

Abstract
Neutrophils can release extracellular traps (NETs) in infectious, inflammatory, and thrombotic diseases. NETs have been detected in deep vein thrombosis, atherothrombosis, stroke, disseminated intravascular coagulation, and trauma. We have previously shown hemodynamic forces trigger rapid NETosis within sterile occlusive thrombi in vitro. Here, we tested the effects of thrombin, fibrin, and fibrinolysis on shear-induced NETosis by imaging NETs with Sytox-green during microfluidic perfusion of Factor XIIa-inhibited or thrombin-inhibited human whole blood over fibrillar collagen (± tissue factor). For perfusions under venous pressure drops (19 mmHg/mm-clot), thrombin generation did not alter the near-zero level of NET generation. In contrast, production of thrombin/fibrin led to a 2-fold reduction in neutrophil accumulation and a 6-fold reduction in NET generation after 30 min of arterial perfusion (163 mmHg/mm-clot). Exogenously added tissue type plasminogen activator (tPA) drove robust fibrinolysis, however tPA did not trigger NETosis under venous flow. In contrast, tPA did enhance NET generation in clots subjected to arterial pressure drops. After 45 min of arterial perfusion, clots treated with 30 nM tPA had a 3-fold increase in total NET production and a 2-fold increase in normalized NET generation (measured as DNA:Neutrophil) compared to fibrin-rich clots. Blocking fibrin polymerization resulted in similar level of NET release seen in tPA-treated clots, whereas εACA abolished the NET-enhancing effect of tPA. Therefore, fibrin suppresses NET generation and the absence of fibrin promotes NETs. We demonstrated that fibrin was strongly inversely correlated with shear-induced NETosis in sterile occlusive clots.
Keywords: Extracellular Traps, hemodynamics, fibrin, neutrophil, platelet
Introduction
Neutrophils are known to release extracellular traps (NETs) made of DNA and protein components to trap and kill bacteria (1–4). Since the initial discovery of NETosis, a variety of other inducers have been identified (5–9). For example, phorbol 12-myristate 13-acetate (PMA), lipopolysaccharides (LPS), and calcium ionophore can all induce NET release through different mechanisms (10–14). While originally thought to be beneficial, NETs can be pathological, capable of causing direct injuries to tissue and vessels. NETosis has been implicated in the pathogenesis of various diseases including infectious diseases, thrombosis, atherosclerosis, and autoimmune diseases (15). NETs have been found in sepsis, venous and arterial thrombosis, disseminated intravascular coagulation (DIC), and trauma-induced coagulopathy (TIC) (5,14,16–22). In these diseases, NETs can provide physical scaffolds for clot growth by catching platelets, erythrocytes, fibrin, and von Willebrand factor (23,24). Individual NET components may also contribute to thrombus formation through their interaction with the coagulation pathway. DNA and histone have been reported to activate factor XII and amplify tissue factor (TF)-dependent thrombin generation (25). Neutrophil elastase and myeloperoxidase can inactivate tissue factor pathway inhibitor and thrombomodulin. However, intact NETs may behave differently from DNA or purified histone proteins (26).
NETs and their individual components also modulate clot lysis. In a plasma environment, DNA and histones increase fibrin fiber diameter, stability, rigidity and permeability (27,28). Histones have been shown to delay clot lysis, and the combination with DNA further prolongs lysis time (27). Histones also inhibit thrombin inactivation by antithrombin, and DNA reduces plasminogen activation on plasma clots (28). NETs can also inhibit fibrinolysis by promoting tPA inactivation by PAI-1, and potentiate fibrinolysis by stimulating fibrin-independent plasminogen activation (29). DNase accelerated tPA-induced lysis in clots obtained from acute ischemic stroke and acute coronary syndrome patients (20,30).
Our group previously showed that large intrathrombus hemodynamic forces driven by transthrombus pressure drop (ΔP/L > 70 mm Hg/mm-clot) trigger rapid NET release within sterile occlusive clots (31). Shear-induced NETs are DNase I sensitive and stain positive for common NET markers such as myeloperoxidase and citrullinated histones. Shear induced NETosis (SIN) can occur with or without thrombin/fibrin. How and whether fibrin generation or fibrinolysis influence NET production still remains unclear.
To investigate the effect of fibrin polymerization/dissolution on NETosis, NET production was measured in clots with varying amount of fibrin. Whole blood was perfused over prothrombotic surfaces in microfluidic channels to deposit platelet/fibrin mass. The degree of fibrin accumulation was controlled through changing the dosage of tPA, εACA or Gly-Pro-Arg-Pro (GPRP). At occlusion, clots can be viewed as a porous media, through which the transport of soluble and insoluble blood constituents is driven by the prevailing pressure drop (ΔP/L). Blood cells can become physically trapped and migrate locally within clots. Large pressure gradients can result in high interstitial fluid shear stress and high membrane stress (31), which then lead to SIN. In this study, we found that the extent of shear-induced NET release is inversely correlated with the amount of fibrin in occlusive clots in vitro. Furthermore, a fibrin network reduces NETs within thrombi at high ΔP/L. Conversely, fibrinolysis or GPRP enhances SIN when ΔP/L is high.
Materials and Methods
Reagents
Reagents were obtained as follows: Alexa Fluor (AF) 647 conjugated anti-human CD41 (Bio-Rad, Raleigh, NC, USA), Anti-human CD18 antibody (clone TS1/18), PE anti-human CD11a (BioLegend, San Diego, CA, USA), AF647 conjugated human fibrinogen (ThermoFisher Scientific, Grand Island, NY, USA), Sytox-green (Life Technologies, Grand Island, NY, USA), fibrillar collagen (type I, Chrono-log, PA, USA), Dade Innovin lipidated tissue factor (TF, Siemens, Malvern, PA, USA), D-Phe-Pro-Arg-CMK (PPACK, Santa Cruz Biotechnology, Dallas, TX, USA), corn trypsin inhibitor (CTI, Haematologic Technologies, Essex Junction, VT, USA), tPA (Abcam, Cambridge, MA, USA), H-Gly-Pro-Arg-Pro-OH acetate salt (GPRP, Bachem Americas, Torrance, CA, USA), ε-aminocaproic acid (εACA), ethylenediaminetetraacetic acid (EDTA), and Sigmacote (Sigma, St. Louis, MO, USA).
Blood collection and Preparation
Whole blood (WB) was collected in 40 μg/mL corn trypsin inhibitor (high CTI or HCTI), 4 μg/mL CTI (low CTI or LCTI), 100 μM D-Phe-Pro-Arg-CMK (PPACK), or 50 mM ethylenediaminetetraacetic acid (EDTA) from healthy donors who self-reported to be free of alcohol and medication for at least 48 hr prior to phlebotomy. All donors consented under IRB approval (Univ. Penn.) Platelets were labeled with anti-human CD41 antibody, neutrophils were labeled with anti-human CD11a antibody, and DNA was labeled with Sytox-green (or Hoechst 33342 when indicated). AF647 conjugated fibrinogen was added to blood to monitor thrombin/fibrin generation.
