

Abstract
Immune checkpoint blockers (ICB) reinvigorate the immune system by removing the molecular brakes responsible for the scarce activity of immune phenotypes against malignant cells. After having proven their remarkable role as monotherapy, combinations of anti-Programmed cell death 1 (PD-1)/Programmed death-ligand 1 (PD-L1) agents with cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) antibodies, chemotherapy and/or anti-angiogenic compounds provide unprecedented results and durable responses across a variety of tumour types. Nevertheless, the main drawbacks of ICB are represented by primary and acquired resistance, translating into disease progression, as well as by immune-related toxicities. In this sense, novel strategies to foster the immune system through its direct stimulation are being tested in order to provide additional clinical improvements in patients with cancer. In this scenario, the co-stimulatory molecule OX40 (CD134) belongs to the next generation of immune therapeutic targets. Preliminary results of early clinical trials evaluating OX40 stimulation by means of different agents are encouraging. Here we review the rationale of OX40 targeting, highlighting the combination of OX40-directed therapies with different anticancer agents as a potential strategy to foster the immune system against malignant phenotypes.
Keywords: OX40, OX40L, T cells, cancer, immunotherapy
Introduction
A dramatic paradigm shift in cancer immunotherapy came from the demonstration that drugs targeting immune checkpoint signalling are able to restore immune anticancer activity reinforcing the biological and clinical significance of immune system/tumour interactions. The treatment strategies involving immune checkpoint blockers (ICB, ie, anti-CTLA-4 and anti-Programmed cell death 1 (PD-1)/Programmed death-ligand 1 (PD-L1) agents) are standard of care in several metastatic settings and have shown their role in earlier disease stages and adjuvant setting, with particular regard to melanoma and non-small cell lung cancer.1–3
However, a proportion of the patients does not benefit from ICB, experiencing primary resistance. Moreover, besides their prolonged activity in responding cases, their efficacy is limited by the onset of acquired resistance, turning out in clinical progression. Among the resistance mechanisms, there are tumour-intrinsic pathways leading to diminished infiltration and function of immune cells in tumour microenvironment (TME): (i) genomic defects in interferon-gama (IFN-γ) signalling, like mutations occurring in JAK1/2,4 5 (ii) expression of T-cell inhibitory surface ligands (including PD-L1) by the tumour, (iii) altered tumour antigen presentation,6 (iv) signalling through Wnt/β-catenin pathway,7 (v) Phosphatase and TENsin homolog (PTEN) loss,8 and (vi) induction of indoleamine 2,3-dioxygenase.9 Regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs) and tumour-associated macrophages (TAMs) release factors (interleukin 4 (IL-4), IL-10, transforming growth factor beta, vascular endothelial growth factor and arginase) in TME that suppress immune cells implicated in anti-tumoural response.10 To prevent and to overcome the onset of resistance, new strategies envisaging combination therapies with chemotherapy, anti-angiogenic treatments, radiotherapy and new immunomodulatory agents are promising.
The signalling induced by antigenic MHC/peptide interaction with T-cell receptor is a prerequisite to T cell activation, but insufficient to initiate T cell responses by itself. Further signalling by co-stimulatory molecules is crucial to optimal priming, expansion and differentiation of T-cells. These molecules are primarily classified into two groups: immunoglobulin superfamily (IgSF) and tumour necrosis factor receptor superfamily (TNFRSF).11 12 IgSF includes CD28, inducible co-stimulator (ICOS) and CD226. TNFRSF is composed of CD27, OX40 (CD134) and its ligand OX40L (CD252), 4-1BB (CD137), glucocorticoid-induced tumour necrosis factor receptor (TNFR)-related protein, death receptor 3, CD40 and CD30. Differently from standard ICB that blocks surface receptors in tumour and T cells that are responsible for inhibition of anti-tumoural immune response, drugs targeting OX40 act by direct activation and modulation of immune response.
Altogether, targeted therapies to these co-stimulatory molecules combined to standard ICB are an interesting option to overcome primary resistance by enhancing immune response.13 Here we will review the rationale that has supported the development of clinical trials with drugs targeting OX40 and we will discuss the current understanding of the mechanism of action of compounds designed to potentiate the immune system.
Molecular characteristics
OX40, a type 1 transmembrane glycoprotein, is predominantly expressed by T cells (constitutively by regulatory T phenotypes and, after activation, by effector T cells). Figure 1 shows human cells expressing OX40 and OX40L and its potential interactions, allowing migration of activated T-cells into tissues following inflammatory signals.11 12 14–16 OX40 has a cytoplasmic tail that binds to molecules implicated in signal transduction pathways, namely TNFR-associated factor-2, -3, and -5,17 18 mediating nuclear factor-kappa B (NF-kB) activation.
Figure 1.
