

Abstract
CD19-targeted chimeric antigen receptor T-cell (CAR-T) therapy is effective in refractory/relapsed (R/R) B-cell acute lymphoblastic leukemia (B-ALL). This review focuses on achievements, current obstacles, and future directions in CAR-T research. A high complete remission rate of 68% to 93% could be achieved after anti-CD19 CAR-T treatment for B-ALL. Cytokine release syndrome and CAR-T-related neurotoxicity could be managed. In view of difficulties collecting autologous lymphocytes, universal CAR-T is a direction to explore. Regarding the high relapse rate after anti-CD19 CAR-T therapy, the main solutions have been developing new targets including CD22 CAR-T, or CD19/CD22 dual CAR-T. Additionally, some studies showed that bridging into transplant post-CAR-T could improve leukemia-free survival. Some patients who did not respond to CAR-T therapy were found to have an abnormal conformation of the CD19 exon or trogocytosis. Anti-CD19 CAR-T therapy for R/R B-ALL is effective. From individual to universal CAR-T, from one target to multi-targets, CAR-T-cell has a chance to be off the shelf in the future.
Keywords: Chimeric antigen receptor T-cell, B-cell acute lymphoblastic leukemia, Complete remission, Cytokine release syndrome, Relapse, Transplantation
Introduction
Chimeric antigen receptor T (CAR-T)-cell therapy is a promising immunotherapy that has been a landmark innovation in the treatment of malignant tumors. CAR-T cells are produced by transducing a genetically engineered CAR fusion protein into T cells by means of a retrovirus or lentivirus. Currently approved CAR-T cells utilize CAR constructs consisting of a single-chain variable fragment (scFV) antigen-recognition domain, a CD3-derived T-cell activation domain, and a costimulatory domain (CD28 or 4-1BB). The CAR-T cells are then infused into patients, typically following a lymphodepleting conditioning regimen, such as fludarabine and cyclophosphamide.[1,2]
For the costimulatory domain, 4-1BB CAR appears to favor persistence and memory T cell formation, while CD28 CAR presents with more potent cytotoxic activity and early tumor eradication.[3] The CD28 costimulatory component has been used by the National Cancer Institute (Rockville, MD, USA),[4] Memorial Sloan Kettering Cancer Center (MSKCC; New York, NY, USA),[5] and Baylor College of Medicine (Houston, Texas, USA),[6] while the 4-1BB costimulatory component has been incorporated by the University of Pennsylvania (Philadelphia, PA, USA)[7] and the Fred Hutchinson Cancer Research Center (Seattle, WA, USA).[8]
However, CAR-T is not effective for all kinds of cancers. At present, the efficacy of CAR-T treatment is greatest for hematological malignancies.[1] CD19-targeted CAR-T therapy has demonstrated high efficacy in refractory/relapsed (R/R) B-cell acute lymphoblastic leukemia (B-ALL). As a result, those patients who had only a 20% to 40% complete remission (CR) rate from traditional chemotherapy could achieve as high as 68% to 93% CR rate through CAR-T treatment.[1–12] CD19 CAR-T therapy has also been shown to be effective in treating B-cell lymphoma, achieving a CR rate of 53% to 67%.[13–15] In addition, anti-BCMA CAR-T therapy could achieve a CR rate of 60% to 90% for multiple myeloma.[16,17] Multiple targets in development for directed CAR-T therapy in acute myeloid leukemia (AML) include CD33, CD123, FRβ, CLL1 or CLEC12A, FLT3, B7H6, NKG2D, and Lewis Y.[18–24] While CD123-targeted CAR-T therapy showed a high CR rate of 86% in blastic plasmacytoid dendritic cell neoplasm (BPDCN), a relatively low CR rate of 26% was observed in AML because CD123 expression on BPDCN cells was significantly higher than that on AML blasts.[25]
In this review paper, we focus on CAR-T-cell therapy for B-ALL. Although a CR rate as high as 68% to 93% was achieved in B-ALL after CAR-T treatment, many problems persist. First, there is lack of large sample analysis on the CAR-T prognosis of each high-risk sub-group, including B-ALL patients with MLL-AF4 fusion gene (+), TP53 gene mutation (+), and extramedullary disease (EMD), especially in patients with central nervous system leukemia (CNSL). Second, although the CR rate of CD19 CAR-T is high, the side effects can be fatal. Management of CAR-T-related adverse side effects such as cytokine release syndrome (CRS) and neurotoxicity is critical to clinicians. Third, it is difficult to collect effective T cells from patients with high tumor burden. Furthermore, it is difficult to carry out quality inspection of product preparation due to individual differences. Fourth, although CAR-T therapy is effective, relapse remains a significant problem. In recent years, there have been some studies on how to prolong leukemia-free survival (LFS) after CAR-T therapy, how to select new targets after relapse, and whether bridging into allogeneic hematopoietic stem cell transplantation (Allo-HSCT) is necessary after CAR-T. Finally, there are still 10% to 30% of B-ALL patients who do not respond to CAR-T therapy and the potential etiology and risk factors remain unclear. This article will elaborate on the above issues.