Microfluidic assay
For coating collagen or collagen/lipidated tissue factor (TF), a single 1000-μm wide channel polydimethylsiloxane (PDMS) patterning device was vacuum-sealed to a Sigmacote-treated glass slide, as previously described (32,33). A total of 5 μL collagen solution (or collagen followed by 5 μL infusion of 20 nM TF) was perfused through the channel to create a prothrombotic surface trigger on the glass. The patterning device was then replaced by an 8-channel PDMS device (Figure S1). The 8 parallel channels were positioned perpendicular to the patterned collagen to form 8 uniformly distributed prothrombotic patches (250 μm wide × 1000 μm long). Thrombi were formed under pressure-relief (P relief) mode at a constant withdrawal flowrate of 2 μL/min or 20 μL/min per pair of channels, respectively. EDTA was added to blood to abolish clotting in four alternating channels. As thrombi grew in the other four matched channels perfused with FXIIa-inhibited WB (with CTI) or thrombin-inhibited WB (with PPACK), flow was diverted to the EDTA-treated WB channels. At channel occlusion, pressure drops across the clots have been measured to be 19 mm Hg/mm for venous perfusion and 163 mm Hg/mm for arterial perfusion.
A fixed device outlet flowrate sets a well-defined initial venous or arterial wall shear rate. As a clot continues to grow and occupy the luminal space of a microfluidic channel, the flow-facing side of the thrombus experiences increased fluid shear rate and shear stress. Upon occlusion, the pressure drop regulated by the paired flowing EDTA-blood lane drives a Darcy’s flow (permeation) of solutes and blood cells through the porous clot. The rate of this interstitial flow (or the superficial velocity) is influenced by the clot permeability, which can vary significantly depending on the clot composition. The transthrombus pressure drop also generates intrathrombus hemodynamic shear stresses on the physically entrapped neutrophils and dictates the extent of shear-induced NETosis.
Imaging
Platelets, fibrin, neutrophils, and extracellular DNA were detected by an epifluorescence microscopy (IX81, Olympus America Inc., Center Valley, PA, USA) and a CCD camera (Hamamatsu, Bridgewater, NJ, USA). 4X objective/0.16NA was used in the experiments. ImageJ (NIH) was used to analyze acquired images. The mean fluorescent intensity was measured over the entire collagen patch region. This signal represents total NETs detected per collagen patch-driven thrombotic occlusion.
Results
Reduced NET release in the presence of thrombin/fibrin generation
Whole blood (WB) treated with 100 μM PPACK was perfused over collagen surfaces to deposit platelets. Minimal fibrin(ogen) was detected as the concentration of PPACK used was enough to inhibit the majority of the thrombin generated (Figure 1A). We also perfused CTI-treated (40 μg/ml) WB over collagen+TF surfaces to enable a full thrombotic response involving platelet accumulation, thrombin generation, and fibrin deposition. After 30 min perfusion under arterial pressure drops, fewer neutrophils accumulated when thrombin/fibrin generation was present (Figure 1B). The difference was non-significant under venous conditions. Minimal levels of extracellular DNA signal were detected during venous perfusion (± thrombin/fibrin) and no significant differences were seen between the near-zero DNA content of thrombin-poor and thrombin-rich clots (Figure 1C, D). In contrast, the presence of thrombin/fibrin generation resulted in a 6-fold reduction to the NET production under arterial pressure drops. Total NET generation was normalized to the number of neutrophils within clots (DNA:Neutrophil) to measure the extent of NETosis on a per neutrophil basis. The results showed that essentially no neutrophils formed NETs under venous flow, thus the presence/absence of thrombin did not make a difference (Figure 1D). In contrast, substantial amount of NETs were observed at arterial pressure drop and thrombin production significantly reduced normalized NET generation. Therefore, thrombin/fibrin generation reduced the extent of NETosis during arterial clot formation, as measured by total DNA or by total DNA/neutrophils.
Figure 1. Thrombin and fibrin generation suppressed NET release after 30 min of arterial perfusion.
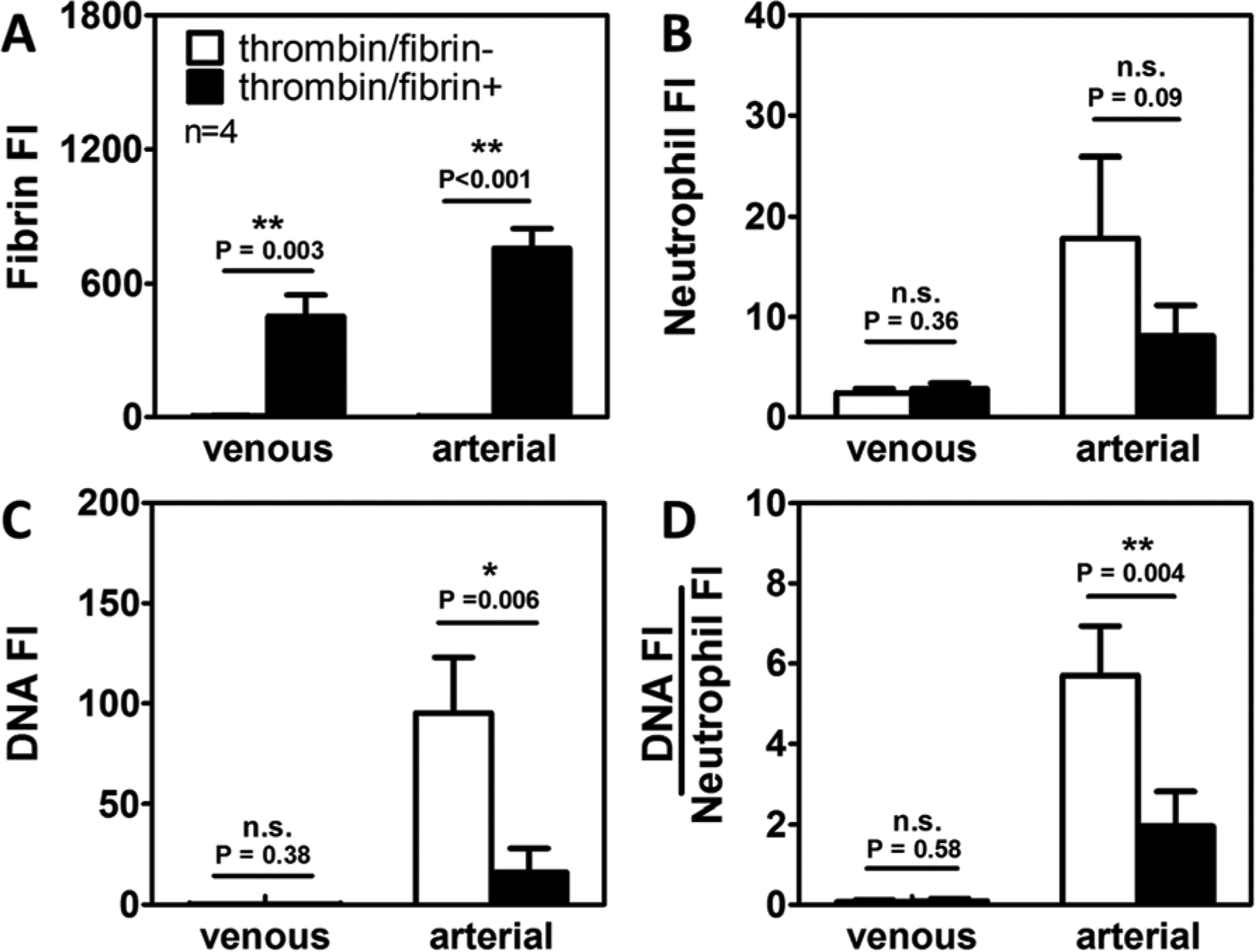
PPACK WB was perfused over collagen surfaces (⬜, thrombin/fibrin-) and CTI WB was perfused over collagen+TF surfaces (⬛, thrombin/fibrin+) under both venous and arterial pressure drops. Fibrin polymerization occurred (A) in thrombin-rich clots but not in thrombin-inhibited clots. Neutrophil accumulation (B), NET generation (C), and DNA:Neutrophil ratio (D) were significantly reduced in the presence of thrombin under arterial conditions. No significant differences were observed under venous conditions. (Venous: ΔP/L=19 mm Hg/mm-clot; arterial: ΔP/L = 163 mmHg/mm-clot; n.s.; not significant; *: p<0.05; **: p<0.005).
