

Abstract
Background
Ferroptosis is a newly discovered form of iron-dependent oxidative cell death characterized by lethal accumulation of lipid-based reactive oxygen species (ROS). It is distinct from other forms of cell death including apoptosis, necrosis, and autophagy in terms of morphology, biochemistry and genetics.
Discussion
Ferroptosis can be induced by system xc- inhibitors or glutathione peroxidase 4 (GPx4) inhibitors, as well as drugs such as sorafenib, sulfasalazine (SAS), and artesunate (ART). Ferroptosis has been recently shown to be critical in regulating growth of tumors, such as hepatocellular carcinoma (HCC), renal cell carcinoma (RCC), non-small cell lung cancer (NSCLC), ovarian cancer, pancreatic carcinoma, and diffuse large B cell lymphoma (DLBCL). Ferroptosis is also associated with resistance to chemotherapeutic drugs and the anti-tumor efficacy of immunotherapy.
Conclusion
This review summarizes the mechanism of ferroptosis and its relationship with different types of tumors, to advance our understanding of cell death and to find a novel approach for clinical cancer management.
Keywords: Ferroptosis; iron metabolism; ROS; cancer; chemotherapeutic drug resistance,therapeutic approach
1. INTRODUCTION
Regulated Cell Death (RCD) plays a critical role in the normal development and maintenance of homeostasis in multicellular organisms [1]. Ferroptosis, a unique form of RCD, was first coined by Stockwell et al. in 2012 to describe a non-apoptotic form of iron-dependent oxidative cell death [2, 3]. This unique RCD was first discovered in 2003 when a small molecule called erastin was found to possess the ability to trigger a non-apoptotic cell death process specifically in RAS-mutated tumor cells [4]. Later in 2008, another compound named RAS-selective lethal small molecular-3 (RSL-3) was also suggested to possess similar properties as erastin through a high-throughput small molecule-screening study [5]. This newly discovered mechanism of iron-dependent cell death is characterized by cellular iron-dependent aberrant accumulation of reactive oxygen species (ROS) and is morphologically, biochemically, and genetically distinct from apoptosis, necrosis, and autophagy (Table 1) [2].
Table 1. Basic features and characteristics of ferroptosis.
| Morphological Feature | Biochemical Feature | Genetic Feature | Inducers | Inhibitors |
|---|---|---|---|---|
| Smaller mitochondria; increased membrane density; Reduced mitochondrial crista |
Can not be modulated by compounds known to be inhibitors of regular cell death, such as caspase, cathepsin or calpain proteases (z-VAD-fmk, E64d or ALLN),RIPK1 (necrostatin-1), cyclophilin D (cyclosporin A | Required for erastin-induced ferroptosis: RPL8, IREB2, ATP5G3, CS,TTC35, ACSF2 |
Class 1 ferroptosis inducers: erastin, erastin derivatives (aldehyde erastin, morpholine erastin II, and piperazine erastin),buthionine sulfoximine, DPI2, glutamate, lanperisone, sulfasalazine, SRS13-45, SRS13-60); Class 2 ferroptosis inducers: DPI7, DPI10, DPI12, DPI13, DPI17, DPI18, DPI19, ML162, and RSL3; Drugs: Sorafenib and artemisinin derivatives (artesunate) |
Antioxidants: Vitamin E, Trolox, U0126; inhibit ROS formation: Ferroptatin-1, SRS8-72, SRS11-92, SRS12-45, SRS13-35, SRS13-37, SRS16-86; Iron chelators: Desferoxamine, 2,2-bipyridy1, Ciclopirox olamine; Protein synthesis inhibitor: Cycloheximide) Transaminase inhibitor: Aminooxyacetie acid |
Excessive iron-dependent oxidative stress has been shown to partially contribute to carcinogenesis [6]. In addition, cancer cells exhibited persistently high levels of ROS-related oxidative stress due to genetic alterations and aberrant proliferation [7]. Thus, agents affecting ROS metabolism can influence tumor cell growth. Erastin and RSL-3-termed ferroptosis inducers-have demonstrated the ability to kill rat sarcoma viral oncogene (RAS)-mutated cancer cells by ferroptosis [4, 5]. Therefore, it is rational to apply inducers of ferroptosis to cancer management. Manz et al. classified the inducers of ferroptosis into three types based on their specific targets: class 1 ferroptosis inducers, class 2 ferroptosis inducers, and drugs including sorafenib and artemisinin derivatives. Moreover, inhibitors of ferroptosis are categorized into five types, which include antioxidants, ROS inhibitors, iron chelators, protein synthesis inhibitors, and transaminase inhibitors [6]. Furthermore, an investigation of 114 cancer cell lines showed that diffuse large B cell lymphomas (DLBCLs) and renal cell carcinomas (RCCs) were highly vulnerable to erastin [8]. Kim et al. revealed that ultra-small nanoparticles can induce ferroptosis of nutrient-deprived cancer cells and suppress tumor growth, which further validates the role of ferroptosis inducers in killing tumor cells and inhibiting tumor growth [9].
