

Abstract
The great gerbil (Rhombomys opimus) is a social rodent living in permanent, complex burrow systems distributed throughout Central Asia, where it serves as the main host of several important vector-borne infectious pathogens including the well-known plague bacterium (Yersinia pestis). Here, we present a continuous annotated genome assembly of the great gerbil, covering over 96% of the estimated 2.47-Gb genome. Taking advantage of the recent genome assemblies of the sand rat (Psammomys obesus) and the Mongolian gerbil (Meriones unguiculatus), comparative immunogenomic analyses reveal shared gene losses within TLR gene families (i.e., TLR8, TLR10, and the entire TLR11-subfamily) for Gerbillinae, accompanied with signs of diversifying selection of TLR7 and TLR9. Most notably, we find a great gerbil-specific duplication of the MHCII DRB locus. In silico analyses suggest that the duplicated gene provides high peptide binding affinity for Yersiniae epitopes as well as Leishmania and Leptospira epitopes, putatively leading to increased capability to withstand infections by these pathogens. Our study demonstrates the power of whole-genome sequencing combined with comparative genomic analyses to gain deeper insight into the immunogenomic landscape of the great gerbil and its close relatives.
Keywords: great gerbil, genome assembly, plague resistance, immune gene evolution, MHC gene duplication, comparative genomics
Introduction
The recent advancement within whole-genome sequencing technologies accompanied with rapid developments of bioinformatical and analytical tools has led to unprecedented opportunities and genomic insight into nonmodel organisms (Ellegren 2014). Access to genome assemblies from numerous organisms has facilitated genome comparisons both within and across species (Bean et al. 2013; Sironi et al. 2015; Meadows and Lindblad-Toh 2017). Pathogens are one of the main selective drivers of local adaptation (Fumagalli et al. 2011) and thus, genomic footprints of past and present selection pressure from pathogens are readily detectable in host genomes (Corona et al. 2018). Advances in statistical methods to locate such footprints combined with the increasing amounts of genomic data across the tree of life have armed scientists to investigate such events at large scales and in a diversity of species (Vitti et al. 2013).
The great gerbil (Rhombomys opimus) is a key reservoir species for several vector-borne diseases in Central Asia (Anisimov et al. 2004), and its habitat stretches from Iran to Kazakhstan to North Eastern China. This diurnal, fossorial rodent lives in arid and semiarid deserts, and forms small family groups that reside in extensive and complex burrow systems with a large surface diameter and multiple entrances, food storage, and nesting chambers (Addink et al. 2010). Where great gerbil communities coincide with human settlements and agriculture, they are often viewed as pests through the destruction of crops and as carriers of vector-borne diseases (Nowak 1999; Zhang et al. 2003; Gage and Kosoy 2005). Plague, caused by the Gram-negative bacterium Yersinia pestis, is a common disease in wildlife rodents living in semiarid deserts and montane steppes, as well as in tropical regions (Stenseth et al. 2008; Bramanti et al. 2016). It is predominantly transmitted between rodents such as the great gerbil by fleas living on rodents or in rodent burrows and nests (Hinnebusch et al. 2017) and regularly spills over into human populations (Samia et al. 2011), leading to individual cases and sometimes localized plague outbreaks (Nguyen et al. 2018). Other zoonotic diseases transmitted by the great gerbil include cutaneous leishmaniasis that is widespread in certain areas of Iran and is caused by protozoan parasites of Leishmania spp. transmitted by phlebotomine sandflies (Rassi et al. 2008; Akhavan et al. 2010). Once the primary physical barriers of the mammalian immune defense have been breached, the pathogens encounter a diverse community of innate immune cells and proteins evolved to recognize and destroy invasive pathogens. Here, Toll-like receptors (TLRs) and other pattern recognition receptors (PRRs) are at the forefront and have a vital role in the recognition and initiation of innate immune responses. Stimulation of adaptive immunity is in turn governed by the major histocompatibility complex (MHCs). MHC class I (MHCI) and class II (MHCII) proteins present antigens to CD8+ and CD4+ T lymphocytes, respectively. In particular, the CD4+ T lymphocytes are master activators and regulators of adaptive immune responses (Neefjes et al. 2011; Murphy and Weaver 2016).
In host–pathogen interactions, both sides evolve mechanisms to overpower the other engaging in an evolutionary arms race that shapes the genetic diversity on both sides (Brockhurst et al. 2014; Sironi et al. 2015). For instance, pathogenic Gram-negative bacteria such as Y. pestis has evolved to subvert the host immune system evoking a specialized and complex attack to evade detection and destruction by the mammalian immune system to establish infection (Dyer et al. 2010; Xu and Liu 2014). Upon entering a mammalian host, the change in temperature to 37 °C initiates a change in Y. pestis bacterial gene expression switching on a wealth of virulence genes whose combined action enables the bacterium to evade both extracellular and intracellular immune defenses (Chung and Bliska 2016) throughout the course of infection (Sebbane et al. 2006; Nham et al. 2012; Shannon et al. 2013, 2015; Gonzalez et al. 2015). The host, in addition to standard immune responses, will have to establish counter measures to overcome the Y. pestis strategy of suppressing and delaying the innate immune responses (Comer et al. 2010; Yang et al. 2017). This includes recognition of the pathogen, resisting the bacterial signals that induce apoptosis of antigen-presenting cells and successfully producing an inflammatory response that can overpower the infection while avoiding hyperactivation.
Like all main reservoir hosts, great gerbils cope remarkably well with the diseases they serve as reservoir for and have been shown to handle plague infections with only a minor increase in mortality levels compared with the natural mortality (see Begon et al. 2006; Samia et al. 2011 for details). Despite many years of research, the genetic basis of plague resistance is still unclear. The adaptive immune system requires several days to respond to an infection and Y. pestis progresses so rapidly that it can kill susceptible hosts within days. Consequently, the genetic background of the innate immune system could potentially play a pivotal part in plague survival (Casanova and Abel 2013). For a successful response, the innate immune system must keep the infection in check while properly activating the adaptive immune system (Murphy and Weaver 2016), which can then mount an appropriate immune response leading to a more efficient and complete clearance of the pathogen. Moreover, previous studies investigating plague and/or leishmania resistance have implicated components of both innate and adaptive immunity (Tollenaere et al. 2008, 2012, 2013; Sakthianandeswaren et al. 2009; Blanchet et al. 2011; Busch et al. 2011, 2013; Demeure et al. 2012; Vladimer et al. 2012; Cobble et al. 2016), although, none of these studies has involved wild reservoir hosts in combination with whole-genome sequencing.
Two of the latest studies presenting rodent genome assemblies include the closest relatives of the great gerbil, the sand rat (Psammomys obesus), and the Mongolian gerbil (Meriones unguiculatus) (Hargreaves et al. 2017; Zorio et al. 2019). Both species are model systems for human diseases such as diabetes and neurological disorders. The Mongolian gerbil is also considered a host of plague and like the great gerbil lives in family groups in burrow systems in Mongolia as well as in sympatry with the great gerbil in parts of China. However, the Mongolian gerbil differs from the great gerbil in being consistently sensitive to plague (Gage and Kosoy 2005). Contrastingly, the sand rat lives solitary in burrows in North African deserts and parts of the Arabian Peninsula, and is rarely infected with plague but is a major host of leishmania (Fichet-Calvet et al. 2003). Access to close relatives of the great gerbil in addition to other well-established rodent genomes allows for thorough comparative genomic investigations of immune gene evolution and putative mechanisms for disease resistance. Here, we present the first de novo whole-genome sequence assembly of the major plague host, the great gerbil (R. opimus). We use this genome resource to investigate the genomic landscape of innate and adaptive immunity with focus on immune genes relevant for disease resistance such as TLRs and MHC, through genomic comparative analyses with the closely related Mongolian gerbil and sand rat, as well as other mammals.
Materials and Methods
Sampling and Sequencing
A male great gerbil weighing 180 g was captured in the Midong District outside Urumqi in Xinjiang Province, China in October 2013. The animal was humanely euthanized and tissue samples of liver were conserved in ethanol prior to DNA extraction. Blood samples from the individual were screened for F1 “capsular” antigen (Caf1) and anti-F1 as described in Zhang et al. (2012, 2015) to confirm plague-negative status. The DNA used in the library construction was extracted from liver tissue using Gentra Puregene Tissue Kit (Qiagen Inc.). Use of great gerbil tissue was approved by the Committee for Animal Welfares of Xinjiang CDC, China. The sampling is not legislated by the Nagoya Protocol and was conducted prior to China signed the membership on September 6, 2016. The sampled species have a “least concern” status in the IUCN Red List of Threatened Species.
The sequence strategy was tailored toward the ALLPATHS-LG assembly software (Broad Institute, Cambridge, MA) following their recommendations for platform choice and fragment size resulting in the combination of one short paired-end (PE) library with an average insert size of 220 bp (150 bp read length) and two mate-pair (MP) libraries of 3- and 10 kb insert size (100 bp read length). See supplementary table S1, Supplementary Material online, for a list of libraries and sequence yields. Sequencing for the de novo assembly of the great gerbil reference genome was performed on the Illumina platform using HiSeq 2500 instruments at the Norwegian Sequencing Centre at the University of Oslo for the PE library (https://www.sequencing.uio.no) and using HiSeq 2000 instruments at Génome Québec at McGill University for the MP libraries (http://gqinnovationcenter.com/index.aspx? l=e).
Genome Assembly and Maker Annotation
The Illumina sequences were quality checked using FastQC v0.11.2 and SGA-preqc (downloaded June 25, 2014) with default parameters. Both MP libraries were trimmed for adapter sequences using Cutadapt v1.5 with option -b and a list of adapters used in MP library prep (Martin 2011) and the trimmed reads were used alongside the PE short read as input for ALLPATHS-LG v48639 generating a de novo assembly. This combination of short-read sequencing technology combined with the ALLPATHS-LG assembly algorithm is documented to perform well in birds and mammals (Gnerre et al. 2011; Elgvin et al. 2017; Pujolar et al. 2018). File preparations were conducted according to manufacturer’s recommendation and the option TARGETS=submission was added to the run to obtain a submission prepared assembly version.