Enhanced NET generation during fibrinolysis
At high ΔP/L, thrombin and/or fibrin inhibition could have contributed to the reduction in NET production (Figure 1C–D). To help explore the role of fibrin, whole blood was treated with different concentrations of exogenous tPA and perfused over collagen+TF surfaces under arterial pressure drops. After 15 min (Figure 2A) of clot growth, tPA resulted in a dose-dependent lysis of fibrin. Whereas thrombi composition was unaltered in the presence of low dose tPA (3 nM), higher doses almost completely dissolved the fibrin network within 30 min (Figure 2B). With or without added tPA, platelets (Figure 2C) rapidly accumulated and occluded channels. In the presence of medium doses of tPA (15 nM), enhanced fibrinolysis led to slower net fibrin deposition initially. The rate of fibrinolysis exceeded fibrin polymerization rate after 18 min and clots started experiencing net fibrin loss (Figure 2D). At high doses of tPA (30 nM), fibrin deposition rate was similar to what we observed at 15 nM tPA early on, and fibrin content reached a lower peak value at an earlier time point (9min). Most importantly, the medium and high doses of tPA both caused a 2.5 fold increase in NET production (Figure 2B, E).
Figure 2. Tissue type plasminogen activator dose dependently lysed fibrin clots and promoted NETosis.
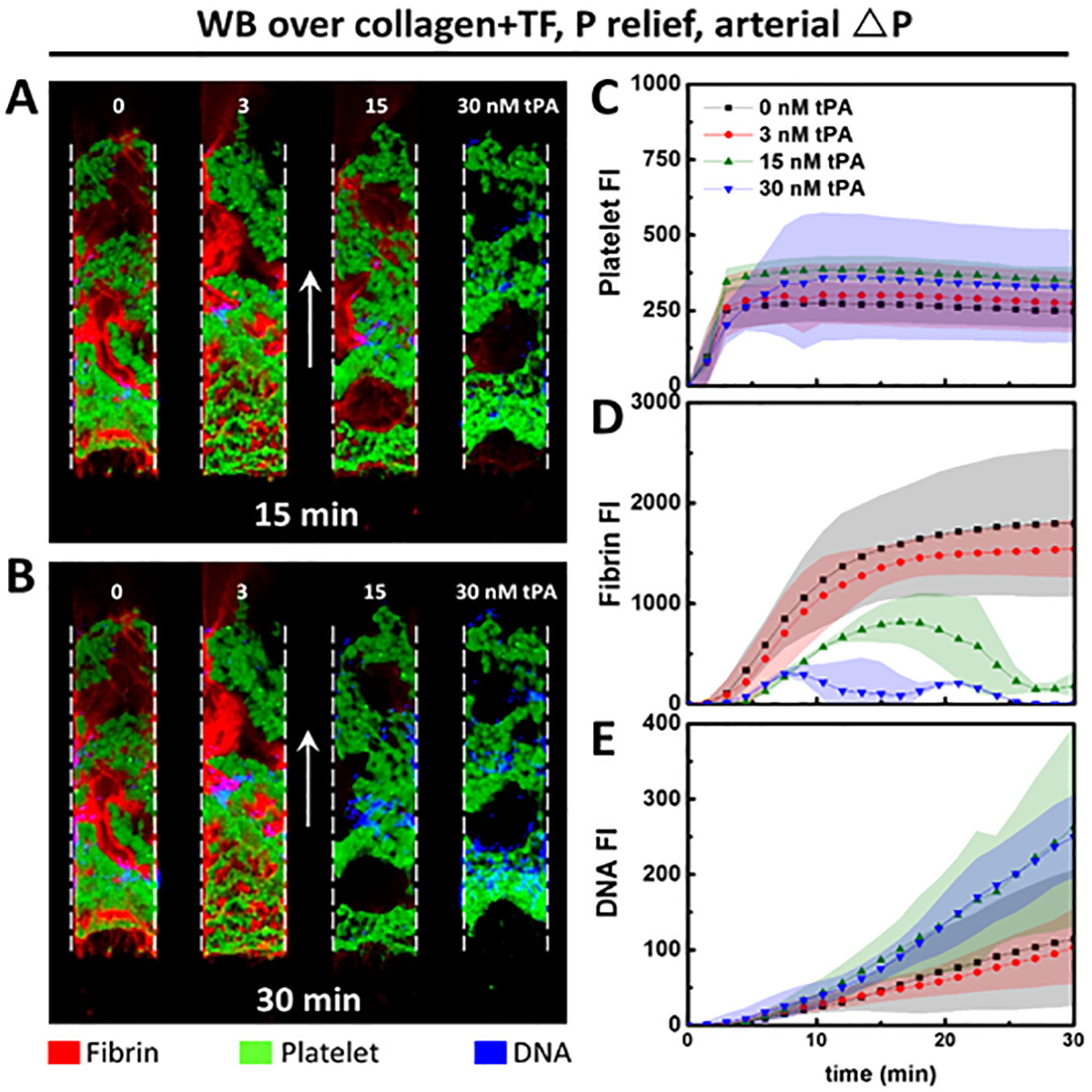
CTI treated whole blood dosed with different concentrations of tPA were perfused over collagen+TF surfaces under constant arterial pressure drops to form clots with varying degree of fibrin. Dose dependent increase in fibrin degradation was evident after 15 min of perfusion (A). At 30 min (B), both 15 nM and 30 nM tPA almost completely removed fibrin network. Platelet deposition (C) was similar among all clots regardless of the different fibrin growth dynamics (D). Medium and high doses of tPA that were sufficient for fibrin degradation also resulted in enhanced NET generation. (Shaded area: SD; arrows: flow direction).
