All 3 mL of cells were centrifuged (5000 xg, 1 min, 25°C), washed once with 200 μL of TSB, and resuspended in 3 mL of TSB without D-serine supplemented. Cultures were then incubated for 20 min on a shaker at room temperature to allow incorporation of D-alanine into the cell wall. One to two mL of culture were centrifuged (4000 xg, 1 min, 25°C), and the cell pellet was resuspended in 500 μL of TSB. To the resuspended cells, 0.5 μL of a 1:1 (v/v) mixture of 0.15 mg/mL fluorescent vancomycin (Van-FL) and 0.15 mg/mL vancomycin-hydrochloride was added. The cells were labeled for 5 min at room temperature. The cells were centrifuged (4000 xg, 1 min, 25°C), washed once with 200 mL 1X PBS, pH 7.4, and resuspended in 50 mL of 1X PBS, pH 7.4.
Visualization of fluorescent fusion proteins.
To assess sGFP-PBP2 localization, the strains PBF001, PBF002, and PBF003 were used. To assess FtsZ55–56sGFP localization, the strains TD268, TD269, and TD282 were used. To assess sGFP-PBP2 and FtsZ-mCherry colocalization, the strains TD303 and TD304 were used. All strains were grown in TSB overnight; 10 μg/mL erythromycin was added for FtsZ55–56sGFP strains to maintain the plasmid. Overnight cultures were diluted to a starting OD600 of 0.02 in 3 mL TSB. For strains TD303 and TD304, the medium was supplemented with 0.4 μM of the inducer anhydrotetracycline. For FtsZ55–56sGFP strains3, the medium was supplemented with 10 μg/mL erythromycin and 0.1 μM of the inducer CdCl2. Cultures were grown at 37°C with aeration to mid-log phase. One to two mL of cells were centrifuged (4000 xg, 1 min, 25°C), washed once with 200 μL 1X PBS, pH 7.4, and resuspended in 50 μL of 1X PBS, pH 7.4. For the strains PBF001, PBF002, and PBF003, cultures were grown in TSB to an OD600 of 0.5, and one mL of cells was pelleted and resuspended in the same medium.
Wide-field epifluorescence microscopy.
Cells were spotted onto a thin 2% agarose pad prepared using 1X PBS, pH 7.4. A 1.5 cover slip was used and Valap sealant (equal weight of petroleum jelly, lanolin, and paraffin) was applied to seal all sides of the cover slip. Cells were imaged using brightfield, phase-contrast, and wide-field epifluorescence microscopy at the MicRoN (Microscopy Resources On the North Quad) facility, Harvard Medical School. Images were obtained using a Nikon Ti inverted microscope fitted with a custom-made cage incubator set at 30°C, a Nikon motorized stage with an OkoLab gas incubator and a slide insert attachment, an Andor Zyla 4.2 Plus sCMOS camera, Lumencore SpectraX LED Illumination, Plan Apo lambda 100x/1.45 Oil Ph3 DM objective lens, and Nikon Elements 4.30 acquisition software. The microscope was fitted with Chroma ET filter cubes in a motorized filter turret: DAPI (49000), CFP (49001), GFP (49002), YFP (49003), and mCherry (49008). The following exposure times were used: 10–30 ms (Nile red labeling), 50 ms (FDL labeling), 100–200 ms (Van-FL labeling), 800 ms (DAPI), 100 ms (FtsZ-mCherry), 2 s (sGFP-PBP2) and 5 s (FtsZ55–56sGFP). Images were analyzed using FIJI and MATLAB scripts developed in-house.
The strains PBF001, PBF002, and PBF003 were mounted on 1.2% agarose pads prepared using 1X PBS. Images were acquired using a Zeiss Axio Observer microscope equipped with a Photometrics CoolSNAP HQ2 camera (Roper Scientific) and Metamorph 7.5 software (Molecular Devices). Images of PBF001 and PBF002 were analyzed by a line scan across the major axis of the cell to acquire the fluorescence at the cell periphery and septum along this trajectory. Images for PBF003 were analyzed using eHooke software3 previously developed in-house. To calculate the septal to fluorescence membrane signals, eHooke was used to define the septum and periphery of the cell. Only cells with a closed septum were selected for analysis.
Semi-automated image analysis.
S. aureus cell volumes were estimated using StaphSizer, a home-built MATLAB code, which was also used to categorize cellular phenotypes. Segmentation of cell clusters was performed on brightfield images using a watershedding algorithm55. First, brightfield images were used to create a binary image, to which a Euclidean distance transform function56–58 was applied to obtain an image whose centers corresponded to the cell regions within clusters of cells. Local minima that are not centered within the cells in the distance transform image were filtered out to prevent oversegmentation. Finally, the watershed algorithm was applied to the modified distance transform image to obtain the ridge lines that define the borders between neighboring cells. From this segmented image output, the x-y coordinates of each pixel on the cell boundary were obtained.
To sort cells based on phenotypes, the calculated cell boundary was overlaid on top of the fluorescence image and paired with its corresponding brightfield image to enable visual inspection of each cell. A 2-dimensional ellipse function was fitted to each cell boundary to calculate the lengths of the major and minor axes of the ellipse. To estimate the volume, S. aureus geometry was modeled as a prolate spheroid54 with the following volume equation:
Where V is the volume, a is half of the major (longer) axis, and b is half of the minor (shorter) axis of each fitted cell. Cell volumes were calculated using only phase 1 cells (cells without a visible septum).
The ratio of fluorescence intensity at the septum vs. periphery (Fseptum/Fperiphery) for selected cells was calculated using FreehandPeriSep, a home-built MATLAB code. After background correction, the coordinates of the boundary of a cell with a single complete septum were defined by the user manually tracing on a phase-contrast image. The coordinates of the user-defined region of interest (ROI) were used to create a binary mask that encompasses the entire cell (outer ROI). To obtain the fluorescence signal at the septum (septum ROI) in the same cell, the user manually traces the septum on the corresponding fluorescence image. To obtain the fluorescence signal at the cell periphery (periphery ROI), an inner ROI containing the septum and cytoplasm in the cell was first obtained by eroding the original binary mask such that the peripheral region is excluded; this inner ROI was then subtracted from the outer ROI. Finally, the periphery and septum ROIs were used to retrieve all pixel values at the cell periphery and septum, respectively. Only 50% of the brightest pixels at the septum and at the cell periphery were considered to avoid inclusion of misidentified pixels. The coordinates of the outer ROI were also used to model cells as prolate spheroids in order to calculate cell volume. Fluorescence ratios3 were calculated as follows:
The absolute fluorescence intensity at the septum and at the periphery between two different strains in a mixed population was calculated using TriPeriSep, a home-built MATLAB code. This script is similar to FreehandPeriSep, with a modification enabling the user to assign cells in the mixed population to the correct strain background. Prior to defining the peripheral and septal ROIs, the user is asked to distinguish if the chosen cell belongs to strain A or B based on the presence or absence of DAPI signal in the cell. DAPI signal was shown in a separate fluorescence image. Only cells with a single complete septum were considered in the analysis. Septal and peripheral activity was calculated as the median of the 50% brightest pixels at the septum and periphery, respectively. The septal and peripheral activities between strains A and B were compared within each image acquired. The signals from different images were also visualized on the same plot. The p-values for the combined signals were determined by two-sided Mann-Whitney U tests after signal normalization: for cells within the same image, the septal (or peripheral) signal of each cell was divided by the mean of the WT septal (or peripheral) signal.