Assembly completeness was assessed by analyzing the extent of conserved eukaryotic genes present using CEGMA v2.4.010312 and BUSCO v1.1.b (Parra et al. 2007, 2009; Simão et al. 2015). Gene mining for the highly conserved Homeobox (HOX) genes was also conducted as an additional assessment of assembly completeness (see supplementary note S1 and fig. S1, Supplementary Material online).
All reads were mapped back to the assembly using BWA-MEM v0.7.5a and the resulting bam files were used alongside the assembly in REAPR v1.0.17 to evaluate potential scaffolding errors as well as in Blobology to inspect the assembly for possible contaminants, creating taxon-annotated-GC-coverage plots of the results from BLAST searches of the NCBI database (Kumar et al. 2013).
The genome assembly was annotated using the MAKER2 pipeline v2.31 run iteratively in a two-pass fashion (as described in https://github.com/sujaikumar/assemblage/blob/master/README-annotation.md, last accessed April 13, 2016) (Holt and Yandell 2011). Multiple steps are required prior to the first pass though MAKER2 and include creating a repeat library for repeat masking and training three different ab initio gene predictors. Firstly, construction of the repeat library was conducted as described in Varadharajan et al. (2018). In brief, a de novo repeat library was created for the assembly by running RepeatModeler v1.0.8 with default parameters, and sequences matching known proteins of repetitive nature were removed from the repeat library through BlastX against the UniProt database. Next, GeneMark-ES v2.3e was trained on the genome assembly using default parameters with the exception of reducing the –min-contig parameter to 10.000 (Lomsadze et al. 2005). SNAP v20131129 and AUGUSTUS v3.0.2 was trained on the genes found by CEGMA and BUSCO, respectively. The generated gene predictors and the repeat library were used in the first pass alongside proteins from UniProt/SwissProt (downloaded February 16, 2016) as protein homology evidence and Mus musculus cDNA as alternative EST evidence (GRCm38 downloaded from Ensembl, March 15 2016). For the second pass, SNAP and AUGUSTUS were retrained with the generated MAKER2 predictions and otherwise performed with the same setup. The resulting gene predictions had domain annotations and putative functions added using InterProScan v5.4.47 and BLASTp against the UniProt database with e-value 1e-5 (same methodology as Tørresen et al. 2018; Varadharajan et al. 2018). Finally, the output was filtered using the MAKER2 default filtering approach only retaining predictions with annotation edit distance score (AED) <1.
Genome Mining and Gene Alignments
We searched for TLR genes, associated receptors, and adaptor molecules as well as genes of the MHC region (complete list of genes can be found in supplementary table S2, Supplementary Material online) collected from UniProt and Ensembl. Throughout, we performed TBlastN searches, manual assembly exon by exon in MEGA7, and verified annotations through reciprocal BlastX against the NCBI database and phylogenetic analysis including orthologs from human (Homo sapiens), mouse (Mus musculus), rat (Rattus norvegicus), and all three members of the Gerbillinae subfamily. For details on the phylogenetic analyses, we refer to descriptions in sections below. In the TLR analyses, Algerian mouse (Musspretus), Ryukyu mouse (Muscaroli), Chinese hamster (Cricetulus griseus), and Chinese tree shrew (Tupaia belangeri chinensis) were also included.
Sand rat and Mongolian gerbil genome assemblies were downloaded from NCBI (September 12, 2017). The genome assemblies of the great gerbil, sand rat, and Mongolian gerbil were made into searchable databases for gene mining using the makeblastdb command of the BLAST+ v2.6.0 program. Local TBlastN searches, using protein sequences of mouse and occasionally rat, human, and Mongolian gerbil as queries, were executed with default parameters including an e-value cutoff of 1e + 1. The low e-value was utilized to capture more divergent sequence homologs. Hits were extracted from assemblies using bedtools v2.26.0 and aligned with orthologs in MEGA v7.0.26 using MUSCLE with default parameters. In cases where annotations for some of the TLRs for a species were missing in Ensembl and could not be located in either the NCBI nucleotide database or in UniProt, the Ensembl BLAST Tool (TBlastN) was used with default parameters to find the genomic region of interest using queries from mouse.
Synteny Analyses of MHC Regions
A combination of the Ensembl Genome Browser v92 and comparisons presented in Hurt et al. (2004) and TBlastN searches, as described earlier, were used in synteny analyses of the MHCI and II regions of human, rat, and mouse with great gerbil. Synteny of MHCII genes of sand rat and Mongolian gerbil was also investigated, however, for simplicity and visualization purposes, they are not included in figure 1.
Fig. 1.

—Synteny of genes in the major histocompatibility (MHC) region of human, rat, mouse, and gerbil. Genomic synteny of genes in the MHC regions of human, mouse, rat, and great gerbil mapped onto a cladogram. Genes are represented by arrow-shaped boxes indicating the genomic orientation. The boxes are colored by class region and for class I by classical (Ia) or nonclassical (Ib) subdivision: framework (FW) genes (light blue), MHCII (dark blue), MHCIa (blue), MHCIII (yellow), and MHCIb (green). Square brackets indicate multiple gene copies not displayed for practical and visualization purposes, but copy number is indicated outside in superscript. Due to limitations in space and to emphasize the conserved synteny of FW genes across lineages, the genes are placed in between the general syntenies and their respective locations are indicated by light blue lines. The light blue brackets surrounding the Psmb and TAP genes indicate their constitutive organization. Putative pseudogenes are denoted with ψ. For visualization purposes, genes of the DP (termed H in rat) and DO (termed O in mouse) loci are excluded. The location of all great gerbil MHCII genes including Rhop-DP and Rhop-DO can be found in table 3. (A) Synteny of all MHC regions detailing MCHI and II. Panel (B) further details the genomic locations of great gerbil MHCI genes as indicated by the presence of FW genes located on the scaffolds and inferred from synteny comparisons with human, rat, and mouse regions and phylogenetic analysis (see fig. 3A). The overall synteny of the MHCI and II regions is very well conserved in great gerbil displaying the same translocation of MHCI genes upstream of MHCII as seen in mouse and rat and resulting in the separation of the MHCI region into two. Most notably, for MHCII, there is a duplication of a β gene of the DR locus in great gerbil (highlighted by a red asterisk) whose orientation has changed and is located downstream of the FW gene Btnl2 that normally represents the end of the MHCII region.
Alignment and Phylogenetic Reconstruction of TLR and MHC
Sequences were aligned with MAFFT (Katoh and Standley 2013) using default parameters: for both nucleotides and amino acid alignments the E-INS-i model was utilized. The resulting alignments were edited manually using Mesquite v3.4 (Maddison and Maddison 2018). See supplementary tables S3, S9, and S10, Supplementary Material online, for accession numbers. Ambiguously aligned characters were removed from each alignment using Gblocks (Talavera et al. 2007) with the least stringent parameters for codons and proteins.
Maximum likelihood (ML) phylogenetic analyses were performed using the “AUTO” parameter in RAxML v8.0.26 (Stamatakis 2006) to establish the evolutionary model with the best fit. The general time reversible model was the preferred model for the nucleotide alignments, and JTT for the amino acid alignments. The topology with the highest likelihood score of 100 heuristic searches was chosen. Bootstrap values were calculated from 500 pseudoreplicates. Taxa with unstable phylogenetic affinities were prefiltered using RogueNaRok (Aberer et al. 2013) based on evaluation of a 50% majority rule consensus tree, in addition to exclusion of taxa with >50% gaps in the alignment.
Bayesian inference was performed using a modified version of MrBayes v3.2 (Huelsenbeck and Ronquist 2001) (https://github.com/astanabe/mrbayes5d, last accessed June 14, 2018). The data set was executed under a separate gamma distribution. Two independent runs, each with three heated and one cold Markov Chain Monte Carlo (MCMC) chain, were started from a random starting tree. The MCMC chains were run for 20,000,000 generations with trees sampled every 1,000th generation. The posterior probabilities and mean marginal likelihood values of the trees were calculated after the burn-in phase (25%), which was determined from the marginal likelihood scores of the initially sampled trees. The average split frequencies of the two runs were <0.01, indicating the convergence of the MCMC chains.
Selection Analyses
All full-length TLRs located in the genomes of great gerbil, sand rat, and Mongolian gerbil along with other mammalian TLRs (supplementary table S3, Supplementary Material online) were analyzed in both classic Datamonkey and Datamonkey 2.0 (datamonkey.org) testing for signs of selection with a phylogeny-guided approach (Delport et al. 2010; Weaver et al. 2018). For each TLR gene alignment, a model test was first run prior to the selection test and the proposed best model was used in the analyses. The mixed effects model of evolution (MEME) and adaptive branch-site random effects model (aBSREL) were used to test for site-based and branch level episodic selections, respectively (Kosakovsky Pond et al. 2011; Murrell et al. 2012; Smith et al. 2015). aBSREL was iterated three times per gene alignment; initially, running an exploratory analysis where all branches were tested for positive selection and subsequently, in a hypothesis mode by which first, the Gerbillinae clade and secondly, the great gerbil was selected as “foreground” branches to test for positive selection. All TLR alignments are available in the Github repository (https://github.com/uio-cels/Nilsson_innate_and_adaptive).
TLR Protein Structure Prediction
Translated full-length great gerbil TLR sequences were submitted to the Phyre2 structure prediction server for modeling (Kelley et al. 2015). All sequences were modeled against human TLR5 (c3j0aA) and the resulting structures were colored for visualization purposes using Jmol (Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/). Colors were used to differentiate between helices, sheets, and loops as well as the transmembrane domain, linker, and Toll/interleukin-1 receptor (TIR) domain. Sites found in the MEME selection analysis were indicated in pink and further highlighted with arrows (supplementary fig. S2, Supplementary Material online). All great gerbil PDB files are available in the GitHub repository (https://github.com/uio-cels/Nilsson_innate_and_adaptive).
As TLR4 is the prototypical PRR for lipopolysaccharide (LPS) which are found in all Gram-negative bacteria including Y. pestis, we subjected the sequence alignment to additional investigation of certain residues indicated in the literature to have an impact on signaling (Sironi et al. 2015). These were the residues at position 367 and 434, which in mouse are both basic and positively charged, enabling the mouse TLR4 to maintain some signaling even for hypoacetylated LPS (Sironi et al. 2015). Hypoacetylated LPS is a common strategy for Gram-negative bacteria to avoid recognition and strong stimulation of the TLR4–MD2–CD14 receptor complex (Rebeil et al. 2004; Raetz et al. 2007; Steimle et al. 2016).