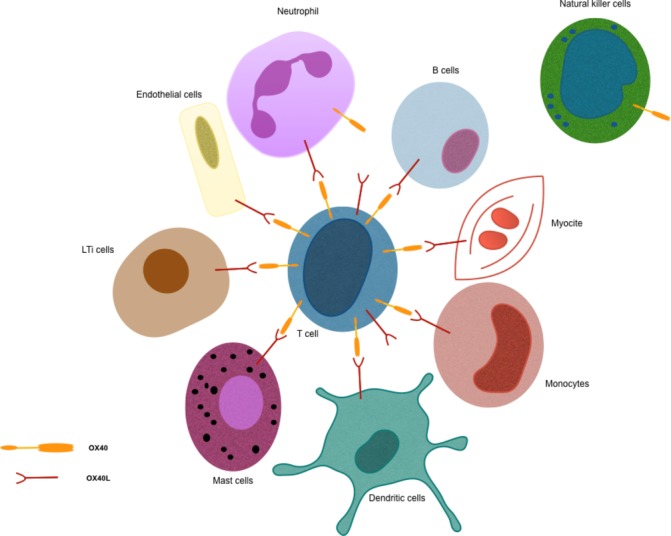
Human cells expressing OX40 and/or OX40L.
Immune response mediated by OX40 signalling
T-cell activation
OX40 induces expression of proteins with anti-apoptotic (Bcl-2, Bcl-xl and Bfl-1) and cell-cycle progression (Survivin) properties.19 20 OX40 counterbalance the inhibition of immune cells (including T lymphocytes CD4 +and CD8+, NK cells and B lymphocytes) while directly stimulating effector T cells (figure 2).
Figure 2.
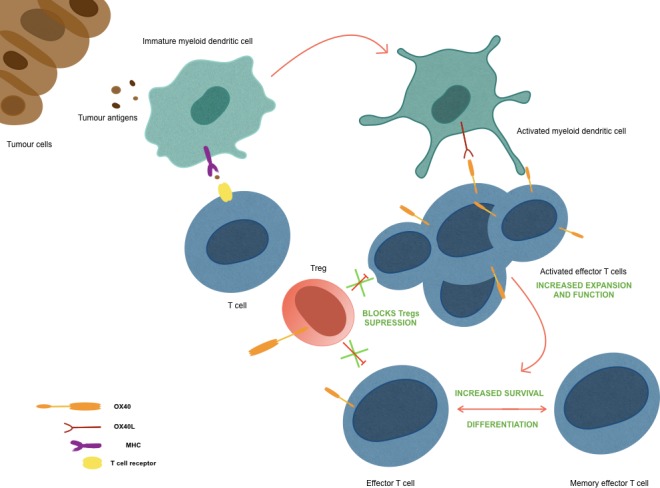
Mechanism of action of OX40.
Depletion of Treg cells
For anti-CTLA-4 therapy, there is evidence of a selective depletion of Treg cells in TME via Fcγ receptor-expressing macrophages, suggesting that OX40-directed antibodies can also deplete OX40+Tregs in TME without reducing effector T cells expressing the receptor.21 Zhang et al showed that OX40 co-stimulation leads to inhibition of FOXP3 gene expression, crucial to Treg differentiation, by two independent mechanisms1: enhancing the expression of the activator protein 1 transcription factors BATF, BATF3 and2 activating AKT- mammalian target of rapamycin (mTOR) pathway.22 There is evidence in preclinical models of reduction in IL-10 production by tumour-infiltrating Treg cells after treatment with anti-OX40 monoclonal antibody (mAb), allowing dendritic cells (DC) maturation, probably by downregulation of transcription factor interferon regulatory factor 1 mRNA expression.23 So, it creates a permissive immune status and leads to myeloid cell accumulation and development of innate and adaptive immunity, important steps to anti-tumoural effect of anti-OX40.24 25
Other immune pathways
Whether OX40 influences B cell response is controversial. Nevertheless, initial data suggest that, although it is not crucial for generating humoral response, OX40 activates ICOS pathway and can favour Th2 response by stimulating a profile of high immunoglobulin-producing cells.26–29 Expressed by DC, OX40L signalling via OX40 T-cell plays a role in antigen-presenting cell (APC) activation.30 31
OX40 expression in tumour immune microenvironment
In a preclinical study conducted by Marabelle et al, the analysis of tumour tissues in B cell lymphoma line-bearing mouse model and humans with mantle cell and follicular lymphomas showed a high expression of OX40 and CTLA-4 on the surface of tumour-specific Tregs (CD4+Foxp3+),32 with higher levels than lymphoid tissues. This strengthened the idea of using these drugs as targets of lymphocytes in TME. Burocchi et al found higher levels of OX40-expressing Tregs in murine colon carcinoma CT26 than in dLNs.23
Role of OX40 as a biomarker
Ramser et al analysed the positivity for OX40+infiltrating immune cells and tumour tissue from biopsies of primary and recurrent stages III and IV ovarian cancer (OC) in humans.33 Chemosensitivity was associated with high expression of OX40 on immune cells for primary OC and on tumour cells in recurrent OC; the patients who were OX40 negative in immune and tumour cells had the worse recurrence-free survival. In primary colon cancer, the higher expression of OX40 in tumour infiltrating lymphocytes was significantly associated with better survival, with a difference of 11 months between high and low OX40 expression.34
Although relying on a small number of patients, the study by Martins and colleagues showed that patients with gastric cancer (GC) had higher levels of T cells, monocytes and neutrophils with OX40 expression in peripheral blood when compared with healthy controls. Moreover, the percentage of OX40+T cells resulted in reduced more advanced stages, with a median of 3.0% in stages I–II and 1.4% in stages III–IV GC.35 In a cohort of 20 patients with advanced GC, the expression of OX40 on CD4+/CD8+T cells prior to Nivolumab therapy positively correlated with progression-free survival.36 Among cutaneous melanoma patients, the expression of OX40 in sentinel lymph node T cells inversely correlated with poor prognostic features such as tumour size, presence of ulceration and nodal infiltration.37