The Pearson correlation coefficient (PCC)3 between two fluorescence channels was calculated using FreehandPCC, a home-built MATLAB code. A higher PCC indicates a greater degree of colocalization between sGFP-PBP2 and FtsZ-mCherry. Only cells showing FtsZ signal at the septum were considered for PCC analysis. The coordinates of the boundary of a cell were defined by the user manually tracing on a phase-contrast image. These coordinates were used to isolate images of chosen cells from the two background-corrected fluorescence channels. The PCC between the channels were obtained using the built-in MATLAB function corr2, which calculates the 2-D correlation coefficient of two images of size m-by-n via the following equation:
Where Amn and Bmn are the pixel intensities for the two fluorescence channels, and and are the mean intensities of those channels.
Extended Data
Extended Data Fig. 1. Deletion of lytH sensitizes cells to oxacillin and causes growth defects.
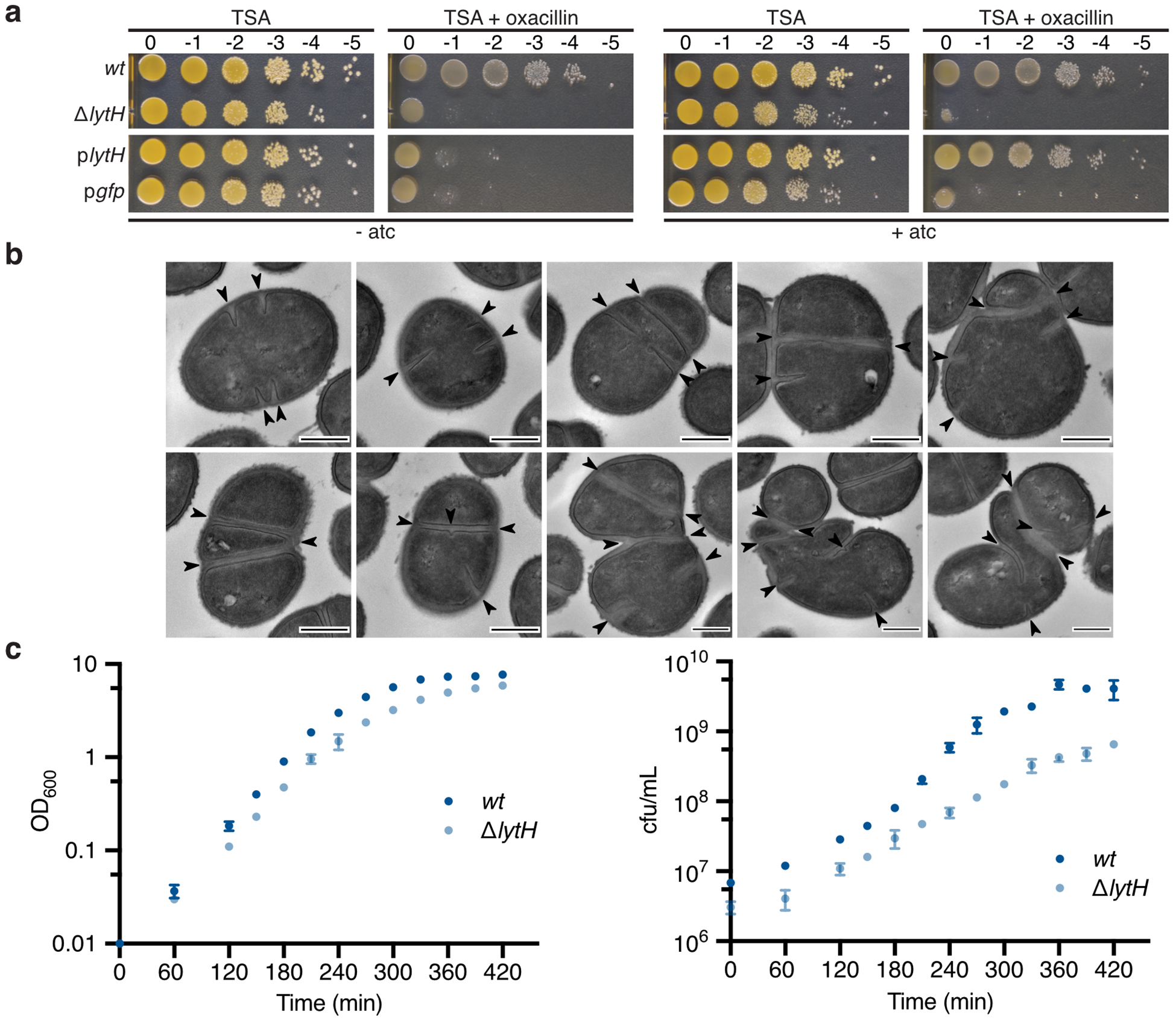
a, HG003 strains were spotted on TSA containing 0.1 μg/mL oxacillin and 0.4 μM of the inducer, anhydrotetracycline (atc), when indicated. Strains expressing lytH or gfp from an atc-inducible promoter in a ∆lytH background are denoted by plytH or pgfp, respectively. b, Transmission electron micrographs: Additional examples of defects in placement of nascent septa observed for the ∆lytH mutant. Scale bars, 500 nm. c, Growth was assessed by measuring optical density (left graph) and colony-forming units (right graph). Data represent mean ± SD. Error bars that are smaller than the dots are not shown. Data are representative of two (a–b) and three (c) independent experiments.
Extended Data Fig. 2. Muropeptide analysis identifies a tetrasaccharide-monopeptide.