Tissue plasminogen activator does not directly promote NET formation under venous shear
Because tPA has been previously reported to cause neutrophil degranulation (34), we also evaluated its potential to directly induce NETosis. Clots were formed in the presence/absence of 30 nM tPA under venous shear. Low concentrations of CTI (4 μg/ml) was used to reduce any non-specific effects of CTI on tPA (35). Early on (<7.5 min), clots formed from blood with/without added tPA were indistinguishable in terms of fibrin accumulation (Figure 3A). Fibrin content then started to decrease in tPA-treated clots, but continued to increase in the absence of added tPA. Neutrophil accumulation reached the maximum approximately at 7.5 min and stayed relatively constant for the remaining time of the experiments (Figure 3B). With or without tPA, only near-zero baseline levels of extracellular DNA signal (Figure 3C) and DNA:Neutrophil ratio (Figure 3D) were detected at low ΔP/L. This demonstrated that tPA and/or plasmin, on their own, do not trigger NETosis. Rather, the NET-promoting effect of tPA and fibrinolysis was shear-dependent.
Figure 3. Tissue type plasminogen activator is not a direct NET inducer under venous flow.
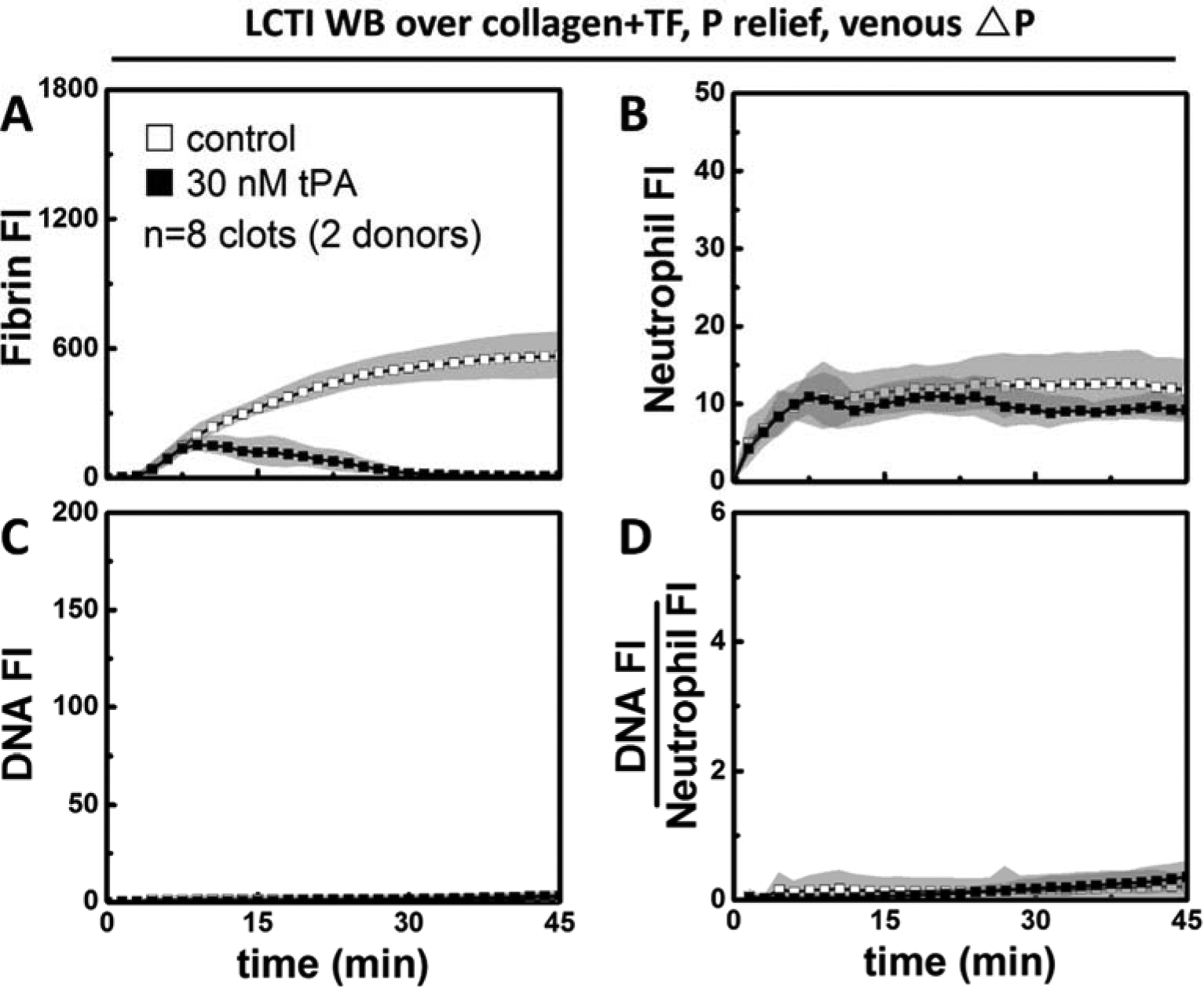
In the presence of low level CTI, 30 nM tPA was still enough to completely remove fibrin (A) after 45 min of perfusion under venous conditions. The presence of tPA did not alter the trend in neutrophil accumulation (B), NET release (C), or DNA:Neutrophil ratio (D).
More neutrophils undergo shear-induced NETosis upon fibrinolysis at high ΔP/L
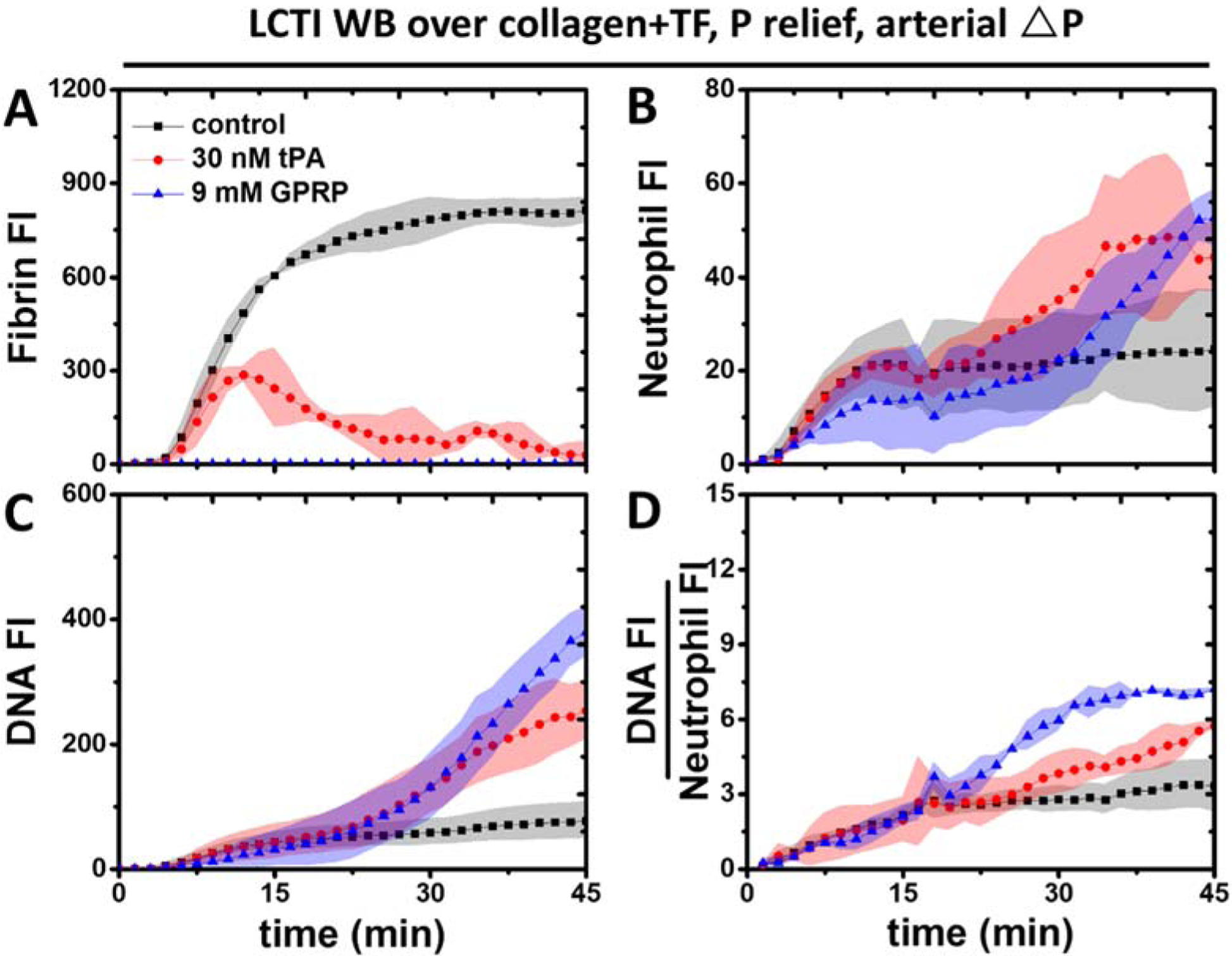
