

Cholinergic inputs originating from the peripheral nervous system regulate the inflammatory immune responses of macrophages during clearance of blood-based pathogens. Because microglia are involved in clearing amyloid and tau pathology from the central nervous system, we hypothesized that cholinergic input originating from the basal forebrain might similarly regulate inflammatory immune responses to these pathologies in the aging brain.
Keywords: amyloid, basal forebrain cholinergic system, complement C3, phosphorylated tau, preclinical Alzheimer's disease, soluble TREM2
Abstract
Cholinergic inputs originating from the peripheral nervous system regulate the inflammatory immune responses of macrophages during clearance of blood-based pathogens. Because microglia are involved in clearing amyloid and tau pathology from the central nervous system, we hypothesized that cholinergic input originating from the basal forebrain might similarly regulate inflammatory immune responses to these pathologies in the aging brain. To explore this hypothesis, we leveraged the Alzheimer's Disease Neuroimaging Initiative dataset. Cognitively normal older male and female human adults were differentiated according to the relative concentration of phosphorylated tau and amyloid in their cerebrospinal fluid, yielding neurotypical and preclinical, cognitively healthy, subgroups. We then tracked these two groups longitudinally with structural MRI and biomarkers of inflammation, including soluble sTREM2 levels in the CSF and complement C3 expression in the blood transcriptome. Longitudinal loss of basal forebrain volume was larger in the preclinical compared with the neurotypical subgroup. Across preclinical adults, loss of basal forebrain volume was associated with greater longitudinal accumulation of sTREM2 and higher peripheral blood C3 expression. None of these relationships were attributable to degeneration in the whole-brain gray matter volume. Preclinical APOE e4 carriers exhibited the largest loss of basal forebrain volume and highest C3 expression. Consistent with the known anti-inflammatory influence of the peripheral cholinergic pathways on macrophages, our findings indicate that a loss of central cholinergic input originating from the basal forebrain might remove a key check on microglial inflammation induced by amyloid and tau accumulation.
SIGNIFICANCE STATEMENT In the peripheral nervous system, cholinergic modulation holds the reactivity of macrophages to blood-based pathogens in check, promoting clearance while preventing runaway inflammation and immune-triggered cell death. Microglia are the brain's resident macrophages and play an important role in clearing accumulated amyloid and tau from neurons. Here, we demonstrate that a loss of cholinergic integrity in the CNS, indexed by longitudinal decreases of basal forebrain volume, interacts with multiple biomarkers of inflammation in cognitively normal older adults with abnormal amyloid and tau pathology. These interactions were not detected in cognitively normal older adults with “neurotypical” levels of amyloid and tau. An age-related loss of cholinergic neuromodulation may remove key checks on microglial reactivity to amyloid and tau.
Introduction
Progressive neuronal accumulation of misfolded amyloid and tau proteins is observed in virtually all older adults >70 years (Kok et al., 2009; Nelson et al., 2012). Yet, the progressive neuronal degeneration which causes the cognitive prodrome of AD occurs in only ∼10% of this population. The pathogenic mechanism that links age-related “proteinopathies” to neurodegeneration in the earliest phases of Alzheimer's disease (AD) remains ambiguous.
Recent evidence from rodent models of AD points to microglia, part of the brain's innate immune defense system against neuronal injury and infection, as a potential link. Microglia play a central role in sensing and clearing accumulated amyloid and tau proteins from neurons in the aging brain. Some microglia perform these activities with little apparent inflammation (M2 profile), whereas others are proinflammatory (M1 profile) and can cause neurodegeneration (Zamanian et al., 2012; Martinez and Gordon, 2014; Heppner et al., 2015). The M2 transcriptional profile is associated with upregulation of TREM2, an innate immune receptor protein located on the microglial cell membrane, which is linked to phagocytosis (clearance) of accumulated amyloid and tau debris with little apparent inflammation (Kleinberger et al., 2014; Ewers et al., 2019; Parhizkar et al., 2019). By contrast, the M1 transcriptional profile is associated with upregulation of multiple proinflammatory cytokines in response to neuronal amyloid and tau accumulation (Hong et al., 2016; Liddelow et al., 2017; Shi et al., 2017a). Proinflammatory M1 microglia are causally linked to proliferation of neurotoxic A1-type reactive astrocytes (Liddelow et al., 2017). In particular, complement component C3, the activating gene of the complement cascade, is one of the most characteristic and highly upregulated genes in the cross talk between M1 microglia and A1 astrocytes (Liddelow et al., 2017). Strikingly, genetic KO of C3 spares synapse loss and cognitive impairment in mouse models of AD, despite the fact that this KO partially impairs the capacity of microglia to clear accumulated amyloid (Shi et al., 2017a) and tau pathology (Wu et al., 2019). These studies suggest that, in the presence of amyloid and tau: (1) unchecked inflammation from microglial-astrocyte cross talk causes neurodegeneration in mouse models of AD, as opposed to amyloid and tau alone; and (2) the proliferation of inflammatory M1 microglia might be regulated by an endogenous mechanism, which is susceptible to age-related dysfunction.
Here we examined whether the cholinergic system might constitute one such endogenous mechanism. Both the peripheral (Wang et al., 2003) and CNS (Shytle et al., 2004; De Simone et al., 2005; Lehner et al., 2019) are endowed with cholinergic pathways that regulate the magnitude of innate immune responses to pathogens, preventing excessive inflammation. In the periphery, cholinergic modulation through the vagus nerve can reduce systemic inflammation through nicotinic modulation of blood-based macrophages (Wang et al., 2003). In the CNS, nicotinic modulation also reduces inflammatory responses of microglia, indicating that the cholinergic anti-inflammatory pathway is highly conserved (Shytle et al., 2004; De Simone et al., 2005). Dysregulation of cholinergic modulation (e.g., due to an early age-related decline of cholinergic basal forebrain [BF] integrity) (Schmitz and Spreng, 2016; Fernández-Cabello et al., 2020) may therefore remove a key check on the proliferation of proinflammatory M1 microglia. However, this hypothesis has yet to be evaluated in living humans.
We examined whether the integrity of the central cholinergic system, indexed by longitudinal gray matter volume changes in the BF, is related to biomarkers of inflammation in preclinical AD. To do so, we differentiated a large group (n = 268) of cognitively normal older adults into neurotypical (NTYP) and preclinical (PREC) subgroups according to their CSF biomarkers of amyloid and tau pathology (see Fig. 1). We then examined whether loss of gray matter volume in the cholinergic BF, which is the major supplier of the brain's cholinergic input (Mesulam and Geula, 1988; Mesulam, 2013), is associated with variation in multiple biomarkers of inflammation, including soluble (s)TREM2 in the CSF and expression of TREM2 and C3 in the peripheral blood.
Figure 1.
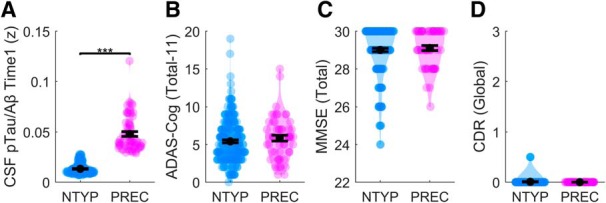
Staging the preclinical progression of AD according to CSF biomarkers of neuropathology and cognitive status. A, The NTYP phenotype is defined by CSF pTau/Aβ < 0.028 and ADNI diagnosis of clinically normal (nonconverter). The PREC CSF phenotype is defined by CSF pTau/Aβ ≥ 0.028 and normal cognitive function. B, We detected an additional effect of APOE genotype (y axis) orthogonal to the CSF grouping strategy (dotted line), characterized by higher CSF pTau/Aβ in ε4+ adults. C, D, All groups were clinically indistinguishable from one another on the Alzheimer's Disease Assessment Scale (y axis: ADAS). Violin plots represent kernel densities for each dataset. Each colored point represents a subject. Black circles represent means. Horizontal black lines indicate standard error of the mean.
Materials and Methods
Participant inclusion criteria
We selected Alzheimer's Disease Neuroimaging Initiative (ADNI) subjects who provided a CSF sample via lumbar puncture and at least two time points of structural MRI data. We then cross-referenced these individuals with their neuropsychological status, excluding those with a mild cognitive impairment (MCI) or AD diagnosis. Subjects who changed neuropsychological status (e.g., CN→MCI, or MCI→CN, MCI→AD, AD→MCI) over the course of ADNIGO or ADNI2 study phases were excluded. After these exclusion criteria were applied, 268 cognitively normal male and female human (Homo sapiens) participants remained. See Fig. 1-1), Table 1-1, and Table 1-2).
Grouping variables and dataset composition for the N=268 sample. Download Table 1-1, TIF file (53KB, docx)
Demographic characteristics of the N=268 sample. Download Table 1-2, TIF file (13.2KB, docx)
CSF biomarkers
CSF Aβ and pTau.