MHCII Promoter Investigation
The region 400 bp upstream of human HLA-DRB, mouse H2-Eb, and rat RT-Db genes were retrieved from Ensembl (GRch38.p12, GRCm38.p6, and Rnor_6.0). Similarly, the region 400 bp upstream of the start codon of DRB genes in the three Gerbillinae were retrieved using bedtools v2.26.0. Putative promoter S–X–Y motifs, as presented for mouse in Péléraux et al. (1996), were manually identified for each gene in MEGA7 and all sequences were subsequently aligned using MUSCLE with default parameters (Péléraux et al. 1996).
Peptide Binding Affinity
The functionality of MHCII genes is defined by the degree of expression of the MHC genes themselves and the proteins’ ability to bind disease-specific peptides to present to the immune system. The ability of an MHCII protein to bind particular peptides can with some degree of confidence be estimated by MHC prediction algorithms, even for unknown MHCII molecules, as long as the alpha- and beta-chain protein sequences are available (Jensen et al. 2018). We here use the NetMHCIIpan predictor v3.2 (Jensen et al. 2018) to estimate the peptide binding affinities of the novel Rhop-DRB3 MHCII molecule and compare it to various other MHCII molecules from great gerbil, mouse, sand rat, and Mongolian gerbil. The program was run with default settings and provided with the relevant protein sequences of alpha and beta chains. We compared the predicted binding affinity of these MHCII molecules for 17 known Y. pestis epitopes derived from positive ligand assays of Y. pestis (https://www.iedb.org/, last accessed August 17, 2019). Specifically, we tested against 16 ligands derived from the F1 capsule antigen of Y. pestis, and 1 ligand from the virulence-associated low calcium V antigen (LcrV) of Y. pestis. In addition, we compared the binding affinity of these MHCII molecules against the superantigen Yersiniapseudotuberculosis-derived mitogen precursor (YPm) (Monzón-Casanova et al. 2016). Finally, we compared the predicted binding affinity profiles of the five Rhop-MHCII molecules for three other pathogens of great gerbils (Rabiee et al. 2018), for which, known epitopes derived from positive ligand assays were available at https://www.iedb.org/, last accessed August 17, 2019, namely Leishmania donovani, Leishmania major, and Leptospira interrogans. The threshold for binders was set to <500 nM (Jensen et al. 2018).
RNA Sampling and Sequencing
Two additional great gerbils were captured in the Midong District outside Urumqi in Xinjiang Province, China, in September 2014. The animals were held in captivity for 35 days before being humanely euthanized and liver tissue samples were conserved in RNAlater at −20 °C prior to RNA extraction. RNA was extracted using standard chloroform procedure (Chomczynski and Sacchi 2006). Library prep and sequencing were conducted at the Beijing Genomics Institute (BGI, https://www.bgi.com/us/sequencing-services/dna-sequencing/) using Illumina TruSeq RNA Sample Prep Kit and PE sequencing on the HiSeq 4000 instrument (150 bp read length).
The reads were trimmed using Trimmomatic v0.36 and mapped to the genome assembly using HISAT2 v2.0.5 with default parameters. A raw count matrix was created by using HTSeq v0.7.2 with default parameters to extract the raw counts from the mapped files.
Results
Genome Assembly and Annotation
We sequenced the genome of a wild-caught male great gerbil, sampled from the Xinjiang Province in China, using the Illumina HiSeq 2000/2500 platform (supplementary tables S1 and S4, Supplementary Material online). The genome was assembled de novo using ALLPATHS-LG resulting in an assembly consisting of 6,389 scaffolds with an N50 of 3.6 Mb and a total size of 2.376 Gb (table 1), thus covering 96.4% of the estimated genome size of 2.47 Gb. Assembly assessment with CEGMA and BUSCO, which investigates the presence and completeness of conserved eukaryotic and vertebrate genes, reported 85.88% and 87.5% gene completeness, respectively (table 1). We were also able to locate all 39 HOX genes conserved in four clusters on four separate scaffolds through gene mining (supplementary fig. S1, Supplementary Material online). Further genome assessment with Blobology, characterizing possible contaminations, demonstrated a low degree of contamination, reporting that >98.5% of the reads/bases had top hits of Rodentia. Thus, no scaffolds were filtered from our assembly.
Table 1.
Great Gerbil Genome Assembly Statistics
| Assembly Metrics | |
| Total size of scaffolds (bp) | 2,376,008,858 |
| Estimated genome size (bp) | 2,464,792,293 |
| Number of scaffolds | 6,389 |
| Scaffold N50 (bp) | 3,610,217 |
| Longest scaffold (bp) | 16,185,803 |
| Total size of contigs (bp) | 2,16,488,676 |
| Number of contigs | 106,018 |
| Contig N50 (bp) | 56,880 |
| Assembly Validation | |
| Complete CEGMAa genes | 85.88% (213/248) |
| Partial CEGMA genes | 95.16% (236/248) |
| Complete single-copy BUSCOsb | 2,114 (69.9%) |
| Complete duplicated BUSCOs | 21 (0.69%) |
| Fragmented BUSCOs | 533 (17.6%) |
| Missing BUSCOs | 377 (12.5%) |
| Total BUSCOs searched | 3,023 |
note.—The table details scaffold and contig assembly statistics as well as results from the assembly validation on genic completeness with CEGMA and BUSCO.
Based on 248 highly conserved eukaryotic genes (CEGs).
Based on 3,023 vertebrate-specific BUSCO genes.
Annotation was performed using the MAKER2 pipeline and resulted in 70,974 predicted gene models of which 22,393 protein coding genes were retained based on default filtering on AED < 1.
Reduced TLR Repertoire in Great Gerbil and Gerbillinae
We characterized the entire TLR genetic repertoire in the great gerbil genome and found 13 TLRs: TLR1-13 (fig. 2A). Of these, TLR1-7 and TLR9 were complete with signal peptide, ectodomain, transmembrane domain, linker, and TIR domain that phylogenetically clustered well within each respective subfamily (table 2 and fig. 2B). For the remaining five TLRs, we were only able to retrieve fragments of TLR8 and TLR10 genes and although sequences of TLR11-13 were near full length, all three members of the TLR11 subfamily are putative nonfunctional pseudogenes as they contain numerous point mutations that creates premature stop codons and frameshift-causing indels. In addition, TLR12 contains a large deletion of 78 residues (supplementary fig. S3D, Supplementary Material online). For TLR8, the recovered sequence almost exclusively covers the conserved TIR domain. Relative synteny of TLR7 and TLR8 on chromosome X is largely conserved in both human and published rodent genomes, as well as in the great gerbil with the fragments of TLR8 being located upstream of the full-length sequence of TLR7 on scaffold00186 (supplementary fig. S4, Supplementary Material online). The great gerbil TLR10 fragments are located on the same scaffold as full-length TLR1 and TLR6 (scaffold00357), in a syntenic structure comparable to other mammals (supplementary fig. S4, Supplementary Material online). In addition to being far from full-length sequences, the pieces of TLR8 and TLR10 in the great gerbil genome have point mutations that create premature stop codons and frameshift-causing indels (supplementary fig. S3A and B, Supplementary Material online). The same TLR repertoire is seen in great gerbils’ closest relatives, Mongolian gerbil, and sand rat, with near full-length sequences of TLR12 and TLR13 and shorter fragments of TLR8 and TLR10. Interestingly, for TLR11 only shorter fragments were located for these two species, which is in contrast to the near full-length sequence identified in great gerbil. Moreover, also in these two species, premature stop codons and indel-causing frameshifts were present in both the near full-length and the fragmented genes (fig. 2 and supplementary fig. S3, Supplementary Material online).
Fig. 2.

—TLR repertoire and ML phylogeny of investigated Gerbillinae, Rodentia, human, and Chinese tree shrew. (A) TLR repertoire of the investigated Gerbillinae, Rodentia, human, and Chinese tree shrew mapped onto a composite cladogram (see supplementary fig. S12, Supplementary Material online). The lineage-specific loss of TLR8 and all members of the TLR11-subfamily in Gerbillinae and other lineage-specific TLR losses are marked by arrows. Depicted in boxes colored by the six major subfamilies are the individual species’ TLR repertoires: TLR1-subfamily (dark pink), TLR3-subfamily (pink), TLR4-subfamily (light pink), TLR5-subfamily (light green), TLR7-subfamily (green), and TLR11-subfamily (dark green). Teal-colored boxes represent established pseudogenes, empty (white) boxes indicate putative pseudogenes, yellow boxes indicate complete absence of genes, and gray boxes represent missing information. (B) A maximum likelihood (ML) phylogeny of nucleotide sequences all full-length TLRs was created using RAxML with 100× topology and 500× bootstrap replicates. A MrBayes phylogeny with 20,000,000 generations and 25% burn-in was also created and the posterior probabilities added to the RAxML phylogeny. BS/PP; *, BS 100 or PP > 0.96; **, BS of 100 and PP>0.97; <, support values below 50/0.8; -, node not present in Bayesian analysis. Great gerbil genes are marked in bold and by orange circles. The six major TLR subfamilies are marked with colored bars and corresponding names. All investigated TLRs, including great gerbils, cluster well within each subfamily as well as being clearly separated into each TLR subfamily member.
Table 2.