Development of drugs targeting OX40
Given the biological rationale to use co-stimulatory receptors as target therapy for enhancing immune response against tumours and based on in vitro results, many drugs that stimulate OX40 signalling have been developed. OX40 signalling can be triggered by OX40-specific agonistic antibodies, OX40L-Fc fusion proteins, transfection of DC with OX40L mRNA and tumour cells engineered to express OX40L on the surface.10 38 39 Furthermore, the development of a single antibody targeting both OX40 as a T cell co-stimulatory receptor and CTLA-4 as an ICB is ongoing.40 Table 1 shows drugs tested in in vitro studies either in human clinical trials and its technology.
Table 1.
OX40-targeted drugs
| Type | Drug |
| Humanised IgG1 agonist mAb | ABBV-368 |
| GSK3174998 | |
| MEDI0562 | |
| MOXR0916 (vonlerolizumab) | |
| Fully human IgG1 agonist mAb | INCAGN01949 |
| IBI101 | |
| BMS-986178 | |
| Fully human IgG2 agonist Ab | PF-04518600 |
| Murine IgG1 agonist mAb | MEDI6469P |
| 9B12 | |
| Human IgG1 CTLA-4 × OX40 bispecific Ab | ATOR-1015 |
| Lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23 and IL-36γ | mRNA-2752 |
| Human OX40L IgG4P Fc fusion protein | MEDI6383 |
| Dual-sided Fc fusion protein PD1-Fc-OX40L | SL-279252 |
Ab, antibodies; IL, interleukin; mAb, monoclonal antibodies.
Anti-tumoural activity of OX40 in animal models
The modulation of immune cells and anti-tumour activity of agents targeting OX40 has been shown in several preclinical cancer models. In a B cell lymphoma line-bearing mouse model, a TLR9 agonist, which stimulates APC, were administrated intratumourally in combination with OX40 murine mAb and/or anti-CTLA4, drugs that can modulate Tregs in TME.32 The intratumoural administration of TLR9 agonist with either OX40 murine mAb or anti-CTL4 was effective in eradicating most of systemic and central nervous system (CNS) metastases and reducing tumour-specific Tregs in injected site, even with low doses than in systemic therapy. These results were more impressive with the combination of the three drugs, with depletion of tumour-specific Tregs and cured most of the mice. In this study, the intratumoural injection generated complete and prolonged responses when compared with systemic infusion and seemed to improve immunologic memory, since mice treated locally did not relapse and were resistant to the development of CNS metastases after a new infusion of lymphoma cell line in the brain.
Oberst et al showed that MEDI6383, the human OX40L IgG4P Fc fusion protein, induced activation of T cells in vitro and in vivo models and overcome suppression mediated by Tregs. Its anti-tumoural efficacy was dependent on T cells in mouse models injected with A375 melanoma cells41 and has been tested in phase 1 trials enrolling advanced malignancy patients (table 2, see section 6). In a murine sarcoma model (MCA205), Moran and colleagues showed that anti-OX40 mAb treatment increased of T cells with strong T cell receptor signalling in the TME and a smaller increase in CD8 +T cells in tumour-draining lymph node (dLN). When used in combination to adoptive T cell therapy, anti-OX40 mAb improved cure rates from 9% to 70%, with greater tumour regressions and longer survival in this MCA205 tumour-bearing mice.13 Data generated by Weinberg and colleagues show that OX40 signalling is associated with enhanced specific anti-tumoural immune response.42 43 In mice bearing a colon cancer model (CT26), treatment with murine OX40L:Ig (mOX40L:Ig) prolonged tumour-free survival (cured mice) and these mice resisted to a second CT26 inoculation, remaining tumour free. However, they had to be sacrificed when challenged to a renal cell origin tumour because of tumour burden.42
Table 2.