CSF samples were acquired at the baseline visit. CSF was measured using the Elecsys CSF immunoassays for Aβ and pTau on a cobase 601 analyzer (software version 05.02) at the Clinical Neurochemistry Laboratory, University of Gothenburg, Gothenburg, Sweden (BioFINDER) or at the Biomarker Research Laboratory, University of Pennsylvania (ADNI), according to the preliminary kit manufacturer's instructions and as described in previous studies (Bittner et al., 2016). The analyses and data are a revision in which CSF Aβ1–42 values exceeding the Elecsys protocol calibration (>1700 pg/ml) were computed using an extrapolation of the calibration curve (Schindler et al., 2018). The Elecsys Aβ1–42 protocol is currently under development, and the performance beyond this upper technical limit has not been formally established. These values are restricted to research purposes and excluded for clinical decision making. The technical report and datafile UPENNBIOMK9_04_19_17_NEW.csv are available at http://adni.loni.usc.edu. Units are in pg/ml.
CSF pTau/Aβ grouping strategy.
Head-to-head comparisons of AD biomarkers demonstrate that CSF Aβ1–42 is among the earliest to differentiate abnormal from normal age-related changes in neurophysiology (Trojanowski et al., 2010; Jack et al., 2013). However, the ratio of the pTau and Aβ1–42 Elecsys assays has been shown to supersede the individual biomarkers to distinguish PET-amyloid status (Schindler et al., 2018). Moreover, neuronal deposition of Aβ is closely (de Calignon et al., 2012; Ahmed et al., 2014; Khan et al., 2014; Pooler et al., 2015), and perhaps causally (Jacobs et al., 2018), related to pTau, accelerating its formation in neurons, its accumulation into neurofibrillary tangles, and its propagation to anatomically connected brain areas. For these reasons, the CSF ratio of pTau/Aβ1–42 has been proposed to have greater diagnostic utility, as it combines two different but interdependent pathological processes into a single measure (Fagan et al., 2011; Hansson et al., 2018).
CSF sTREM2.
The CSF sTREM2 measurements were acquired from the baseline and follow-up visits. Baseline visit codes were cross-checked and aligned with the same CSF samples used to quantitate Aβ and pTau. CSF sTREM2 was measured by an ELISA immunoassay using the meso-scale discovery (MSD) platform. The procedure is documented extensively in Kleinberger et al. (2014) and Suárez-Calvet et al. (2016a,b). Briefly, a subset of the samples from the same participants were run at two different centers to allow for cross-validation of the MSD protocol. Cross-site correlation was high (r = ∼77). Of the 1005 individuals assessed, 25 fell outside the predicted 98% tolerance level in cross-site covariation. These individuals were removed from subsequent analyses. Additionally, at each site, four internal standards were run on each ELISA plate. The internal standards were used to produce a mean correction factor accounting for interplate variability. Raw values were then multiplied by this correction factor, producing MSD-corrected values. These values were uploaded to the ADNI repository for each site. We used the MSD-corrected CSF TREM2 values from the larger Washington University sample. The technical report and datafile ADNI_HAASS_WASHU_LAB.csv are available at http://adni.loni.usc.edu. Units are in pg/ml. Annual percent changes in CSF TREM2 were computed as follows:
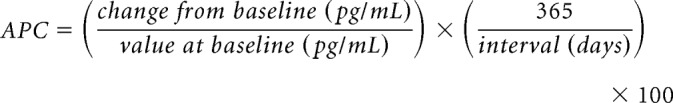 |
APOE genotyping
The ε4 allele (ε4) of APOE is the strongest known genetic risk factor for AD with a twofold to threefold increased risk for AD in people with one ε4 allele rising to ∼12-fold in those with two alleles (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993; Poirier et al., 1995). APOE genotyping was performed at the time of participant enrollment and included in the ADNI database. The two SNPs (rs429358, rs7412) that define the ε2, ε3, and ε4 alleles were genotyped using DNA extracted by Cogenics from a 3 ml aliquot of blood, with EDTA as in vitro anticoagulant. Individuals with either one or two ε4 alleles were classified as ε4+. The APOE genotypes for all individuals of the 268 cohort were obtained from the APOERES.csv spreadsheet available at http://adni.loni.usc.edu.
Serum gene expression data
Individuals in ADNI-GO/ADNI-2 contributed blood samples, which were submitted to microarray-based gene expression analysis. Only a single time point is currently available. Transcriptional profiles obtained from tissues in the CNS and from peripheral blood have been found to be correlated with one another (Liew et al., 2006; Sullivan et al., 2006; Maes et al., 2007; Grünblatt et al., 2009; Soreq et al., 2014). Moreover, anti-inflammatory cholinergic modulation of blood-based macrophages (Wang et al., 2003) and brain-based microglia (Shytle et al., 2004; De Simone et al., 2005) occurs via nicotinic receptors located on both cell types, indicating that this pathway is highly conserved. We therefore cross-referenced the unique 4-digit research identification (RID) numbers for all individuals of the 268 cohort with their gene expression data. The technical report and datafile ADNI_Gene_Expression_Profile.csv are available at http://adni.loni.usc.edu. Briefly, the quality of gene expression data, including sample quality and hybridization and overall signal quality, was analyzed using Affymetrix Expression Console software and Partek Genomic Suite 6.6, according to standard QC criteria provided by each software package. Raw expression values obtained directly from CEL files were preprocessed using the Robust Multichip Average (RMA) normalization method. The Affymetrix HG U219 Array contains 530,467 probes for 49,293 transcripts. All Affymetrix U219 probe sets were mapped and annotated with reference to the human genome (hg19). Using the RMA-normalized gene expression data, the Genetics Core completed additional QC steps. First, the gender of donors was checked using gender-specific gene expression data, such as XIST and USP9Y (Vawter et al., 2004). Second, sample identity was verified on the basis of gene expression profiling and Illumina Omni2.5M genotypes using a Bayesian method to predict individual SNP genotypes from only gene expression data (Schadt et al., 2012); that is, the 1000 most significant SNP-transcript cis-eQTL pairs from quality-controlled gene expression and genotype data were used to estimate a posterior probability for a match between gene expression and genotype data. Three questionable subjects were identified from the additional QC steps and removed. RMA-normalized gene expression data for our two a priori genes of interest, TREM2 and C3, were extracted from this dataset.
Longitudinal structural MRI
Data acquisition.
The unique 4-digit RID numbers for all individuals of the 268 cohort were entered as query terms into the ADNI IDA imaging database to obtain their structural MRI data. Additional search parameters were used to filter the search results. These included T1-weighted images that were (1) in native space, (2) acquired in the sagittal plane, (3) corrected for image distortion (GradWarp), and (4) B1 corrected for image nonuniformity. After download, we computed for each individual the maximum time intervals between their scans. We applied a bounded interval of a mean = 1.5 years ± 12 months.
High-resolution 3D T1-weighted MR images were acquired on 3.0T scanners using the ADNI-GO/ADNI-2 scan acquisition protocol: 8-channel coil, TR = 400 ms, TE = min full, flip angle = 11°, slice thickness = 1.2 mm, resolution = 256 × 256 mm, and FOV = 26 cm. The data were acquired in the sagittal plane using either an MPRAGE pulse sequence (Siemens and Philips scanners) or an IR-FSPGR pulse sequence (GE scanners), depending on the acquisition site. More information on the scan protocols for ADNIGO/2 are described by Jack et al. (2010).
Preprocessing.
For the longitudinal analysis (mean = 1.5 year ± 12 months), T1-weighted scans for both time points for a given subject were registered to one another using the symmetric diffeomorphic registration incorporated in the serial longitudinal anatomical MRI package in SPM12 (Ashburner and Ridgway, 2012). This procedure is optimized for longitudinal analysis by correcting for intensity inhomogeneities and creating an average T1-weighted image for each subject, avoiding asymmetric bias that can result from using a particular time point (e.g., the baseline image) as the reference image. Default parameters were used for warping regularization and bias regularization. The midpoint (average) map was scaled by the interscan interval. The longitudinal alignment step also produces Jacobian determinant maps for the baseline volume (Time1; J1) and follow-up scan (Time2; J2). The average (midpoint) scan was then bias-corrected to correct for regional variation in signal intensities before segmentation. We then segmented the average (midpoint) volume image into gray and white matter images with light regularization, a 60 mm bias FWHM cutoff, and Gaussians per tissue type of [2, 2, 2, 3, 4, 2]. The segmented gray matter volume (c1) was multiplied by the Jacobian deformation maps (J1 and J2) to produce c1*J1 and c1*J2 volumes. We then computed a population average template with DARTEL using all midpoint (average) c1 (gray) and c2 (white) matter segmentations (Ashburner, 2007). The deformation maps produced for each subject in this template-building step were then used to warp their c1*J1 and c1*J2 volumes into the population template space using DARTEL. This step used linear elastic regularization and “preserve amount,” with default parameters for the number of inner iterations, regularization parameters, and time steps. The intracranial volume (ICV) index was computed using the “Tissue Volumes” Utility in SPM12 (Malone et al., 2015) and the “*seg8.mat” segmentation file corresponding to the bias-corrected average (midpoint) volume produced by the longitudinal pipeline.