Efficacy Analysis on CD19-targeted CAR-T Treatment of R/R B-ALL
R/R B-ALL is associated with extremely poor prognosis and remains a leading cause of death for pediatric and young adult leukemia patients.[10–12] The development of CD19-targeted CAR-T cell therapy has been a milestone for these patients. Since 2011, several CD19-targeted CAR-T clinical trials have demonstrated excellent therapeutic efficacy for R/R B-ALL, with a CR rate between 68% and 93%.[4–9,26–29] Furthermore, a relatively high minimal residual disease (MRD)-negative CR ranging from 75% to 93% was also achieved.[5,30] Until now, there have been no published studies including large samples of high-risk B-ALL sub-groups, such as patients with the MLL-AF4 fusion gene, BCR-ABL fusion gene, TP53 gene mutations, or R/R B- ALL with the EMD.[31–33]
From April 2017 to March 2019, 254 patients with R/R B-ALL received CD19-targeted CAR-T therapy at our single-center from five clinical trials (https://clinicaltrials.gov, NCT03173417; NCT02546739; NCT03671460; www.chictr.org.cn, ChiCTR-ONC-17012829, and ChiCTR1800016541). On day 30 post-CAR-T infusion, 90.6% (230/254) patients achieved CR, and 89.4% (227/254) had MRD-negative CR. We focused on the analysis of patients with several independent adverse prognostic factors according to the National Comprehensive Cancer Network guidelines.[34] The results are shown in Table 1. For the patients with the high-risk fusion gene of MLL-AF4, the CR rate after CD19 CAR-T therapy was 80%, which was inferior to those with other or no fusion genes (P = 0.041). There were no differences between patients with or without other fusion genes, including BCR-ABL (+) vs. BCR-ABL (−) groups, E2A-PBX1 (+) vs. E2A-PBX1 (−) groups, E2A-HLF (+) vs. E2A-HLF (−) groups. For the patients with gene mutations, a lower CR rate after CD19 CAR-T therapy was observed in patients with TP53 mutation compared to those with other or no mutations (72.73% vs. 92.11% vs. 94.39%, P = 0.004). Patients with bone marrow (BM) blasts >20% had lower odds of achieving CR compared to those with BM blasts ≤20%. The CR rate of patients who received prior treatment with either CAR-T or blinatumomab was lower than those who had not (50.00% vs. 91.53%, P = 0.01). There were no differences between groups with or without complex chromosomes, with or without EMD, or with or without prior HSCT history. There were no differences in CR between patients aging 1 to 14 years and >14 years.
Table 1.
Univariate analysis on CR rates post CAR-T therapy of patients’ characteristics.
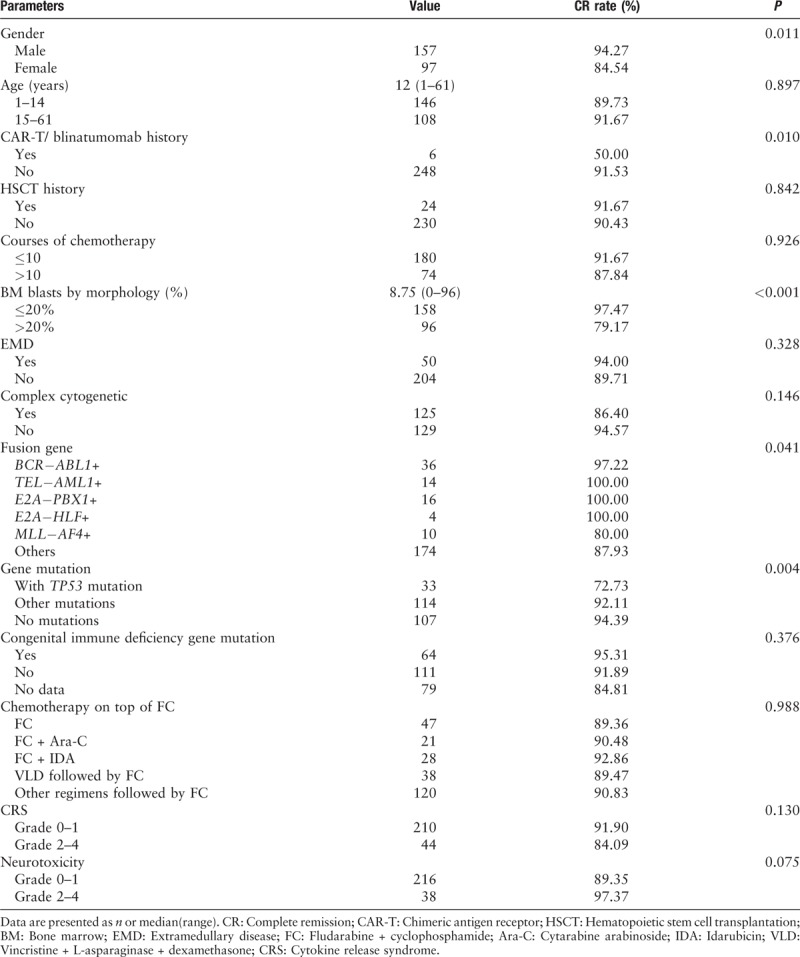
For the patients relapsed post-Allo-HSCT, the CR rate was as high as those without prior Allo-HSCT before CAR-T. Davila et al[35] found there was no clinical evidence of graft-versus-host disease (GVHD) after CAR-T therapy, despite the fact that the infused 19-28Z CAR-T cells were of donor origin. In our 110 published cases, two of 15 patients developed acute GVHD (grade I and grade III), and two patients developed extensive chronic GVHD, having been treated mostly with 4-1BB CAR-T cells.[36]
Diagnostic Criteria and Management of the Main Side Effects of CD19-targeted CAR-T: CRS and Neurotoxicity
There are two main severe adverse events (SAE) after CAR-T cell therapy. One is CRS, and the other is CAR-T related neurotoxicity. CRS has been reported in 18% to 100% of patients, with severe CRS, noted in 27% to 53% of patients; encephalopathy in 25% to 47% of patients.[3,37] Interleukin (IL)-6 and IL-1 increased significantly after CAR-T cells infusion, Human monocytes are the major source of IL-1 and IL-6 during CRS. Therefore, CRS could theoretically be prevented by monocyte depletion or by blocking the IL-6 receptor with tocilizumab (Initial U.S. Approval: 2010). Nonetheless, tocilizumab failed to protect mice from delayed lethal neurotoxicity. Rather, blocking IL-1 abolished both CRS and neurotoxicity. The IL-1 receptor antagonist anakinra has been shown to protect against severe CRS and reduce the severity of CAR-T cell-related neurotoxicity.[38,39] In addition, products with CD28 costimulatory domains are typically associated with a greater incidence of severe neurotoxicity relative to CRS, while the opposite for those using 4-1BB.[2] In our published data, we found that BM blasts ≥5% and CAR-T with CD28 costimulatory domain were significantly associated with a higher incidence of grade III/IV CRS and more severe neurotoxicity.[36]
