

Abstract
Subtype selectivity and functional bias are vital in current drug discovery for G protein-coupled receptors (GPCRs) as selective and biased ligands are expected to yield drug leads with optimal on-target benefits and minimal side-effects. However, structure-based design and medicinal chemistry exploration remain challenging in part because of highly conserved binding pockets within subfamilies. Herein, we present an affinity mass spectrometry approach for screening herbal extracts to identify active ligands of a GPCR, the 5-HT2C receptor. Using this method, we discovered a naturally occurring aporphine 1857 that displayed strong selectivity for activating 5-HT2C without activating the 5-HT2A or 5-HT2B receptors. Remarkably, this novel ligand exhibited exclusive bias toward G protein signaling for which key residues were identified, and it showed comparable in vivo efficacy for food intake suppression and weight loss as the antiobesity drug, lorcaserin. Our study establishes an efficient approach to discovering novel GPCR ligands by exploring the largely untapped chemical space of natural products.
Short abstract
An affinity mass spectrometry approach is developed for screening herbal extracts to identify active ligands for a GPCR. A novel aporphine ligand with a unique pharmacological profile was discovered.
Introduction
Belonging to the superfamily of G protein-coupled receptors (GPCRs), the serotonin (5-hydroxytryptamine, 5-HT) receptors mediate a plethora of physiological processes in the brain and the periphery.1 The human genome encodes 13 5-HT receptors that exert the biological effects of serotonin, and eight are drug targets for the treatment of obesity, migraine, anxiety, depression, and hypertension.1,2 Among them, the serotonin 2C receptor (5-HT2C) is recognized as a promising therapeutic target for obesity and central nervous system (CNS) disorders, such as epilepsy, schizophrenia, and drug abuse.2−4 The value of 5-HT2C in antiobesity medication development is manifested by the FDA-approved drug lorcaserin, a 5-HT2C selective agonist.1 Moreover, the efficacy of lorcaserin in the treatment of nicotine addiction is currently being evaluated clinically.5
The development of 5-HT2C agonists as potential antiobesity and antipsychotic medications requires high selectivity over other subfamily members, the 5-HT2A and 5-HT2B receptors, whose activation is associated with hallucination6 and cardiac valvulopathy.7,8 For example, due to their off-target activities at 5-HT2B, the nonselective serotoninergic drugs, fenfluramine and pergolide, were withdrawn from markets, and the drug cabergoline has been restricted.8−10 Even the safety of lorcaserin has been questioned due to its moderate selectivity (∼100-fold) over 5-HT2B.11,12 However, developing subtype-selective agonists for 5-HT2C is challenging owing to the highly conserved ligand-binding pockets among the three 5-HT2 members.13−15 To date, only a handful of scaffolds have been disclosed as selective 5-HT2C agonists, all of which were obtained through extensive medicinal chemistry exploration.16−20
The concept of signaling bias or functional selectivity has recently reshaped our understanding of GPCR signaling and shifted the paradigm for GPCR drug discovery.21,22 Signaling bias refers to a process whereby GPCR ligands can either activate G proteins or recruit β-arrestins to mediate specific downstream signaling pathways for a given receptor.23,24 Biased GPCR ligands, which can trigger a specific pathway responsible for a given therapeutic effect while not activating other pathways that are implicated in side-effects, possess significant potential to become drug leads with optimal on-target benefits.25,26 For example, G protein-biased μ-opioid receptor agonists are potentially analgesic but have reduced side-effects (e.g., respiratory depression and constipation).27,28 Although an increasing number of biased ligands have been discovered for different GPCRs,26,29−32 very few for 5-HT2C have been reported. Recently, a class of compounds based on the (2-phenylcyclopropyl)methylamine scaffold synthesized by Chen et al. and Zhang et al. exhibited functional selectivity at 5-HT2C with preference to Gq-mediated calcium flux.16,19 However, synthesizing compounds with both signaling bias and subtype selectivity remains a major obstacle for medicinal chemists.