Quality control.
Modulated gray matter images were inspected using a two-step procedure that proceeded as follows: The first step used the “check sample homogeneity” tool in the SPM-VBM8 toolbox (version r435; http://dbm.neuro.uni-jena.de/vbm/check-sample-homogeneity/). This tool calculates the SD by the sum of the squared distance of each modulated gray matter 3D image from the sample mean. The second step involved visual inspection of all images at a common reference slice to cross-reference gray matter segmentation, signal intensity, and nonlinear warping across all subjects. Two subjects were removed from further analysis due to low-quality T1 scans.
ROI analysis
The BF ROI was produced using the probabilistic atlases (Zaborszky et al., 2008) used by the SPM Anatomy Toolbox (Eickhoff et al., 2005). The BF ROI was restricted to the nucleus basalis of Meynert (NbM) due to its high concentration of large projection cholinergic neurons (Mesulam, 2004). The NbM ROI is equivalent to areas Ch4 and Ch4p (Mesulam et al., 1983a,b; Mesulam and Geula, 1988). The Ch4p area is also referred to as the nucleus subputaminalis of Ayala, and it represents the rostrolateral extension of the NbM. Research by others on the subregional degeneration within the distributed nuclei of the BF has highlighted the Ch4p region as particularly vulnerable to early degeneration in amnestic MCI (Teipel et al., 2005, 2014; Grothe et al., 2012; Kilimann et al., 2014). Because of their neuroanatomical proximity to one another and higher susceptibility to AD neurodegeneration, we have considered the Ch4p and Ch4 regions as a single unit.
To minimize the amount of deformation needed to register this advanced age cohort to a common template, the NbM ROI was warped from MNI space into their DARTEL population template space (Grothe et al., 2012, 2013; Schmitz and Spreng, 2016). The registration of the NbM ROI to the population DARTEL template, and single-subject segmentations are displayed in Fig. 1-2.
Estimates of modulated GM volume (in units of milliliters) in the NbM and whole-brain gray matter (WBGM) ROIs at each time point were obtained by summing the total intensity of voxels falling within the ROI masking region divided by the number of voxels and a scaling factor for GM concentration (http://www0.cs.ucl.ac.uk/staff/g.ridgway/vbm/get_totals.m). Annual percentage change (Cavedo et al., 2017) of the NbM and WBGM ROIs was computed as follows:
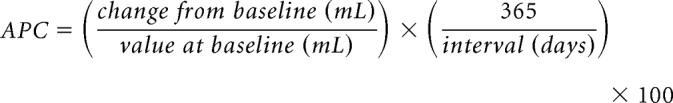 |
Statistical analyses
All primary ANOVAs and regression analyses included age, sex, education, and APOE genotype (ε4+, ε4−) as model covariates. Analyses modeling gray matter volume additionally covaried for ICV. Regression modeling was performed using the MATLAB function fitlm (version 9.4.0 R2018a, The MathWorks). All model covariates were z-scored to a common scale.
In all moderation analyses, the moderator variable (M) was the dichotomous group factor for CSF pTau/Aβ (NTYP vs PREC), which specified the conditions under which a given predictor is related to an outcome. The moderator M explains “when” a DV and IV are related, and therefore represents an interaction effect because the moderating variable changes the direction or magnitude of the relationship between two variables. A moderation effect could be (1) enhancing, where increasing M would increase the effect of the IV on the DV; (2) buffering, where increasing M would decrease the effect of the IV on the DV; or (3) antagonistic, where increasing M would reverse the effect of the IV on the DV. Moderation analyses were computed using a hierarchical multiple regression strategy (Hayes, 2012). Age, sex, education, and APOE genotype (ε4+, ε4−) were included as model covariates. Moderation analyses assessing gray matter volume additionally covaried for ICV. All moderation analyses used a heteroscedasticity consistent SE and covariance matrix estimator. All variables in each moderation analysis were first mean centered. Then, the direct linear relationships of the IV and M variables with the DV were assessed (block 1 model). The interaction term assessing the effect of the IV on the DV at each level of M is then added (block 2 model). If the R2 of the block 2 model is significant, then moderation is occurring.
Results
Partitioning cognitively normal older adults by CSF biomarker
Baseline CSF and neuropsychological data were acquired from the ADNI (Mueller et al., 2005), focusing on the ADNI-GO and ADNI-2 study phases (see Materials and Methods; Fig. 1; Fig. 1-1; Table 1-1 and Table 1-2). Older adults (mean age = 71.1 ± 6.71 years, 121 males, 147 females) were staged into distinct phenotypic groups of brain aging by integrating their CSF biomarker and neuropsychological information (Schmitz and Spreng, 2016; Schmitz et al., 2018; Fernández-Cabello et al., 2020).
Final cohort characteristics. A proportionally scaled Venn diagram illustrating overlap of the primary grouping variables (magenta) with the two primary measures of cholinergic BF degeneration: Longitudinal structural MRI (sMRI; blue), and Gene expression (orange). The green area indicates the component of our sample with fully overlapping datasets. Download Figure 1-1, TIF file (180.8KB, tif)
(Top) Coronal slice showing the registration of the NbM to the population DARTEL template. (Bottom) The registration of the NbM ROI in 2 example single subject brains after high-dimensional nonlinear warping from native to population template space. Download Figure 1-2, TIF file (530.1KB, tif)
We evaluated concentrations of the amyloid 1–42 peptide (Aβ) and tau phosphorylated at threonine 181 (pTau) using the values produced by the automated Elecsys protocol (Bittner et al., 2016; Hansson et al., 2018) (see Materials and Methods). Reduced CSF concentrations of Aβ index neuronal accumulation of insoluble amyloid-β plaques, whereas increased CSF concentrations of pTau index the release of tau proteins from damaged and dying neurons harboring tau neurites and neurofibrillary tangles (Shaw et al., 2009). The ratio of the pTau and Aβ (pTau/Aβ) was recently shown to supersede the individual biomarkers to distinguish abnormal PET-amyloid status (Schindler et al., 2018). Individuals falling above a pTau/Aβ ratio cutpoint of 0.028 are highly likely to develop AD. This cutpoint achieves ∼90% sensitivity and specificity to differentiating abnormal PET-amyloid status, and was recently cross-validated between two large independent cohorts (Hansson et al., 2018). We therefore differentiated cognitively normal adults with neurotypical (NTYP) age-related neuropathology (pTau/Aβ < 0.028, n = 212) from cognitively normal adults with “preclinical” (PREC) age-related neuropathology (pTau/Aβ ≥ 0.028, n = 56) (see Fig. 1A).
All groups were confirmed as cognitively normal across a range of neuropsychological batteries, including the Alzheimer's Disease Assessment Scale, Mini-Mental, or Clinical Dementia Rating Scale (Fig. 1B–D). On all measures, the NTYP and PREC groups were within cognitively normal ranges.
Longitudinal NbM degeneration interacts with blood C3 expression in PREC adults
We first examined baseline volume and follow-up gray matter volume in the BF NbM ROI (Fig. 2A). To do so, we computed a 2 (PREC, NTYP) × 2 (Time1, Time2) mixed ANOVA, covarying for age, sex, APOE genotype, education, and ICV. The model revealed a trend-level interaction suggestive of larger longitudinal decreases in the PREC compared with the NTYP group (F(1,211) = 3.48, p = 0.06; Fig. 2B). To assess this effect further, we computed within-subject indices (Time1 − Time2) of annual percent change relative to baseline volume, which additionally adjusts for variance in the longitudinal interval (see Materials and Methods), and entered these values into a nonparametric group comparison using the Kolmogorov–Smirnov (KS) test, which is more robust to unequal sample sizes than standard parametric t tests. The KS test significantly differentiated magnitudes of NbM degeneration between the NTYP and PREC groups (asymptotic p = 0.016). Moreover, one-sample t tests examining annual percent decreases in NbM gray matter relative to baseline volume were significantly >0 (no-change) in both the NTYP (t(172) = 5.39, p < 0.001) and PREC groups (t(44) = 5.00, p < 0.001). These findings are consistent with an accelerated early longitudinal loss of cholinergic BF neurons in PREC adults, which we previously demonstrated in an independent sample from the ADNI-1 cohort (Schmitz and Spreng, 2016).
Figure 2.