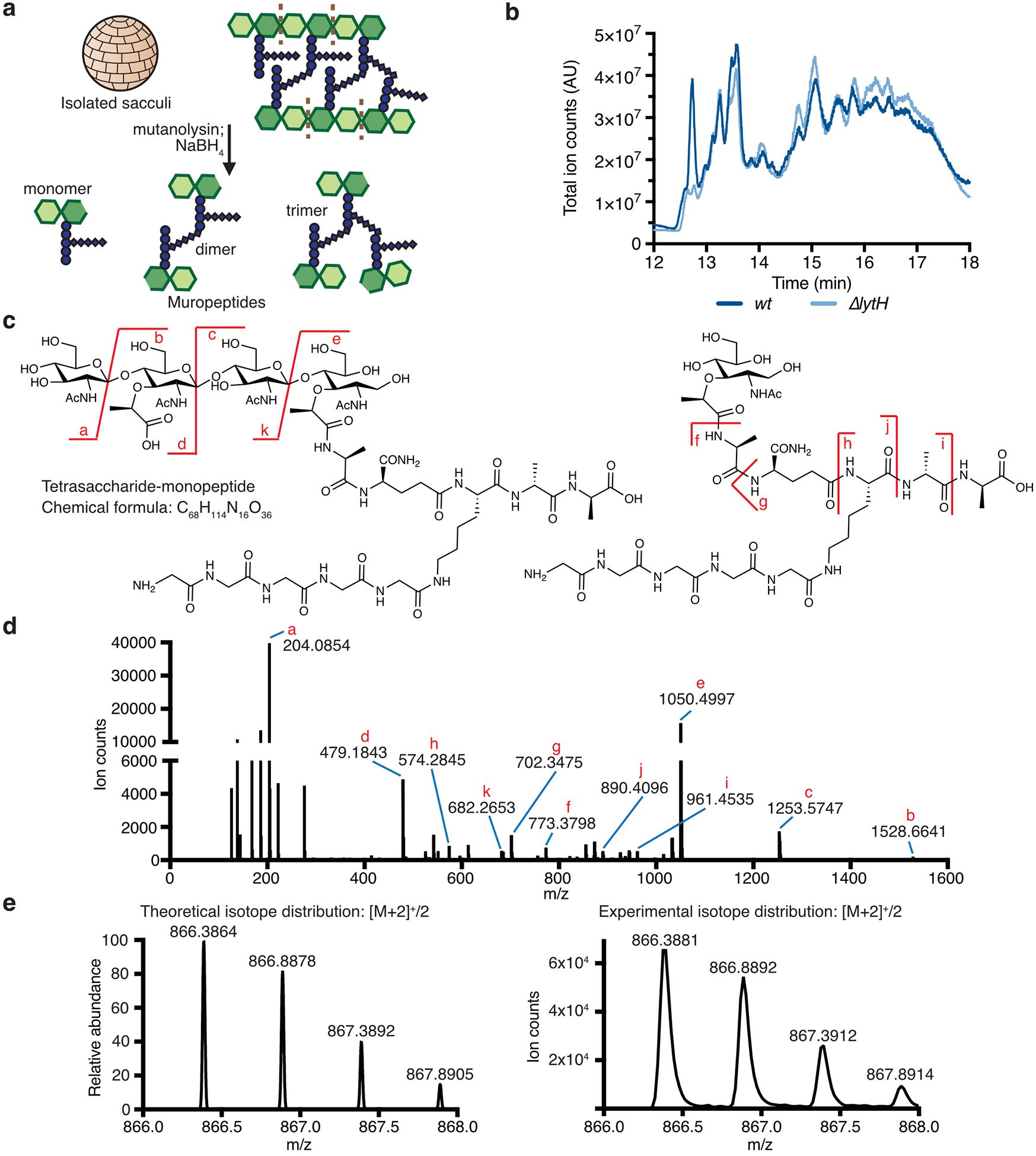
a, Schematic of sacculi isolation and mutanolysin/NaBH4 treatment to generate muropeptides for LC-MS analysis. b, Total ion chromatograms comparing the muropeptide species isolated from WT vs. ∆lytH sacculi. Muropeptide species eluted between 12–18 min using this analytical method. The XCMS online platform was used to identify species that were enriched or depleted between the two strains. c, The tetrasaccharide-monopeptide species was enriched in sacculi isolated from cells expressing lytHWT. A chemical structure consistent with this species, based on an [M]+2 ion with experimental m/z = 866.3881, is shown. d, The [M]+2 ion was targeted for fragmentation to confirm the species was a tetrasaccharide-monopeptide. e, The theoretical and experimental isotope distributions for the tetrasaccharide-monopeptide species are shown.
Extended Data Fig. 3. LytH pulled down ActH, a previously uncharacterized polytopic membrane protein.

a, A homology model of LytH highlights the predicted catalytic residues identified by sequence alignment of S. aureus LytH with E. coli AmiC. b, TMHMM topology prediction suggests that LytH is anchored in the cytoplasmic membrane. c, Schematic representation of the BDL-labeling assay to assess LytH hydrolytic activity in vitro. Short glycan strands were synthesized from Lipid II using S. aureus SgtBY181D. E. faecalis PBPX was used to exchange the terminal amino acid of stem peptides for biotin-D-Lys (BDL, yellow spheres), enabling visualization of glycan strands by western blotting with streptavidin. d, Glycan strands synthesized in vitro were treated with LytH. No difference in the distribution or intensity of polymers was observed between the three lanes. Blot is representative of two independent experiments. e, SDS-PAGE analysis of the co-immunoprecipitation experiment to identify proteins that bound to full-length LytH. Purple asterisks mark the protein bands corresponding to the baits. A red box is drawn around the ActH protein band. ActH is encoded by the gene saouhsc_01649. The fractions are: (FT) flowthrough, (W1–W2) washes, and (E1–E2) elutions. The schematics for LytH constructs are shown with the predicted transmembrane (TM), SH3, and amidase catalytic domains annotated. The predicted domain structure of ActH is also shown. f, A cladogram based on 16S rRNA alignment of bacterial species that encode ActH homologs with a similar domain structure was generated. The tree shows 196 bacterial species. For ease of visualization, only some species are labeled. Firmicutes are highlighted in purple and Deltaproteobacteria are highlighted in red.
Extended Data Fig. 4. The LytH-ActH complex has peptidoglycan hydrolase activity.
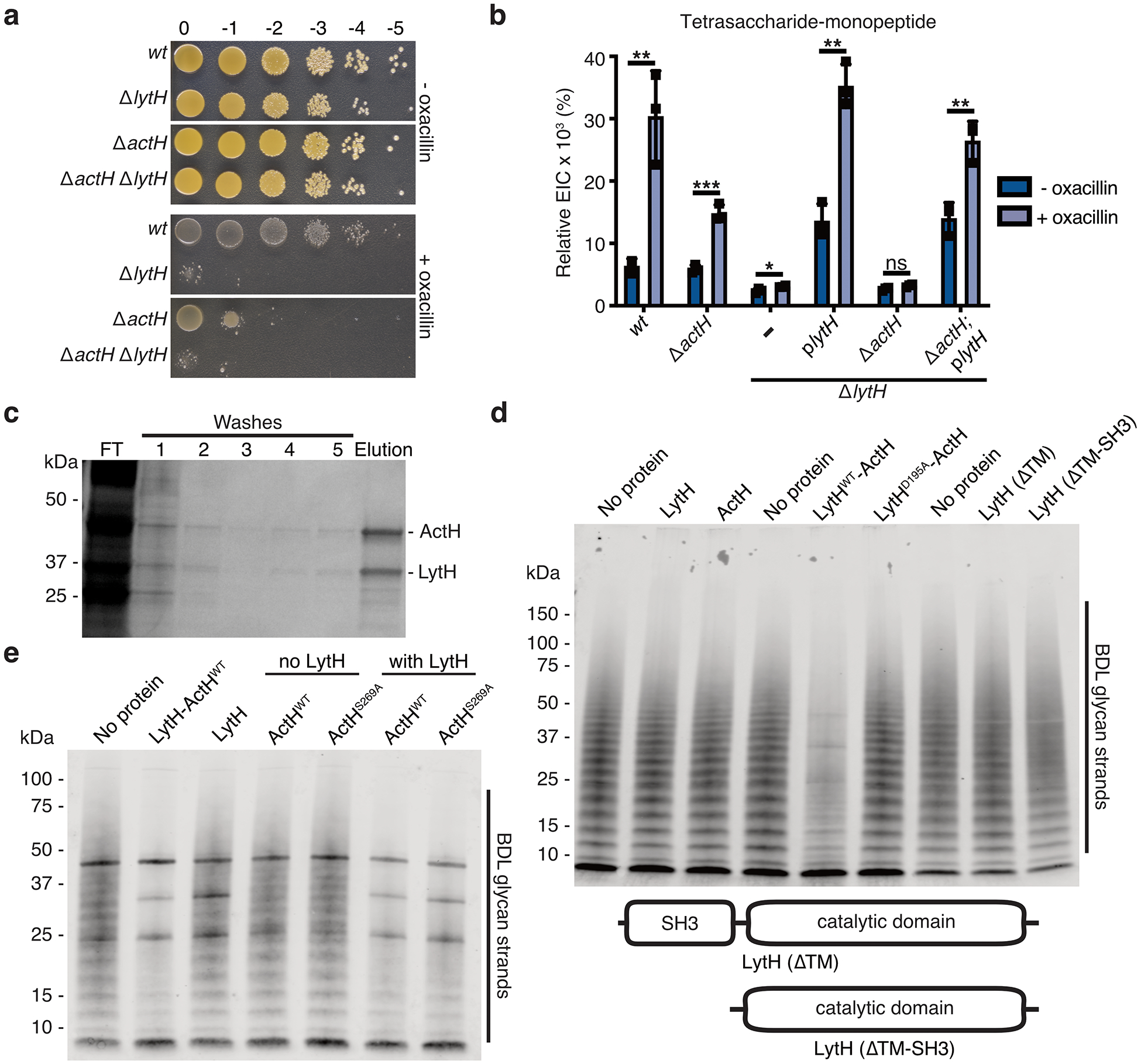