This review is an overview of ferroptosis summarizing the mechanisms and signaling pathways of ferroptosis and the relationship between inducers of ferroptosis with diverse tumors, so as to provide novel prospects for cancer management.
2. MECHANISM OF FERROPTOSIS
Mechanistically, ferroptosis is caused by intracellular iron overload and lethal accumulation of ROS. Yang et al. have identified two main targets of ferroptosis induced by erastin and RSL3 [10].
2.1. Inhibition of System xc- Induces Ferroptosis by Erastin
Cystine/glutamate exchange system (system xc-)-a sodium-dependent antiporter composed of 4F2hc (SLC3A2) and xCT (SLC7A11)-has been identified as a mediator for the 1:1 exchange of extracellular cystine and intracellular glutamate, and for the conversion of intracellular cystine into cysteine, which is required for the synthesis of glutathione (GSH) [11, 12]. GSH is essential for restoring intracellular redox balance upon generation of ROS, and the depletion of GSH would lead to ROS accumulation, which can impede cellular antioxidant defense mechanism [13]. In 2012, Dixon and his colleagues demonstrated that in NRAS mutant HT-1080 fibrosarcoma cells, erastin acted as a system xc- inhibitor to impede cysteine-dependent GSH synthesis by decreasing cystine uptake, eventually inducing ferroptosis via lethal accumulation of cytosolic and lipid ROS [2]. Thus, system xc- is required for erastin-induced ferroptosis. Moreover, β-mercaptoethanol (β-ME) has been found to strongly inhibit erastin, sulfasalazine (SAS), and glutamate activity, but not RSL3-induced cell death in HT-1080 cells, by promoting cystine uptake through another pathway, which further confirms system xc- function in erastin-induced ferroptosis [2, 14, 15]. Other similar ferroptosis inducers that can trigger ferroptosis via the inhibition of system xc- were discovered later. Dixon et al. found that SAS and sorafenib (BAY 43-9006, Nexavar) can selectively trigger iron-dependent cell death by blocking system xc- (SLC7A11 + SLC3A2) mediated cystine uptake in HT-1080 and Calu-1 cells [15]. Moreover, a glutamate release assay in HT-1080 and Calu-1 cells revealed that erastin is approximately 2500 times more potent than SAS as an inhibitor of system xc-, suggesting that erastin acts as a highly potent inducer of ferroptosis [15]. However, erastin-induced cell death and ROS increase are suppressed by the iron chelator deferoxamine (DFO, 100μm), and this process could be potentiated by exogenous iron, both of which confirmed the requirement of iron for ferroptosis [2]. The function of iron in ferroptosis has been identified through the Fenton reaction [16], and the iron status is suggested to be related to the sensitivity of cancer cells to ferroptosis [17]. Similar ferroptosis inhibitors including ferrostatin-1 (Fer-1) and the MEK inhibitor U0126 have also been discovered [18]. In addition, Yang et al. demonstrated that GSH depletion is essential for erastin lethality because supplementing the culture medium with GSH or N-acetylcysteine (NAC), a biosynthetic precursor to GSH, could prevent erastin-induced cell death [10]. However, they found that the four BJ-derived cell lines treated with antioxidant inhibitors including an SOD inhibitor (DETC), a thiol-reactive reagent (DIA), a thioredoxin reductase inhibitor (DCNB), or a catalase inhibitor (ATZ), displayed selective lethality without GSH depletion, suggesting that unique biochemical and metabolic changes downstream of GSH depletion are likely to be responsible for the selective induction of ferroptosis [10]. In BJeLR cells treated with GSH-depleting reagents (erastin or BSO), GPXs are inactivated due to GSH depletion, resulting in excessive generation of cytoplasmic and lipid ROS [10]. Studies further demonstrated that CHAC1 mRNA is upregulated in response to system xc- inhibitors such as erastin analogs, and CHAC1 upregulation can be completely reversed by co-treatment with β-ME, indicating that CHAC1 upregulation may specifically predict system xc- function inhibition [10, 15, 19]. Sun et al. have demonstrated that heat shock protein beta-1 (HSPB1) is a negative regulator of ferroptosis in cancer cells and erastin induces the expression of heat shock factor 1 (HSF1)-dependent HSPB, the overexpression of which inhibits erastin-induced ferroptosis in cancer cells [20].
It is well known that system xc- is widely expressed in a variety of cells in the Central Nervous System (CNS) and cancer cells. The blockage of system xc- can not only inhibit cystine intake, but also inhibit intracellular glutamate release, which can lead to an alternative oxidative iron-dependent cell death, termed oxytosis or oxidative glutamate toxicity [21]. Ferrostatin-1, a small molecule, inhibits ferroptosis in cancer cells and glutamate-induced cell death in organotypic rat-brain slices, suggesting similarities between the above two processes [2]. Previous studies revealed that oxytosis can be initiated either by calcium influx after glutamate receptor activation or competitive inhibition of system xc-, indicating the involvement of ferroptosis in glutamate-induced toxicity [22, 23]. However, Wolpaw et al. demonstrated that calcium chelators are unable to prevent erastin-induced cell death, suggesting that glutamate receptor activation is not involved in ferroptosis [24].