CRS can involve many organs and can be graded into 0 to 5 grades.[40–42] The CRS grading system was originally determined by Lee et al[40] and common terminology criteria for adverse events,[41] but the system was complicated for clinicians, so current CRS grading systems follow the diagnostic criteria put forth by the American Society of Transplantation & Cellular Therapy (ASTCT), which are simplified and use high fever, hypotension, hypoxia as bases for classification.[42] Generally, patients with grade I-II-CRS only need to be treated symptomatically, such as with antipyretics, oxygen inhalation, and liquid supplements. CRS of grade 3 or higher is considered to be severe. The Food and Drug Administration (FDA) has approved tocilizumab for the treatment of CAR-T-cell-induced CRS. Glucocorticoid is promptly administered if the patient does not have a rapid response to the interleukin-6 receptor blockade.[28,40–42]
CAR-T related neurotoxicity, also called “CAR T cell-related encephalopathy syndrome,” is another important SAE.[42–44] In 2018, CAR-T related neurotoxicity was defined as immune effector cell-associated neurotoxicity syndrome (ICANS) by ASTCT.[42] A phase II clinical trial by Juno Therapeutics had been put on hold due to unexpected lethal cerebral edema.[45] Researchers have identified anti-CD19 CAR-T cells in the cerebrospinal fluid (CSF), confirming the ability of these cells to cross the blood-brain barrier.[44,46] Due to the possible lethality of CAR-T related neurotoxicity, clinicians have been fearful about safety especially for patients with CNSL. Maude et al[7] suggested that the presence of CNS-3 disease should constitute an exclusion criterion for CAR-T. Subsequently; however, several articles have shown that CNSL does not increase the risk of CAR-T related neurotoxicity. It is usually fully reversible and not related to the spread of cancer to the CNS.[30,46] However, it is recommended that the lumbar puncture and intrathecal chemotherapy should be injected as fast as possible before the infusion of CAR-T to reduce the tumor load in the CSF.[7]
ICANS is divided into five grades.[42–44] In phase II and single-cohort clinical trial of Novartis, 75 patients received an infusion of tisagenlecleucel (Kymriah), and neurologic events occurred in 30 patients (40%). The most common neurologic events were encephalopathy, confusional state, delirium, tremor, agitation, and somnolence; one patient had seizure.[47] Of the 254 patients in our clinical trials, 19 patients (7.5%) suffered from ≥grade 3 ICANS, whose main manifestations were convulsions and loss of consciousness. Novartis study showed severe neurologic events occurred more frequently in patients with higher-grade CRS.[47] The study from MSKCC found that a higher disease burden (≥5% BM blasts or EMD) was associated with a higher risk of severe CRS and neurotoxic effects, and the disease burden and peak CAR-T-cell expansion were independent predictors of severe neurotoxic effects.[9] Similarly, in our cohort of 254 patients, the occurrence and intensity of CRS and ICANS were highly consistent, and the peak value of CAR-T in vivo was closely related to CRS and ICANS. Among them, 12/149 (8.0%) patients were children aged ≤14 years and 7/105 (6.6%) of adults. No patient died of ICANS. The management of ICANS follows the ASTCT guidelines in our center.[36,42] If loss of consciousness or convulsions occur, glucocorticoids are administered immediately accompanied by sedative and maintained for several days until convulsions stop for more than 24 to 48 h and consciousness recovers, after which glucocorticoid and sedative can be gradually tapered.