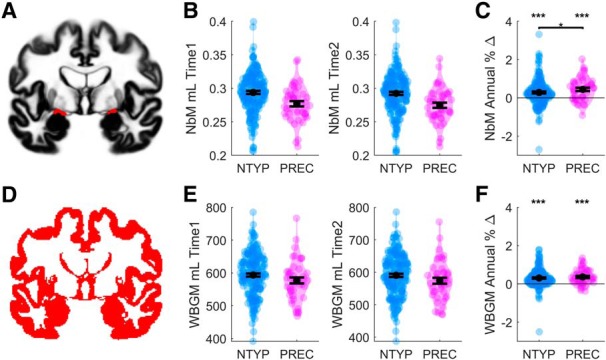
Longitudinal changes in NbM and WBGM gray matter volume in the NTYP and PREC groups. A, The NbM ROI is superimposed in red on a gray matter segmentation of the population DARTEL template space. B, Gray matter volume in the NbM (y axis: ml = milliliters) at baseline (Time1) and follow-up scans (Time2). C, Differences in NbM gray matter volume over time (Time1 − Time2) expressed as annualized % change relative to the baseline volume. D–F, Corresponding data for the WBGM ROI. Error bars indicate SEM. Violin plots represent kernel densities for each dataset. Each colored point represents a subject. Black circles indicate means. Black horizontal lines indicate standard error of the mean. ***p < 0.001, *p < 0.05.
Are the observed effects of NbM degeneration localized, or do they instead reflect patterns of WBGM volume loss, for example, due to cortical thinning in large spatially distributed brain areas? To evaluate this possibility, we also measured longitudinal degeneration in the WBGM (Fig. 2D) for comparison with NbM degeneration, again covarying for age, sex, APOE genotype, education, and ICV. We observed no group × time interaction (F(1,211) < 1; Fig. 2E), nor any group difference between annualized percent change (KS asymptotic p > 0.1; Fig. 2F). However, annual percent losses in the WBGM were significantly >0 (all p < 0.001), indicating a general background effect of aging equivalent to ∼0.3% per year in WBGM loss.
We then tested our first core hypothesis that increased damage to the cholinergic BF neurons (and their cortical cholinergic projections) would interact with biomarkers of inflammation in the presence of elevated amyloid and tau pathology. To do so, we surveyed a priori genes of interest TREM2 (noninflammatory marker) and C3 (proinflammatory marker) from the participant's blood transcriptome data. Although blood-based markers of TREM2 and C3 are not a direct assay of cortical tissue, microglia form a key component of the blood–brain barrier and participate in cross talk with perivascular macrophages and vascular endothelial cells (Bell and Zlokovic, 2009; Ryu and McLarnon, 2009; da Fonseca et al., 2014; Bekris et al., 2018).
We first examined whether there was a difference in the mean C3 expression between the NTYP and PREC groups. However, we observed no effect of group (KS asymptotic p > 0.1; Fig. 3A). We therefore examined whether variation in C3 at the level of individual difference within each group tracks with indices of gray matter degeneration. Regression modeling was accomplished in two steps: first examining the linear relationship of C3 with WBGM degeneration by itself, and then with NbM degeneration after accounting for WBGM degeneration. The multiple linear regressions in the NTYP and PREC groups covaried for age, sex, APOE genotype, education, and ICV. No relationship was detected between WBGM degeneration and C3 in either the NTYP (b = 0.01, t(104) < 1) or PREC (b = 0.08, t(22) < 1) groups. We therefore next assessed the unique contribution of degeneration in the NbM by adding this covariate to the regression model. Here we observed a significant positive relationship between magnitudes of NbM degeneration and C3 expression in the PREC group (b = 0.68, t(21) = 2.97, p = 0.007; Fig. 3B), but not the NTYP group (b = 0.12, t(103) < 1). Although NbM and WBGM degeneration was positively correlated (NTYP: r = 69, PREC: r = 0.46), variance inflation factors (VIFs) for all model covariates were low (<4), indicating that neither model was severely affected by multicollinearity (NTYP: VIF mean = 1.52, NbM = 1.98; PREC: VIF mean = 1.92, NbM = 1.69). Hence, consistent with a disruption of cholinergic regulation on inflammation, larger annual decreases in NbM volume selectively predicted higher C3 expression, over and above WBGM degeneration.
Figure 3.

C3 and TREM2 gene expression in the NTYP and PREC groups. A, There were no differences in the mean expression of C3 between NYTP and PREC adults. B, In PREC adults, increasing magnitudes of NbM gray matter loss (x axis: Time1 − Time2 expressed as annualized % change relative to the baseline volume) were associated with increasing expression of proinflammatory C3 (y axis: normalized gene expression at Time1), over and above longitudinal degeneration in the WBGM. A, B, Violin plots represent kernel densities for each dataset. Each colored point represents a subject. Black circles indicate means. Black horizontal lines indicate standard error of the mean. C, Moderation analysis revealed that the observed relationship between NbM degeneration and C3 in PREC adults was significantly different from NTYP adults. D–F, There were no differences in the mean expression of TREM2 between NYTP and PREC adults. The relationships between NbM degeneration and TREM2 were nonsignificant in the NTYP and PREC groups. All regression model covariates were z-scored to a common scale before splitting into PREC and NTYP subgroups. **p < 0.005, *p < 0.05.
We next evaluated with moderation analysis whether the strength of the relationship between NbM degeneration and C3 expression changed significantly as a function of amyloid and tau burden. To do so, we conducted a moderation analysis on this relationship by adding a dichotomous interaction term for the PREC versus NTYP subgroups, and again covarying for WBGM degeneration, as well as age, sex, APOE genotype, education, and ICV. The moderation model revealed a small but significant interaction effect (t(130) = 2.0, p = 0.047; Fig. 3C), demonstrating that the observed selective relationship between NbM degeneration and C3 expression in the PREC group was significantly strengthened by abnormal CSF pTau/Aβ.
We found no differences in the mean TREM2 expression level between the NTYP and PREC groups (KS asymptotic p > 0.1; Fig. 3D). Across individuals of each group, WBGM degeneration did not significantly covary with TREM2 expression (NTYP: b = 0.03, t(104) < 1; PREC: b = 0.14, t(22) < 1). Adding NbM degeneration as a covariate did not improve model fitting in this case: neither group exhibited a significant relationship between NbM degeneration and TREM2 (NTYP: b = 0.04, t(103) < 1; PREC: b = −0. 43, t(21) = 1.5, p > 0.1). Nevertheless, the patterns of covariation with TREM2 showed an inverted symmetry with those observed in the analyses of C3 expression (e.g., Fig. 3E vs Fig. 3B), suggestive of an opponent relationship between C3 and TREM2 expression. We therefore assessed the direct relationship between C3 and TREM2, independent of gray matter degeneration. Consistent with opponent relationships between proinflammatory and noninflammatory markers, we observed a significant negative relationship between TREM2 and C3 in the PREC adults, after covarying for age, sex, APOE genotype, and education (b = −0. 49, t(23) = 2.50, p = 0.02; Fig. 4A). No relationship was detected in the NYTP group (b = 0.12, t(109) = 1.29, p > 0.1). Moderation analyses revealed that the observed negative relationship in PREC adults was significantly different from NTYP adults (t(136) = 2.74, p = 0.007; Fig. 4B), indicating that this effect changes significantly as a function of abnormal CSF pTau/Aβ.
Figure 4.
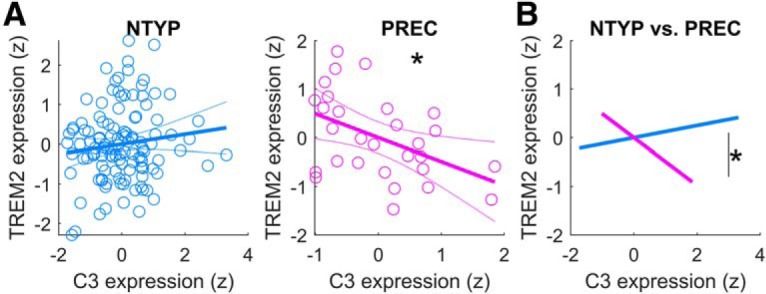
Direct interrelationships between C3 and TREM2 gene expression. A, In the PREC adults, increasing magnitudes of proinflammatory C3 expression (x axis: normalized expression at Time1) were associated with decreasing expression of noninflammatory TREM2 expression (y axis: normalized gene expression at Time1). B, Moderation analysis revealed that the observed relationship between C3 and TREM2 expression in PREC adults was significantly different from NTYP adults. All model covariates were z-scored to a common scale before splitting the PREC and NTYP subgroups. **p < 0.005, *p < 0.05.
Longitudinal NbM degeneration interacts with longitudinal CSF biomarkers of TREM2 shedding
Thus far, our findings indicate that in cognitively normal adults, increased NbM degeneration selectively interacts with biomarkers of increased inflammation in a molecular background of abnormal amyloid and phosphorylated tau accumulation, consistent with a loss of cholinergic anti-inflammatory signaling in preclinical AD. Moreover, TREM2 and C3 expression were negatively correlated, indicating that a loss of anti-inflammatory signaling may induce a tradeoff in the pool of noninflammatory and proinflammatory microglia (Liddelow and Barres, 2017).