Mechanistically, ferroptosis induced by system xc- inhibitors such as erastin, SAS, or sarefanib causes a significant decrease in the intracellular GSH levels and promotes lethal accumulation of ROS and iron-dependent cell death due to disruption of cellular redox balance (Fig. 1).
Fig. (1).
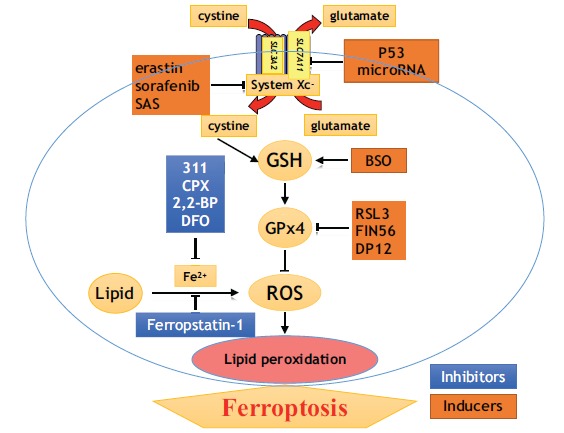
Inducers and inhibitors of ferroptosis.
2.2. Direct Inhibition of Glutathione Peroxidase 4 (GPx4) by RSL3
Erastin and RSL3 share common ferroptotic features [25]. However, in contract to erastin, RSL3-induced ferroptosis does not decrease GSH levels or inhibit Na+-independent uptake of cystine [2, 10], which suggests that RSL3 may demonstrate a different ferroptosis-initiating mechanism. In addition, affinity-based chemoproteomics revealed that selective lethality in the BJ cell system is effected only when the (1S, 3R)-RSL3 diastereomer is covalently bound to one or more proteins. Moreover, three different LC-MS/MS analyses showed GPX4 as the best candidate protein target for (1S, 3R)-RSL3 due to its well-characterized lipid peroxidation inhibiting protein (PIP), which can degrade H2O2 and small hydroperoxides [26]. Treatment of a clone of the COH BR1 breast cancer cell line overexpressing GPX4 (L7G4) with (1S, 3R)-RSL3 did not reduce 7a-cholesterol-OOH (a GPX4-specific substrate) levels [10]. These findings indicate that GPX4 is directly inhibited by (1S, 3R)-RSL3. Furthermore, a large screening experiment identified 10 other compounds that exhibit selective lethality for HRAS in the four BJ-derived cell lines [10]. Treatment of BJeLR cells with any of the eight FIN compounds (DPI7, DPI10, DPI12, DPI13, DPI17, DPI18, DPI19, and RSL3) exhibited GPX4 inactivation without GSH depletion, and subsequently resulted in cell ferroptosis [10]. However, RNAi-mediated GPX4 knockdown in HT-1080 cells led to lipid ROS accumulation-related-cell death that can be inhibited by RSL3 suppressors, including an iron chelator (DFO), a MEK inhibitor (U0126), and an antioxidant (vitamin E). These findings suggest that inhibition of GPx4 activity is the primary inducer of ferroptosis [10]. Recent studies have demonstrated GPx4 to function as a key regulator for a variety of inducer-initiated ferroptosis, including erastin and RSL3 [10].
Furthermore, GPX4 was found to essentially regulate membrane peroxide levels that, in turn, affect lipoxygenase (LOX) activity [27]. Several studies have demonstrated the efficacy of LOX inhibitors in preventing erastin-induced ferroptosis in MEFs and pancreatic cancer cells, suggesting that LOX can influence chemical-induced ferroptosis [28, 29]. Shintoku et al. have also demonstrated the ability of ALOX15 to regulate cell ferroptosis by localizing to the cell membrane, indicating that tumor ferroptosis is promoted by LOX-catalyzed lipid hydroperoxide production in cellular membranes [30].
2.3. Other Associated Mechanisms
Yagoda et al. have identified VDAC2/3 as a target for erastin. RNAi-mediated knockdown of VDAC2 or VDAC3 confers resistance to erastin. Moreover, erastin changed the permeability of the outer mitochondrial membrane. These findings suggest that the tumors harboring activating mutations can be targeted at VDAC proteins and induce ferroptosis in the RAS-RAF-MEK pathway [5].
Recent investigations have demonstrated that accumulation of the mutant p53 (a tumor suppressor protein) can downregulate SLC7A11 expression through binding to NRF2 (negative regulator of ferroptosis) and thus promote ferroptosis in tumor cells by reducing cystine intake [31, 32]. In addition, Wang et al. found that K98 acetylation of p53 can facilitate p53-mediated ferroptosis in a xenograft mouse model by downregulating SLC71A11 expression [33]. A recent study has also demonstrated GLS2-a p53 metabolic target-to induce ferroptotic cell death by promoting glutaminolysis [34]. However, the role of the p53 gene in the regulation of ferroptosis remains unknown.