Ongoing clinical trials targeting OX40
| Monotherapy | ||||
| Population | OX40 target | Phase | NCT number | Endpoints |
| Advanced solid tumours | MEDI0562 | 1 | 02318394 | Safety and DLTs |
| ATOR-1015 | 1 | 03782467 | Safety | |
| INCAGN01949 | 1/2 | 02923349 | Safety | |
| ABBV-368 | 1 | 03071757 | Safety, pharmacokinetics and preliminary efficacy | |
| Advanced malignancies | SL-279252 | 1 | 03894618 | Safety and DLTs |
| CRC | MEDI6469 | 1 | 02559024 | Safety |
| HNSCC | MEDI6469 | 1 | 02274155 | Safety |
| HNSCC or melanoma | MEDI0562 | 1 | 03336606 | Activation of immune response |
HNSCC, head and neck squamous cell carcinoma.
| Combination therapy | |||||
| Population | OX40 target | Combination therapy | Phase | NCT number | Endpoints |
| Advanced solid tumours | PF-04518600 | Avelumab (anti-PD-L1) Utomilumab (4-1BB agonist mAb) PD 0360324 (anti-CSF1 mAb) |
2 | 02554812 | DLTs and ORR |
| MEDI6383 | Durvalumab (anti-PD-L1) | 1 | 02221960 | Safety | |
| PF-04518600 | Utomilumab (4-1BB agonist mAb) | 1 | 02315066 | Safety and DLTs | |
| GSK3174998 | GSK1795091(TLR4 agonist) GSK3359609 (anti-ICOS agonist) Pembrolizumab (anti-PD-1) |
1 | 03447314 | Safety and DLTs | |
| MEDI0562 | Durvalumab (anti-PD-L1) Tremelimumab (anti-CTLA-4) |
1 | 02705482 | Safety and DLTs | |
| MOXR0916 | Atezolizumab (anti-PD-L1) | 1b | 02410512 | Safety and DLTs | |
| GSK3174998 | Pembrolizumab (anti-PD-1) | 1 | 02528357 | Safety and DLTs | |
| IBI101 | Sintilimab (anti-PD-1) | 1 | 03758001 | Safety | |
| INCAGN01949 | Nivolumab (anti-PD-1) Ipilimumab (anti-CTLA-4) |
1/2 | 03241173 | Safety and ORR | |
| BMS-986178 | Nivolumab (anti-PD-1) Ipilimumab (anti-CTLA-4) |
1/2a | 02737475 | Safety | |
| BMS-986178 | TLR9 Agonist SD-101 | 1 | 03831295 | Safety | |
| Advanced malignancies | MEDI6469 | Durvalumab(anti-PD-L1) Tremelimumab (anti-CTLA-4) Rituximab (anti-CD20) |
1b/2 | 02205333 | Safety and DLTs |
| Anti-OX40 | Biological vaccines tetanus toxoid and KLH | 1 | 01644968 | DLTs | |
| mRNA-2752 | Durvalumab(anti-PD-L1) Tremelimumab (anti-CTLA-4) |
1 | 03739931 | Safety and DLTs | |
| Lymphomas | PF-04518600 | Utomilumab (4-1BB agonist mAb) Rituximab (anti-CD20) Avelumab (anti-PD-L1) |
1 | 03636503 | Recommended phase 2 dosing and complete response rate |
| BMS-986178 | TLR9 agonist SD-101 Radiotherapy |
1 | 03410901 | DLTs | |
| AML | PF-04518600 | Avelumab (anti-PD-L1) Azacitidine Venetoclax (anti-Bcl-2) Gemtuzumab ozogamicin (recombinant humanised IgG4 kappa Ab conjugated with calicheamicin derivative) |
1/2 | 03390296 | Safety and composite complete response |
| RCC | PF-04518600 | Axitinib | 2 | 03092856 | PFS |
| CRPC | MEDI6469 | Cyclophosphamide Radiotherapy |
1b | 01303705 | MTD |
| BC | MEDI6469 | Radiotherapy | 1/2 | 01862900 | Safety and MTD |
| Ovarian, fallopian tube or peritoneal cancers | MEDI0562 | Durvalumab(anti-PD-L1) Tremelimumab (anti-CTLA-4) Oleclumab (anti-CD73) |
2 | 03267589 | Disease control rate |
| Urothelial carcinoma | MOXR0916 | Atezolizumab (anti-PD-L1) | 2 | 03029832 | PFS and overall survival |
AML, acute myeloid leukaemia; BC, breast cancer; CRC, colorectal cancer; CRPC, castration-resistant prostate cancer;DLTs, dose limiting toxicities; ICOS, inducible co-stimulator; mAb, monoclonal antibodies; MTD, maximum tolerated dose; NCT, ClinicalTrials.gov identifier; ORR, objective response rate; PD-1, Programmed cell death 1; PD-L1, Programmed death-ligand 1; PFS, progression-free survival; RCC, renal cell carcinoma.