Overview of TLRs in Great Gerbil and Their Location in the Genome Assembly
| Gene | Scaffold | Strand | Start | End | Size (aa) | Full-Length Coding Sequence |
|---|---|---|---|---|---|---|
| TLR1 | scaffold00357 | + | 837,967 | 840,348 | 794 | Yes |
| TLR2 | scaffold00513 | + | 45,595 | 47,946 | 784 | Yes |
| TLR3 | scaffold00205 | − | 1,713,605 | 1,703,075 | 905 | Yes |
| TLR4 | scaffold00158 | + | 3,555,106 | 3,573,424 | 838 | Yes |
| TLR5 | scaffold00165 | − | 2,379,076 | 2,376,503 | 858 | Yes |
| TLR6 | scaffold00357 | + | 820,802 | 823,186 | 795 | Yes |
| TLR7 | scaffold00186 | − | 3,421,186a | 3,418,040 | 1,049 | Yesb |
| TLR8 | scaffold00186 | − | 130 | Noc | ||
| TLR9 | scaffold00044 | + | 7,083,426a | 7,086,521 | 1,032 | Yesb |
| TLR10 | scaffold00357 | + | 341 | No | ||
| TLR11 | scaffold00001 | + | 917 | Yesd | ||
| TLR12 | scaffold00071 | − | 4,008,732 | 4,006,236 | 817 | Nod |
| TLR13 | scaffold00845 | − | 899 | Yesd |
note.—The table details which scaffolds and in what orientation each TLR is located as well as coordinates for the start and end of each gene (except for the pseudogenes) on the respective scaffolds. Information on the size of the translated amino acid sequence and whether it is complete is also shown.
Start of second codon in sequence.
Missing start codon.
Close to complete TIR domain plus c-terminal.
Contains multiple point mutations and indels causing frameshifts.
Diversifying Selection of TLRs
To explore possible variations in selective pressure across the species in our analysis, we ran the adaptive branch-site random effects model (aBSREL) on all full-length TLRs. Evidence of episodic positive selection was demonstrated for the Gerbillinae lineage for TLR7 and TLR9 and for the Mongolian gerbil TLR7 specifically (supplementary figs. S5 and S6, Supplementary Material online). Additionally, all full-length great gerbil TLRs were analyzed for sites under selection using phylogeny-guided MEME, from the classic datamonkey and datamonkey version 2.0 websites. Reported sites common between both analyses for all full-length TLRs at P value 0.05 and their distribution among each domain of the proteins are listed in supplementary table S5, Supplementary Material online. Overall, the sites under selection were almost exclusively located in the ectodomains with a few sites located in the signal peptide (TLR3, TLR6, and TLR9) and in the Linker and TIR domains (TLR1, TLR2, TLR4, and TLR5). The 3D protein structure of TLR4, TLR7, and TLR9 modeled onto the human TLR5 structure further demonstrated that the sites are predominantly located in loops interspersed between the leucine-rich repeats (supplementary fig. S2A–C, Supplementary Material online).
Scrutiny of the TLR4 amino acid sequence alignment revealed drastic differences in the properties of the residues at two positions reported to be important for maintaining signaling of hypoacetylated LPS. In rat (Rattus norvegicus) and all mouse species used in this study, the residues at position 367 and 434 are basic and positively charged, whereas for the remaining species in the alignment including all Gerbillinae, the residues are acidic and negatively charged.
Characterization of the Great Gerbil Class I MHC Region
The overall synteny of the MHCI region is well conserved in great gerbil, displaying the same translocation of some MHCI genes upstream of the MHCII region as demonstrated in mouse and rat, that is, with a distinct separation of the MHCI region into two clusters (fig. 1). Four great gerbil copies identified were not included in the phylogeny due to missing data (>50% gaps in the alignment), which hindered their annotation. Additionally, the annotation was obstructed either by the copies being located on scaffolds not containing framework (FW) genes or due to variation in the microsynteny of those particular loci of MHCIa and MHCIb between mouse, rat, and great gerbil (fig. 1). From the synteny, it appears that MHCI genes are missing in the region between FW genes Trim39 and Trim26 and possibly between Bat1 and Pou5f1 in the great gerbil. For full gene names for these and other FW genes mentioned below, see supplementary table S2, Supplementary Material online.
We were able to identify seven scaffolds containing MHCI genes (fig. 1 and supplementary table S6, Supplementary Material online). Four of the scaffolds contained FW genes that enabled us to orient them. In total, we located 16 MHCI copies, of which, we were able to obtain all three α domains for 10 of the copies. Three copies contain two out of three domains, whereas for the last three copies, we were only able to locate the α3 domain. In one instance, the missing α domain was due to an assembly gap. Reciprocal BLAST confirmed hits as MHCI genes. Due to high similarity between different MHCI lineages, annotation of identified sequences was done through phylogenetic analyses and synteny. Our phylogeny reveals both inter- and intraspecific clustering of the great gerbil MHCI genes with other rodent genes with decent statistical support (i.e., bootstrap and/or posterior probabilities) of the internal branches (fig. 3A). Five great gerbil MHCI genes (RhopA1-5) cluster together in a main monophyletic clade, whereas the remaining copies cluster with mouse and rat MHCIb genes. Two of the copies (Rhop-A3ψ and Rhop-M6ψ) appear to be pseudogenes as indicated by the presence of point mutations and frameshift-causing indels. Additionally, our phylogeny displays a monophyletic clustering of human MHCI genes (fig. 3A). The clade containing five of the great gerbil MHCI genes (Rhop-A1-5) possibly include a combination of both classical (MHCIa) and nonclassical (MHCIb) genes as is the case for mouse and rat, where certain MHCIb genes cluster closely with MHCIa genes (figs. 1 and 3A). In addition, due to the high degree of sequence similarity of rodent MHCI genes, the phylogenetic relationship between clades containing nonclassical M and TMHCI genes could not be resolved by sufficient statistical support.
Fig. 3.
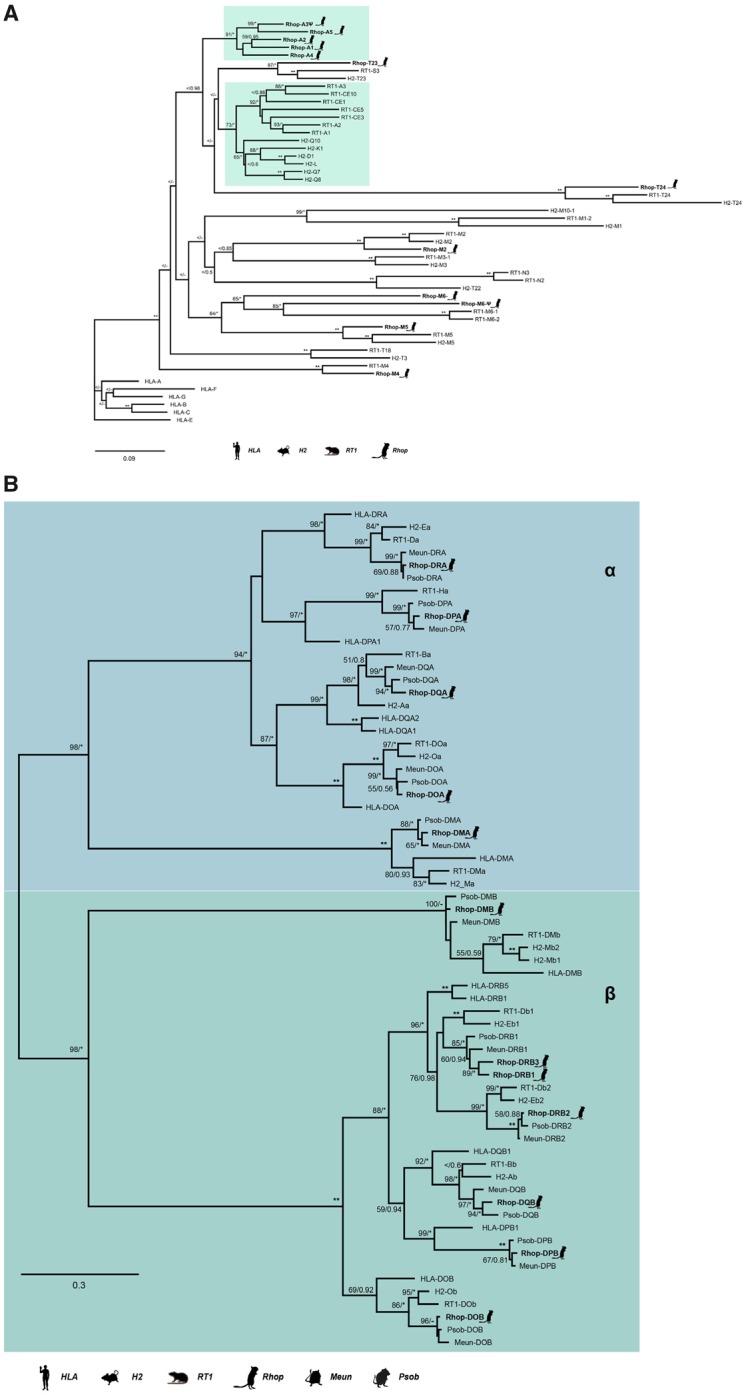
—ML phylogenies of MHCI and MHCII genes. (A) A maximum likelihood (ML) phylogeny of nucleotide sequences containing the three α domains of MHCI was created using RAxML with 100× topology and 500× bootstrap replicates. A MrBayes phylogeny with 20,000,000 generations and 25% burn-in was also created and the posterior probabilities added to the RAxML phylogeny. About 12 of the 16 great gerbil sequences were used in the analysis and are marked with a gerbil silhouette and in bold lettering. The remaining four MHCI sequences were excluded from the phylogenetic analyses due to missing data exceeding the set threshold of 50%. The clusters containing MHCIa (classical MHCI) and the closest related MHCIb genes are marked by teal boxes. Putative pseudogenes are denoted with ψ. (B) A ML phylogeny of nucleotide sequences containing the α and β domains of MHCII α and β genes was created using RAxML with 100× topology and 500× bootstrap replicates. A MrBayes phylogeny with 20,000,000 generations and 25% burn-in was also created and the posterior probabilities added to the RAxML phylogeny. Great gerbil genes are indicated with bold lettering and by silhouettes. The 12 great gerbil MHCII genes located in the genome assembly cluster accordingly with the orthologs of human, mouse, rat, sand rat, and Mongolian gerbil. The Rhop-DRB duplication (Rhop-DRB3) cluster closely with the Rhop-DRB1 and other DRB1 orthologs with good support. The nomenclature of MHCII genes in Gerbillinae is in concordance with the recommendations of the MHC Nomenclature report (Ballingall et al. 2018). BS/PP; *, BS 100 or PP > 0.96; **, BS of 100 and PP>0.97; <, support values below 50/0.8; -, node not present in Bayesian analysis.