Long non-coding RNA (lncRNA) has been reported to be important for the regulation of ferroptosis. Mao et al. have revealed the cytosolic P53RRA-G3BP1 interaction as a novel mechanism of ferroptosis, in which transcription of several metabolic genes (positive correlation with RIMBP3C, TIGAR, GPR162, and TIAI; negative correlation with SLC1A5, SLC7A11, HNRNPC, and BATF3) is affected. This effect resulted in an increase in the intracellular concentrations of iron and lipid ROS and eventually led to ferroptosis in lung cancer cells [35]. Another recent study demonstrated that lncRNA LINC00336 functions as a competing endogenous RNA and could inhibit ferroptosis in lung cancer via binding to RNA-binding protein ELAVL1 (ELAV-like RNA-binding protein 1) using nucleotides 1901–2107 of LINC00336 and the RRM interaction domain and key amino acids (aa) of ELAVL1 (aa 101-213) [36]. Many recent studies have revealed microRNAs as important mediators for ferroptosis. Different microRNAs have different targets. For example, miR-9 can inhibit ferroptosis by downregulating glutamic-oxaloacetic transaminase 1 (GOT1) expression, thereby resulting in ROS accumulation and ferroptosis [37]. SLC7A11 is a target for miR-375, miR-27a, miR-26b, and a reduction in its expression can promote ferroptosis [38-40]. Furthermore, microRNAs can regulate ferroptosis by targeting Nrf2, iron metabolism-related genes (transferrin (TF), transferrin receptor 1 (TFR1), ferroportin (FPN), divalent metal transporter 1 (DMT1), ferritin heavy chain 1 (FTH1), and ferritin light chain (FTL)), acyl-CoA synthetase long-chain family member 4 (ACSL4) (ROS generation-associated gene), and ROS [41-49].
3. RELATIONSHIP BETWEEN FERROPTOSIS INDUCERS AND TUMOR CELLS
Erastin and RSL3 inducers have been previously shown to trigger ferroptosis in diverse tumor cells in vitro by promoting inhibition of system xc- and GHx4. Therefore, ferroptosis inducers have been used reasonably for cancer therapy. The following context contains all the current data for ferroptosis inducers used in the management of tumorous diseases. Type 1 molecules, including erastin, sorafenib (elastin analogue), and safasalazine (SASP), inhibit extracellular cystine uptake by targeting system xc-; Type 2 molecules bind directly to GHx4, a precursor of GSH, to impede the metabolism of lipid peroxidation and results in lethal accumulation of ROS, such as RSL3 [25] (Table 2).
Table 2. Inducers of ferroptosis in diverse tumor cells.
| Inducers | Tumor Types | Inhibitors | Decrease Drugs Resistant | Refs. |
|---|---|---|---|---|
| erastin | AML(HL-60 cells) | ferrostatin-1/necrostatin-1 c | cytarabine/ara-C and doxorubicin/adriamycin | [9] |
| erastin | GC cells | Suppression of CDO1 | N/A | [7] |
| Erasin or SASP | GC cells | cisplatin | [50] | |
| dm-Erastin | Pancreatic cancer patient-derived xenograft | N/A | N/A | [51] |
| erastin | Wt EGFR nsclc cells | N/A | Cisplatin | [52] |
| Erastin and SASP | HNC cells | N/A | Cisplatin | [53] |
| erastin | GBM cells | N/A | TMZ | [54] |
| erastin | RMS13 cells | Ferrostatin-1 | PI103 | [55] |
| sorafenib | HCC cells | ferrostatin-1 | N/A | [56] |
| sorafenib | HCC, kidney cancer, melanoma, lung carcinoma, pancreatic adenocarcinoma and colon carcinoma cells | N/A | N/A | [57] |
| sorafenib | HCC cells | N/A | N/A | [59, 60-62] |
| SASP | lymphoma cells | N/A | N/A | [63, 64] |
| Artemisinin derivatives | 60 tumor cell lines | N/A | N/A | [65] |
| ART | ovarian cancer cells,PDAC cell | ferrostatin-1 | N/A | [66] |
| RSL3 | DLBCL cells | ferrostatin-1 | N/A | [68] |
3.1. Erastin
Of the 117 cancer cell lines originating from different tissues including hematopoietic and lymphoid tissues as well as those from the large intestine, lung, ovary, and skin that were tested, erastin was found to be particularly sensitive to DLBCL cells [8]. Furthermore, ferroptosis inducers-erastin and BSO-could enhance sensitivity of acute lymphoblastic leukemia (ALL) cells to LCL161, suggesting a novel mechanism for ALL therapy [50]. Yu et al. demonstrated that erastin can sensitize acute myeloid leukemia (AML) cells to chemotherapy and low dose of erastin could significantly enhance the anticancer activity of chemoagents (cytarabine/ara-C and doxorubicin/adriamycin) in HL-60 cells. Moreover, erastin could inhibit the growth of HL-60 cells by inducing ferroptosis and necroptosis [51]. The major MAPKs-c-JUN N-terminal kinase (JNK) and p38- were found to participate in erastin-induced cell death in HL-60 cells, which was consistent with the findings of Yagoda et al. who showed that erastin selectively induced ferroptosis of selected tumor cells through the RAS-RAF-MEK pathway [18, 51]. An investigation of gastric cancer(GC) cells revealed that erastin can induce ferroptosis in GC cells and the silencing of cysteine dioxygenase 1 (CDO1) can inhibit ferroptosis of erastin-induced GC cells both in vivo and in vitro [52]. Wang et al. have