a, HG003 strains were spotted on TSA ± 0.125 μg/mL oxacillin. b, The relative ion count (extracted ion count/total ion count) for the tetrasaccharide-monopeptide was calculated. Data represent the mean ± SD. P-values were determined using unpaired, two-tailed t-tests (*P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant). From left to right: P = 0.0048, 0.0005, 0.0387, 0.0010, 0.0580, and 0.0056. c, Full-length LytH and ActH were co-expressed in E. coli and purified from detergent-solubilized membranes as a stable complex. SDS-PAGE analysis of purification fractions: (FT) flowthrough. d, Western blot analysis of LytH reactions with glycan strand substrate, showing a distinct difference for the WT complex. Glycan strands were made in vitro from native S. aureus Lipid II; the strands were labeled with BDL for visualization. e, Western blot analysis of LytH reactions with glycan strand substrate, showing the catalytic serine of ActH is dispensable for activating LytH. The bold bands appearing in all lanes in the blot correspond to purified proteins that were added to the reactions and non-specifically labeled. Data are representative of three (b–c) and two (a, d–e) independent experiments.
Extended Data Fig. 5. The LytH-ActH complex removes stem peptides from uncrosslinked glycan strands.
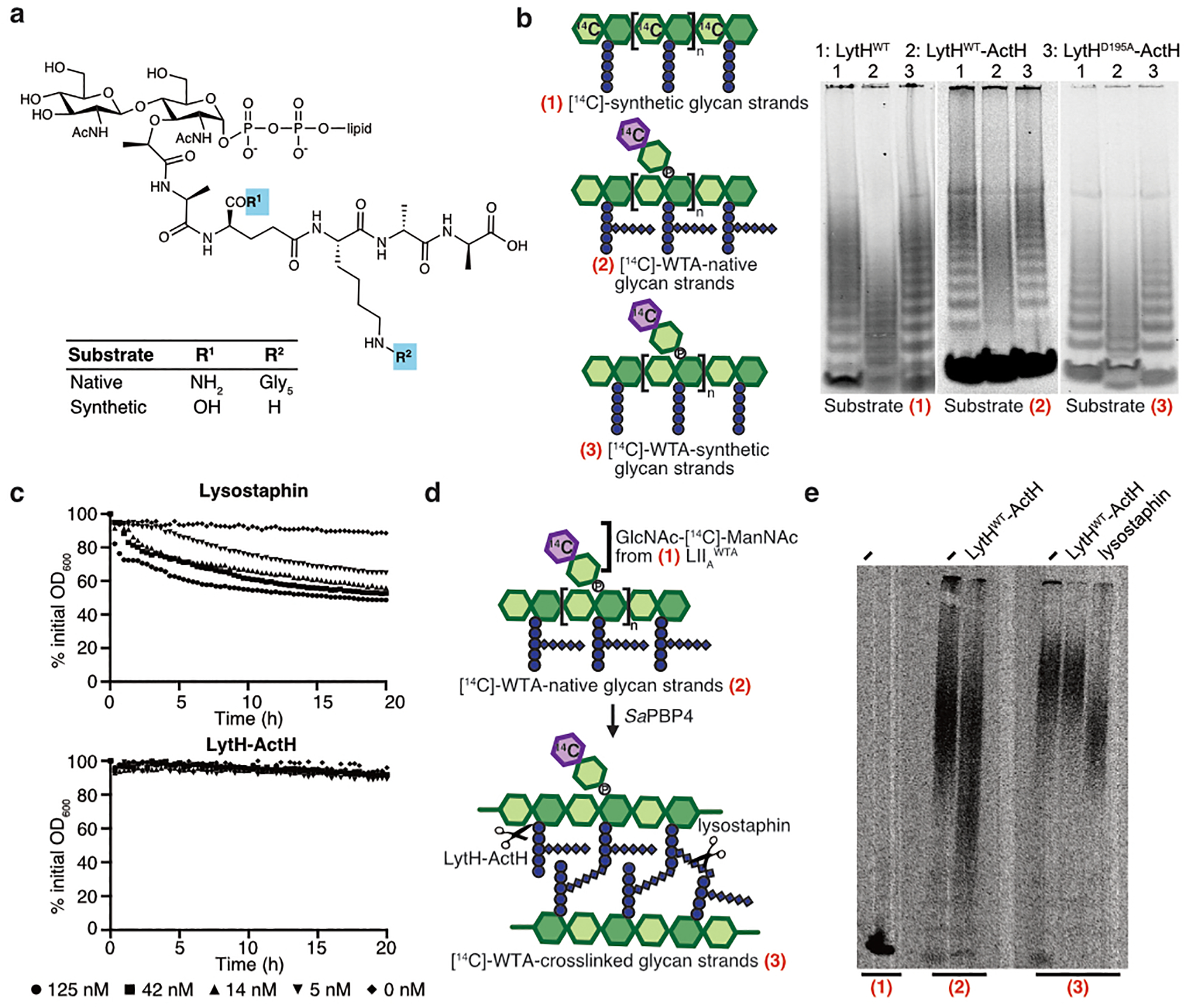
a, Chemical structure highlighting the differences between synthetic and S. aureus native Lipid II. b, PAGE autoradiographs of reactions with uncrosslinked glycan strand substrates. The glycopolymers for substrates (1) and (3) were made from synthetic Lipid II, while the glycopolymers for substrate 2 were generated from native Lipid II. For substrate (1), the radiolabel was found in the [14C]-GlcNAc residues of the glycan backbone. For substrates (2) and (3), the radiolabel was provided by a short wall teichoic acid (WTA) disaccharide branch ([14C]-LIIAWTA) attached to the hydroxyl group at the C6 position. c, Purified S. aureus sacculi were treated with lysostaphin and LytHWT-ActH. Hydrolysis of sacculi was monitored over time as a decrease in OD600. Experiments were performed in triplicate for each concentration of protein; each line represents the mean, plotted as percent of initial absorbance. d, Schematic for a PAGE assay to assess if LytH-ActH cleaves crosslinked peptidoglycan. Uncrosslinked glycan strands (substrate 2) were made from native Lipid II and radiolabeled with a short [14C]-LIIAWTA disaccharide branch (substrate 1) at the C6 position. The radiolabeled glycan strands were crosslinked by S. aureus PBP4 to generate crosslinked peptidoglycan (substrate 3). e, Peptidoglycan substrates were treated with LytHWT-ActH or lysostaphin. Reaction products were resolved by PAGE. Lysostaphin cleaves crosslinks, generating smaller species that migrate faster in the gel. Molecular weight ladders were not run on these PAGE gels. Data are representative of three independent experiments (b–c, e).