Characterization of the Great Gerbil Class II MHC Region
A single scaffold (scaffold00896) of 471,076 bp was identified to contain all genes of the MHCII region, flanked by the reference FW genes Col11a2 and Btnl2. We were able to obtain orthologs of α and β genes of the classical MHCII molecules DP, DQ, and DR as well as for the “nonclassical” DM and DO molecules (table 3). The antigen-processing genes for the class I presentation pathway, Psmb9, TAP1, Psmb8, and TAP2 also maps to scaffold00896 (fig. 1). Synteny of the MHCII region was largely conserved in great gerbil when compared with mouse, rat, and human regions except for a single duplicated copy of Rhop-DRB (Rhop-DRB3) that was located distal to the Btnl2 FW gene representing the border between classes II and III of the MHC region (fig. 1). The duplicated copy of the Rhop-DRB gene has an antisense orientation in contrast to the other copies of the Rhop-DRB genes in great gerbil. In rodents, the DR locus contains a duplication of the β gene and the two copies are termed β1 and β2, with the β2 gene being less polymorphic than the highly polymorphic β1 gene. The relative orientation of the β and α genes of the DR locus is conserved in most eutherian mammals studied to date with the genes facing each other, as is the case for Rhop-DRB1, Rhop-DRB2, and Rhop-DRA (fig. 1). Sequence alignment and a ML phylogeny based on coding regions establish Rhop-DRB3 to be a duplication of Rhop-DRB1 (fig. 3B). If Rhop-DRB3 is not a true ortholog to Rhop-DRB1, but rather an allele erroneously assembled as a separate, duplicated gene we would expect them to be almost identical. Comparing the complete sequences from Rhop-DRB3 and Rhop-DRB1, including introns, we uncover several indels and single point mutations (p-distance = 0.038), indicating that they are true copies and not the result of an assembly error. This is visualized by a dot plot of scaffold00896 against itself, showing the similarity of the two genes by counter-diagonals above and below the main diagonal (supplementary note S2 and fig. S7, Supplementary Material online), further, the mean read coverage and SD of read coverage of scaffold00896 is similar to other scaffolds with immune genes and the assembly as a whole (supplementary table S6, Supplementary Material online). The average per base coverage across all MHCII genes on scaffold00896 is also similar with no apparent decrease in coverage in the region covering the Rhop-DRB genes further supporting Rhop-DRB3 to be a true copy (supplementary fig. S8, Supplementary Material online). Rhop-DRB1 and Rhop-DRB3 are separated by the Rhop-DRB2, Rhop-DRA genes, and five assembly gaps (table 3).
Table 3.
Overview of Great Gerbil MHCII Genes and Their Location in the Genome Assembly
| Gene | Scaffold | Strand | Start | End | Size (aa) | Full-Length Coding Sequence |
|---|---|---|---|---|---|---|
| Rhop-DPB | scaffold00896 | − | 116,069 | 105,406 | 264 | Yes |
| Rhop-DPA | scaffold00896 | + | 117,514 | 120,092 | 252 | Yes |
| Rhop-DOA | scaffold00896 | + | 125,319 | 127,406 | 241 | Yesa |
| Rhop-DMa | scaffold00896 | + | 166,162 | 168,926 | 265 | Yesb |
| Rhop-DMb | scaffold00896 | + | 175,763 | 182,090 | 257 | Yes |
| Rhop-DOB | scaffold00896 | + | 239,915 | 245,006 | 172 | Noc |
| Rhop-DQB | scaffold00896 | + | 271,007 | 278,482 | 231 | Nod |
| Rhop-DQA | scaffold00896 | − | 294,074 | 290,301 | 255 | Yes |
| Rhop-DRB1 | scaffold00896 | + | 310,644 | 319,368 | 265 | Yes |
| Rhop-DRB2 | scaffold00896 | + | 326,384 | 347,931 | 272 | Yese |
| Rhop-DRA | scaffold00896 | − | 355,351 | 351,425 | 254 | Yes |
| Rhop-DRB3 | scaffold00896 | − | 403,768 | 395,068 | 265 | Yes |
note.—The table details the assigned great gerbil gene names, which scaffold and in what orientation they are located as well as genomic location on the scaffold for start and end of the genes. Information on the size of the translated amino acid sequence and whether it is complete is also shown. In addition, see supplementary figure S7, Supplementary Material online.
Missing final residue and stop codon due to conserved overlapping splice site.
Unable to locate a stop codon. The structure of the final two exons is conserved among human, mouse, and rat with the ultimate residue overlapping the splice site. The final coding exon therefore only contains 2 bp and the stop codon making it hard to determine the location of the final exon in the gerbil without supporting RNA information.
Missing 99 residues (121–219) due to assembly gap.
Start location is that of exon 2 as exon 1 is missing due to an assembly gap.
The cytoplasmic tail of β2 genes are encoded by an exon with no known homology to other exons in the MHCII genes and has a low degree of homology between mouse and rat (Monzón-Casanova et al. 2016). There is therefore some uncertainty related to the completeness of the final exon of Rhop-DRB2.
Any similar duplication of the Rhop-DRB1 gene is not seen in either of the two close family members of the Gerbillinae subfamily used in our comparative analyses. BLAST searches of the sand rat genome returned a single full-length copy of the β1 gene and a near full-length copy of the β2 gene (fig. 3B and supplementary table S7, Supplementary Material online). According to the annotations of the Mongolian gerbil genome provided by NCBI, this species contains two copies of the DR locus β genes. A manual TBlastN search using the protein sequences of Mongolian gerbil DRB genes to search the genome assembly did not yield additional hits of β genes in this locus that could have been missed in the automatic annotation process. The phylogeny confirms the copies found in Mongolian gerbil to be β1 and β2 genes (fig. 3B).
MHCII DRB Promoters
MHCII genes each contain a proximal promoter with conserved elements termed S–X–Y motifs that are crucial for the efficient expression of the gene. We aligned the proximal promoter of the β genes of the DR locus in great gerbil and the other investigated species to establish if the integrity of the promoter was conserved as well as examining similarities and potential dissimilarities causing the previously reported differences in transcription and expression of β1 and β2 genes in rodents (Braunstein and Germain 1986; Monzón-Casanova et al. 2016). The alignment of the promoter region reveals the conserved structure and similarities within β1 and β2 genes as well as characteristic differences (fig. 4 and supplementary table S11, Supplementary Material online). Clear similarities are seen for the proximal promoter regions of Rhop-DRB1 and Rhop-DRB3 to the other rodent and human β1 promoters, as illustrated by high sequence similarity and the presence of a CCAAT box just downstream of the Y motif in all investigated rodent β1 promoters. Notably, the CCAAT box is missing in β2 promoters. The crucial distance between the S and X motifs is conserved in all β genes and the integrity of the S–X–Y motifs is observable for Rhop-DRB1 and DRB3 promoters. However, both the S and the X boxes of DRB2 are compromised by deletions in great gerbil. The deletion in the X box severely disrupts the motif and reduces its size by half. An identical deletion in the X box is seen in Mongolian gerbil, whereas the sand rat X box sequence appears complete with parts of it being highly divergent from the conserved sequence found in the rest of the promoters (fig. 4). Furthermore, for the β2 genes, two deletions downstream of the motifs are shared among all rodents in the alignment as well as an additional insertion observed in Gerbillinae members.
Fig. 4.

—Alignment of the DR locus β1 and β2 proximal promoters. Sequences of the proximal promoters of β1 and β2 genes of the DR locus (E locus in mouse and D locus in rat) were aligned in MEGA7 (Kumar et al. 2016) using MUSCLE with default parameters. The resulting alignment was edited manually for obvious misalignments and transferred and displayed in Jalview (Waterhouse et al. 2009). For visualization purposes only, the alignment was further edited in Adobe Illustrator (CS6), changing colors of the bases and adding boxes to point out the S–X–Y motifs. The three copies of DRB genes located in the great gerbil genome are marked with gray boxes. The alignment shows clear similarities of the proximal promoter region of Rhop-DRB1 and Rhop-DRB3 to the other rodent and human β1 promoter sequences. For the DRB2 genes, two deletions are shared among all rodents in the alignment as well as additional indels observed in Gerbillinae members. Most notably, both great gerbil and Mongolian gerbil have deletions of half the X box, whereas sand rat X box sequences in that same position are highly divergent from the otherwise conserved sequence seen in the alignment.
Peptide Binding Affinity Predictions and Expression of Rhop-DR MHCII Molecules
Mouse and rat β2 molecules have been shown to have an extraordinary capacity to present the YPm (Monzón-Casanova et al. 2016). We therefore investigated the peptide binding affinities of the Rhop-DR molecules by running translations of Rhop-DRA in combinations with each of the three Rhop-DRB genes through the NetMHCIIpan 3.2 server (Jensen et al. 2018) along with peptide/protein sequences of YPm, Y. pestis F1 “capsular” antigen, and LcrV antigen. Universally, the Rhop-DRB3 shows an affinity profile identical to that of Rhop-DRB2 displaying high affinity toward both Y. pseudotuberculosis and Y. pestis epitopes, whereas Rhop-DRB1 does not (fig. 5 and Github repository). The translated great gerbil MHCII from DP and DQ loci were also tested for peptide binding affinity but only Rhop-DP displayed affinity to one of the epitopes tested. Furthermore, analyses of the translated amino acid sequences of sand rat DR (Psob-DR) molecules as well as published protein sequences of Mongolian gerbil DR (Meun-DR) molecules and the mouse ortholog H2-E confirmed the high affinity of β2 molecules to Y. pseudotuberculosis and Y. pestis (supplementary fig. S9, Supplementary Material online and Github repository). The equal capacity of Rhop-DRB2 and Rhop-DRB3 to putatively present Yersiniae combined with the proximal promoter investigations lead us to question the expression of DRB genes in great gerbil. Searching a set of raw counts of great gerbil-expressed genes reveals that Rhop-DRB1 and Rhop-DRB3 are both expressed at similar levels, whereas Rhop-DRB2 is not expressed or at undetectable levels (3,936 and 2,279 vs. 14). The similarity in epitope binding affinity profiles of Rhop-DRB2 and Rhop-DRB3 are not limited to Yersiniae—both DRB2 and DRB3 are predicted to present the same 11 out of 12 putative Leptospira interrogans MHC Class II epitopes, and can present many of the known Leishmania epitopes (15 out of 20 in the case of DRB2, and 9 out of 20 in the case of DRB3—supplementary figs. S10 and S11, Supplementary Material online and Github repository).