demonstrated that repeated cisplatin treatment in gastric cancer cells can induce cellular resistance to cisplatin via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway, and GC cells can be sensitized to cisplatin by xCT inhibitors (sulfasalazine or erastin), xCT siRNA, or a GSH synthesis inhibitor (buthionine sulphoximine, BSO) [53]. Ohman et al. treated syngeneic and pancreatic cancer patient-derived xenograft models with a drug named SW V-49 (consists of sigma-2 ligand SV119 and an erastin derivative, des-methyl Erastin (dm-Erastin)) and showed a decrease in the tumor size and double extension of the median survival of xenograft, which indicated a new therapeutic utility of erastin for the treatment of patients with pancreatic cancer [54]. In the wild-type EGFR non-small cell lung cancer, erastin enhanced the effect of cisplatin [55]. An NCI60 originating from eight tissues demonstrated increased sensitivity of erastin to renal cell carcinomas [8]. Another study revealed that erastin and SAS can inhibit head and neck cancer cell (HNC) growth by increasing lipid ROS. In addition, they were shown to enhance the effect of cisplatin to resistant HNC cells [56]. Chen et al. found that GSH and ROS levels were closely associated with the sensitivity of glioblastoma (GBM) cells to temozolomide (TMZ) and erastin was able to sensitize TMZ-resistant GBM cells to TMZ, indicating that the combination of TMZ and erastin may be a better therapeutic strategy for patients with GBM [57]. A study also revealed the role of erastin in inducing ferroptotic cell death in rhabdomyosarcoma (RMS13) cells; however, the process was found to be inhibited by ferrostatin-1. Besides, they also reported that overexpression of RAS can attenuate erastin- or RSL3-induced ferroptosis, which was related to the resistance of PI3K/mTOR inhibitor(PI103) in RMS13 cells [58].
In summary, erastin can not only induce ferroptosis in diverse tumor cells, but also re-sensitize chemoresistance (especially cisplatin and TMZ) of cancer cells. However, the mechanism requires further investigation.
3.2. Sorafenib
Sorafenib is the only effective medicine that is able to kill HCC cells by triggering ferroptosis. However, DFX (an iron chelator), pharmacological inhibitors (ferrostatin-1), and genetic procedures (RNA interference against IREB-2) can significantly prevent cytotoxicity of sorafenib in HCC cell lines [59]. Louandre et al. revealed the ability of sorafenib to induce ferroptosis in different cancers including HCC, kidney cancer, melanoma, lung carcinoma, pancreatic adenocarcinoma, and colon carcinoma. They also found that the increased sensitivity of sorafenib and erastin was independent of oncogenic mutations including RAS, RAF, PIK3CA, and TP53 genes of cancer cells [60]. Another study demonstrated that the expression of the metallothionein-1 (MT1) gene family members was increased in the HCC cell line Huh7 when exposed to sorafenib and resulted in an upregulated MT1G mRNA expression, suggesting that an increase of serum MTI may be predictive of poor prognosis of HCC patients treated with sorafenib [61]. Sun et al. also found that metallothionein (MT)-1G is associated with sorafenib resistance in human HCC cells that could be overcome via genetic and pharmacological inhibition of MT-1G [61]. Both studies emphasized on the necessity of the activation of the transcription factor NRF2 (nuclear factor erythroid 2-related factor 2) for inducing MT-1G expression following sorafenib treatment [61, 62]. Sun et al. demonstrated that the p62-Keap1-NRF2 pathway plays a critical role in ferroptosis in HCC cells [61]. Several studies have suggested that NRF2 partly contributes to ferroptosis resistance by increasing the expression of genes associated with heme, iron, and ROS metabolism, such as the NRF2 target gene quinone oxidoreductase-1 (NQO1), heme oxygenase-1 (HO), and ferritin heavy chain 1 (FTH1). NRF2 is a key regulator for resistance to sorafenib- and erastin-induced ferroptosis in HCC cells [63]. Bai et al. showed that sorafenib and erastin increase the expression of Sigma 1 receptor (S1R) protein, which was related to oxidative stress metabolism. They also demonstrated that an SIR antagonist, haloperidol, can significantly promote sorafenib- and erastin-induced ferroptosis in HCC cells [64]. Thus, the combination of drugs may achieve a better outcome of HCC patients. The retinoblastoma (Rb) protein is important for hepatic tumorigenesis. Louandre et al. found that HCC cells with reduced levels of Rb protein present a two- to three-fold increase in cell death when exposed to sorafenib compared to that with the control. Moreover, Balb/c HCC xenografts mice characterized by reduced levels of Rb showed complete tumor regression in 50% of sorafenib-treated animals, which is consistent with the in vitro findings [65]. Therefore, the Rb status might be considered as another sorafenib response prediction marker.