During arterial perfusion, fibrin accumulation (Figure 4A) continued to rise in the tPA-free channels. Similar to the venous condition, treatment with 30 nM tPA was enough to cause net fibrin loss in the presence of 4 μg/ml CTI. The number of neutrophils reached the peak around 15 min within fibrin-rich clots (no tPA added). In contrast, neutrophil deposition kept increasing in tPA-treated clots likely due to the increased porosity with ongoing fibrinolysis (Figure 4B). Extracellular DNA production continued to grow both in the presence and absence of added tPA (Figure 4C), but the slope was steeper when fibrinolysis occurred. Whereas NET generation started to slow down in fibrin-rich clots, it actually accelerated in tPA-treated clots at later time points. DNA:Neutrophil ratios were similar between the two conditions early on but diverged after 15 min of arterial perfusion (Figure 4D). In the absence of added tPA, the ratio started to plateau as both neutrophil accumulation and DNA release began to slow down. In tPA-treated clots, NET generation was faster than neutrophil accumulation as the percentage of neutrophils undergoing NETosis continued to rise.
Figure 4. Tissue type plasminogen activator promoted NETs per neutrophil at arterial pressure drop.
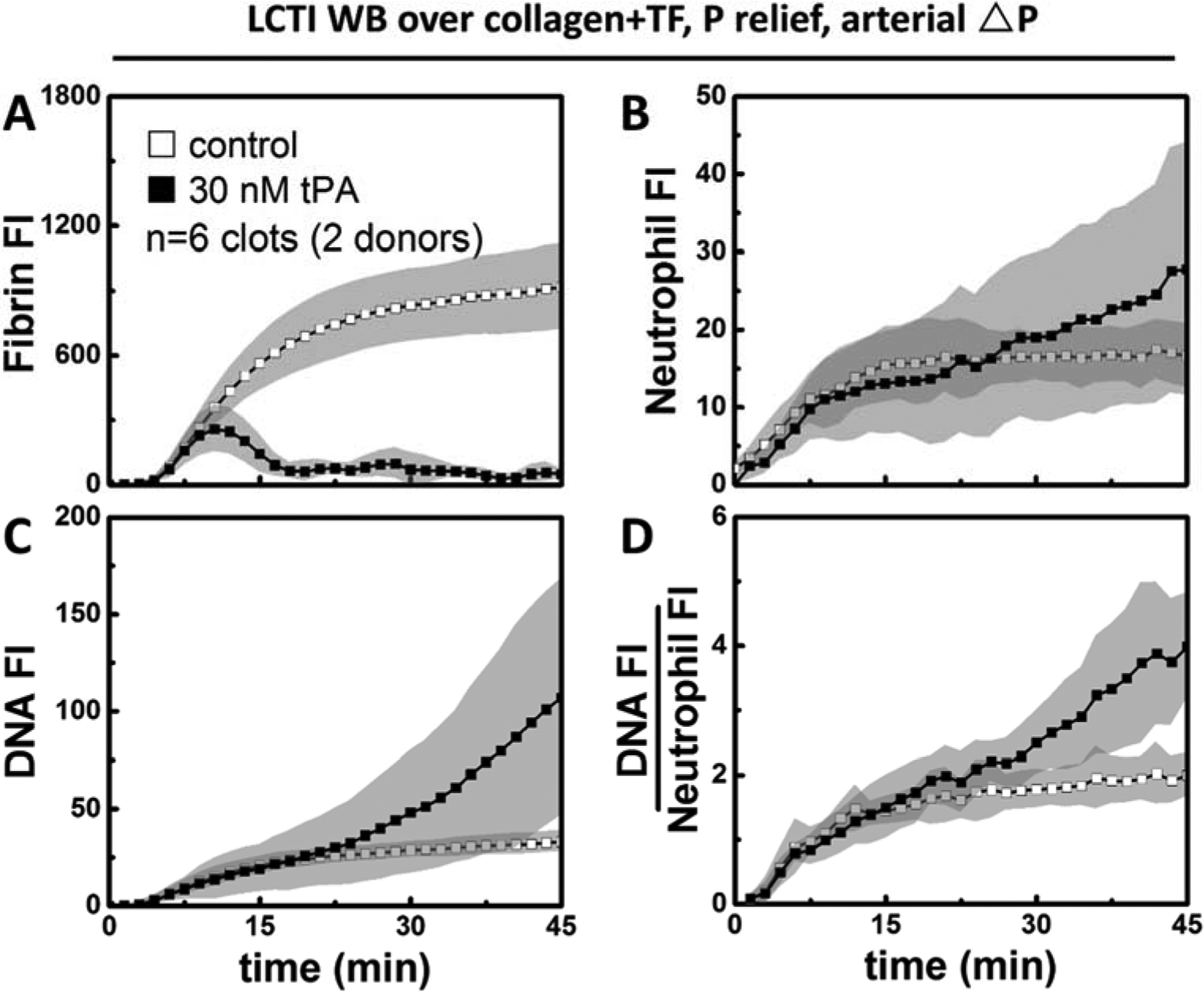
In the presence of low level CTI, 30 nM tPA was sufficient for complete fibrin degradation (A) under arterial pressure drops as well. Neutrophil deposition (B) slowed down in untreated clots, but continued to rise in tPA-treated clots. NET generation (C) was significantly faster in tPA-treated clots at later time points. After normalizing total NET generation to neutrophil accumulation, the NET generation per neutrophil (D) was still greater with tPA present.