Fig. 5.

—Affinity predictions of great gerbil MHCII molecules. Affinity predictions of great gerbil MHC class II molecules represented as a heatmap. For the known Yersinia pestis antigens, all are from the F1 capsule precursor except ID16182 (red asterisk) which is from the V antigen. Strong binders are defined as <500 nM and depicted in orange, whereas weak or nonbinders are represented in blue.
Discussion
Here, we present a highly contiguous de novo genome assembly of the great gerbil covering over 96% of the estimated genome size and almost 88% of the gene space, which is equivalent to the genic completeness reported in the recently published and close relative sand rat genome (Hargreaves et al. 2017) (supplementary table S8, Supplementary Material online). By comparative genomic analyses where we include genome data from its close relatives within the Gerbillinae, we provide novel insight into the innate and adaptive immunological genomic landscape of this key plague host species.
TLRs are essential components of PRRs and the innate immune system as they alert the adaptive immune system of the presence of invading pathogens (Kawai and Akira 2010). The detailed characterization of TLRs did not uncover any species-specific features for the great gerbil. However, a shared TLR gene repertoire for the Gerbillinae lineage (i.e., the great gerbil, sand rat, and Mongolian gerbil), with gene losses of TLR8, TLR10, and all members of the TLR11-subfamily was revealed. This finding could indicate quite similar selective pressures on these species, at least in regard to their function of TLRs, all being desert dwelling, burrowing rodents living in arid or semiarid ecosystems and being capable of carrying plague. Thus, it is possible that the members of this clade have reduced the TLR repertoire in a cost-benefit response to environmental constraints or due to altered repertoire of pathogen exposure (Salazar Gonzalez et al. 2014). These results are in line with the fairly conserved TLR gene repertoire reported within the vertebrate lineage (Roach et al. 2005), although the repertoire of TLR genes present within vertebrate groups can show major differences (Roach et al. 2005; Temperley et al. 2008; Solbakken et al. 2016), presumably in response to presence or lack of certain pathogen or environmental pressures (Barreiro et al. 2009; Babik et al. 2015). Outside of Gerbillinae, the presence of TLR11-subfamily appears to be universal in Rodentia, however, functionally lost from the human repertoire (Roach et al. 2005; Solbakken et al. 2016). The TLR11-subfamily recognizes parasites and bacteria through profilin, flagellin, and 23S ribosomal RNA (Mathur et al. 2012; Oldenburg et al. 2012) and it is possible that cross-recognition of these patterns by other TLR members or other PRRs might have made the TLR11-subfamily redundant in Gerbillinae and humans (Salazar Gonzalez et al. 2014). The varying degree of point mutations, frameshift-causing indels, and in some cases, almost complete elimination of sequence in TLR8, TLR10, and TLR11-13 in Gerbillinae suggest successive losses of these receptors, where a shared pseudogenization of TLR12-13 across all species investigated were recorded. For TLR11 however, the pseudogenization seems to have occurred in multiple steps, that is, with a more recent event in the great gerbil, where a near full-length sequence was identified compared with the shorter fragments identified for Mongolian gerbil and sand rat (supplementary fig. S3C, Supplementary Material online). Furthermore, the high degree of shared disruptive mutations among all three species of Gerbillinae indicates that the initiation of pseudogenization predates the speciation estimated to have occurred ∼5.5 Ma (Chevret and Dobigny 2005).
In the context of plague susceptibility, the branch-specific diversifying selection reported here for TLR7 and TLR9 in Gerbillinae is intriguing, as both receptors have been implicated to affect the outcome of plague infection in mice and humans (Saikh et al. 2004; Amemiya et al. 2009; Dhariwala et al. 2017). For instance, the study by Dhariwala et al. (2017) showed, in a murine model, that TLR7 recognizes intracellular Y. pestis and is important for defense against disease in the lungs but was detrimental to septicemic plague (Dhariwala et al. 2017). Moreover, recognition of Y. pestis by TLR9 was also demonstrated by Saikh et al. (2004) in human monocytes (Saikh et al. 2004). All but one of the residues under site-specific selection seen in TLR7 and TLR9 were located in the ectodomain, which may suggest possible alterations in ligand recognition driven by selection pressure from Y. pestis or other shared pathogens. Stimulation of TLR7 and TLR9 has also been reported to regulate antigen presentation by MHCII in murine macrophages (Celhar et al. 2016). For TLR4, the selection tests and sequence alignment analysis did not reveal any branch-specific selection for great gerbil nor Gerbillinae, whereas we did detect signs of site-specific selection in the ectodomain, often with derived substitutions in the great gerbil or Gerbillinae. TLR4 is the prototypical PRR for detection of LPS found in the outer membrane of Gram-negative bacteria like Y. pestis. As part of the arms race, however, it is well known that Gram-negative bacteria, including Y. pestis, alter the conformation of their LPS in order to avoid recognition and strong stimulation of the TLR4–MD2–CD14 receptor complex (Rebeil et al. 2004; Raetz et al. 2007; Steimle et al. 2016). Despite this, in mice at least, some inflammatory signaling still occurs through this receptor complex but require particular residues in TLR4 not found to be conserved in the Gerbillinae lineage. Whether other mutations in TLR4 in Gerbillinae have a similar functionality as the residues that allow mice to respond to Y. pestis LPS is not known. However, if such functionality is missing in Gerbillinae, the loss of responsiveness to the hypoacetylated LPS (Sironi et al. 2015) could perhaps defer some protection from pathologies caused by excessive initiation of inflammatory responses (Foster and Medzhitov 2009), and thus, TLR4 is not likely directly involved in the resistance of plague in great gerbils.
Cumulatively, our investigations of the great gerbil innate immune system, focusing on the TLR gene repertoire, reveal shared gene losses within TLR gene families for the Gerbillinae lineage, all being desert dwelling species capable of carrying plague. The evolutionary analyses conducted did not uncover any great gerbil-specific features that could explain their resistance to Y. pestis, indicating that other PRRs (not investigated here) could be more directly involved during the innate immune response to plague infection in the great gerbil (Vladimer et al. 2012).
MHCI and II proteins are crucial links between the innate and adaptive immune system continuously presenting peptides on the cell surface for recognition by CD8+ and CD4+ T cells, respectively, and MHC genes readily undergo duplications, deletions, and pseudogenization (Nei et al. 1997). For MHCI, the discovery of 16 copies in great gerbil is in somewhat agreement with what has earlier been reported in rodents, where the MHCI region is found to have undergone extensive duplication followed by sub- and neofunctionalization with several genes involved in nonimmune functions (Amadou et al. 2003; Ohtsuka et al. 2008). Indeed, the great gerbil seems to have undergone species-specific duplications of MHCI genes as well as additional losses of some MHCI genes compared with mouse and rat (fig. 1). However, not all copies could be confidently placed in the gene maps of the MHC regions as some scaffolds lacked colocalizing FW genes, which is needed to confirm these findings for synteny analyses and exact copy number estimation.
For MHCII, we discovered a gerbil-specific duplication that is not present in other closely related plague hosts or in other rodents investigated. The phylogeny established the duplication’s (Rhop-DRB3) relationship to Rhop-DRB1 and other mammalian β1 genes and reflects the orthology of mammalian MHCII genes (Hughes and Nei 1990). The localization of Rhop-DRB3 outside of the generally conserved FW of the MHCII region (i.e., distal to the FW gene Btnl2) and not directly in tandem with the other β genes of the DR locus is unusual and not generally seen for eutherian mammals. For instance, major duplication events with altered organization and orientation of DR and DQ genes has been reported for the MHCII region in horse (Equus caballus), however, all genes are found within the FW genes (Viļuma et al. 2017). Duplications tend to disperse within the genome with age (Katju and Lynch 2003), thus, the reversed orientation and translocation of the great gerbil copy might indicate that the duplication event is not a recent event, and occurred sometime after the species split from a shared ancestor ∼5 Ma. However, it should also be noted that the observed assembly gaps located between Rhop-DRB1 and Rhop-DRB3 may be indicative of the translocation being a result of an assembly error, yet, the significant differences in nucleotide substitutions and indels between the two genes makes this less likely.
Predictions of the affinity of the β1, β2, and β3 MHCII molecules to Y. pestis and Y. pseudotuberculosis antigens matched the reported high affinity of rodent β2 molecules for Yersiniae epitopes (Monzón-Casanova et al. 2016). Rhop-DRB3 had an equally high affinity and largely identical affinity profile as Rhop-DRB2. A high affinity for Y. pestis epitopes is important in the immune response against plague, as the initiation of a T cell response is more efficient and requires fewer antigen-presenting cells and T cells when high-affinity peptides are presented by MHCII molecules (Gregers et al. 2003). In the early stages of an infection where presence of antigen is low, there will be fewer MHCII molecules presenting peptides and affinity for those peptides is paramount to fast initiation of the immune response against the pathogen. Individuals presenting MHCII molecules with high affinity for pathogen epitopes are able to raise an immune defense more quickly and have a better chance of fighting off the rapidly progressing infection than individuals that are fractionally slower. This fractional advantage could mean the difference between death or survival.