In shortly, sorafenib was mainly focused on the management of advanced HCC.
3.3. Sulfasalazine (SASP), Artemisinin Derivatives and ART
Lymphoma cells treated with SASP demonstrated reduced lymphoma cell proliferation in vitro. Moreover, an in vivo experiment conducted on system xc- deficient Nb2-U17 rat transplanted with lymphoma demonstrated that SASP can inhibit cysteine secretion by tumor-associated somatic cells (macrophages and dendritic cells) and cause cysteine starvation of tumor cells and apoptosis [66, 67]. Artemisinin derivatives (artesunate, artemether, arteether, artenimol, artemisitene, arteanuin B, another monomeric artemisinin derivative, and three artemisinin dimer molecules) have been reported to induce ferroptosis in a panel of 60 tumor cell lines (NCI60) and a panel of genes associated with iron metabolism (transferrin (TF), transferrin receptors 1 and 2 (TFRC, TFR2), cerulopasmin (CP), lactoferrin (LTF)) have been identified as predictive biomarkers of tumor sensitivity to artemisinin drugs [68]. Artesunate (ART) is a well-tolerated anti-malarial drug. A study confirmed that ART not only inhibits the growth of ovarian cancer cell lines, but also patient-derived ovarian cancer cells in vitro [69]. Moreover, an in vivo research study on a mouse model of ovarian cancer showed reduced tumor growth [54]. In another study, ART specifically induced ROS- and lysosomal iron-dependent cell death in PDAC cell lines and this process was inhibited by ferrostatin-1. Furthermore, ductal pancreatic cancer with KRAS mutation was found more sensitive to ferroptosis [70].
In conclusion, SASP induces ferroptosis in lymphoma cells and ART in ovarian cancer and pancreatic cancer cells.
3.4. RSL and Other Agents
Yang et al. demonstrated that RSL3 and erastin can inhibit tumor growth in athymic nude mice implanted with subcutaneous (s.c.) xenograft tumors derived from BJeLR cells. They also confirmed its lethality in two DLBCL cell lines including SU-DHL-8 and WSU-DLCL-2 [8]. A previous study has revealed that SLCA711 is overexpressed in approximately 70% of human cancers, including colon, liver, and kidney tumors [31]. Agent P53 targeted at SLCA711 inhibited ROS-induced ferroptosis and blocked p533KR-mediated tumor growth suppression in xenograft models [31]. Another study revealed that MON (induces Fenton reaction) encapsulated with p53 plasmid (MON-p53) not only induces ferroptosis in cancer cells, but can also reduce metastasis of blood, lung, and liver cells via the intracellular oxidative stress pathway, suggesting an association between ferroptosis and tumor metastasis [71].
In addition, some clinical drugs including lapatinib (tyrosine kinase inhibitor), BAY87-2243 (inhibitor of NADH-coenzyme Q oxidoreductase), lanperisone, and antibiotics including salinomycin and ironomycin have been found to induce ferroptosis in diverse cancer cell lines [72-75].