TREM2 is selectively expressed by microglia in the CNS. Increased TREM2 on the microglial cell surface facilitates phagocytosis of accumulated amyloid (Kleinberger et al., 2014; Ewers et al., 2019; Parhizkar et al., 2019), a process that results in the cleavage of TREM2 and its release into the CSF as soluble (s)TREM2 (Kleinberger et al., 2014; Heslegrave et al., 2016; Piccio et al., 2016; Suárez-Calvet et al., 2016a,b). Although in AD this process is thought to be neuroprotective during the earliest stages of amyloid pathology (Keren-Shaul et al., 2017), the subsequent onset of tau pathology is associated with a large increase in sTREM2 (Nordengen et al., 2019; Suárez-Calvet et al., 2019), which may reflect the transition of microglia to a proinflammatory transcriptional profile (Leyns et al., 2017). Given the co-occurrence of abnormal amyloid and phosphorylated tau pathology in the PREC group, we predicted a larger longitudinal increase in sTREM2 in PREC compared with the NYTP group. Moreover, if cholinergic modulatory input imposes a check on microglial inflammation, we predicted that larger longitudinal decreases in NbM volume in the PREC adults would be associated with larger longitudinal increases in sTREM2.
To explore the first prediction, we examined baseline volume and follow-up CSF concentrations of sTREM2 in subsets of the same groups of NTYP and PREC individuals (where longitudinal data were available). A 2 (PREC, NTYP) × 2 (Time1, Time2) mixed ANOVA, covarying for age, sex, APOE genotype, and education revealed a trend-level interaction suggestive of larger longitudinal increases in sTREM2 in the PREC compared with the NTYP group (F(1,89) = 3.90, p = 0.05; Fig. 5A). Despite a pattern of larger increases in sTREM2 over time in the PREC compared with the NTYP group (Fig. 5A), when the within-subject differences (Time1 − Time2) of annual percent change relative to baseline concentration were computed and adjusted for the intersample interval (see Materials and Methods), the group difference was no longer statistically significant (asymptotic p > 0.1; see Fig. 5B). Annual percent increases in sTREM2 were significantly >0 (no-change) in both the NTYP (t(73) = 3.42, p = 0.001) and PREC groups (t(20) = 2.97, p = 0.008).
Figure 5.
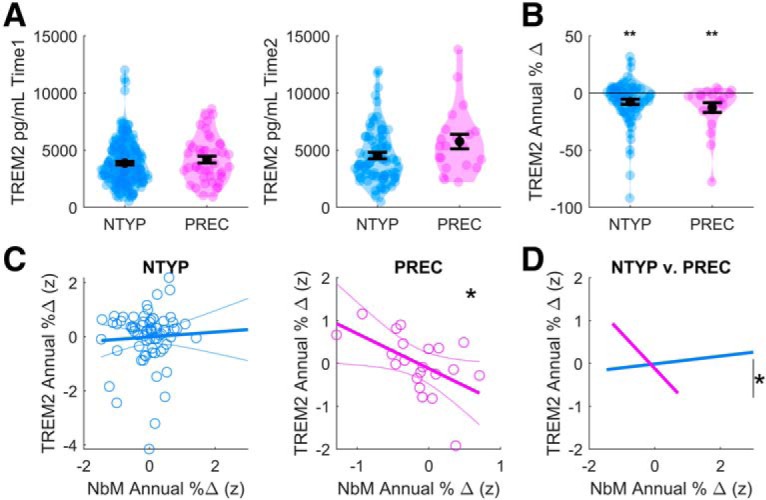
Longitudinal indices of CSF sTREM2. A, CSF sTREM2 concentrations (y axis: pg/ml) at baseline (Time1) and follow-up visits (Time2). B, Differences in CSF sTREM2 concentration over time (y axis: Time1 − Time2 expressed as annualized % change relative to the baseline concentration). A, B, Violin plots represent kernel densities for each dataset. Each colored point represents a subject. Black circles indicate means. Black horizontal lines indicate standard error of the mean. C, In the PREC adults, increasing magnitudes of NbM degeneration (x axis: Time1 − Time2 expressed as annualized % change relative to the baseline volume) were associated with larger increases in longitudinal CSF sTREM2 (Time1 − Time2 expressed as annualized % change relative to the baseline concentration), over and above WBGM degeneration. D, Moderation analysis revealed that the observed relationship between NbM degeneration and CSF sTREM2 proliferation in PREC adults was significantly different from NTYP adults. All model covariates were z-scored to a common scale before splitting the PREC and NTYP subgroups. *p < 0.05, **p < 0.005.
We next examined whether increased damage to the cholinergic BF (and its cortical cholinergic projections) would predict increased sTREM2 biomarkers of microglial TREM2 shedding in the CNS. As before, we stepped through two successive regression models to examine the unique contributions of WBGM and NbM degeneration to longitudinal changes in sTREM2. Covarying for age, sex, APOE genotype, education, and ICV, we found no relationship between WBGM degeneration and sTREM2 in either the NTYP (b = 0.10, t(62) < 1) or PREC groups (b = 0.09, t(14) < 1). We therefore next assessed the unique contribution of degeneration in the NbM by adding this covariate to the regression model. We observed a significant negative linear relationship (Fig. 5C), whereby larger annual decreases in NbM volume predicted larger annual increases in sTREM2 in the PREC group (b = −0.82, t(13) = 2.3, p = 0.037), over and above WBGM degeneration. No such relationship was detected in the NTYP group (b = 0.09, t(62) < 1). Moreover, we confirmed with moderation analysis that the strength of this relationship in PREC adults was significantly different from NTYP adults (t(81) = 2.32, p = 0.02). These results are consistent with the hypothesis that a loss of anti-inflammatory cholinergic afferent input might also disrupt neuroprotective microglial phagocytic activity, as indexed by larger longitudinal increases in sTREM2 in the presence of abnormal pTau and Aβ pathology.
The APOE ε4 genotype increases NbM degeneration and inflammation in PREC adults
Because the cholinergic neurons of the BF are exceptionally vulnerable to early degeneration in the APOE ε4 genetic background (Poirier, 1994; Poirier et al., 1995), we also leveraged genomic information to differentiate “high risk” ε4 carriers (ε4+) from lower risk noncarriers (ε4−) in the PREC group. We then conducted exploratory post hoc comparisons, surveying for group differences between APOE ε4+ and ε4− PREC adults across the multimodal measures used in the current study. These included the following: CSF amyloid 1–42 and pTau-181 (Fig. 6A), neuropsychological tests of general cognitive function (Fig. 6B), longitudinal structural MRI indices of annual percent change in NbM and WBGM volume (Fig. 6C), blood-based transcriptional markers of C3 and TREM2 gene expression (Fig. 6D), and longitudinal CSF indices of annual percent change in sTREM2 (Fig. 6E). Each exploratory two group ANOVA covaried for age, sex, education, and, where gray matter volumes were tested, ICV. All comparisons were nonsignificant with the exception of two: NbM degeneration (F(1,39) = 5.23, p = 0.028) and C3 expression (F(1,24) = 6.45, p = 0.018). These provisional findings suggest that a loss of anti-inflammatory cholinergic modulation over microglia, which we have observed in preclinical AD, is exacerbated by genetically imposed deficiencies in lipid metabolism.
Figure 6.

Effects of the APOE ε4 genotype on biomarker indices in the PREC group. A, No effects of APOE were observed in the CSF indices of Aβ 1–42 or phosphorylated tau-181 (y axes: units are in picograms over milliliters). B, No effects of APOE were observed in the neuropsychological tests for the ADAS cognitive total subscore, MMSE total score, or CDR global scores. C, NbM degeneration (y axis: Time1 − Time2 expressed as annualized % change relative to the baseline volume) was significantly larger in the PREC ε4+ compared with PREC ε4− adults. No effects of APOE were observed in the WBGM. D, C3 expression (y axis: normalized gene expression values) was significantly higher in the PREC ε4+ compared with PREC ε4− adults. No effects of APOE were observed in TREM2 expression. E, No effects of APOE were observed in longitudinal changes in CSF sTREM2 (y axis: Time1 − Time2 expressed as annualized % change relative to the baseline concentration). Violin plots represent kernel densities for each dataset. Each colored point represents a subject. Black circles indicate means. Black horizontal lines indicate standard error of the mean. *p < 0.05.
Discussion
Our findings indicate that BF damage interacts with biomarkers of inflammation in cognitively normal older adults with preclinical levels of pTau/Aβ. We observed increased longitudinal degeneration of the NbM in PREC compared with NTYP adults. We then demonstrated with moderation analyses three relationships that increased significantly as a function of PREC pTau/Aβ. First, abnormal pTau/Aβ strengthened the relationship between longitudinal NbM degeneration and biomarkers of C3 expression in the blood transcriptome, over and above degeneration of the WBGM volume. Second, TREM2 expression, which is associated with neuroprotective microglial functions, was negatively correlated with C3 expression. Third, we found that longitudinal decreases in NbM volume were associated with longitudinal increases in CSF sTREM2, over and above WBGM degeneration. Finally, we examined the impact of the APOE ε4 genotype on the preclinical aging phenotype, surveying all multimodal measures in our data fusion. The APOE ε4 genotype exacerbated markers of NbM degeneration and C3 expression, consistent with the vulnerability of the magnocellular cholinergic BF neurons to disruptions in lipid metabolism (Poirier, 1994; Poirier et al., 1995).