Combinations of agents targeting OX40 with other therapies are promising and under evaluation. In a preclinical model, combined therapy with anti-OX40 and anti-CTLA-4 resulted in significant increase in proliferation and activity of CD4 +and CD8+T cells that was translated into better outcomes compared with anti-OX40 monotherapy.44 The upregulation of PD-L1 on TAMs and macrophages and of PD-1 on T cells induced by OX40 targeted therapy can explain the resistance.45 When given in combination anti-PD-1 and/or anti-PD-L1, anti-OX40 significantly increased the expansion and effector properties of differentiated T cells in the dLN and tumour itself, with an increase in CD8+/Treg ratio, that was translated in rapid tumour shrinkage and durable responses.45 46 In another murine model, the combined therapy anti-OX40 and a drug targeting CD73 (responsible for immunosuppression and pro-angiogenesis in TME),47 resulted in longer survival, increased immune response and tumour response than controls. Better ascites fluid control was obtained when compared with anti-PD-1 and anti-OX40 combination.48 ATOR-1015 administration resulted in prolonged survival, tumour shrinkage and complete response rates when compared with anti-OX40 or anti-CTLA-4 monotherapy in a murine model of bladder cancer, by enhancing CD8+T cells infiltrate and reducing Treg in TME.40 In this study, ATOR-1015 improved the outcomes also when combined to anti-PD1 therapy: all bladder cancer-harbouring mice were cured and colon carcinoma models experienced tumour shrinkage and longer survival.
Depending on the pathway of immune stimulation, the timing of differential drug administration in combinatorial strategies can be crucial. Shrimali et al showed that, in a murine model injected with TC-1 tumour cells (mouse lung epithelial cells cotransformed by human papillomavirus strain 16 early proteins 6 and 7 and activated RAS oncogene), simultaneous administration of OX40 costimulation and anti-PD-1 had a negative effect on OX40-directed drug, reducing survival and tumour inhibition.49 They showed that simultaneous infusion lead to apoptosis of antigen-specific T cells, reducing TME-infiltrating CD8+. Although increased CD8+T cells apoptosis was not seen with sequential administration of anti-PD-1 (delay of 7 days), combined therapy did not have negative or additive effects to anti-OX40. Using a different tumour model, Messenheimer et al also showed a diminished efficacy of OX40 costimulation when simultaneously administrated with anti-PD-1. Simultaneous infusion increased acute cytokine release (TNF-α, IFN-γ, IL-4, IL-10) and expression of inhibitory markers (eg, CTLA-4 and TIM-3) by tumour-infiltranting CD4+ and CD8+T cells in mammary tumour-bearing mices.50 In this study, sequential treatment of OX40-targeted followed anti-PD-1 or anti-PD-L1 (delay of 6 days) resulted in better outcomes (tumour control and survival) with reduced T-cell exhaustion. Administration of anti-PD-1 with delayed OX40 did not improve outcomes.
Targeting OX40 in clinical trials
Preliminary data about the utilisation of OX40-targeting drugs in humans come from initial trials, which most included advanced and pretreated tumours. Whether these trials included patients that had progressed to ICB and the type of resistance (primary or acquired) is not specified. Initial data from clinical trials evaluating OX40-directed therapy in advanced tumours showed satisfactory safety profiles and signs of clinical activity. The phase 1 clinical trial published by Curti et al showed a good tolerance for 9B12, with grades 1 and 2 lymphopenia, fatigue, fever/chills, and rashes and a transient lymphopenia as the only grade 3 and 4 toxicity.51 Stable disease (SD) was the best response for 20% of the 30 patients with metastatic solid tumours refractory to conventional therapy. During an observation period of 57 days, there was a significant increase of proliferation markers in lymphocytes and activation of CD8+T cells in patients treated with 9B12 compared with controls.51 Among 48 patients with solid tumours (melanoma, hepatocellular carcinoma, head and neck squamous cell and renal cell carcinoma) treated with PF-8600 in a phase 1 study, grade 1–2 fatigue, nausea and vomiting were the most common adverse events (AE). Out of the 48 treated patients, 4% and 52% experienced, respectively, partial response (PR) and SD as best response, but there is no published data about duration of response.52 In a phase 1 dose-escalation study of MEDI0562, which included 55 patients with advanced solid tumours, the treatment was well tolerated and showed clinical activity. AE were mostly grade 1 or 2, including fatigue in 31% of patients and infusion reactions in 15%. Fever occurred in 4% of the patients and was the most common grade 3 event. Two patients (3,6%) carriers of head and neck squamous cell carcinoma (HNSCC) and bladder cancer experienced PR, with an overall survival of 13.8 and 10.2+months, respectively. SD was observed in 22 patients (44%) with a duration of response lasting more than 3 months in 20 of these patients.53 Preliminary data from a phase 1 dose-escalation trial sustain the safety of OX40-targeted mABs. ABBV-368 was well tolerated when given as monotherapy for patients with advanced or metastatic tumours and, though further evaluation is ongoing, they observed initial tumour activity.54