SUMMARY AND CONCLUSION
Numerous studies have demonstrated that ferroptosis inducers play an important role in suppressing tumor growth and killing tumor cells. In addition, inducers such as erastin and its analogues as well as sorafenib were able to re-sensitize chemoresistant cancer cells including glioblastoma cells, non-small cell lung cancer cells, and head and neck cancer cells [55-57]. Therefore, the combination of chemotherapeutic drugs and ferroptosis inducers can be prospectively used to improve chemoefficiency and achieve a better outcome of cancer patients. However, further studies are required to investigate the specific mechanism. Studies have shown that sensitivity to ferroptosis differs among different tissue cells. Wang et al. have demonstrated that the anti-tumor efficacy of immunotherapy is partially caused by ferroptosis that is induced by downregulated SLC3A2 and SLC7A11 expression and impairment of cystine uptake by interferon gamma released from immunotherapy-activated CD8+ T cells. Thus, T cell-promoted tumor ferroptosis is a novel anti-tumor mechanism and targeting this pathway in combination with checkpoint blockade is a potential therapeutic approach [76]. Moreover, some biomarkers may predict tumor sensitivity to sorafenib and artemisinin derivatives such as MT1 and iron-related genes (TF, TFRC, and TFR2) [61, 68]. However, the role of ferroptosis in tumors is not fully understood due to limitations in the number of experiments and lack of clinical trials. Ferroptosis has been gaining renewed interest from researchers and we believe that it will potentially serve as a novel strategy for cancer treatment.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by research grants from the New Xiangya Talent Project of the Third Xiangya Hospital of Central South University (JY201524).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Gao M., Monian P., Jiang X., et al. Metabolism and iron signaling in ferroptotic cell death. Oncotarget. 2015;6(34):35145–35146. doi: 10.18632/oncotarget.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon S.J., Lemberg K.M., Lamprecht M.R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J.Y., Dixon S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016;73(11-12):2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolma S., Lessnick S.L., Hahn W.C., et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 5.Yang W.S., Stockwell B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toyokuni S., Ito F., Yamashita K., et al. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 7.Cramer S.L., Saha A., Liu J., Tadi S., et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017;23(1):120–127. doi: 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WS. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S.E., Zhang L., Ma K., et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016;11(11):977–985. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang WS. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato H., Tamba M., Ishii T., et al. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 12.Conrad M., Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42(1):231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 13.Lewerenz J., Hewett S.J., Huang Y., et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013;18(5):522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishii T., Bannai S., Sugita Y., et al. Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. Role of the mixed disulfide of 2-mercaptoethanol and cysteine. J. Biol. Chem. 1981;256(23):12387–12392. [PubMed] [Google Scholar]
- 15.Dixon S.J., Patel D.N., Welsch M., et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e2523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torti S.V., Torti F.M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer. 2013;13(5):342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manz D.H., Blanchette N.L., Paul B.T., et al. Iron and cancer: recent insights. Ann. N. Y. Acad. Sci. 2016;1368(1):149–161. doi: 10.1111/nyas.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yagoda N., von Rechenberg M., Zaganjor E., et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon S.J., Patel D.N., Welsch M., et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e2523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X., Ou Z., Xie M., et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan S., Schubert D., Maher P., et al. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001;1(6):497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 22.Bridges R.J., Natale N.R., Patel S.A., et al. System xc (-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012;165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehta A., Prabhakar M., Kumar P., et al. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013;698(1-3):6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 24.Wolpaw A.J., Shimada K., Skouta R., et al. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc. Natl. Acad. Sci. USA. 2011;108(39):E771–E780. doi: 10.1073/pnas.1106149108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang W.S., Stockwell B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brigelius-Flohe R., Maiorino M. Glutathione peroxidases. Biochim. Biophys. Acta. 2013;1830(5):3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Friedmann Angeli J.P., Schneider M., Proneth B., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai H., Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 29.Xie Y., Song X., Sun X., et al. Identification of baicalein as a ferroptosis inhi- bitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016;473:775–780. doi: 10.1016/j.bbrc.2016.03.052. [DOI] [PubMed] [Google Scholar]
- 30.Shintoku R., Takigawa Y., Yamada K., et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017;108:2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L., Kon N., Li T., et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu D.S., Duong C.P., Haupt S., et al. Inhibiting the system xC (-) /glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017;8:14844. doi: 10.1038/ncomms14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S.J., Li D.W., Ou Y., et al. Acetylation is crucial for p53-mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17(2):366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao M., Monian P., Quadri N., et al. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao C., Wang X., Liu Y., et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78(13):3484–3496. doi: 10.1158/0008-5472.CAN-17-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M., Mao C., Ouyang L., et al. Long. noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019;••• doi: 10.1038/s41418-019-0304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K., Wu L., Zhang P., et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018;57:1566–1567. doi: 10.1002/mc.22878. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y., Sun X., Song B., et al. MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017;6:1686–1697. doi: 10.1002/cam4.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drayton R.M., Dudziec E., Peter S., et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin. Cancer Res. 2014;20:1990–2000. doi: 10.1158/1078-0432.CCR-13-2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X.X., Li X.J., Zhang B., et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011;585:1363–1367. doi: 10.1016/j.febslet.