Extended Data Fig. 6. Mass spectrometry confirms LytH-ActH amidase activity.

a, Glycan strands were made from native Lipid II using SgtBWT protein and treated with the LytH complex prior to LC-MS analysis. In the presence of LytHWT-ActH, the tetrasaccharide-monopeptide was observed, but only the monomer (starting material) was observed in the LytHD195A-ActH reaction. We could not detect the released stem peptide in these reactions with native glycan strands likely due to the poor ionization efficiency of the released pentaglycine-containing stem peptide. b, The chemical structure and isotope distributions for the disaccharide peptide species (blue peak) shown in Fig. 2f. c, The chemical structure and isotope distributions for the synthetic stem peptide product (red peak) shown in Fig. 2f.
Extended Data Fig. 7. PBP2 suppressor mutants showed reduced peptidoglycan synthesis activity.
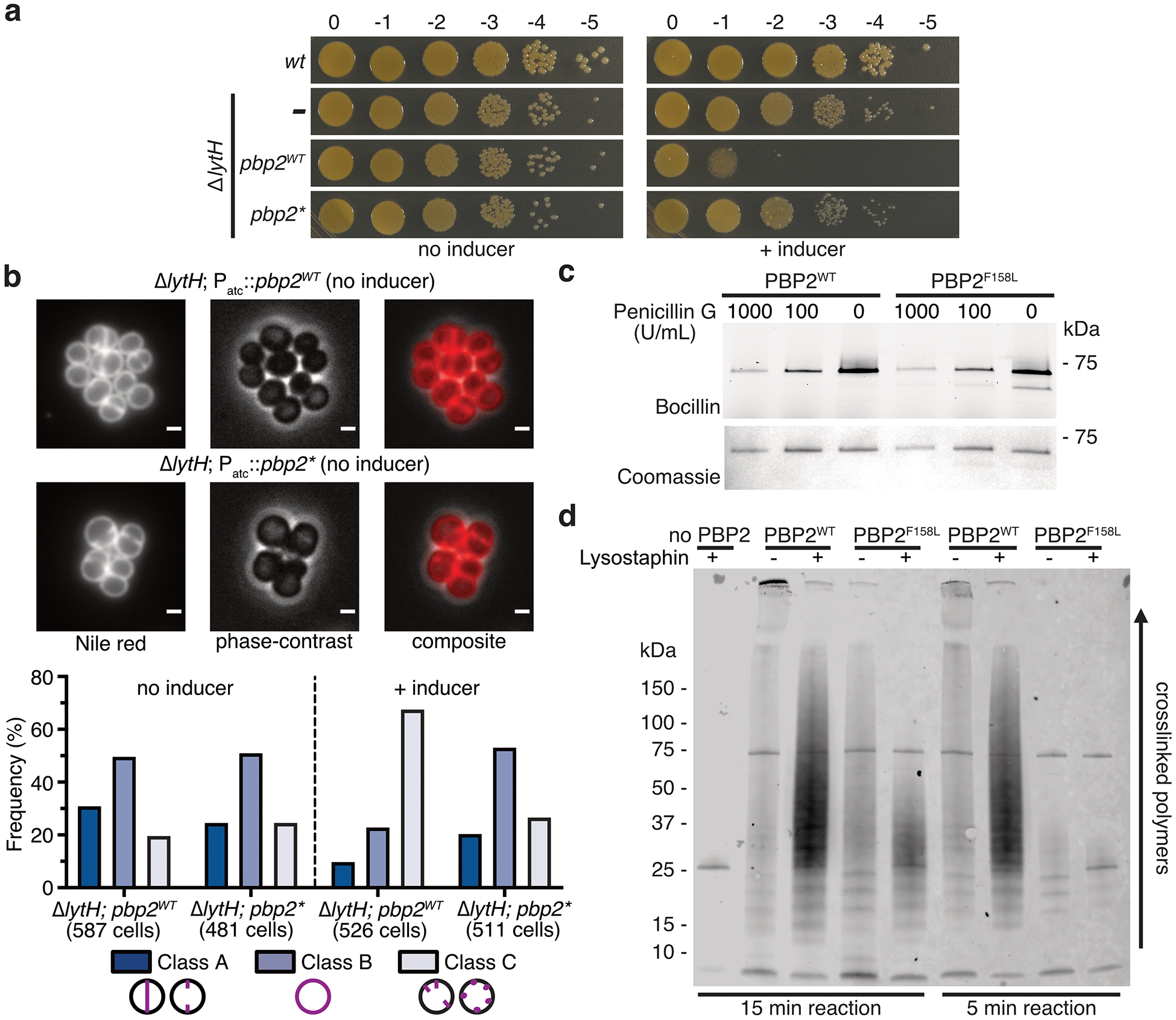
a, Spot dilutions showing that excess PBP2 is toxic in the absence of LytH. Wild-type pbp2 (pbp2WT) or a pbp2* suppressor allele (pbp2N220→KDLN) was expressed from an inducible promoter in strain HG003 ΔlytH. In these strains, the native pbp2WT allele was still present. b, Fluorescence images of Nile red-stained PBP2 overexpression strains. The controls in which the inducer was withheld to prevent PBP2 overexpression are shown. Quantitation of cellular phenotypes is summarized in the graph below: cells with a nascent or complete septum at midcell (Class A), cells showing only membrane fluorescence (Class B), and cells showing fluorescent punctate foci or multiple septa (Class C). Scale bars, 1 µm. c, A PBP2 suppressor variant was purified. Bocillin-labeling of purified proteins showed that PBP2F158L was properly folded. The Coomassie gel was a control for amounts of protein loaded. The experiment was performed once. d, Western blot comparing PBP2WT and PBP2F158L reactions with Lipid II substrate. PBP2 polymerizes Lipid II into glycan strands and crosslinks the glycan strands; PBP2 also incorporates BDL to enable visualization of crosslinked peptidoglycan. The PBP2 reactions were incubated for 5 or 15 min. When indicated, lysostaphin was added to cleave peptidoglycan crosslinks so that highly-crosslinked material could enter into the gel. Data are representative of two independent experiments (a–b, d).