The dosage of infused anti-CD19 CAR-T cells has varied in prior B-ALL CAR-T trials.[3,37] The majority exceeded 1 × 106/kg.[9,28] The study reported by Turtle et al was designed to evaluate the safety of 3 dose levels (DLs) (2 × 105/kg; 2 × 106/kg; and 2 × 107/kg, respectively) of CAR-T cells administered. The first two patients treated at dose level 3 (DL3) developed severe toxicities, including one death. DL3 was deemed too toxic, and no further patients were treated at 2 × 107/kg.[8] We have previously demonstrated that a low median dose of 3 × 105/kg of CAR-T cells could achieve high CR rates with much less toxicity, even in patients with high-risk features.[36,48,49]
In addition to the above two main side effects, B cell aplasia (BCA) after CAR-T and immunoglobulin deficiency are the main causes of post-CAR-T infection. Intravenous immunoglobulin replacement therapy is useful for treatment. Furthermore, B-cell aplasia rapidly reverses after CAR-T cells disappear.[28]
Solving the Problems in Standardized Preparation and Manufacture of CAR-T Cells
Although autologous CAR-T-cell therapy has immense therapeutic potential, many problems in autologous T-cell therapy make it difficult for broader applications. If the patient's tumor burden is high, or if they have undergone strong chemotherapy before CAR-T infusion, they will have a significant reduction of normal T lymphocytes or poor quality of collected cells. Thus, high-quality CAR-T cells cannot be well-prepared for some patients. Therefore, allogeneic CAR-T cells are needed. Our clinical practice has found that if an human leukocyte antigen (HLA)-matched sibling donor is used, the short-term efficacy of CAR-T-cells can be maintained. However, if an HLA haploidentical matched or HLA unmatched donor is used, with the basic fludarabine + cyclophosphamide preconditioning regimen, the donor's CAR-T cells will be quickly rejected by the immune cells in the patient's body and cannot remove the tumor cells successfully. Besides the impact of CAR-T cell origin (ie, autologous or allogeneic), many other factors also influence CAR-T treatment outcomes, such as individual differences in the number and quality of cells collected, the infection of pathogenic microorganisms. As a result, it is difficult to prepare CAR-T cells or evaluate the activity of CAR-T cells in a standardized way, rendering it even less likely to be similar to a drug manufactured off-the-shelf.
The development of “universal” CAR-T (UCAR) cells is the hope to produce an off-the-shelf product derived from allogeneic healthy donor T cells. The greatest barrier to implementation of this approach is the prevention of GVHD or host vs. graft disease, and thus innovative gene-editing technologies are being investigated.[50] Some UCAR productions have been successful in a few children with pre-B-ALL with a high degree of immunosuppression.[51] Novel gene-editing technologies like zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regularly interspaced short palindromic repeat (CRISPR-Cas9) allow facile editing of specific genes within the genome, by knocking out the endogenous T cell receptor (TCR) or HLA β chain to avoid allograft rejection.[50] The ideal universal CAR-T product should have the following characteristics: (i) a lack of naïve TCRs, to avoid GVHD; (ii) matched or absent HLA to avoid rejection; (iii) natural killer (NK) inhibitory strategies (non-classical HLA or siglec-7/-9 ligands); and (iv), a significant amount of naïve and stem cell memory T cells to ensure adequate T cell expansion and persistence.[52] While ZFN, TALEN, and CRISPR technologies are used, the CRISPR/Cas9 system is one of the most promising ways to develop off-the-shelf CAR-T products and to advance T cell immunotherapy because of the high specificity of this technology and its relative ease and limited cost.[50–55]
This system demonstrated high fidelity in the disruption of up to four genes T cell receptor (TCR), human leukocyte antigen (HLA), programmed death-ligand 1 (PD-1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) to generate universal CAR-T cells resistant to two inhibitory pathways.[50,52,56] Investigators from the Cellectis company reported a TCR-negative CART19 product (UCART19) wherein the TALEN technology was used to disrupt TCRα and CD52 genes.[57] The CRISPR/Cas9 technology together with an adeno-associated virus vector repair matrix was recently employed to directly insert the CAR encoding DNA into the TCR alpha chain locus, simultaneously generating a TCR-negative CAR-positive T cell. These cells were shown to be more potent than conventional lentivirally transduced CAR-T cells because of a more physiological TCR-like regulation of CAR expression.[58,59] Activation of NK cells through “missing self” recognition would be circumvented by enforcing the expression of non-classical HLA molecules such as HLA-E and HLA-G, which can protect universal CAR-T from NK-cell-mediated lysis.[60,61]
Solving Relapse After CD19-targeted CAR-T Therapy
Despite the early high CR rate after CD19-targeted CAR-T therapy, relapse occurred in a large subset of patients. The 53 cases study from MSKCC showed that the 1-year event-free survival (EFS) was 50%.[9] In a study of 75 patients treated with tisagenlecleucel from Novartis, the EFS was 73% at 6 months and 50% at 12 months.[47]
Many researchers showed that CAR-T-cell therapy had favorable long-term remission rates in a population of patients with a low disease burden, who had significantly longer EFS with a lower incidence of toxic effects than did those with a high disease burden.[9,30] Rebecca A's team[30] found that subsequent remission durability was correlated with increased frequencies of TNF-α-secreting CAR CD8+ T cells, and was dependent on a sufficiently high CD19+ antigen load at the time of infusion to trigger CAR-T cell proliferation. CD19 antigen burden was the only independent predictive factor, and more than 15% of CD19 positive blast cells before CAR-T cell infusion was associated with longer BCA after CAR-T therapy. BCA can be used as a measure of the persistence of functional CD19-targeted T cells. Continued BCA was seen in all patients who had a sustained remission, and none of the patients with BCA had a CD19-positive relapse.[7,30]
About half of B-ALL relapse following CD19 CAR-T therapy was CD19-positive relapse, and the other half was CD19-negative relapse.[9,29,36]
CD19-positive relapses appear to occur exclusively in patients who do not have CAR-T persistence due to a premature CAR-T loss. Immune-mediated rejection of CAR-T has been demonstrated as a mechanism for limited CAR-T persistence in some patients who experience a relapse.[8,62] The inclusion of mouse sequences can trigger rejection of the CAR-T cells by the host immune system, and many studies suggest that lack of immunogenicity, and hence persistence of CAR-T cells, is associated with improved relapse-free survival after CAR-T. Thus, CAR designs that are composed of fully human sequences have become preferred.[28] To treat CD19-positive relapse, it is possible to re-infuse anti-CD19 CAR-T cells with a humanized sequence of scFV. Zhao et al[63] showed five humanized selective (hs) CAR-T cells were infused into five patients who had relapsed after receiving murine CAR-T treatment. Subsequent hsCAR-T treatments proved effective in all five patients and achieved complete molecular remission in four.