The “cholinergic anti-inflammatory pathway” was initially characterized in the peripheral nervous system after the discovery that systemic inflammatory responses of blood-based macrophages to pathogens are regulated by cholinergic afferent input originating from the vagus nerve (Borovikova et al., 2000; Tracey, 2002). The identity of the specific macrophage cholinergic receptor was subsequently localized to the α7 nicotinic acetylcholine receptor (α7 nAChR, CHRNA7), when α7 nAChR-deficient mice exhibited unchecked inflammation in response to Escherichia coli, despite vagal nerve stimulation (Wang et al., 2003). It was then found that inflammatory responses of microglia to lipopolysaccharides could also be suppressed by pharmacological modulation of α7 nAChR-mediated signaling, indicating that the anti-inflammatory cholinergic pathway is highly conserved (Shytle et al., 2004; De Simone et al., 2005). In both activated macrophages and microglia, increased cholinergic modulation of α7 nAChRs therefore appears to suppress release of proinflammatory cytokines, including TNFα, IL-1β, and IL-6, suggestive of a potential endogenous neuroprotective mechanism in the context of aging and AD (Carnevale et al., 2007; Egea et al., 2015). In more recent work, activated M1 microglia expressing precisely this proinflammatory molecular profile were found to induce proliferation of immunoreactive A1 reactive astrocytes, which trigger the complement cascade via upregulation of C3 and profound neurodegeneration (Liddelow et al., 2017). In the context of brain aging and AD, a core outstanding question concerns what factors lead to proliferation of the immunoreactive M1 and A1 transcriptional profile in the presence of age-related neuronal amyloid and tau accumulation, as opposed to more immunosuppressive M2 and A2 profiles (Zamanian et al., 2012; Martinez and Gordon, 2014; Heppner et al., 2015; Liddelow and Barres, 2017; Clarke et al., 2018). Our findings suggest that loss of afferent cholinergic input originating from the BF might constitute a key mechanism for the tradeoff between M2/A2 and M1/A1 trajectories.
The early pathogenesis of AD may therefore result from two interrelated events in the aging brain. Initially, damage to cholinergic neurons in the aging brain might result from disruptions in lipid metabolism, due, for example, to the APOE4 genetic background (Poirier, 1994; Poirier et al., 1995; Liu et al., 2013; Kanekiyo et al., 2014). In APOE knock-in mouse models and cell culture studies, Apoe4 glia secrete cholesterol and fatty acids, which are less lipidated and exhibit reduced neuronal transport compared with Apoe2/Apoe3 glia (Kanekiyo et al., 2014; Hu et al., 2015; Zhao et al., 2017). This loss of lipid transport function disrupts key pathways which neurons rely on to build, repair, and maintain their axons and synapses. Neurons encumbered with supporting large and complex axons, as well as the need to continually reshape and grow these axons throughout the adult lifespan, would theoretically be among the first affected by dysfunctions in the molecular pathways that enable axonal and synaptic remodeling. The cortically projecting cholinergic neurons of the NbM are severely constrained by both of these properties. Complete morphologies of individual cholinergic BF neurons have revealed >1000 cortical synapses per cell, with length estimates for the full arborization of a single neuron >100 m in humans (Wu et al., 2014). Moreover, due to the essential role of acetylcholine in cortical learning and memory, these neurons also maintain a high propensity for neuroplasticity throughout adulthood (Mesulam, 1999). Consistent with the vulnerability of cholinergic neurons to disrupted lipid metabolism in an APOE ε4 genetic background, we observed a significantly larger progression of NbM degeneration in the ε4+ PREC adults compared with ε4− PREC adults, despite equivalent levels of CSF Aβ and pTau (Fig. 6A,C). With a progressive loss of afferent cholinergic input in the brain, a progressive weakening of anti-inflammatory cholinergic signaling may enable M1 microglia to more freely proliferate in the presence of age-related Aβ and pTau accumulation. This would potentiate the second stage of runaway M1/A1 glial proliferation, inflammation and degeneration established in recent mouse models of AD (Liddelow et al., 2017; Shi et al., 2017b).
Despite contending with multiple vulnerabilities in the aging brain, the cholinergic BF neurons likely do not passively degenerate in AD. Cholinergic neurons can react to neuropathology through pretranscriptional and post-transcriptional regulation of ChAT, and possibly ApoE (Soreq and Seidman, 2001; Soreq, 2015). Recent RNA-sequencing work identified upregulated lipid processing transcripts in the temporal lobe of patients with AD neuropathology but no cognitive decline at the time of death (Barbash et al., 2017), suggesting possible involvement of ApoE in other brain regions vulnerable to early AD. With intact lipid metabolism (e.g., in an APOE ε4− genetic background), these endogenous mechanisms might sustain central cholinergic integrity and thus anti-inflammatory cholinergic signaling. Consistent with this possibility, in PREC ε4− adults, we observed that annual rates of NbM degeneration as well as C3 expression were indistinguishable from the NTYP group, despite significantly elevated CSF Aβ and pTau.
If anti-inflammatory cholinergic signaling is disrupted in AD due a loss of afferent cholinergic BF input, then restoration of anti-inflammatory cholinergic input (e.g., with pharmacological intervention) represents a promising therapeutic intervention. AChE inhibitors have been shown to significantly modulate innate immunity, possibly as a result of the increased availability of acetylcholine activating the cholinergic anti-inflammatory pathway (Pavlov et al., 2009; Pohanka, 2014). Moreover, accumulating evidence suggests that donepezil, a selective noncompetitive inhibitor of AChE, directly affects cellular functions via a mechanism that is independent of its AChE inhibition (Akasofu et al., 2008; Takada-Takatori et al., 2008). For instance, Hwang et al. (2010) reported that donepezil's anti-inflammatory effects on microglia cell lines occur in the absence of AChE activity, and suggested that donepezil directly stimulates α7 nAChRs. Our findings suggest that the anti-inflammatory effects of these drugs could potentially be substantially leveraged by integrating them with diagnostic screens of microglial function in cognitively normal older adults (e.g., using CSF sTREM2) as well as their APOE genotype (Poirier, 1994, 2008; Poirier et al., 1995). By identifying and treating individuals at earlier stages of disease, AChE inhibitors would potentially exert their suppressive effects on microglia at preventative stages of disease, before rampant cortical neurodegeneration.
The current study has limitations. We surveyed a priori genes of interest C3 and TREM2 in the blood transcriptome as surrogate in vivo markers of microglial reactivity to amyloid and tau pathology. Because microglia are an integral component of the neurovascular unit, cross talk between blood cells and microglia is thought to occur at the blood–brain barrier (Bell and Zlokovic, 2009; Ryu and McLarnon, 2009; da Fonseca et al., 2014). However, our findings require cross-validation from cell-type-specific transcriptomic data (e.g., from microglial cells derived in culture from human induced pluripotent stem cells or isolated from tissue biopsies in living patients). Moreover, future studies using next-generation RNA-sequencing techniques, which provide improved quantification of gene expression (Wang et al., 2009) as well as analyses of the recently discovered noncoding RNAs related to cholinergic function (Hanin et al., 2014; Zhou and Xu, 2015; Simchovitz et al., 2019), are needed to corroborate and extend the current microarray-based data. Finally, quantification of CSF C3 is possible with ELISAs, and has been successfully implemented in the ADNI-1 cohort (Bonham et al., 2016; Hu et al., 2016), but not the ADNI-GO/ADNI-2 cohort used for the current study. Conversely, blood transcriptomic data have been analyzed in the ADNI-GO/ADNI-2 cohort, but not the ADNI-1 cohort. Filling these gaps within ADNI, as well as cross-referencing homologous measures between ADNI and other large longitudinal in vivo human aging datasets (Meyer et al., 2018), will enable key multimodal analyses in much larger samples.
Together, our findings demonstrate that a loss of anti-inflammatory cholinergic inputs originating from the BF may represent a key mechanistic trigger for unchecked microglial reactivity to Aβ and pTau, setting the stage for runaway inflammation, cortical degeneration, and the cognitive prodrome of AD.
Footnotes
This work was supported in part by National Institute on Aging Grant R03 RAG060263A to T.W.S. and R.N.S., the Canada First Research Excellence Fund to T.W.S. and R.N.S., and Israel Science Foundation Grant 1016/18 to H.S. Data used in preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu/). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
The authors declare no competing financial interests.