In the neoadjuvant setting, the results from a phase 1b clinical trial conducted by Bell and colleagues showed that MEDI6469 induced proliferation and activity of T cells in the TME in 17 treatment-naïve patients with resectable HNSCC (stage III to IVA). In this trial, treatment was well tolerated, without grade 3 or 4 AE, and did not delay curative surgery.55
Published data from phase 1 trials showed that the safety profile seems to be maintained even in combination strategies. In a phase 1 trial that included patients with advanced solid tumours, Infante et al showed a safety tolerance profile with vonlerolizumab combined with atezolizumab and evidence of PD-L1 induction and immune activation in tumour paired biopsies.56 In the same population, another ongoing phase 1 trial of GSK3174998 administered as monotherapy or combined with pembrolizumab (anti-PD-1 Ab) showed no dose-limiting toxicities.57 The combination of agonistic mAb against OX40 (PF-8600) and 4-1BB (utomilumab) increased the expression of markers and genes related to immune activation in paired biopsies after 6 weeks of treatment when compared with baseline.58 Data about efficacy are not yet published.
Conclusive remarks
Currently, the biggest lesson from clinical trials using OX40-targeted drugs is its safety when used as monotherapy or combined with ICB. Although OX40-targeted therapy showed impressive results in tumour bearing mice, preliminary clinical data show that its efficacy as monotherapy in humans is modest. OX40 costimulation is a promising strategy when used in combination with immunotherapies targeting inhibitory receptors such as anti-PD-1 and anti-PD-L1. It would be an interesting strategy for tumours benefiting from these treatments, in advanced or localised settings. Following biological rationale and preclinical data, OX40-targeted drugs should be administered as sequential treatment followed by anti-PD-1 or anti-PD-L1. It is important to test combination approaches, evaluating timing and sequential strategies. More studies should be performed in order to find some predictors of response, better comprehension of resistance mechanism and immunological dynamics in order to trigger immune activation and enhance clinical activity of OX40-targeted drugs against tumours, especially in combination strategies.
Footnotes
BR and LD contributed equally.
Contributors: LD and BR contributed equally to this work.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1. Eggermont AMM, Blank CU, Mandala M, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med Overseas Ed 2018;378:1789–801. 10.1056/NEJMoa1802357 [DOI] [PubMed] [Google Scholar]
- 2. Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 2017;377:1824–35. 10.1056/NEJMoa1709030 [DOI] [PubMed] [Google Scholar]
- 3. Durvalumab after chemoradiotherapy in stage III Non–Small-Cell lung cancer. N Engl J Med 2019;380:989–90. 10.1056/NEJMc1900407 [DOI] [PubMed] [Google Scholar]
- 4. Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404. 10.1016/j.cell.2016.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 2017;7:188–201. 10.1158/2159-8290.CD-16-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 2017;7:1420–35. 10.1158/2159-8290.CD-17-0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. 10.1038/nature14404 [DOI] [PubMed] [Google Scholar]
- 8. Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019;25:462–9. 10.1038/s41591-019-0349-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer 2017;76:167–82. 10.1016/j.ejca.2017.01.011 [DOI] [PubMed] [Google Scholar]
- 10. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, et al. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer 2016;52:50–66. 10.1016/j.ejca.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 11. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018;131:39–48. 10.1182/blood-2017-07-741025 [DOI] [PubMed] [Google Scholar]
- 12. Willoughby J, Griffiths J, Tews I, et al. Ox40: structure and function – what questions remain? Mol Immunol 2017;83:13–22. 10.1016/j.molimm.2017.01.006 [DOI] [PubMed] [Google Scholar]