Extended Data Fig. 8. Reducing PBP2 activity suppresses lytH deletion defects.

a, Fluorescence images of Nile red-stained HG003 strains. The suppressor allele pbp2F158L (pbp2*) mitigates ∆lytH division defects. Quantitation of cellular phenotypes is shown in the graph to the right: cells with a nascent or complete septum at midcell (Class A), cells showing only membrane fluorescence (Class B), and cells showing multiple septa (Class C). Scale bars, 1 µm. b, Deletion of lytH leads to larger cell size. The distribution of cell volume was calculated for cells at different phases of growth: cells that have not initiated septal synthesis (phase 1), cells with a nascent septum (phase 2), and cells with a complete septum prior to splitting into two daughter cells (phase 3). Each dot represents a single cell (from left to right): n = 566, 516, 615, 259, 160, 201, 215, 145, and 241 cells. The median of each spread is indicated by a black line. P-values were determined by two-sided Mann-Whitney U tests (***P < 0.001). From left to right: P = 4.58×10−50, 8.10×10−86, 2.21×10−26, 1.94×10−30, 2.44×10−21, and 2.70×10−25. c, Transmission electron micrograph of the suppressor strain HG003 ∆lytH pbp2F158L is shown. Scale bar, 500 nm. The experiment was performed once at 30°C and 37°C. d, The number of cells with septal defects was counted. Class A cells showed normal placement of a nascent or complete septum at midcell, while Class B cells showed misplaced septa. Data are representative of two independent experiments (a–b).
Extended Data Fig. 9. Loss of LytH reduces peptidoglycan synthesis at the septum relative to the periphery.
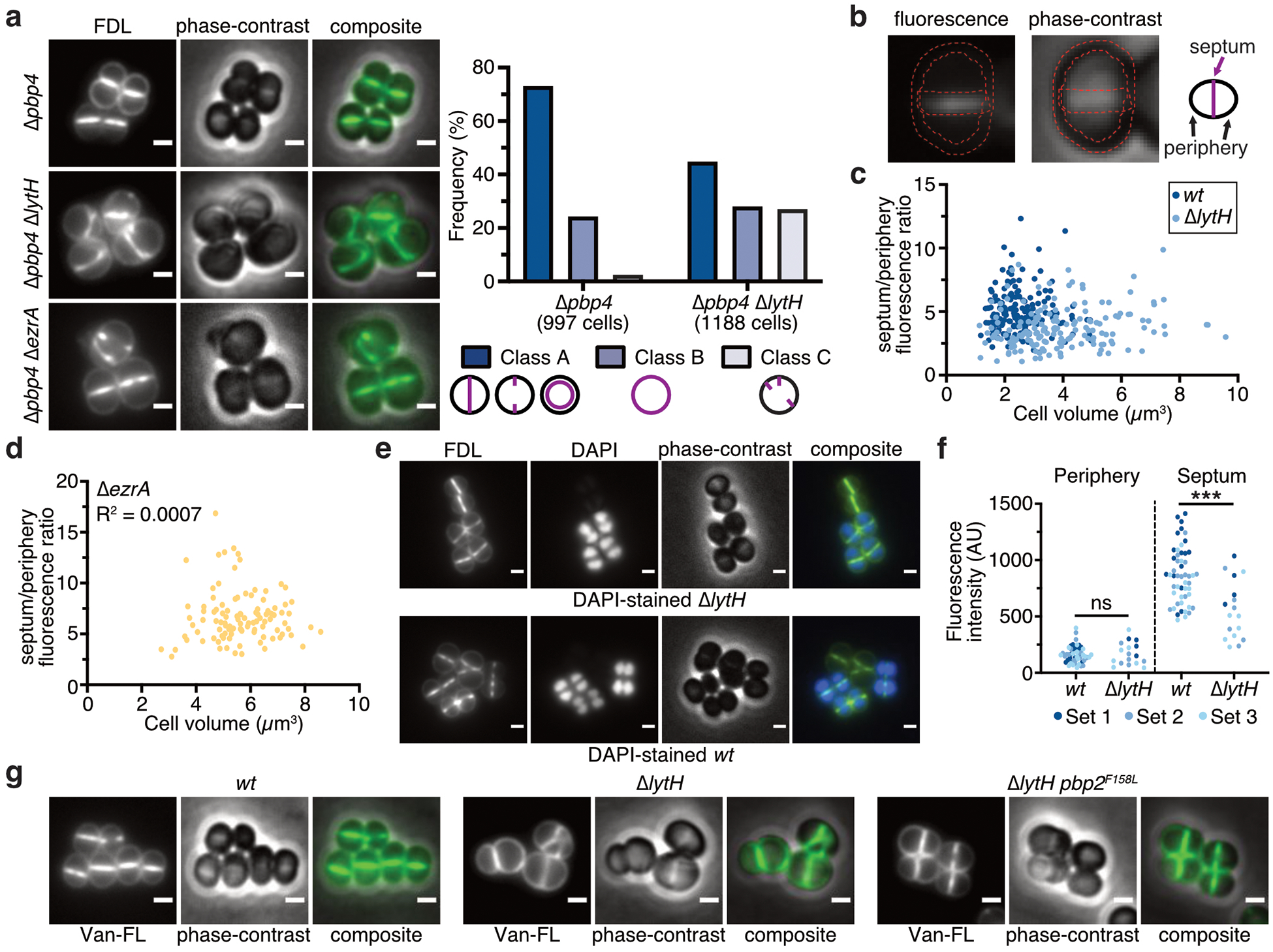
a, Fluorescence images of fluorescein-D-lysine (FDL)-labeled cells. All FDL labeling was performed in strains lacking PBP4 to reduce background. Cells were sorted into different classes according to the pattern of FDL signal: at the septum (Class A), around the membrane (Class B), or at misplaced septa and/or peripheral puncta (Class C). Scale bars, 1 µm. b, The ratio of fluorescence intensity at the septum vs. periphery was calculated for cells with a single complete septum. Two regions of interests, outlined in red, were defined to retrieve pixel values at the periphery and the septum. c, Deletion of lytH leads to reduced septal to peripheral FDL signal independently of cell size. Each dot represents a single cell: n = 159 (wt) and 167 (∆lytH) cells. d, FDL fluorescence ratio is not correlated with cell size (n = 105 cells). Deletion of ezrA causes division defects and larger cell size. e, Fluorescence images of an FDL-labeled, mixed population of WT and ∆lytH cells. To distinguish WT from ∆lytH cells, one strain was stained with DAPI prior to mixing the two strains and labeling with FDL. Reciprocal labeling of DAPI was performed. Scale bars, 1 µm. f, FDL signal for mixed populations containing DAPI-stained WT cells. Fluorescence intensity measurements correspond to the median intensity at the periphery or septum. Different colored dots represent cells analyzed from different fields of view (sets 1–3): n = 52 (wt) and 17 (∆lytH) cells. P-values were determined by two-sided Mann-Whitney U tests (***P < 0.001; ns, not significant). From left to right: P = 0.9389 and 7.02×10−5. g, Fluorescence images of cells labeled with fluorescent vancomycin (Van-FL) to detect newly synthesized peptidoglycan. Scale bars, 1 μm. Data are representative of two independent experiments (a–g).