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, Bose S, Clavaguera F, Tolnay M, Lavenir I, Goedert M, Hutton ML, O'Neill MJ (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127:667–683. 10.1007/s00401-014-1254-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasofu S, Kimura M, Kosasa T, Sawada K, Ogura H (2008) Study of neuroprotection of donepezil, a therapy for Alzheimer's disease. Chem Biol Interact 175:222–226. 10.1016/j.cbi.2008.04.045 [DOI] [PubMed] [Google Scholar]
- Ashburner J. (2007) A fast diffeomorphic image registration algorithm. Neuroimage 38:95–113. 10.1016/j.neuroimage.2007.07.007 [DOI] [PubMed] [Google Scholar]
- Ashburner J, Ridgway GR (2012) Symmetric diffeomorphic modeling of longitudinal structural MRI. Front Neurosci 6:197. 10.3389/fnins.2012.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash S, Garfinkel BP, Maoz R, Simchovitz A, Nadorp B, Guffanti A, Bennett ER, Nadeau C, Türk A, Paul L, Reda T, Li Y, Buchman AS, Greenberg DS, Seitz A, Bennett DA, Giavalisco P, Soreq H (2017) Alzheimer's brains show inter-related changes in RNA and lipid metabolism. Neurobiol Dis 106:1–13. 10.1016/j.nbd.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Khrestian M, Dyne E, Shao Y, Pillai JA, Rao SM, Bemiller SM, Lamb B, Fernandez HH, Leverenz JB (2018) Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease. J Neuroimmunol 319:19–27. 10.1016/j.jneuroim.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer's disease. Acta Neuropathol 118:103–113. 10.1007/s00401-009-0522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Zetterberg H, Teunissen CE, Ostlund RE Jr, Militello M, Andreasson U, Hubeek I, Gibson D, Chu DC, Eichenlaub U, Heiss P, Kobold U, Leinenbach A, Madin K, Manuilova E, Rabe C, Blennow K (2016) Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta-amyloid (1–42) in human cerebrospinal fluid. Alzheimers Dement 12:517–526. 10.1016/j.jalz.2015.09.009 [DOI] [PubMed] [Google Scholar]
- Bonham LW, Desikan RS, Yokoyama JS (2016) The relationship between complement factor C3, APOE epsilon4, amyloid and tau in Alzheimer's disease. Acta Neuropathol Commun 4:65. 10.1186/s40478-016-0339-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458–462. 10.1038/35013070 [DOI] [PubMed] [Google Scholar]
- Carnevale D, De Simone R, Minghetti L (2007) Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets 6:388–397. 10.2174/187152707783399193 [DOI] [PubMed] [Google Scholar]
- Cavedo E, Grothe MJ, Colliot O, Lista S, Chupin M, Dormont D, Houot M, Lehéricy S, Teipel S, Dubois B, Hampel H (2017) Reduced basal forebrain atrophy progression in a randomized donepezil trial in prodromal Alzheimer's disease. Sci Rep 7:11706. 10.1038/s41598-017-09780-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA (2018) Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 115:E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, Lima FR (2014) The impact of microglial activation on blood–brain barrier in brain diseases. Front Cell Neurosci 8:362. 10.3389/fncel.2014.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73:685–697. 10.1016/j.neuron.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L (2005) Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation 2:4. 10.1186/1742-2094-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97:463–472. 10.1016/j.bcp.2015.07.032 [DOI] [PubMed] [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005) A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25:1325–1335. 10.1016/j.neuroimage.2004.12.034 [DOI] [PubMed] [Google Scholar]
- Ewers M, Franzmeier N, Suárez-Calvet M, Morenas-Rodriguez E, Caballero MAA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M, Trojanowski JQ, Shaw LM, Weiner MW, Haass C (2019) Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Sci Transl Med 11:eaav6221. 10.1126/scitranslmed.aav6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Shaw LM, Xiong C, Vanderstichele H, Mintun MA, Trojanowski JQ, Coart E, Morris JC, Holtzman DM (2011) Comparison of analytical platforms for cerebrospinal fluid measures of beta-amyloid 1–42, total tau, and p-tau181 for identifying Alzheimer disease amyloid plaque pathology. Arch Neurol 68:1137–1144. 10.1001/archneurol.2011.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Cabello S, Kronbichler M, Van Dijk KR, Goodman JA, Spreng RN, Schmitz TW (2020) Basal forebrain volume reliably predicts the cortical spread of Alzheimer's degeneration. Brain, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel SJ (2012) Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer's disease. Biol Psychiatry 71:805–813. 10.1016/j.biopsych.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel S (2013) Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer's disease. Neurobiol Aging 34:1210–1220. 10.1016/j.neurobiolaging.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünblatt E, Bartl J, Zehetmayer S, Ringel TM, Bauer P, Riederer P, Jacob CP (2009) Gene expression as peripheral biomarkers for sporadic Alzheimer's disease. J Alzheimers Dis 16:627–634. 10.3233/JAD-2009-0996 [DOI] [PubMed] [Google Scholar]
- Hanin G, Shenhar-Tsarfaty S, Yayon N, Yau YH, Hoe YY, Bennett ER, Sklan EH, Rao DC, Rankinen T, Bouchard C, Geifman-Shochat S, Shifman S, Greenberg DS, Soreq H (2014) Competing targets of microRNA-608 affect anxiety and hypertension. Hum Mol Genet 23:4569–4580. 10.1093/hmg/ddu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, Lifke V, Corradini V, Eichenlaub U, Batrla R, Buck K, Zink K, Rabe C, Blennow K, Shaw LM (2018) CSF biomarkers of Alzheimer's disease concord with amyloid-beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 14:1470–1481 10.1016/j.jalz.2018.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes AF. (2012) PROCESS: a versatile computational tool for observed variable mediation, moderation, and conditional process modeling [White paper]. http://www.afhayes.com/public/process2012.pdf.
- Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372. 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Öhrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H (2016) Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener 11:3. 10.1186/s13024-016-0071-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G (2015) Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener 10:6. 10.1186/s13024-015-0001-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WT, Watts KD, Tailor P, Nguyen TP, Howell JC, Lee RC, Seyfried NT, Gearing M, Hales CM, Levey AI, Lah JJ, Lee EK (2016) CSF complement 3 and factor H are staging biomarkers in Alzheimer's disease. Acta Neuropathol Commun 4:14. 10.1186/s40478-016-0277-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Hwang H, Lee HW, Suk K (2010) Microglia signaling as a target of donepezil. Neuropharmacology 58:1122–1129. 10.1016/j.neuropharm.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, Dale AM, Carmichael OW, Tosun D, Weiner MW (2010) Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement 6:212–220. 10.1016/j.jalz.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HI, Hedden T, Schultz AP, Sepulcre J, Perea RD, Amariglio RE, Papp KV, Rentz DM, Sperling RA, Johnson KA (2018) Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat Neurosci 21:424–431. 10.1038/s41593-018-0070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G (2014) ApoE and abeta in Alzheimer's disease: accidental encounters or partners? Neuron 81:740–754. 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I (2017) A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169:1276–1290.e17. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, Small SA (2014) Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci 17:304–311. 10.1038/nn.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilimann I, Grothe M, Heinsen H, Alho EJ, Grinberg L, Amaro E Jr, Dos Santos GA, da Silva RE, Mitchell AJ, Frisoni GB, Bokde AL, Fellgiebel A, Filippi M, Hampel H, Klöppel S, Teipel SJ (2014) Subregional basal forebrain atrophy in Alzheimer's disease: a multicenter study. J Alzheimers Dis 40:687–700. 10.3233/JAD-132345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, et al. (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6:243ra286. 10.1126/scitranslmed.3009093 [DOI] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ (2009) Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 65:650–657. 10.1002/ana.21696 [DOI] [PubMed] [Google Scholar]
- Lehner KR, Silverman HA, Addorisio ME, Roy A, Al-Onaizi MA, Levine Y, Olofsson PS, Chavan SS, Gros R, Nathanson NM, Al-Abed Y, Metz CN, Prado VF, Prado MA, Tracey KJ, Pavlov VA (2019) Forebrain cholinergic signaling regulates innate immune responses and inflammation. Front Immunol 10:585. 10.3389/fimmu.2019.00585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM (2017) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A 114:11524–11529. 10.1073/pnas.1710311114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity 46:957–967. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA (2006) The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med 147:126–132. 10.1016/j.lab.2005.10.005 [DOI] [PubMed] [Google Scholar]
- Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM (2007) Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging 28:1795–1809. 10.1016/j.neurobiolaging.2006.08.004 [DOI] [PubMed] [Google Scholar]
- Malone IB, Leung KK, Clegg S, Barnes J, Whitwell JL, Ashburner J, Fox NC, Ridgway GR (2015) Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. Neuroimage 104:366–372. 10.1016/j.neuroimage.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M. (2004) The cholinergic lesion of Alzheimer's disease: pivotal factor or side show? Learn Mem 11:43–49. 10.1101/lm.69204 [DOI] [PubMed] [Google Scholar]
- Mesulam MM. (1999) Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron 24:521–529. 10.1016/S0896-6273(00)81109-5 [DOI] [PubMed] [Google Scholar]
- Mesulam MM. (2013) Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. J Comp Neurol 521:4124–4144. 10.1002/cne.23415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Geula C (1988) Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol 275:216–240. 10.1002/cne.902750205 [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI (1983a) Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience 10:1185–1201. 10.1016/0306-4522(83)90108-2 [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH (1983b) Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 214:170–197. 10.1002/cne.902140206 [DOI] [PubMed] [Google Scholar]