- 13. Moran AE, Polesso F, Weinberg AD. Immunotherapy expands and maintains the function of high-affinity tumor-infiltrating CD8 T cells in situ. J.i. 2016;197:2509–21. 10.4049/jimmunol.1502659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Souza HS, Elia CCS, Spencer J, et al. Expression of lymphocyte-endothelial receptor-ligand pairs, alpha 4beta 7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut 1999;45:856–63. 10.1136/gut.45.6.856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imura A, Hori T, Imada K, et al. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med 1996;183:2185–95. 10.1084/jem.183.5.2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taylor L, Bachler M, Duncan I, et al. In vitro and in vivo activities of OX40 (CD134)-IgG fusion protein isoforms with different levels of immune-effector functions. J Leukoc Biol 2002;72:522–9. [PubMed] [Google Scholar]
- 17. Kawamata S, Hori T, Imura A, et al. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem 1998;273:5808–14. 10.1074/jbc.273.10.5808 [DOI] [PubMed] [Google Scholar]
- 18. Arch RH, Thompson CB. 4-1BB and Ox40 Are Members of a Tumor Necrosis Factor (TNF)-Nerve Growth Factor Receptor Subfamily That Bind TNF Receptor-Associated Factors and Activate Nuclear Factor κB. Mol Cell Biol 1998;18:558–65. 10.1128/MCB.18.1.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rogers PR, Song J, Gramaglia I, et al. Ox40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001;15:445–55. 10.1016/S1074-7613(01)00191-1 [DOI] [PubMed] [Google Scholar]
- 20. Song J, So T, Cheng M, et al. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity 2005;22:621–31. 10.1016/j.immuni.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 21. Chao JL, Savage PA. Unlocking the complexities of tumor-associated regulatory T cells. J.i. 2018;200:415–21. 10.4049/jimmunol.1701188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Xiao X, Lan P, et al. Ox40 costimulation inhibits FOXP3 expression and Treg induction via BATF3-Dependent and independent mechanisms. Cell Rep 2018;24:607–18. 10.1016/j.celrep.2018.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burocchi A, Pittoni P, Gorzanelli A, et al. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol 2011;41:3615–26. 10.1002/eji.201141700 [DOI] [PubMed] [Google Scholar]
- 24. Bulliard Y, Jolicoeur R, Zhang J, et al. Ox40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol 2014;92:475–80. 10.1038/icb.2014.26 [DOI] [PubMed] [Google Scholar]
- 25. Piconese S, Valzasina B, Colombo MP. Ox40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med 2008;205:825–39. 10.1084/jem.20071341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stüber E, Strober W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J Exp Med 1996;183:979–89. 10.1084/jem.183.3.979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stüber E, Neurath M, Calderhead D, et al. Cross-Linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity 1995;2:507–21. 10.1016/1074-7613(95)90031-4 [DOI] [PubMed] [Google Scholar]
- 28. Kopf M, Ruedl C, Schmitz N, et al. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL responses after virus infection. Immunity 1999;11:699–708. 10.1016/S1074-7613(00)80144-2 [DOI] [PubMed] [Google Scholar]
- 29. Metzger TC, Long H, Potluri S, et al. ICOS Promotes the Function of CD4 + Effector T Cells during Anti-OX40–Mediated Tumor Rejection. Cancer Res 2016;76:3684–9. 10.1158/0008-5472.CAN-15-3412 [DOI] [PubMed] [Google Scholar]
- 30. Chen AI, McAdam AJ, Buhlmann JE, et al. Ox40-ligand has a critical costimulatory role in dendritic cell:T cell interactions. Immunity 1999;11:689–98. 10.1016/S1074-7613(00)80143-0 [DOI] [PubMed] [Google Scholar]
- 31. Ohshima Y, Tanaka Y, Tozawa H, et al. Expression and function of OX40 ligand on human dendritic cells. J Immunol 1997;159:3838–48. [PubMed] [Google Scholar]
- 32. Marabelle A, Kohrt H, Sagiv-Barfi I, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest 2013;123:2447–63. 10.1172/JCI64859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramser M, Eichelberger S, Däster S, et al. High OX40 expression in recurrent ovarian carcinoma is indicative for response to repeated chemotherapy. BMC Cancer 2018;18:425 10.1186/s12885-018-4339-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petty JK, He K, Corless CL, et al. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134). Am J Surg 2002;183:512–8. 10.1016/S0002-9610(02)00831-0 [DOI] [PubMed] [Google Scholar]
- 35. Martins MR, d SRL. Could OX40 agonist antibody promote activation of the anti-tumor immune response in gastric cancer? J Surg Oncol 2018;117:840–4. 10.1002/jso.25001 [DOI] [PubMed] [Google Scholar]
- 36. Ohmura H, Yamaguchi K, Hanamura F, et al. Activation of central/effector memory T cells in advanced gastric cancer patients treated with antiprogrammed death-1 antibody. JCO 2019;37:54 10.1200/JCO.2019.37.4_suppl.54 [DOI] [Google Scholar]