In both venous and arterial perfusions, 30 nM tPA was sufficient to dissolve fibrin (Figure 5A) and the differences in neutrophil accumulation were non-significant between the control and tPA conditions at the end time point (45 min). In contrast to clots formed under venous shear, which contained similar levels of NETs (Figure 5C, E) with/without tPA, clots formed under arterial shear had a 3-fold increase in total NET generation when tPA was present (Figure 5C, F). Under arterial pressure drops, tPA also caused a 2-fold increase in DNA:Neutrophil ratio (Figure 5D, E). The difference was non-significant under venous shear (Figure 5D, F).
Figure 5. The effects of tPA on clot composition mimicked the effects of PPACK.
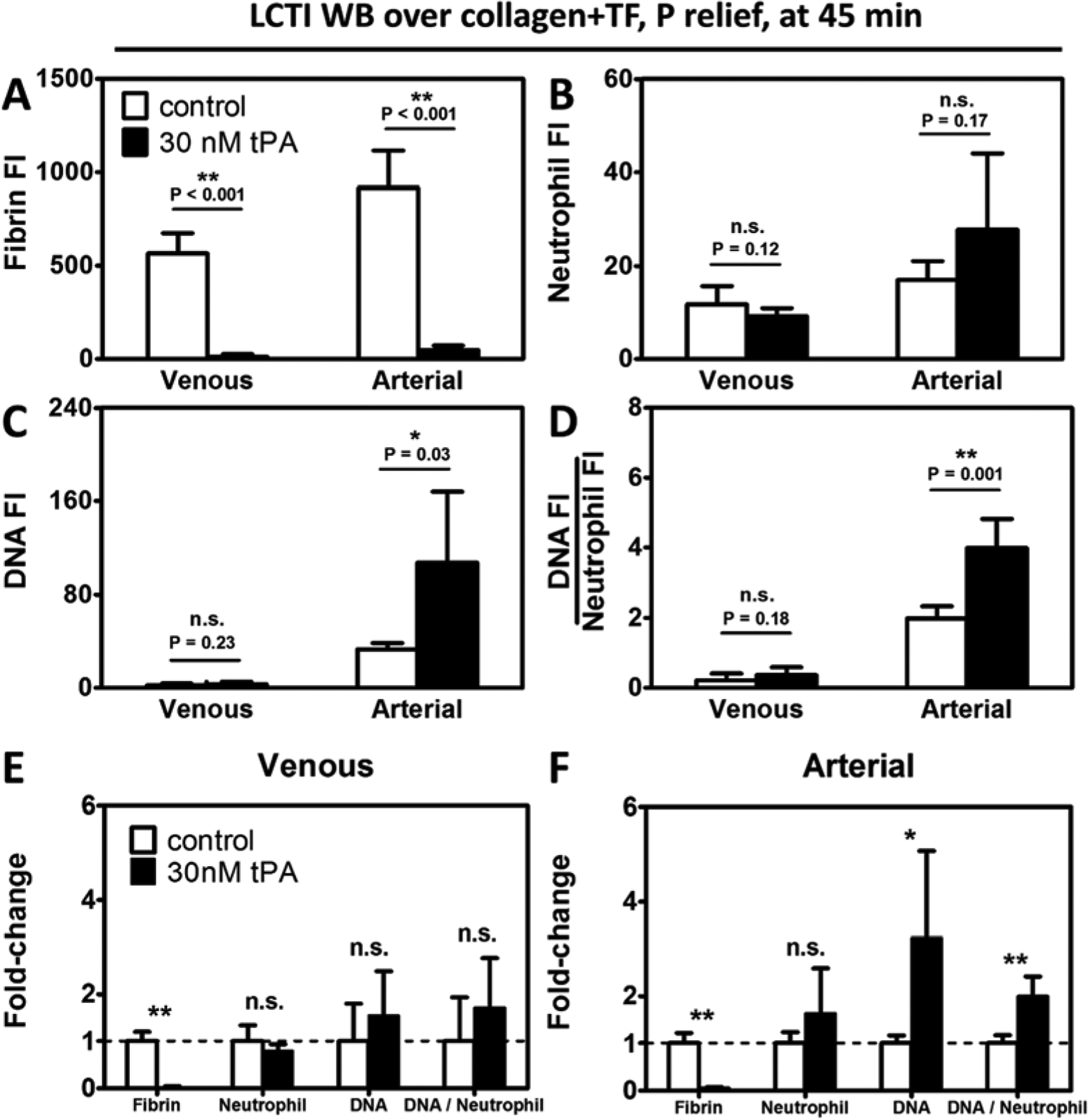
30 nM tPA eliminated essentially all fibrin (A) under both venous and arterial pressure drops. Neutrophil accumulation (B) was not significantly different between tPA-treated and untreated clots, regardless of the shear conditions. Extracellular DNA release (C) and normalized NET level (D) were similar between fibrin-rich and fibrin-depleted clots under venous flow, whereas a significant increase was observed in both measurements in the presence of tPA. The plasminogen activator did not significantly alter clot composition except the fibrin content under venous pressure drops (E). In contrast, the fibrinolytic agent caused a 3-fold reduction to total NET production and a 2-fold reduction to normalized NET generation under arterial pressure drops (F). (n≥6 clots. n.s.; not significant; *: p<0.05; **: p<0.005).
Because tPA only promoted NET formation in clots with ongoing fibrinolysis, we hypothesized that fibrinolysis may be responsible for the enhanced NET production. However, tPA may also promote NET generation through other mechanisms. To test this, we added the inhibitor εACA to tPA-treated blood. εACA blocked fibrin degradation and reduced NET generation to the tPA-free level (Figure S2). We also tested tPA in arterial clots formed with PPACK treated blood, and it did cause any significant differences in NET generation (Figure S3). Therefore, fibrinolysis was indispensable for the NET-enhancing effect of tPA. A closer look also revealed that NETs formed in fibrin-rich clots were mostly retained in their original position, whereas NETs inside fibrin-poor thrombi translocated through clots in the direction of flow (Video S1). Fibrin network is known to stabilize platelet aggregation and decrease clot permeability. The perceived motion of NETs was likely a result of increased thrombus porosity upon fibrinolysis.
Fibrin content modulates the extent of NETosis
When we treated blood with GPRP to inhibit fibrin polymerization while still preserving thrombin generation, a similar increase in NET production was observed. In contrast, adding GPRP to arterial clots formed with PPACK treated blood did not affect NET generation (Figure S3). As shown in Figure 6A, fibrin was not made throughout the experiment with GPRP present. GPRP also resulted in an upward trend in neutrophil deposition and NET generation (Figure 6B, C) presumably due to increases in clot permeability and blood constituent influx. Most importantly, there was a 2.5-fold increase in the normalized NET level (DNA:neutrophil) when fibrin polymerization was inhibited with GPRP (Figure 6D).
Figure 6. Blocking fibrin polymerization also promoted shear-induced NETosis at arterial pressure drop.
GPRP completely abolished fibrin polymerization (A) and resulted in an upward trend of neutrophil accumulation, similar to the one for tPA-treated clots. The presence of GPRP led to even higher levels of total NET generation (C) and NET:Neutrophil ratio (D) compared to tPA-treated clots.