However, CD19-negative relapse is a difficult problem. Deep sequencing has identified that malignant CD19-negative clones were actually present in peripheral blood and marrow as early as day 23 after CAR-T infusion, a time when the patient was initially felt to not have the residual disease.[29,64] Fry et al[65] found that the majority of patients who develop CD19 immune escape retained CD22 surface expression and demonstrated that CD22 remained susceptible to the CAR-T target. A recent phase I clinical trial for CD22-directed CAR-T therapy reported that 17 of 21 patients (81%) had previously received CD19 CAR-T therapy, and ultimately 12/21 (57%) achieved CR. At the first dose level (3 × 105 CD22-CAR-T cells/kg), one of six patients attained CR as compared to 11 of 15 patients (73%) at the dose of ≥1 × 106 cells/kg (P < 0.001).
Although sequential immunotherapeutic targeting of a second antigen resulted in clinical benefit, a high relapse rate was observed associated with diminished CD22 cell-surface expression.[64,65] This phenomenon suggests that simultaneous multi-specific targeting may be a more effective approach to enhance the durability of CAR-T induced remission in B-ALL.[65] Bispecific CAR-T constructs, such as CD19 and CD22, may prevent tumor escape by combining two distinct tumor antigens.[62,65] It has also been shown that bispecific CAR-T cells targeting both CD19 and CD22 can recognize and kill CD19+CD22+, CD19−CD22+, and CD19+CD22− B-ALL, pointing towards a strategy able to overcome anti-CD19 CAR-T cell limitations.[66,67] How to design dual-targeted CAR-T cells is an important consideration. This can be accomplished by one of four different approaches: (a) Generate two or more cell populations expressing different CARs and infuse them together or sequentially (co-administration); (b) Use a bicistronic vector that encodes two different CARs on the same cell; (c) Simultaneously engineer T cells with two different CAR constructs (co-transduction), which will generate three CAR-T subsets consisting of dual and single CAR-expressing cells; or (d) Encode two CARs on the same chimeric protein using a single vector (ie, bi-specific or tandem CARs). These different approaches are highlighted in a recent review article by Majzner and Mackall.[64,68] The methods of mixed input of two kinds of CAR-T cells and continuous CD22 CAR-T cell infusion of CD19 CAR-T cells infusion have been questioned because of the high cost of manufacture.
Another important way to reduce relapse is bridging to Allo-HSCT post-CAR-T. However, it is controversial whether bridging into Allo-HSCT is necessary after CD19-CAR-T. At present, some have shown that proceeding to Allo-HSCT after CD19-CAR-T could improve LFS.[62,69] Davila et al[5] showed that 19-28ζ CAR-T cell expansion in vivo peaked within 12 weeks and persisted for 2 to 3 months post-infusion in most patients, supplying a window of time following transplant; hence researchers defined the 19 to 28ζ CAR-T cell therapy as a “bridge” to transplant. Turtle showed that patients bridging into allo-transplant after CAR-T had a much lower relapse rate of 15.4% (2/13) compared to a non-transplant group of 43.8% (7/16).[8] A meta-analysis of the patients who did and those who did not proceed to transplant following CAR-T infusion in published CD19 CAR-T clinical trials showed that, for the surviving patients who achieved CR from CAR-T therapy and did not proceed to transplant, 54 of 128 patients (42%) eventually relapsed, compared to 8/52 (15%) who relapsed among patients who underwent transplant.[4,5,7,8,47,62] Our previous results from CD19 CAR-T clinical trial spanning 2015 to 2016 (Clinical Trials#: ChiCTR-IIh-16008711) demonstrated that 2/27 of patients bridging to HSCT had a relapse after Allo-HCT, and 9/18 patients who did not receive Allo-HCT had a relapse.[48,49] Results from our CD19 CAR-T clinical trial spanning 2016 to 2018 (www.clinicaltrials.gov, NCT03173417) showed that only 10/75 (13.3%) patients in the transplant group relapsed while 13/27 (48.2%) patients receiving CAR-T alone relapsed. The median time from CAR-T infusion to relapse was 3 months, so the recommended time from CAR-T infusion to transplant was 2 months.[36]
Whether an MRD-negative CR could be achieved before Allo-HSCT was a critical factor affecting LFS after transplantation. Patients who achieved an MRD-negative CR before HSCT experienced fewer relapses compared with patients who were in MRD-positive CR.[70,71] The experience of our center has suggested that transplantation should be carried out if MRD-negative CR is achieved after CAR-T treatment; otherwise, there would be a high recurrence rate after transplantation.[36,48] To predict recurrence, the ratio of CAR-T and BCA in peripheral blood can also be monitored. If the ratio of CAR-T cells in total T cells in peripheral blood is less than 1%, or if the CD19-positive B cells in peripheral blood gradually increase, the effect of CAR-T is weakened and relapse may be imminent, so transplantation should be carried out quickly.[7,30] In addition, for patients with high-risk predictors such as TP53 gene mutation, MLL-AF4, and E2A-HLF fusion genes, early bridging to transplantation is recommended.