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Kabaria S., Choi D.C., Chaudhuri A.D., et al. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015;89:548–556. doi: 10.1016/j.freeradbiomed.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Babu K.R., Muckenthaler M.U. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J. Mol. Med. (Berl.) 2016;94:347–359. doi: 10.1007/s00109-015-1362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshioka Y., Kosaka N., Ochiya T., et al. Micromanaging iron homeostasis: hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J. Biol. Chem. 2012;287:34110–34119. doi: 10.1074/jbc.M112.356717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kindrat I., Tryndyak V., de Conti A., et al. MicroRNA-152-mediated dysregulation of hepatic transferrin receptor 1 in liver carcinogenesis. Oncotarget. 2016;7:1276–1287. doi: 10.18632/oncotarget.6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shpyleva S.I., Tryndyak V.P., Kovalchuk O., et al. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011;126:63–71. doi: 10.1007/s10549-010-0849-4. [DOI] [PubMed] [Google Scholar]
- 46.Andolfo I., De Falco L., Asci R., et al. Regulation of divalent metal transporter 1 (DMT1) non-IRE isoform by the microRNA let-7d in erythroid cells. Haematologica. 2010;95:1244–1252. doi: 10.3324/haematol.2009.020685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu X., Zhi F., Lun W., et al. Baicalin inhibits PDGF-BB-induced hepatic stellate cell proliferation, apoptosis, invasion, migration and activation via the miR-3595/ACSL4 axis. Int. J. Mol. Med. 2018;41:1992–2002. doi: 10.3892/ijmm.2018.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Zheng S., Geng Y., et al. MicroRNA profiling of atrial fibrillation in canines: miR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD1. PLoS One. 2015;10:e0122674. doi: 10.1371/journal.pone.0122674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyrychenko S., Kyrychenko V., Badr M.A., et al. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015;108:324–334. doi: 10.1093/cvr/cvv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hass C., Belz K., Schoeneberger H., et al. Sensitization of acute lymphoblastic leukemia cells for LCL161-induced cell death by targeting redox homeostasis. Biochem. Pharmacol. 2016;105:14–22. doi: 10.1016/j.bcp.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 51.Yu Y., Xie Y., Cao L., et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015;2(4):e1054549. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao S., Yu J., He W., et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia. 2017;19(12):1022–1032. doi: 10.1016/j.neo.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S.F., Chen M.S., Chou Y.C., et al. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2alpha-ATF4-xCT pathway. Oncotarget. 2016;7(45):74132–74151. doi: 10.18632/oncotarget.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohman K.A., Hashim Y.M., Vangveravong S., et al. Conjugation to the sigma-2 ligand SV119 overcomes uptake blockade and converts dm-Erastin into a potent pancreatic cancer therapeutic. Oncotarget. 2016;7(23):33529–33541. doi: 10.18632/oncotarget.9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamaguchi H., Hsu J.L., Chen C.T., et al. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 2013;19(4):845–854. doi: 10.1158/1078-0432.CCR-12-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roh J.L., Kim E.H., Jang H.J., et al. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381(1):96–103. doi: 10.1016/j.canlet.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 57.Chen L., Li X., Liu L., et al. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol. Rep. 2015;33(3):1465–1474. doi: 10.3892/or.2015.3712. [DOI] [PubMed] [Google Scholar]
- 58.Schott C., Graab U., Cuvelier N., et al. Oncogenic RAS Mutants Confer Resistance of RMS13 Rhabdomyosarcoma Cells to Oxidative Stress-Induced Ferroptotic Cell Death. Front. Oncol. 2015;5:131. doi: 10.3389/fonc.2015.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louandre C., Ezzoukhry Z., Godin C., et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer. 2013;133(7):1732–1742. doi: 10.1002/ijc.28159. [DOI] [PubMed] [Google Scholar]
- 60.Lachaier E., Louandre C., Godin C., et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34(11):6417–6422. [PubMed] [Google Scholar]
- 61.Houessinon A., Francois C., Sauzay C., et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer. 2016;15(1):38. doi: 10.1186/s12943-016-0526-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun X., Niu X., Chen R., et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi: 10.1002/hep.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun X., Ou Z., Chen R., et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bai T., Wang S., Zhao Y., et al. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017;491(4):919–925. doi: 10.1016/j.bbrc.2017.07.136. [DOI] [PubMed] [Google Scholar]
- 65.Louandre C., Marcq I., Bouhlal H., et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2 Pt B):971–977. doi: 10.1016/j.canlet.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 66.Gout P.W., Simms C.R., Robertson M.C., et al. In vitro studies on the lymphoma growth-inhibitory activity of sulfasalazine. Anticancer Drugs. 2003;14(1):21–29. doi: 10.1097/00001813-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 67.Gout P.W., Buckley A.R., Simms C.R., et al. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 68.Ooko E., Saeed M.E., Kadioglu O., et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22(11):1045–1054. doi: 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 69.Greenshields A.L., Shepherd T.G., Hoskin D.W., et al. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017;56(1):75–93. doi: 10.1002/mc.22474. [DOI] [PubMed] [Google Scholar]
- 70.Eling N., Reuter L., Hazin J., et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532. doi: 10.18632/oncoscience.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng D.W., Lei Q., Zhu J.Y., et al. Switching Apoptosis to Ferroptosis: Metal-Organic Network for High-Efficiency Anticancer Therapy. Nano Lett. 2017;17(1):284–291. doi: 10.1021/acs.nanolett.6b04060. [DOI] [PubMed] [Google Scholar]
- 72.Ma S., Dielschneider R.F., Henson E.S., et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12:e0182921. doi: 10.1371/journal.pone.0182921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buccarelli M., Marconi M., Pacioni S., et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018;9:841. doi: 10.1038/s41419-018-0864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen Z., Song J., Yung B.C., et al. Emerging strategies of cancer therapy based on ferroptosis. Adv. Mater. 2018;30:e1704007. doi: 10.1002/adma.201704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shaw A.T., Winslow M.M., Magendantz M., et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA. 2011;108:8773–8778. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang W., Green M., Choi J.E., et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]