- Meyer PF, Savard M, Poirier J, Labonté A, Rosa-Neto P, Weitz TM, Town T, Breitner J (2018) Bi-directional association of cerebrospinal fluid immune markers with stage of Alzheimer's disease pathogenesis. J Alzheimers Dis 63:577–590. 10.3233/JAD-170887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, Trojanowski JQ, Toga AW, Beckett L (2005) Ways toward an early diagnosis in Alzheimer's disease: the Alzheimer's disease neuroimaging initiative (ADNI). Alzheimers Dement 1:55–66. 10.1016/j.jalz.2005.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, et al. (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordengen K, Kirsebom BE, Henjum K, Selnes P, Gísladóttir B, Wettergreen M, Torsetnes SB, Grontvedt GR, Waterloo KK, Aarsland D, Nilsson LNG, Fladby T (2019) Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation 16:46. 10.1186/s12974-019-1399-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, Krasemann S, Lichtenthaler SF, Müller SA, Colombo A, Monasor LS, Tahirovic S, Herms J, Willem M, Pettkus N, Butovsky O, et al. (2019) Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22:191–204. 10.1038/s41593-018-0296-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey KJ (2009) Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 23:41–45. 10.1016/j.bbi.2008.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Deming Y, Del-Águila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C (2016) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol 131:925–933. 10.1007/s00401-016-1533-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohanka M. (2014) Inhibitors of acetylcholinesterase and butyrylcholinesterase meet immunity. Int J Mol Sci 15:9809–9825. 10.3390/ijms15069809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier J. (1994) Apolipoprotein E in animal models of CNS injury and in Alzheimer's disease. Trends Neurosci 17:525–530. 10.1016/0166-2236(94)90156-2 [DOI] [PubMed] [Google Scholar]
- Poirier J. (2008) Apolipoprotein E represents a potent gene-based therapeutic target for the treatment of sporadic Alzheimer's disease. Alzheimers Dement 4:S91–S97. 10.1016/j.jalz.2007.11.012 [DOI] [PubMed] [Google Scholar]
- Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S (1995) Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci U S A 92:12260–12264. 10.1073/pnas.92.26.12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, Beier DR, Carlson GA, Spires-Jones TL, Hyman BT (2015) Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta Neuropathol Commun 3:14. 10.1186/s40478-015-0199-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG (2009) A leaky blood–brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med 13:2911–2925. 10.1111/j.1582-4934.2008.00434.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43:1467–1472. 10.1212/WNL.43.8.1467 [DOI] [PubMed] [Google Scholar]
- Schadt EE, Woo S, Hao K (2012) Bayesian method to predict individual SNP genotypes from gene expression data. Nat Genet 44:603–608. 10.1038/ng.2248 [DOI] [PubMed] [Google Scholar]
- Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, Wahl S, Benzinger TLS, Holtzman DM, Morris JC, Fagan AM (2018) Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement 14:1460–1469. 10.1016/j.jalz.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, Spreng RN (2016) Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nat Commun 7:13249. 10.1038/ncomms13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, Mur M, Aghourian M, Bedard MA, Spreng RN (2018) Longitudinal Alzheimer's degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Rep 24:38–46. 10.1016/j.celrep.2018.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ (2009) Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 65:403–413. 10.1002/ana.21610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA (2017a) Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med 9:eaaf6295. 10.1126/scitranslmed.aaf6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, et al. (2017b) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549:523–527. 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89:337–343. 10.1046/j.1471-4159.2004.02347.x [DOI] [PubMed] [Google Scholar]
- Simchovitz A, Hanan M, Niederhoffer N, Madrer N, Yayon N, Bennett ER, Greenberg DS, Kadener S, Soreq H (2019) NEAT1 is overexpressed in Parkinson's disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J 33:11223–11234. 10.1096/fj.201900830R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H. (2015) Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci 38:448–458. 10.1016/j.tins.2015.05.007 [DOI] [PubMed] [Google Scholar]
- Soreq H, Seidman S (2001) Acetylcholinesterase: new roles for an old actor. Nat Rev Neurosci 2:294–302. 10.1038/35067589 [DOI] [PubMed] [Google Scholar]
- Soreq L, Guffanti A, Salomonis N, Simchovitz A, Israel Z, Bergman H, Soreq H (2014) Long non-coding RNA and alternative splicing modulations in Parkinson's leukocytes identified by RNA sequencing. PLoS Comput Biol 10:e1003517. 10.1371/journal.pcbi.1003517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90:1977–1981. 10.1073/pnas.90.5.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C (2016a) Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 8:369ra178. 10.1126/scitranslmed.aag1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, Brendel M, Rominger A, Alcolea D, Fortea J, Lleó A, Blesa R, Gispert JD, Sánchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschütz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, et al. (2016b) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 8:466–476. 10.15252/emmm.201506123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Schlepckow K, Araque Caballero MÁ, Franzmeier N, Capell A, Fellerer K, Nuscher B, Eren E, Levin J, Deming Y, Piccio L, Karch CM, Cruchaga C, Shaw LM, Trojanowski JQ, Weiner M, Ewers M, Haass C (2019) Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related-neurodegeneration but not with amyloid-beta pathology. Mol Neurodegener 14:1. 10.1186/s13024-018-0301-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Fan C, Perou CM (2006) Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 141B:261–268. 10.1002/ajmg.b.30272 [DOI] [PubMed] [Google Scholar]
- Takada-Takatori Y, Kume T, Ohgi Y, Fujii T, Niidome T, Sugimoto H, Akaike A (2008) Mechanisms of alpha7-nicotinic receptor up-regulation and sensitization to donepezil induced by chronic donepezil treatment. Eur J Pharmacol 590:150–156. 10.1016/j.ejphar.2008.06.027 [DOI] [PubMed] [Google Scholar]
- Teipel SJ, Flatz WH, Heinsen H, Bokde AL, Schoenberg SO, Stöckel S, Dietrich O, Reiser MF, Möller HJ, Hampel H (2005) Measurement of basal forebrain atrophy in Alzheimer's disease using MRI. Brain 128:2626–2644. 10.1093/brain/awh589 [DOI] [PubMed] [Google Scholar]
- Teipel SJ, Flatz W, Ackl N, Grothe M, Kilimann I, Bokde AL, Grinberg L, Amaro E Jr, Kljajevic V, Alho E, Knels C, Ebert A, Heinsen H, Danek A (2014) Brain atrophy in primary progressive aphasia involves the cholinergic basal forebrain and Ayala's nucleus. Psychiatry Res 221:187–194. 10.1016/j.pscychresns.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ. (2002) The inflammatory reflex. Nature 420:853–859. 10.1038/nature01321 [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter WZ, Weiner MW, Jack CR Jr, Jagust W, Toga AW, Lee VM, Shaw LM (2010) Update on the biomarker core of the Alzheimer's disease neuroimaging initiative subjects. Alzheimers Dement 6:230–238. 10.1016/j.jalz.2010.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter MP, Evans S, Choudary P, Tomita H, Meador-Woodruff J, Molnar M, Li J, Lopez JF, Myers R, Cox D, Watson SJ, Akil H, Jones EG, Bunney WE (2004) Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsychopharmacology 29:373–384. 10.1038/sj.npp.1300337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388. 10.1038/nature01339 [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M (2009) RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J (2014) Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife 3:e02444. 10.7554/eLife.02444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, Hedehus M, Barck KH, Stark M, Ngu H, Foreman O, Meilandt WJ, Elstrott J, Chang MC, Hansen DV, Carano RA, et al. (2019) Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28:2111–2123.e6. 10.1016/j.celrep.2019.07.060 [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Hoemke L, Mohlberg H, Schleicher A, Amunts K, Zilles K (2008) Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. Neuroimage 42:1127–1141. 10.1016/j.neuroimage.2008.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Davis MD, Martens YA, Shinohara M, Graff-Radford NR, Younkin SG, Wszolek ZK, Kanekiyo T, Bu G (2017) APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet 26:2690–2700. 10.1093/hmg/ddx155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Xu J (2015) Identification of Alzheimer's disease-associated long noncoding RNAs. Neurobiol Aging 36:2925–2931. 10.1016/j.neurobiolaging.2015.07.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Grouping variables and dataset composition for the N=268 sample. Download Table 1-1, TIF file (53KB, docx)
Demographic characteristics of the N=268 sample. Download Table 1-2, TIF file (13.2KB, docx)
Final cohort characteristics. A proportionally scaled Venn diagram illustrating overlap of the primary grouping variables (magenta) with the two primary measures of cholinergic BF degeneration: Longitudinal structural MRI (sMRI; blue), and Gene expression (orange). The green area indicates the component of our sample with fully overlapping datasets. Download Figure 1-1, TIF file (180.8KB, tif)
(Top) Coronal slice showing the registration of the NbM to the population DARTEL template. (Bottom) The registration of the NbM ROI in 2 example single subject brains after high-dimensional nonlinear warping from native to population template space. Download Figure 1-2, TIF file (530.1KB, tif)