Because the effects of tPA on fibrin deposition, neutrophil accumulation, and NET generation almost completely matched the effects of PPACK, we hypothesized the fibrin mass alone was a good predictor of the amount of NETs generated at arterial ΔP/L. Therefore, we plotted DNA and DNA:Neutrophil after 30 minutes of arterial perfusion as functions of clot fibrin content (Figure 7A, B). Clots treated with GPRP and PPACK had minimal fibrin accumulation and highest NET generation. Intermediate levels of fibrin deposition and NET generation were seen in tPA-treated clots at 30 min. Clots made with LCTI/HCTI-treated blood or clots formed in the presence of tPA-εACA combo had most abundant fibrin and least amount of NETs. Both total NET generation (r2=0.858) and normalized NET generation (r2=0.9817) were inversely correlated with fibrin under 6 different pharmacological conditions.
Figure 7. Fibrin modulates the extent of NETosis within platelet-rich clots.
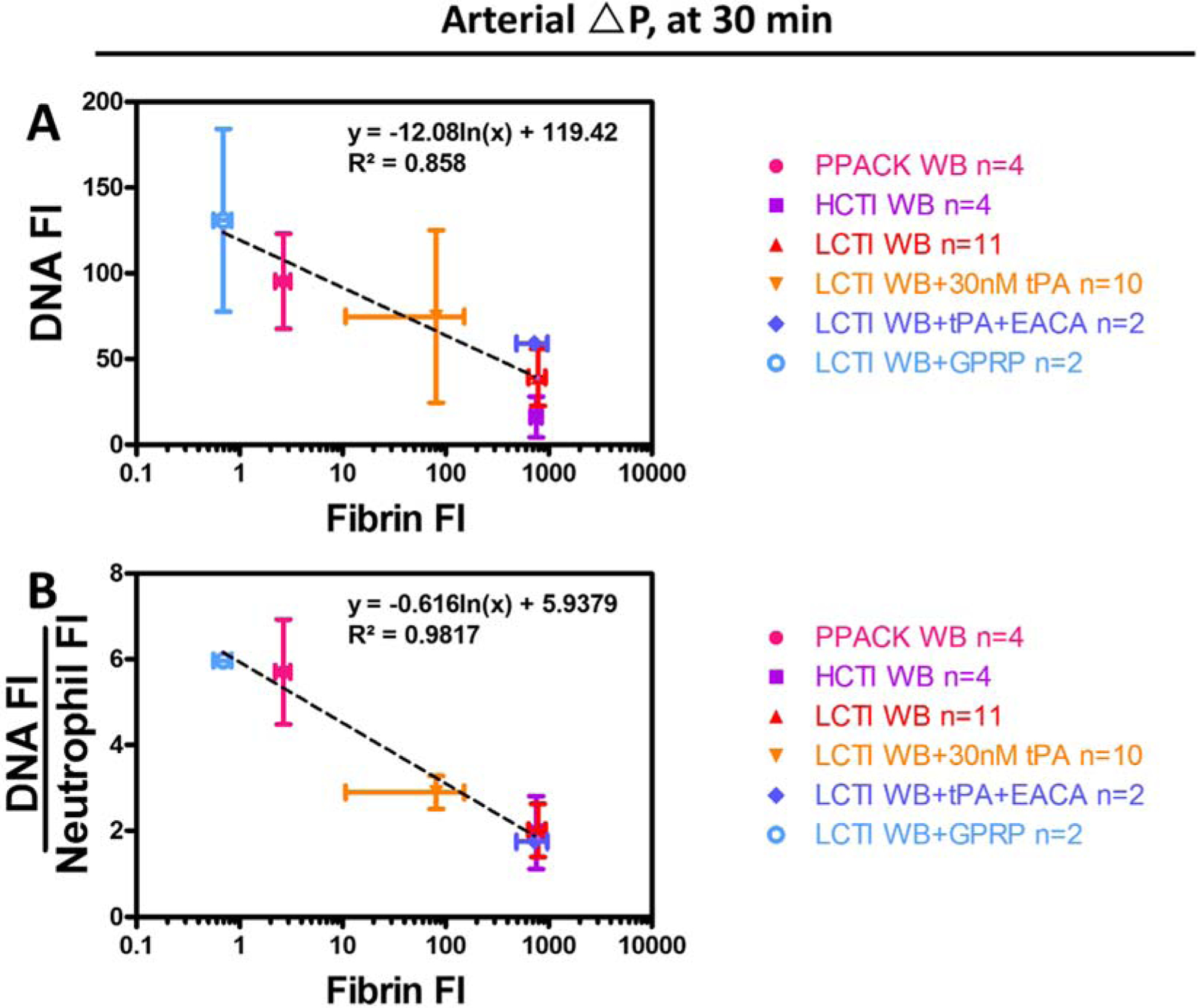
After 30 min of arterial perfusions, Total NET production (A) and normalized NET generation (B) both showed a strong inverse correlation with the level of fibrin deposition across different conditions.
Discussion
Physically entrapped neutrophils can undergo shear-induced NETosis when occlusive thrombi experience >70 mmHg/mm-clot (31). Such phenomena could potentially take place in coronary arterial thrombosis and ischemic strokes, where NETs have been identified in patient thrombus samples (20,30). Previously we have shown the production of NETs does not require thrombin generation. This is in line with a recent study in which the authors reported that NETs could form in chronic neutrophilia in mice treated with Dabigatran, a thrombin inhibitor (36). Here we showed that the formation of shear-induced NETs was actually increased in sterile occlusive thrombi when thrombin generation was inhibited. Forming clots in the presence of high dose exogenous tPA resulted in a similar increase in NET production. Furthermore, the rise in the percentage of NETosing neutrophils was responsible for this increase. Because tPA is known to cause neutrophil degranulation (34), we also examined the possibility of tPA being a direct NET inducer. In contrast to the accelerated NET formation under arterial pressure drop, no significant difference was observed between the fibrin-rich and tPA-treated clots under venous flow. Therefore, tPA does not directly trigger NETosis; instead tPA removes fibrin which then facilitates shear-induced NET formation. Because inhibiting plasminogen activation and plasmin with εACA abolished the NET-promoting effect of tPA, the modulation of NETosis by tPA must rely on fibrinolysis. The enhanced shear-induced NETosis is likely a result of increased clot porosity and enhanced exposure to shear forces when fibrin is removed or absent. It is well established that fibrin network modulates clot permeability, which can increase substantially as fibrin volume fraction decreases in a platelet-rich clot (37). Under the same pressure drop, increased permeability translates to enhanced flow, stresses, and transport. While the neutrophil influx was likely higher as well in fibrin-poor clots, normalized NET generation was also higher on a per neutrophil basis. Similar to tPA, the use of GPRP helped us decouple effects of thrombin generation and fibrin deposition on SIN. Fibrin polymerization was absent within GPRP-treated clots while thrombin generation was left intact. Consistent with the NET-promoting effect of tPA, GPRP also led to enhanced NET generation and a higher DNA:Neutrophil ratio. Together, the aforementioned results suggest a SIN-suppressing role of fibrin.