Extended Data Fig. 10. PBP2 was delocalized in the absence of LytH.
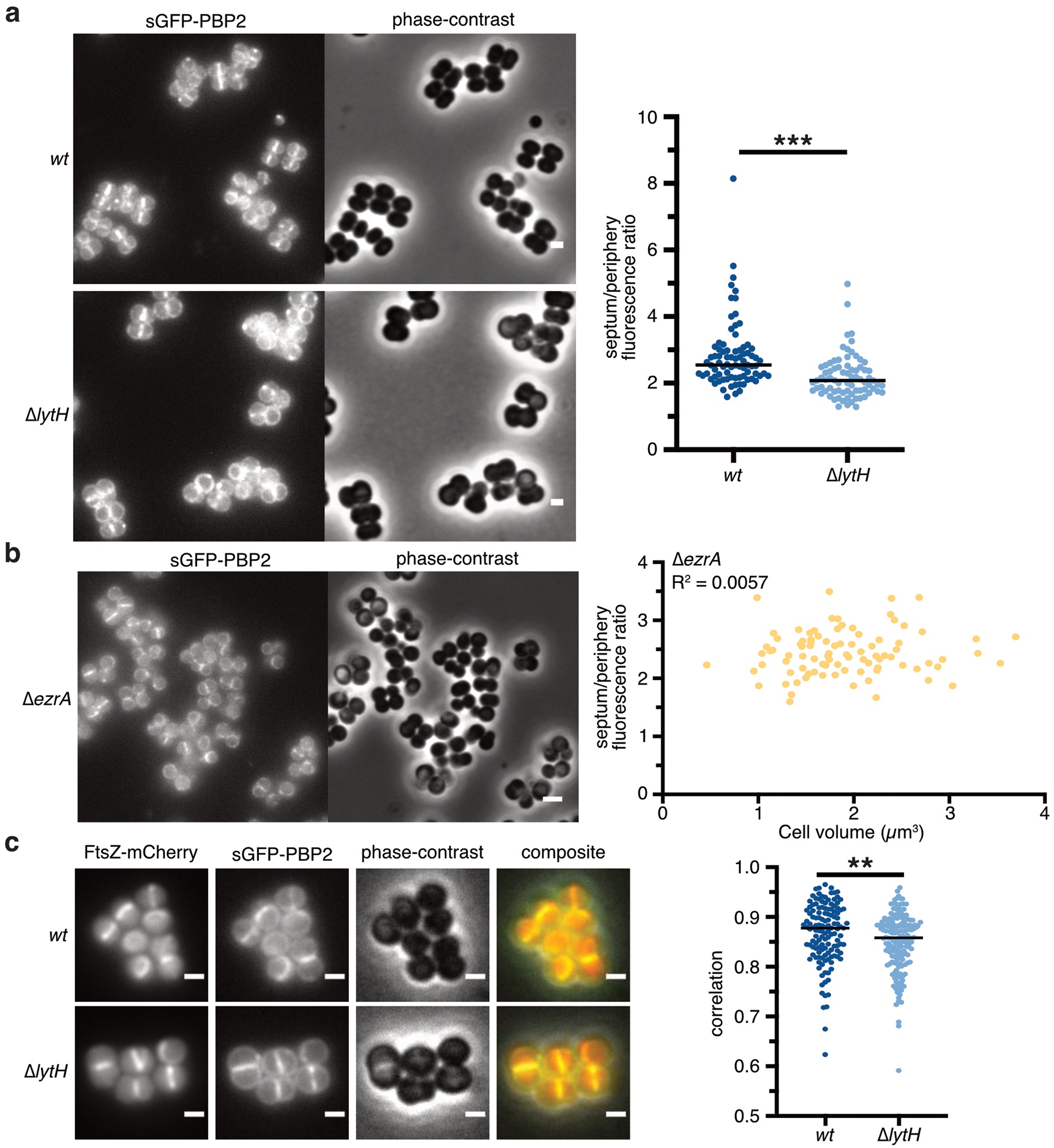
a, Fluorescence images of NCTC8325–4 strains expressing sGFP-PBP2. To compare the spatial distribution of PBP2 signal in WT vs. ∆lytH cells, fluorescence ratios were calculated using cells with a single complete septum. In all plots, each dot represents a single cell, the median of each spread is indicated by a black line, and p-values were determined by two-sided Mann-Whitney U tests. In this plot: n = 81 (wt) and 77 (∆lytH) cells; P = 8.30×10−8 (***P < 0.001). Scale bars, 1 µm. b, Fluorescence images of strain NCTC8325–4 ∆ezrA expressing sGFP-PBP2, showing that delocalization of PBP2 is independent of cell size. Fluorescence ratios of sGFP-PBP2 signal was plotted against cell volume (n = 91 cells). Scale bars, 2 µm. c, Fluorescence images of HG003 strains co-expressing sGFP-PBP2 and FtsZ-mCherry. PBP2 and FtsZ signals showed reduced colocalization in ∆lytH cells. Only cells showing FtsZ signal at the septum were considered for Pearson correlation coefficient analysis to calculate the degree of colocalization: n = 133 (wt) and 166 (∆lytH) cells; P = 0.006 (**P < 0.01). Scale bars, 1 µm. Different fields of view were captured for one biological sample (a–b). Data are representative of two (c) independent experiments.
Supplementary Material
Acknowledgements
We thank M. Welsh and A. Taguchi for help with LC-MS analysis and protein expression. We thank T. Pang from the Bernhardt and Rudner labs for strain TD215. Fluorescence images were partially acquired at the Microscopy Resources on the North Quad (MicRoN) core at Harvard Medical School. We acknowledge support from the NSF (DGE1144152 to T.D.), the NIH (P01AI083214, R01GM076710, R01AI139011, R01AI099144), and the European Research Council (ERC-2017-CoG-771709 to M.G.P).
Footnotes
Data availability
Transposon sequencing data (accession nos. SAMN08025141 and SAMN08025168) can be found in the NCBI BioSample database. Whole-genome sequencing data (accession no. PRJNA579395) can be found in the NCBI BioProject database. All other data are available in the manuscript, extended data, supplementary information, or source data.
Code availability
Code is available at https://github.com/SuzanneWalkerLab/imageanalysis.
Competing interests
The authors declare no competing interests.