- 37. Sarff M, Edwards D, Dhungel B, et al. Ox40 (CD134) expression in sentinel lymph nodes correlates with prognostic features of primary melanomas. Am J Surg 2008;195:621–5. 10.1016/j.amjsurg.2007.12.036 [DOI] [PubMed] [Google Scholar]
- 38. Reuter D, Staege MS, Kühnöl CD, et al. Immunostimulation by OX40 ligand transgenic Ewing sarcoma cells. Front Oncol 2015;5:242 10.3389/fonc.2015.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dannull J, Nair S, Su Z, et al. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood 2005;105:3206–13. 10.1182/blood-2004-10-3944 [DOI] [PubMed] [Google Scholar]
- 40. Kvarnhammar AM, Veitonmäki N, Hägerbrand K, et al. The CTLA-4 X OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer 2019;7 10.1186/s40425-019-0570-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oberst MD, Augé C, Morris C, et al. Potent immune modulation by MEDI6383, an engineered human OX40 ligand IgG4P Fc fusion protein. Mol Cancer Ther 2018;17:1024–38. 10.1158/1535-7163.MCT-17-0200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weinberg AD, Rivera MM, Prell R, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol 2000;164:2160–9. 10.4049/jimmunol.164.4.2160 [DOI] [PubMed] [Google Scholar]
- 43. Maxwell JR, Weinberg A, Prell RA, et al. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol 2000;164:107–12. 10.4049/jimmunol.164.1.107 [DOI] [PubMed] [Google Scholar]
- 44. Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res 2014;2:142–53. 10.1158/2326-6066.CIR-13-0031-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zippelius A, Schreiner J, Herzig P, et al. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res 2015;3:236–44. 10.1158/2326-6066.CIR-14-0226 [DOI] [PubMed] [Google Scholar]
- 46. Polesso F, Weinberg AD, Moran AE. Late-Stage tumor regression after PD-L1 blockade plus a concurrent OX40 agonist. Cancer Immunol Res 2019;7:269–81. 10.1158/2326-6066.CIR-18-0222 [DOI] [PubMed] [Google Scholar]
- 47. Antonioli L, Yegutkin GG, Pacher P, et al. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends in Cancer 2016;2:95–109. 10.1016/j.trecan.2016.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Virani NA, Thavathiru E, McKernan P, et al. Anti-CD73 and anti-OX40 immunotherapy coupled with a novel biocompatible enzyme prodrug system for the treatment of recurrent, metastatic ovarian cancer. Cancer Lett 2018;425:174–82. 10.1016/j.canlet.2018.03.027 [DOI] [PubMed] [Google Scholar]
- 49. Shrimali RK, Ahmad S, Verma V, et al. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol Res 2017;5:755–66. 10.1158/2326-6066.CIR-17-0292 [DOI] [PubMed] [Google Scholar]
- 50. Messenheimer DJ, Jensen SM, Afentoulis ME, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with Anti-OX40. Clin Cancer Res 2017;23:6165–77. 10.1158/1078-0432.CCR-16-2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Curti BD, Kovacsovics-Bankowski M, Morris N, et al. Ox40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res 2013;73:7189–98. 10.1158/0008-5472.CAN-12-4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. El-Khoueiry AB, Hamid O, Thompson JA, et al. The relationship of pharmacodynamics (PD) and pharmacokinetics (pK) to clinical outcomes in a phase I study of OX40 agonistic monoclonal antibody (mAb) PF-04518600 (PF-8600). JCO 2017;35:3027 10.1200/JCO.2017.35.15_suppl.3027 [DOI] [Google Scholar]
- 53. Glisson B, Leidner R, Ferris RL, et al. 1152PSafety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Ann Oncol 2018;29 10.1093/annonc/mdy288.025 [DOI] [PubMed] [Google Scholar]
- 54. Spira A, Chung K, Patnaik A, et al. 1149PSafety, tolerability, and pharmacokinetics of the OX40 agonist ABBV-368 in patients with advanced solid tumors. Ann Oncol 2018;29 10.1093/annonc/mdy288.022 [DOI] [Google Scholar]
- 55. Bell RB, Duhen R, Leidner RS, et al. Neoadjuvant anti-OX40 (MEDI6469) prior to surgery in head and neck squamous cell carcinoma. JCO 2018;36:6011 10.1200/JCO.2018.36.15_suppl.6011 [DOI] [Google Scholar]
- 56. Infante JR, Hansen AR, Pishvaian MJ, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. JCO 2016;34:101 10.1200/JCO.2016.34.15_suppl.101 [DOI] [Google Scholar]
- 57. Infante JR, Ahlers CM, Hodi FS, et al. ENGAGE-1: a first in human study of the OX40 agonist GSK3174998 alone and in combination with pembrolizumab in patients with advanced solid tumors. JCO 2016;34:TPS3107 10.1200/JCO.2016.34.15_suppl.TPS3107 [DOI] [Google Scholar]
- 58. Hamid O, Hu-Lieskovan S, Ros W, et al. 1184PPharmacodynamic (PD) changes in tumors and peripheral blood T cell receptor (TCR) repertoire in a phase I study combining OX40 (PF-04518600) and 4-1BB (utomilumab) agonistic monoclonal antibodies (mAbs). Ann Oncol 2018;29 10.1093/annonc/mdy288.057 [DOI] [Google Scholar]