While NETosis may provide evolutionary advantages of trapping and killing bacteria in infectious diseases (1), they appear to do more harm in sterile inflammation and thrombosis (9). Therefore, the NET-suppressing/sequestering property of fibrin is beneficial as it protects neutrophils (and potentially other blood cells) from large interstitial shear stresses within occlusive thrombi and allows them to proceed with their normal functions.
NETs have been previously shown to act as a platelet/erythrocyte-immobilizing scaffold that is sufficient for vascular occlusion in DNase-deficient mice stimulated with granulocyte colony-stimulating factor (36). NETs can also hinder thrombolysis (20,27,28,30) and contribute to ischemia/reperfusion injuries (38). For those reasons, the DNase/tPA combo proposed by other groups (30,38) is promising as it may facilitate NET digestion and clot resolution during thrombolytic therapies.
In this study, we demonstrated that the extent of shear-induced NETosis is inversely correlated with the amount of fibrin in occlusive platelet-rich clots. Whereas fibrin suppresses shear-induced NETosis and sequesters NETs within clots, fibrinolytic agents promote shear-induced NETosis.
Supplementary Material
Video 1. Enhanced neutrophil extracellular trap (NET) generation after fibrin degradation. The control clot (A) and the thrombus treated with 3 nM tissue plasminogen activator (tPA) (B) were similar in terms of NET activities. In contrast, clots treated with 15 nM tPA (C) and 30 nM tPA (D) both had enhanced NET generation and mobilization.
Summary:
What is known about this topic?
When exposed to various stimuli, neutrophils can release NETs.
Intrathrombus hemodynamic forces trigger rapid NETosis within sterile thrombotic occlusions.
What does this paper add?
Fibrin polymerization suppresses shear-induced NET generation and sequesters NETs within clots
Fibrinolysis promotes shear-induced NETosis.
NET generation is inversely correlated with fibrin content in platelet rich clots, likely by protecting neutrophils from hemodynamic forces.
Acknowledgements
This study was supported by National Institutes of Health grants U01-HL-131053 and R01-HL-103419 to S.L.D.
Footnotes
Disclosure of Conflicts of Interests
None declared.
References
- 1.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–5. [DOI] [PubMed] [Google Scholar]
- 2.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13: 463–9. [DOI] [PubMed] [Google Scholar]
- 3.Urban CF, Reichard U, Brinkmann V, et al. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 2006; 8: 668–76. [DOI] [PubMed] [Google Scholar]
- 4.McDonald B, Urrutia R, Yipp BG, et al. Intravascular Neutrophil Extracellular Traps Capture Bacteria from the Bloodstream during Sepsis. Cell Host Microbe 2012; 12: 324–33. [DOI] [PubMed] [Google Scholar]
- 5.Warnatsch A, Ioannou M, Wang Q, et al. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015; 349: 316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 2012; 122: 2661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenne CN, Kubes P. Virus-Induced NETs – Critical Component of Host Defense or Pathogenic Mediator? Viruses and Neutrophils: An Unlikely Pair. PLoS Pathog 2015; 11: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 2014; 12: 2074–88. [DOI] [PubMed] [Google Scholar]
- 9.Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 2017; 23: 279–87. [DOI] [PubMed] [Google Scholar]
- 10.Pilsczek FH, Salina D, Poon KK, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 2010; 185: 7413–25. [DOI] [PubMed] [Google Scholar]
- 11.Carestia A, Kaufman T, Rivadeneyra L, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol 2016; 99: 153–62. [DOI] [PubMed] [Google Scholar]
- 12.Rochael NC, Guimarães-Costa AB, C Nascimento MT, et al. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci Rep 2015; 5: 18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis HD, Liddle J, Coote JE, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 2015; 11: 189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinod K, Witsch T, Farley K, et al. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost 2016; 14: 551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papayannopoulos V Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol Nature Publishing Group; 2017; 18: 134–47. [DOI] [PubMed] [Google Scholar]
- 16.Mcdonald B, Davis RP, Kim S-J, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017; 129: 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Von Brühl M-L, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012; 209: 819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stark K, Philippi V, Stockhausen S, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016; 128: 2435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savchenko AS, Martinod K, Seidman MA, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost 2014; 12: 860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangold A, Alias S, Scherz T, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res 2015; 116: 1182–92. [DOI] [PubMed] [Google Scholar]
- 21.Delabranche X, Stiel L, Severac F, et al. Evidence of Netosis in Septic Shock-Induced Disseminated Intravascular Coagulation. SHOCK 2017; 47: 313–7. [DOI] [PubMed] [Google Scholar]
- 22.Itagaki K, Kaczmarek E, Lee YT, et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PLoS One 2015; 10:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. PNAS 2010; 107: 15880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinod K, Wagner DD. Review Article Thrombosis : tangled up in NETs. Blood 2014; 123: 2768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noubouossie DF, Whelihan MF, Yu Y-B, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017; 129: 1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noubouossie DF, Whelihan MF, Yu Y-B, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017; 129: 1021–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longstaff C, Varjú I, Sótonyi P, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem 2013; 288: 6946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varjú I, Longstaff C, Szabó L, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost Schattauer GmbH; 2015; 113: 1289–98. [DOI] [PubMed] [Google Scholar]
- 29.Komissarov AA, Florova G, Idell S. Effects of extracellular DNA on plasminogen activation and fibrinolysis. J Biol Chem American Society for Biochemistry and Molecular Biology; 2011; 286: 41949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ducroux C, Di Meglio L, Loyau S, et al. Thrombus Neutrophil Extracellular Traps Content Impair tPA-Induced Thrombolysis in Acute Ischemic Stroke. Stroke 2018; 49: 754–7. [DOI] [PubMed] [Google Scholar]
- 31.Yu X, Tan J, Diamond SL. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J Thromb Haemost 2018; 16: 316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol (Camb) 2010; 2: 183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colace TV, Muthard RW, Diamond SL. Thrombus Growth and Embolism on Tissue Factor-Bearing Collagen Surfaces Under Flow Role of Thrombin With and Without Fibrin. Arterioscler Thromb Vasc Biol 2012; 32: 1466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuadrado E, Ortega L, Hernández-Guillamon M, et al. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol Wiley-Blackwell; 2008; 84: 207–14. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen VG. Corn trypsin inhibitor decreases tissue-type plasminogen activator-mediated fibrinolysis of human plasma. Blood Coagul Fibrinolysis 2009; 20: 191–6. [DOI] [PubMed] [Google Scholar]
- 36.Jiménez-Alcázar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science American Association for the Advancement of Science; 2017; 358: 1202–6. [DOI] [PubMed] [Google Scholar]
- 37.Wufsus AR, Macera NE, Neeves KB. The hydraulic permeability of blood clots as a function of fibrin and platelet density. Biophys J Biophysical Society; 2013; 104: 1812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge L, Zhou X, Ji W-J, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. AJP-Heart Circ Physiol 2014; 308: H500–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Enhanced neutrophil extracellular trap (NET) generation after fibrin degradation. The control clot (A) and the thrombus treated with 3 nM tissue plasminogen activator (tPA) (B) were similar in terms of NET activities. In contrast, clots treated with 15 nM tPA (C) and 30 nM tPA (D) both had enhanced NET generation and mobilization.