References
- 1.Vollmer W, Blanot D & de Pedro MA Peptidoglycan structure and architecture. FEMS Microbiol. Rev 32, 149–167 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Pinho MG, Kjos M & Veening J-W How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat. Rev. Microbiol 11, 601–614 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Monteiro JM et al. Peptidoglycan synthesis drives an FtsZ-treadmilling-independent step of cytokinesis. Nature 554, 528–532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lund VA et al. Molecular coordination of Staphylococcus aureus cell division. Elife 7, 642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Typas A, Banzhaf M, Gross CA & Vollmer W From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol 10, 123–136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egan AJF, Cleverley RM, Peters K, Lewis RJ & Vollmer W Regulation of bacterial cell wall growth. FEBS J 284, 851–867 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Vollmer W, Joris B, Charlier P & Foster S Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev 32, 259–286 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Sauvage E, Kerff F, Terrak M, Ayala JA & Charlier P The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev 32, 234–258 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Adams DW & Errington J Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol 7, 642–653 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Veiga H, Jorge AM & Pinho MG Absence of nucleoid occlusion effector Noc impairs formation of orthogonal FtsZ rings during Staphylococcus aureus cell division. Mol. Microbiol 80, 1366–1380 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Jorge AM, Hoiczyk E, Gomes JP & Pinho MG EzrA contributes to the regulation of cell size in Staphylococcus aureus. PLoS ONE 6, e27542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pang T, Wang X, Lim HC, Bernhardt TG & Rudner DZ The nucleoid occlusion factor Noc controls DNA replication initiation in Staphylococcus aureus. PLoS Genet 13, e1006908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santiago M et al. Genome-wide mutant profiling predicts the mechanism of a Lipid II binding antibiotic. Nat. Chem. Biol 14, 601–608 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomasz A, Albino A & Zanati E Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227, 138–140 (1970). [DOI] [PubMed] [Google Scholar]
- 15.Oshida T et al. A Staphylococcus aureus autolysin that has an N-acetylmuramoyl-L-alanine amidase domain and an endo-beta-N-acetylglucosaminidase domain: cloning, sequence analysis, and characterization. Proc. Natl. Acad. Sci. U.S.A 92, 285–289 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kajimura J et al. Identification and molecular characterization of an N-acetylmuramyl-L-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol. Microbiol 58, 1087–1101 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Lenz JD et al. Amidase Activity of AmiC Controls Cell Separation and Stem Peptide Release and Is Enhanced by NlpD in Neisseria gonorrhoeae. J. Biol. Chem 291, 10916–10933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocaboy M et al. The crystal structure of the cell division amidase AmiC reveals the fold of the AMIN domain, a new peptidoglycan binding domain. Mol. Microbiol 90, 267–277 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Kawata S, Takemura T & Yokogawa K Chacterization of two N-acetylmuramidases from Streptomyces globisporus 1829. Agricultural and Biological Chemistry 47, 1501–1508 (1983). [Google Scholar]
- 20.Qiao Y et al. Lipid II overproduction allows direct assay of transpeptidase inhibition by β-lactams. Nat. Chem. Biol 13, 793–798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebets Y et al. Moenomycin resistance mutations in Staphylococcus aureus reduce peptidoglycan chain length and cause aberrant cell division. ACS Chem. Biol 9, 459–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welsh MA et al. Identification of a functionally unique family of penicillin-binding proteins. J. Am. Chem. Soc 139, 17727–17730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaefer K, Matano LM, Qiao Y, Kahne D & Walker S In vitro reconstitution demonstrates the cell wall ligase activity of LCP proteins. Nat. Chem. Biol 13, 396–401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lupoli TJ et al. Studying a cell division amidase using defined peptidoglycan substrates. J. Am. Chem. Soc 131, 18230–18231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaefer K, Owens TW, Kahne D & Walker S Substrate preferences establish the order of cell wall assembly in Staphylococcus aureus. J. Am. Chem. Soc 140, 2442–2445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lovering AL, de Castro LH, Lim D & Strynadka NCJ Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 315, 1402–1405 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kuru E et al. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew. Chem. Int. Ed. Engl 51, 12519–12523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinho MG & Errington J Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol. Microbiol 50, 871–881 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Tan CM et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med 4, 126ra35 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Ruiz N Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A 105, 15553–15557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sham L-T et al. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345, 220–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinho MG & Errington J Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol. Microbiol 55, 799–807 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Uehara T, Parzych KR, Dinh T & Bernhardt TG Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J 29, 1412–1422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sham L-T, Barendt SM, Kopecky KE & Winkler ME Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc. Natl. Acad. Sci. U.S.A 108, E1061–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartual SG et al. Structural basis of PcsB-mediated cell separation in Streptococcus pneumoniae. Nat Commun 5, 3842 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Rued BE et al. Structure of the Large Extracellular Loop of FtsX and Its Interaction with the Essential Peptidoglycan Hydrolase PcsB in Streptococcus pneumoniae. MBio 10, e02622–18–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meisner J et al. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol. Microbiol 89, 1069–1083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domínguez-Cuevas P, Porcelli I, Daniel RA & Errington J Differentiated roles for MreB-actin isologues and autolytic enzymes in Bacillus subtilis morphogenesis. Mol. Microbiol 89, 1084–1098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rather P Role of rhomboid proteases in bacteria. Biochim. Biophys. Acta 1828, 2849–2854 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Maegawa S, Ito K & Akiyama Y Proteolytic Action of GlpG, a Rhomboid Protease in the Escherichia coli Cytoplasmic Membrane. Biochemistry 44, 13543–13552 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Liew ATF et al. A simple plasmid-based system that allows rapid generation of tightly controlled gene expression in Staphylococcus aureus. Microbiology 157, 666–676 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Kato F & Sugai M A simple method of markerless gene deletion in Staphylococcus aureus. J. Microbiol. Methods 87, 76–81 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Lee W et al. Antibiotic combinations that enable one-step, targeted mutagenesis of chromosomal genes. ACS Infect Dis 4, 1007–1018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charpentier E et al. Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl. Environ. Microbiol 70, 6076–6085 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Jong NWM, van der Horst T, van Strijp JAG & Nijland R Fluorescent reporters for markerless genomic integration in Staphylococcus aureus. Sci Rep 7, 43889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christie GE et al. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α--implications for the specificity of SaPI mobilization. Virology 407, 381–390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiao Y et al. Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J. Am. Chem. Soc 136, 14678–14681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Men H, Park P, Ge M & Walker S Substrate Synthesis and Activity Assay for MurG. J. Am. Chem. Soc 120, 2484–2485 (1998). [Google Scholar]
- 49.Tsukamoto H & Kahne D N-methylimidazolium chloride-catalyzed pyrophosphate formation: application to the synthesis of Lipid I and NDP-sugar donors. Bioorg. Med. Chem. Lett 21, 5050–5053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee W et al. The mechanism of action of lysobactin. J. Am. Chem. Soc 138, 100–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santiago M et al. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16, 252 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kühner D, Stahl M, Demircioglu DD & Bertsche U From cells to muropeptide structures in 24 h: peptidoglycan mapping by UPLC-MS. Sci Rep 4, 7494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barrett D et al. Analysis of glycan polymers produced by peptidoglycan glycosyltransferases. J. Biol. Chem 282, 31964–31971 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monteiro JM et al. Cell shape dynamics during the staphylococcal cell cycle. Nat Commun 6, 8055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer F Topographic distance and watershed lines. Signal Processing 38, 113–125 (1994). [Google Scholar]
- 56.Maurer CR, Qi R & Raghavan V A linear time algorithm for computing exact Euclidean distance transforms of binary images in arbitrary dimensions. IEEE Transactions on Pattern Analysis and Machine Intelligence 25, 265–270 (2003). [Google Scholar]
- 57.Rosenfeld A & Pfaltz JL Sequential Operations in Digital Picture Processing. JACM 13, 471–494 (1966). [Google Scholar]
- 58.Paglieroni DW Distance transforms: Properties and machine vision applications. CVGIP: Graphical Models and Image Processing 54, 56–74 (1992). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.