We find comparable expression levels for Rhop-DRB1 and Rhop-DRB3 but no detectable expression of Rhop-DRB2. These similarities and differences are likely explained by the variations discovered in the proximal promoter of the genes. Integrity of the conserved motifs and the spacing between them is necessary for assembly of the enhanceosome complex of transcription factors and subsequent binding of class II major histocompatibilty complex transactivator (CIITA), and is essential for efficient expression of MHCII genes. The conservation of the proximal promoter of Rhop-DRB3 along with the overall sequence similarity with other β1 genes are indicative of a similar expression pattern. In contrast, the deletion in the X box of Rhop-DRB2 reducing the motif to half the size will likely affect the ability of the transcription factors to bind and could explain the lack of expression. Similar disruptions in the β2 genes of the other Gerbillinae were found along with a major deletion further downstream in all β2 genes that perhaps explains the previously reported low and unusual pattern of transcription for rodent β2 genes (Braunstein and Germain 1986; Monzón-Casanova et al. 2016). The equal affinity profile but different expression levels of Rhop-DRB2 and Rhop-DRB3 could mean that Rhop-DRB3 has taken over the immune function lost by the lack of expression of Rhop-DRB2. The selective pressure for retention and subfunctionalization of the duplicated Rhop-DRB3 gene might have come from Yersinia, but as other great gerbil pathogens (Leishmania and Leptospirosis tested here) are also presented by the Rhop-DRB2 and DRB3 molecules, it would be premature to conclude a single causal relationship. A nonclassical function of MHCII molecules have also been reported where intracellular MHCII interacted with components of the TLR signaling pathway in a way that suggested MHCII molecules are required for full activation of the TLR-triggered innate immune response (Liu et al. 2011). Moreover, in vertebrates, the MHCII DRB genes are identified as highly polymorphic and specific allele variants have frequently been linked to increased susceptibility to diseases in humans (Matzaraki et al. 2017). Intriguingly, in a recent study by Cobble et al. (2016), it was suggested that allelic variation of the DRB1 locus could be linked to plague survival in Gunnison’s prairie dog colonies (Cobble et al. 2016). Thus, investigating how the genetic variation of the DRB1 and DRB3 loci in great gerbil manifests at the population level and the affinity of these allelic variants to Yersiniae epitopes would be the next step to further our understanding of the plague-resistant key host species, the great gerbil, in Central Asia.
From the generation of the great gerbil de novo genome assembly combined with comparative genomic landscape analyses of the adaptive immune system, we uncover the duplication of an MHCII gene in great gerbils with a computed peptide binding profile that putatively would cause a faster initiation of the adaptive immune system when exposed to Yersiniae epitopes as well as epitopes from Leishmania and Leptospirosis. We also find signs of positive selection in TLR7 and TLR9, which have been shown to regulate antigen presentation (Celhar et al. 2016) and could in turn impact the outcome of an infection. Investigations into how the genetic variation of the MHCII locus manifests at the population level are necessary to further understand the role of the gene duplication as part of the pathogen resistance in great gerbils.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
All computational work were performed on the Abel Supercomputing Cluster (Norwegian metacenter for High Performance Computing [NOTUR] and the University of Oslo) operated by the Research Computing Services group at USIT, the University of Oslo IT-department, and the Cod nodes of CEES. Sequencing library creation and high-throughput sequencing were carried out at the Norwegian Sequencing Centre (NSC), University of Oslo, Norway and McGill University and Genome Quebec Innovation Centre, Canada. We would like to thank Morten Skage for assistance in sequence library construction and Srinidhi Varadharajan, Tore O. Elgvin, and Cassandra N. Trier for helpful advice and support during assembly and annotations steps of the genome and Tone F. Gregers for helpful discussions regarding MHCII. For early access to the sand rat genome assembly, we thank John F. Mulley. This project was funded by University of Oslo Molecular Life Science (M.L.S., Allocation No. 152950), the Research Council of Norway (RCN Grant No. 179569 and FRIMEDBIO Grant No. 288551), the European Research Council (ERC-2012-AdG No. 324249—MedPlag), the National Natural Science Foundation of China (No. 31430006), and National Key Research & Development Program of China (2016YFC1200100). The animal used for the reference genome was captured as part of routine plague surveillance conducted by the Xinjiang Centre for Disease Control and Prevention (Xinjiang CDC), and as such, the sampling and use of tissue were approved by the Committee for Animal Welfares of the Xinjiang CDC.
Author Contributions
P.N. created the genome assembly and annotated it, performed all BLAST-based, TLR-based, and promoter analysis and wrote the first draft of the article. M.H.S. conducted the protein model analyses of TLRs and assisted in the BLAST-based and TLR analyses. B.V.S. performed the MHCII affinity analyses. Annotation of MHCI and MHCII genes and investigations of duplications/deletions was done by P.N., H.T.B., and O.K.T. R.J.S.O. performed phylogenetic analysis of TLR, MHCI, and MHCII genes. Y.Z. sampled, acclimatized, and tested individual great gerbil for plague. R.L., Y.C., and Y.S. extracted DNA and RNA for sequencing. P.N., W.R.E., B.V.S., S.J., and K.S.J. designed the sequencing strategy. W.R.E., B.V.S., S.J., K.S.J., N.C.S., and R.Y. oversaw the project. All authors read and approved the final article.
Data deposition: The genome assembly has been deposited at DDBJ/ENA/GenBank under the accession REGO00000000. The version described in this article is version REGO01000000. The genome assembly and annotation are also available from FigShare: https://figshare.com/s/9035ed40f970d0545d06. In the following, GitHub repository contains files of immune gene alignments, PDB files, peptide binding affinity predictions for all MHCII molecules, and more: https://github.com/uio-cels/Nilsson_innate_and_adaptive.
Literature Cited
- Aberer AJ, Krompass D, Stamatakis A.. 2013. Pruning rogue taxa improves phylogenetic accuracy: an efficient algorithm and webservice. Syst Biol. 62(1):162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addink EA, et al. 2010. The use of high-resolution remote sensing for plague surveillance in Kazakhstan. Remote Sens Environ. 114(3):674–681. [Google Scholar]
- Akhavan AA, et al. 2010. Dynamics of Leishmania infection rates in Rhombomys opimus (Rodentia: Gerbillinae) population of an endemic focus of zoonotic cutaneous leishmaniasis in Iran. Bull Soc Pathol Exot. 103(2):84–89. [DOI] [PubMed] [Google Scholar]
- Amadou C, et al. 2003. Co-duplication of olfactory receptor and MHC class I genes in the mouse major histocompatibility complex. Hum Mol Genet. 12(22):3025–3040. [DOI] [PubMed] [Google Scholar]
- Amemiya K, et al. 2009. CpG oligodeoxynucleotides augment the murine immune response to the Yersinia pestis F1-V vaccine in bubonic and pneumonic models of plague. Vaccine 27(16):2220–2229. [DOI] [PubMed] [Google Scholar]
- Anisimov AP, Lindler LE, Pier GB.. 2004. Intraspecific diversity of Yersinia pestis. Clin Microbiol Rev. 17(2):434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babik W, et al. 2015. Constraint and adaptation in newt Toll-like receptor genes. Genome Biol Evol. 7(1):81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballingall KT, et al. 2018. Comparative MHC nomenclature: report from the ISAG/IUIS-VIC committee 2018. Immunogenetics 46 (Database issue):333–338. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, et al. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 5(7):e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean AGD, et al. 2013. Studying immunity to zoonotic diseases in the natural host – keeping it real. Nat Rev Immunol. 13(12):851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begon M, et al. 2006. Epizootiologic parameters for plague in Kazakhstan. Emerg Infect Dis. 12(2):268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet C, et al. 2011. Mus spretus SEG/Pas mice resist virulent Yersinia pestis, under multigenic control. Genes Immun. 12(1):23–30. [DOI] [PubMed] [Google Scholar]
- Bramanti B, Stenseth NC, Walløe L, Lei X.. 2016. Plague: a disease which changed the path of human civilization In: Yang R, Anisimov A, editors. Yersinia pestis: retrospective and perspective. Vol. 918 Dordrecht: Springer; p. 1–26. [DOI] [PubMed] [Google Scholar]
- Braunstein NS, Germain RN.. 1986. The mouse E beta 2 gene: a class II MHC beta gene with limited intraspecies polymorphism and an unusual pattern of transcription. EMBO J. 5(10):2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, et al. 2014. Running with the Red Queen: the role of biotic conflicts in evolution. Proc R Soc B. 281(1797):20141382–20141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch JD, et al. 2011. Population differences in host immune factors may influence survival of Gunnison’s prairie dogs (Cynomys gunnisoni) during plague outbreaks. J Wildl Dis. 47(4):968–973. [DOI] [PubMed] [Google Scholar]
- Busch JD, et al. 2013. The innate immune response may be important for surviving plague in wild Gunnison’s prairie dogs. J Wildl Dis. 49(4):920–931. [DOI] [PubMed] [Google Scholar]
- Casanova J-L, Abel L.. 2013. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet. 14(1):215–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celhar T, et al. 2016. TLR7 and TLR9 ligands regulate antigen presentation by macrophages. Int Immunol. 28(5):223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevret P, Dobigny G.. 2005. Systematics and evolution of the subfamily Gerbillinae (Mammalia, Rodentia, Muridae). Mol Phylogenet Evol. 35(3):674–688. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N.. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 1(2):581–585. [DOI] [PubMed] [Google Scholar]
- Chung LK, Bliska JB.. 2016. Yersinia versus host immunity: how a pathogen evades or triggers a protective response. Curr Opin Microbiol. 29:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobble KR, et al. 2016. Genetic variation at the MHC DRB1 locus is similar across Gunnison’s prairie dog (Cynomys gunnisoni) colonies regardless of plague history. Ecol Evol. 6(8):2624–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer JE, et al. 2010. Transcriptomic and innate immune responses to Yersinia pestis in the lymph node during bubonic plague. Infect Immun. 78(12):5086–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona E, Wang L, Ko D, Patel CJ.. 2018. Systematic detection of positive selection in the human-pathogen interactome and lasting effects on infectious disease susceptibility. PLoS One 13(5):e0196676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL.. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26(19):2455–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeure CE, et al. 2012. Early systemic bacterial dissemination and a rapid innate immune response characterize genetic resistance to plague of SEG mice. J Infect Dis. 205(1):134–143. [DOI] [PubMed] [Google Scholar]
- Dhariwala MO, Olson RM, Anderson DM.. 2017. Induction of type I interferon through a noncanonical Toll-like receptor 7 pathway during Yersinia pestis infection. Infect Immun. 85(11):e00570–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MD, et al. 2010. The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One 5(8):e12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgvin TO, et al. 2017. The genomic mosaicism of hybrid speciation. Sci Adv. 3(6):e1602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2014. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 29(1):51–63. [DOI] [PubMed] [Google Scholar]
- Fichet-Calvet E, Jomâa I, Ben Ismail R, Ashford RW.. 2003. Leishmania major infection in the fat sand rat Psammomys obesus in Tunisia: interaction of host and parasite populations. Ann Trop Med Parasitol. 97(6):593–603. [DOI] [PubMed] [Google Scholar]
- Foster SL, Medzhitov R.. 2009. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 130(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, et al. 2011. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 7(11):e1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage KL, Kosoy MY.. 2005. Natural history of plague: perspectives from more than a century of research. Annu Rev Entomol. 50(1):505–528. [DOI] [PubMed] [Google Scholar]
- Gnerre S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 108(4):1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RJ, Lane MC, Wagner NJ, Weening EH, Miller VL.. 2015. Dissemination of a highly virulent pathogen: tracking the early events that define infection. PLoS Pathog. 11(1):e1004587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregers TF, et al. 2003. MHC class II loading of high or low affinity peptides directed by Ii/peptide fusion constructs: implications for T cell activation. Int Immunol. 15(11):1291–1299. [DOI] [PubMed] [Google Scholar]
- Hargreaves AD, et al. 2017. Genome sequence of a diabetes-prone desert rodent reveals a mutation hotspot around the ParaHox gene cluster. Proc Natl Acad Sci U S A. 114(29):7677–7682. [DOI] [PMC free article] [PubMed]
- Hinnebusch BJ, Jarrett CO, Bland DM.. 2017. “Fleaing” the Plague: adaptations of Yersinia pestis to its insect vector that lead to transmission. Annu Rev Microbiol. 71(1):215–232. [DOI] [PubMed] [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12(1):491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F.. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8):754–755. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M.. 1990. Evolutionary relationships of class II major-histocompatibility-complex genes in mammals. Mol Biol Evol. 7(6):491–514. [DOI] [PubMed] [Google Scholar]
- Hurt P, et al. 2004. The genomic sequence and comparative analysis of the rat major histocompatibility complex. Genome Res. 14(4):631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KK, et al. 2018. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 54(Suppl 12):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, Lynch M.. 2003. The structure and early evolution of recently arisen gene duplicates in the Caenorhabditis elegans genome. Genetics 165(4):1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S.. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 11(5):373–384. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE.. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10(6):845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, et al. 2011. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 28(11):3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M.. 2013. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 4:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, et al. 2011. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol. 12(5):416–424. [DOI] [PubMed] [Google Scholar]
- Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M.. 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33(20):6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. 2018. Mesquite: a modular system for evolutionary analysis. Version 3.4. http://www.mesquiteproject.org.