However, Maude et al[7] reported prolonged persistence of CTL019 cells and BCA for as long as 2 years, implying that CTL019 cells could be a potential treatment alternative for patients who are ineligible for stem-cell transplantation. Park et al[9] found no difference in LFS and OS between patients who underwent allo-HSCT after CAR-T and those who did not. In their study, of the 16 patients who underwent transplantation, six subsequently relapsed, and another six died from transplant-related mortality. Many researchers are investigating how to maintain a longer LFS through CAR-T therapy alone. Whether alternative antigen targets or the combination of CAR-T with checkpoint inhibition, such as PD-1 or CTLA-4 will further enhance CAR-T efficacy and long-term outcomes has yet to be examined and studies are currently ongoing.[72,73]
Reasons for Non-response (NR) to CD19 CAR-T Therapy Among R/R B-ALL Patients
CD19 CAR-T therapy has demonstrated a high CR rate for patients with R/R B-ALL, yet still 10% to 30% of patients have no response and the potential etiology and risk factors remain unclear. In the 254 cases from our center, we found that there were no obvious predictable causes for NR except for in high-risk groups with the MLL-AF4 fusion gene, TP53 mutation, or BM blasts >20%.
Some studies have explored the causes of NR to CD19 CAR-T therapy. A study demonstrated that some of the CD19 isoforms that are pre-existing at diagnosis contribute to CART-19 escape. Spliced CD19 mRNA isoforms affecting exon 2 were expressed in six adult patients with CD19+ B-ALL or even expressed an isoform lacking the CD19 transmembrane and cytosolic domains.[74] when exposed to CART-19, only the full-length CD19 cultures were killed, whereas CD19 Δex2-transduced cells remained fully viable, confirming the loss of the cognate CART-19 epitope.[75] Another remarkable study found that CARs provoked reversible antigen loss through trogocytosis, an active process in which the target antigen was transferred to T cells, thereby decreasing target density on tumor cells and abating T cell activity by promoting fratricide T cell killing and T cell exhaustion.[76]
To solve NR and early relapse after CD19 CAR-T therapy, Cummins et al[25] analyzed some possible reasons and solutions. (1) Failure of the persistence of the CAR-T population, possibly due to cellular immunity against components of the CAR. The solution is to change the CAR-T origin, that is, from murine origin to human origin.[63] (2) B-cell interaction with CAR-T population. A potential solution is combined with small molecule inhibitors of B-cells, such as Ibrutinib in murine models improved response rates to CD19 CAR-T in mantle cell lymphoma and enhanced CD19 CAR-T engraftment and persistence.[77,78] (3) Antigen loss/emergence of antigen-negative leukemic stem cell. Giving a second CAR-T targeting an alternative antigen, for example, CD22 and/or CD20[225] may solve the problem. (4) T-cell suppression/anergy. Checkpoint blockade such as the PD-L1 inhibitor can be added to CAR-T cells. The PD-L1 expression level on tumor cells has been linked to a favorable outcome of patients treated with checkpoint inhibitors.[79,80] Burga et al[72] reported on the combination of CAR-T and anti-PD-L1 antibodies and supported the potential clinical merit of neutralizing myeloid-derived suppressor cells to allow for optimal antitumor efficacy. This approach is being tested in mouse models before phase I clinical trials, so more studies are required to further optimize the dose and timing of combination CAR-T-cells with PD-1 blockade.
Conclusions
In summary, CD19-targeted CAR-T therapy is very effective in R/R B-ALL. The CR rate could be as high as 68% to 93%, and the majority of CR patients achieved MRD-negative CR. The prognosis of some high-risk sub-groups has only been studied in small populations. Our analysis of 254 cases from our single-center showed that CR rates in TP53 mutation and MLL-AF4 groups are slightly lower. Clinicians’ management of CRS and CAR-T-related neurotoxicity, in reference to ASTCT guidelines, has become better. In view of various obstacles in collecting patients’ autologous lymphocytes, universal CAR-T is one of the future directions to explore. At present, the CRISPR/cas9 system is the most effective way to knock out TCR or HLA, but this approach will take some time to be translated from the laboratory to the clinic. To combat the high relapse rate after CAR-T therapy, the main solutions are to develop new targets, such as CD22 CAR-T, CD19/CD22 dual CAR-T, and others. In addition, some studies demonstrated that bridging into transplant after CAR-T therapy could improve LFS and was recommended for high-risk patients. Finally, for the 10% to 30% of patients with no response post-CAR-T, some of the reasons found include the abnormal conformation of CD19 exon or CD19 antigen trogocytosis from B blast cells to CAR-T cell.
Conflicts of interest
None.