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1):10–12. [Google Scholar]
- Mathur R, et al. 2012. A mouse model of Salmonella Typhi infection. Cell 151(3):590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzaraki V, Kumar V, Wijmenga C, Zhernakova A.. 2017. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 18(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows JRS, Lindblad-Toh K.. 2017. Dissecting evolution and disease using comparative vertebrate genomics. Nat Rev Genet. 18(10):624–636. [DOI] [PubMed] [Google Scholar]
- Monzón-Casanova E, et al. 2016. The forgotten: identification and functional characterization of MHC class II molecules H2-Eb2 and RT1-Db2. J Immunol. 196(3):988–999. [DOI] [PubMed] [Google Scholar]
- Murphy K, Weaver C.. 2016. Janeway’s immunobiology. 9th ed. New York (NY): Garland Science.
- Murrell B, et al. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, Jongsma MLM, Paul P, Bakke O.. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 11(12):823–836. [DOI] [PubMed] [Google Scholar]
- Nei M, Gu X, Sitnikova T.. 1997. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci U S A. 94(15):7799–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VK, Parra-Rojas C, Hernandez-Vargas EA.. 2018. The 2017 plague outbreak in Madagascar: data descriptions and epidemic modelling. Epidemics 25:20–25. [DOI] [PubMed] [Google Scholar]
- Nham T, Filali S, Danne C, Derbise A, Carniel E.. 2012. Imaging of bubonic plague dynamics by in vivo tracking of bioluminescent Yersinia pestis. PLoS One 7(4):e34714.CrossRef][10.1371/journal.pone.0034714] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak RM. 1999. Walker’s mammals of the world. Baltimore (MD): JHU Press. [Google Scholar]
- Ohtsuka M, Inoko H, Kulski JK, Yoshimura S.. 2008. Major histocompatibility complex (MHC) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genomics 9(1):178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg M, et al. 2012. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance–forming modification. Science 337(6098):1111–1115. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I.. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23(9):1061–1067. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T, Korf I.. 2009. Assessing the gene space in draft genomes. Nucleic Acids Res. 37(1):289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péléraux A, Karlsson L, Chambers J, Peterson PA.. 1996. Genomic organization of a mouse MHC class II region including the H2-M and Lmp2 loci. Immunogenetics 43(4):204–214. [DOI] [PubMed] [Google Scholar]
- Pujolar JM, Dalén L, Olsen RA, Hansen MM, Madsen J.. 2018. First de novo whole genome sequencing and assembly of the pink-footed goose. Genomics 110(2):75–79. [DOI] [PubMed] [Google Scholar]
- Rabiee MH, Mahmoudi A, Siahsarvie R, Kryštufek B, Mostafavi E.. 2018. Rodent-borne diseases and their public health importance in Iran. PLoS Negl Trop Dis. 12(4):e0006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CRH, Reynolds CM, Trent MS, Bishop RE.. 2007. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem. 76(1):295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassi Y, et al. 2008. Molecular detection of Leishmania major in the vectors and reservoir hosts of cutaneous leishmaniasis in Kalaleh District, Golestan Province, Iran. J Arthropod Borne Dis. 2(2):21–27. [Google Scholar]
- Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ.. 2004. Variation in lipid A structure in the pathogenic Yersiniae. Mol Microbiol. 52(5):1363–1373. [DOI] [PubMed] [Google Scholar]
- Roach JC, et al. 2005. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A. 102(27):9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikh KU, Kissner TL, Sultana A, Ruthel G, Ulrich RG.. 2004. Human monocytes infected with Yersinia pestis express cell surface TLR9 and differentiate into dendritic cells. J Immunol. 173(12):7426–7434. [DOI] [PubMed] [Google Scholar]
- Sakthianandeswaren A, Foote SJ, Handman E.. 2009. The role of host genetics in leishmaniasis. Trends Parasitol. 25(8):383–391. [DOI] [PubMed] [Google Scholar]
- Salazar Gonzalez RM, et al. 2014. Toxoplasma gondii-derived profilin triggers human Toll-like receptor 5-dependent cytokine production. J Innate Immun. 6(5):685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samia NI, et al. 2011. Dynamics of the plague–wildlife–human system in Central Asia are controlled by two epidemiological thresholds. Proc Natl Acad Sci U S A. 108(35):14527–14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ.. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci U S A. 103(14):5526–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon JG, Bosio CF, Hinnebusch BJ.. 2015. Dermal neutrophil, macrophage and dendritic cell responses to Yersinia pestis transmitted by fleas. PLoS Pathog. 11(3):e1004734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon JG, et al. 2013. Yersinia pestis subverts the dermal neutrophil response in a mouse model of bubonic plague. mBio. 4(5):e00170–13–e00170–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Sironi M, Cagliani R, Forni D, Clerici M.. 2015. Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat Rev Genet. 16(4):224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, et al. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 32(5):1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solbakken MH, et al. 2016. Evolutionary redesign of the Atlantic cod (Gadus morhua) Toll-like receptor repertoire by gene losses and expansions. Sci Rep. 6(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690. [DOI] [PubMed] [Google Scholar]
- Steimle A, Autenrieth IB, Frick J-S.. 2016. Structure and function: lipid A modifications in commensals and pathogens. Int J Med Microbiol. 306(5):290–301. [DOI] [PubMed] [Google Scholar]
- Stenseth NC, et al. 2008. Plague: past, present, and future. PLoS Med. 5(1):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J, Kjer K, Page R, Sullivan J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56(4):564–577. [DOI] [PubMed] [Google Scholar]
- Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW.. 2008. Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genomics 9(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaere C, et al. 2008. CCR5 polymorphism and plague resistance in natural populations of the black rat in Madagascar. Infect Genet Evol. 8(6):891–897. [DOI] [PubMed] [Google Scholar]
- Tollenaere C, et al. 2012. Contrasted patterns of selection on MHC-linked microsatellites in natural populations of the malagasy plague reservoir. PLoS One 7(3):e32814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaere C, et al. 2013. Beyond an AFLP genome scan towards the identification of immune genes involved in plague resistance in Rattus rattus from Madagascar. Mol Ecol. 22(2):354–367. [DOI] [PubMed] [Google Scholar]
- Tørresen OK, et al. 2018. Genomic architecture of haddock (Melanogrammus aeglefinus) shows expansions of innate immune genes and short tandem repeats. BMC Genomics 19(1):240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadharajan S, et al. 2018. The grayling genome reveals selection on gene expression regulation after whole genome duplication. Genome Biol Evol. 10(10):2785–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viļuma A, et al. 2017. Genomic structure of the horse major histocompatibility complex class II region resolved using PacBio long-read sequencing technology. Sci Rep. 7(1):45518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitti JJ, Grossman SR, Sabeti PC.. 2013. Detecting natural selection in genomic data. Annu Rev Genet. 47(1):97–120. [DOI] [PubMed] [Google Scholar]
- Vladimer GI, et al. 2012. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ.. 2009. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S, et al. 2018. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 83(17):8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Liu Y.. 2014. Protein secretion systems in bacterial pathogens. Front Biol. 9(6):437–447. [Google Scholar]
- Yang H, et al. 2017. Host transcriptomic responses to pneumonic plague reveal that Yersinia pestis inhibits both the initial adaptive and innate immune responses in mice. Int J Med Microbiol. 307(1):64–74. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. 2012. Dynamics of Yersinia pestis and its antibody response in great gerbils (Rhombomys opimus) by subcutaneous infection. PLoS One 7(10):e46820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. 2015. Transmission efficiency of the plague pathogen (Y. pestis) by the flea, Xenopsylla skrjabini, to mice and great gerbils. Parasites Vectors 8(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhong W, Fan N.. 2003. Rodent problems and management in the grasslands of China In: Singleton GR, Hinds LA, Krebs CJ, Spratt DM, editors. Rats, mice and people: rodent biology and management. Monograph 96, Canberra, Australia: Australian Centre for International Agricultural Research (ACIAR). p. 316–319. Available from: www.aciar.gov.au. [Google Scholar]
- Zorio DAR, et al. 2019. De novo sequencing and initial annotation of the Mongolian gerbil (Meriones unguiculatus) genome. Genomics 111(3):441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.