Footnotes
How to cite this article: Zhang X, Li JJ, Lu PH. Advances in the development of chimeric antigen receptor-T-cell therapy in B-cell acute lymphoblastic leukemia. Chin Med J 2020;133:474–482. doi: 10.1097/CM9.0000000000000638
References
- 1.Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature 2017; 545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sermer D, Brentjens R. CAR T-cell therapy: full speed ahead. Hematol Oncol 2019; 37: Suppl 1: 95–100. doi: 10.1002/hon.2591. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Guo Y, Han W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8:896–925. doi: 10.1007/s13238-017-0400-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25.doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 2011; 121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+composition in adult B cell ALL patients. J Clin Invest 2016; 126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 2018; 378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 2013; 14:e205–e217. doi: 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed] [Google Scholar]
- 11.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood 2013; 121:1077–1082. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun W, Malvar J, Sposto R, Verma A, Wilkes JJ, Dennis R, et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia 2018; 32:2316–2325. doi: 10.1038/s41375-018-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson CA, Farooq U, Ghobadi A. Axicabtagene ciloleucel, an anti-CD19 chimeric antigen receptor T-cell therapy for relapsed or refractory large B-cell lymphoma: practical implications for the community oncologist. Oncologist 2019; pii: theoncologist.2019-0395. doi: 10.1634/theoncologist.2019-0395 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Z, Li L, Wang W, OuYang BS, Cheng S, Wang L, et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell Non-Hodgkin's lymphoma. Clin Cancer Res 2019; 25:6995–7003. doi: 10.1158/1078-0432.CCR-19-0101. [DOI] [PubMed] [Google Scholar]
- 15.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015; 33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016; 128:1688–1700. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kriegsmann K, Kriegsmann M, Cremer M, Schmitt M, Dreger P, Goldschmidt H, et al. Cell-based immunotherapy approaches for multiple myeloma. Br J Cancer 2019; 120:38–44. doi: 10.1038/s41416-018-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Przespolewski A, Szeles A, Wang ES. Advances in immunotherapy for acute myeloid leukemia. Future Oncol 2018; 14:963–978. doi: 10.2217/fon-2017-0459. [DOI] [PubMed] [Google Scholar]
- 19.Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 2015; 125:3466–3476. doi: 10.1182/blood-2014-11-612721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu H, Zhou Q, Deshmukh V, Phull H, Ma J, Tardif V, et al. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for acute myeloid. Leukemia immunotherapy. Angew Chem Int Ed Engl 2014; 53:9841–9845. doi: 10.1002/anie.201405353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saad SK, Marco R, Olga S, Michael K, Miriam YK, Craig S, et al. Leukemia stem cells are characterized by CLEC12A expression and chemotherapy. Refractoriness that can be overcome by targeting with chimeric antigen receptor T cells. Blood 2016; 128:766.doi: 10.1182/blood.V128.22.766.766. [Google Scholar]
- 22.Christopher DC, Christopher TS, Kazusa I, Sang MN, Feng S, Sarah KT, et al. Preclinical development of FLT3-redirected chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia. Blood 2016; 128:1072.doi: 10.1182/blood.V128.22.1072.1072. [Google Scholar]
- 23. Ploch P, Khan S, Bhatti A, Krebs L, Theobald M, Hartwig UF. Exploring chimeric natural killer receptor NKP30 expressing human T lymphocytes for adoptive immunotherapy to acute leukemia. Presented at: 21st Congress of the European Hematology Association. Copenhagen, Denmark, 9-12 June 2016. [Google Scholar]
- 24.Goltz D, Gevensleben H, Grunen S, Dietrich J, Kristiansen G, Landsberg J, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia 2017; 31:738–743. doi: 10.1038/leu.2016.328. [DOI] [PubMed] [Google Scholar]
- 25.Cummins KD, Gill S. Anti-CD123 chimeric antigen receptor T-cells (CART): an evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk Lymphoma 2018; 59:1539–1553. doi: 10.1080/10428194.2017.1375107. [DOI] [PubMed] [Google Scholar]
- 26.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells expressing chimeric receptors establish memory and potent antitumor effects in patients with advanced leukemia. Sci Transl Med 2011; 3:95ra73.doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med 2018; 379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017; 129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stock W. Adolescents and young adults with acute lymphoblastic leukaemia. Hematol Am Soc Hematol Educ Prog 2010; 2010:21–29. doi: 10.1182/asheducation-2010.1.21. [DOI] [PubMed] [Google Scholar]
- 32.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia 2017; 31:705–711. doi: 10.1038/leu.2016.263. [DOI] [PubMed] [Google Scholar]
- 33.Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, et al. TP53 mutations occur in 15. 7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood 2014; 124:251–258. doi: 10.1182/blood-2014-02-558833. [DOI] [PubMed] [Google Scholar]
- 34. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Acute Lymphoblastic leukemia Version 1.2015 NCCN.org. Available from: https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/ped_all.pdf [access date: June 1, 2018] [Google Scholar]
- 35.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224–225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Lu XA, Yang JF, Lv FY, Xiong M, Zhang JP, et al. Efficacy and safety of CD19 chimeric antigen receptor (CAR) T cell therapy for B-cell acute lymphocytic leukemia (B-cell ALL) in a large cohort including patients with extramedullary disease (EMD), high leukemia burden, BCR-ABL (+) mutation, TP53 mutation, and post-transplant relapse. Blood 2018; 132:280.doi: 10.1182/blood-2018-99-115642. [Google Scholar]
- 37.Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol 2016; 13:25–40. doi: 10.1038/nrclinonc.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018; 24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 39.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 2018; 24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 Published: November 27, 2017. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES. National Institutes of Health. National Cancer Institute. Available from: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf [access date: May 1, 2018] [Google Scholar]
- 42.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019; 25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 2017; 7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mei H, Jiang H, Wu Y, Guo T, Xia L, Jin R, et al. Neurological toxicities and coagulation disorders in the cytokine release syndrome during CAR-T therapy. Br J Haematol 2018; 181:689–692. doi: 10.1111/bjh.14680. [DOI] [PubMed] [Google Scholar]
- 45.Hawkes N. Trial of novel leukaemia drug is stopped for second time after two more deaths. BMJ 2016; 355:i6376.doi: 10.1136/bmj.i6376. [DOI] [PubMed] [Google Scholar]
- 46.Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen YB, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N Engl J Med 2017; 377:783–784. doi: 10.1056/NEJMc1704610. [DOI] [PubMed] [Google Scholar]
- 47.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018; 378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan J, Yang JF, Deng BP, Zhao XJ, Zhang X, Lin YH, et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 2017; 31:2587–2593. doi: 10.1038/leu.2017.145. [DOI] [PubMed] [Google Scholar]
- 49.Biping D, Alex HC, Junfang Y, Jing P, Xian Z, Yuehui L, et al. Safety and efficacy of low dose CD19 targeted chimeric antigen receptor T (CAR-T) cell immunotherapy in 47 cases with relapsed refractory B-cell acute lymphoblastic leukemia (B-ALL). Blood 2016; 128:649.doi: 10.1182/blood.V128.22.649.649. [Google Scholar]
- 50.Singh N, Shi J, June CH, Ruella M. Genome-editing technologies in adoptive T cell immunotherapy for cancer. Curr Hematol Malig Rep 2017; 12:522–529. doi: 10.1007/s11899-017-0417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN geneedited CAR T cells. Sci Transl Med 2017; 9:aam9292.doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 52.Ruella M, Kenderian SS. Next generation chimeric antigen receptor T cell therapy: going off the shelf. BioDrugs 2017; 31:473–481. doi: 10.1007/s40259-017-0247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013; 154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017; 8:17002–17011. doi: 10.18632/oncotarget.15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res 2015; 75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 58.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017; 543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hale M, Lee B, Honaker Y, Leung WH, Grier AE, Jacobs HM, et al. Homology-directed recombination for enhanced engineering of chimeric antigen receptor T cells. Mol Ther Methods Clin Dev 2017; 4:192–203. doi: 10.1016/j.omtm.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013; 122:1341–1349. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 2017; 35:765–772. doi: 10.1038/nbt.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taraseviciute A, Broglie L, Phelan R, Bhatt NS, Becktell K, Burke MJ. What is the role of hematopoietic cell transplantation (HCT) for pediatric acute lymphoblastic leukemia (ALL) in the age of chimeric antigen receptor T-cell (CART) therapy? J Pediatr Hematol Oncol 2019; 41:337–344. doi: 10.1097/MPH.0000000000001479. [DOI] [PubMed] [Google Scholar]
- 63.Zhao Y, Liu Z, Wang X, Wu H, Zhang J, Yang J, et al. Treatment with humanized selective CD19CAR-T Cells shows efficacy in highly treated B-ALL patients who have relapsed after receiving murine-based CD19CAR-T therapies. Clin Cancer Res 2019; 25:5595–5607. doi: 10.1158/1078-0432.CCR-19-0916. [DOI] [PubMed] [Google Scholar]
- 64.Shah NN, Maatman T, Hari P, Johnson B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front Oncol 2019; 9:146.doi: 10.3389/fonc.2019.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018; 24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 2019; 20:E1283.doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nat Biomed Eng 2018; 2:377–391. doi: 10.1038/s41551-018-0235-9. [DOI] [PubMed] [Google Scholar]
- 68.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov 2018; 8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 69.Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019; 133:1652–1663. doi: 10.1182/blood-2018-11-883710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eckert C, Henze G, Seeger K, Hagedorn N, Mann G, Panzer-Grümayer R, et al. Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 2013; 31:2736–2742. doi: 10.1200/JCO.2012.48.5680. [DOI] [PubMed] [Google Scholar]
- 71.Coustan-Smith E, Gajjar A, Hijiya N, Razzouk BI, Ribeiro RC, Rivera GK, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia after first relapse. Leukemia 2004; 18:499–504. doi: 10.1038/sj.leu.2403283. [DOI] [PubMed] [Google Scholar]
- 72.Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother 2015; 64:817–829. doi: 10.1007/s00262-015-1692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu J, Zhang Q, Tian K, Wang H, Yin H, Zheng J. Current status and future prospects of the strategy of combining CAR-T with PD-1 blockade for antitumor therapy (Review). Mol Med Rep 2018; 17:2083–2088. doi: 10.3892/mmr.2017.8129. [DOI] [PubMed] [Google Scholar]
- 74.Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother 2017; 40:187–195. doi: 10.1097/CJI.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 2015; 5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019; 568:112–116. doi: 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res 2016; 22:2684–2696. doi: 10.1158/1078-0432.CCR-15-1527. [DOI] [PubMed] [Google Scholar]
- 78.Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016; 127:1117–1127. doi: 10.1182/blood-2015-11-679134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bauer J, Nelde A, Bilich T, Walz JS. Antigen targets for the development of immunotherapies in leukemia. Int J Mol Sci 2019; 20:E1397.doi: 10.3390/ijms20061397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ciombor KK, Goldberg RM. Hypermutated tumors and immune checkpoint inhibition. Drugs 2018; 78:155–162. doi: 10.1007/s40265-018-0863-0. [DOI] [PMC free article] [PubMed] [Google Scholar]