A rich resource for generating tool compounds and drug leads is natural herbs, as their chemical constituents typically possess molecular architectures and bioactivities that are distinct from synthetic molecules.33,34 To expedite ligand discovery for various protein targets from natural products, a number of approaches have been developed, ranging from cell-based activity or biosensor-based binding assays to in silico screening.33,35 Unlike most screening platforms that examine individual pure compounds from a library, affinity mass spectrometry (MS) can directly capture and detect putative ligands from crude natural product extracts toward a protein target.36−40 Although affinity MS has shown great potential in discovering inhibitors or modulators of enzymes and other soluble protein targets,38,39,41−43 it has never been explored in GPCR ligand screening from natural products.
In this study, we adapted the affinity MS technique to discover new ligands for 5-HT2C from a collection of natural product extracts. Emerging from this screen was a unique family of aporphine alkaloids, a rarely investigated chemotype for this target. Guided by the affinity MS screening data, we were able to isolate two novel aporphine ligands for pharmacological characterization. For one ligand (R)-asimilobine (1857) that acted as a selective 5-HT2C agonist with exclusive G protein signaling bias, key residues for 5-HT2C activation were then identified by molecular docking and mutagenesis. Finally, we compared this herb-derived novel agonist against the approved drug lorcaserin for in vivo antiobesity effects.
Results
Selection of Natural Herbs for Affinity MS Screening
To select natural herbs for the affinity MS screen of 5-HT2C agonists, we first assayed bioactivities of crude extracts from 15 different plants using the calcium flux assay that separately measures Gq-coupled activities of the 5-HT2C receptor and two close family members, 5-HT2A and 5-HT2B receptors. The total extracts from eight herbs showed the agonist property at 5-HT2C with potency spanning from nM to μM (assuming an average molecular weight of 500 Da for small molecule constituents) (Figure 1A). Given that major constituents typically account for less than 1% of the total weight, we speculated that agonists with low nanomolar potency might be present in the extracts of Aristolochia debilis (AD) and Tetradium ruticarpum (TR) showing the highest potency at 5-HT2C. Moreover, among the eight herbs with 5-HT2C agonism, five also activated 5-HT2A and 5-HT2B with similar potency (Figure 1B,D). However, this does not exclude the possibility that individual components in the extracts may possess subtype selectivity. Therefore, these five extracts were selected for additional screening with the affinity MS approach.
Figure 1.

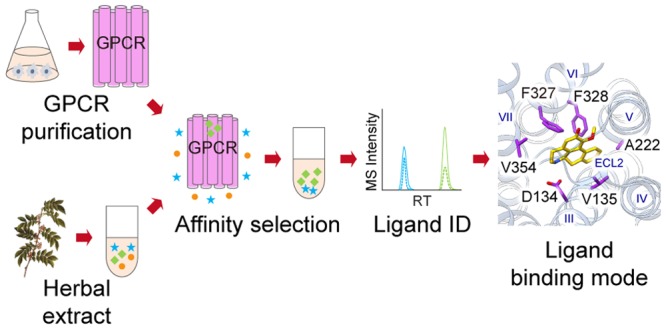
Herbal extracts showing Gq-mediated activity at 5-HT2C were selected for affinity MS screening. (A) Calcium mobilization elicited by three 5-HT2 subtypes treated with different herbal extracts or 5-HT. Full names of the herbs used are listed in the Methods section. NA, not active. (B–D) Dose–response characteristics of calcium mobilization elicited by top 5 extracts or 5-HT at 5-HT2C, 5-HT2B, and 5-HT2A. Crude extract concentrations were calculated from weight, assuming an average molecular weight of 500 Da for small molecule constituents. (E) Workflow of affinity MS-based 5-HT2C ligand screening. A putative ligand (green) and a nonspecific binder (blue) are distinguished based on the MS intensity of each compound detected in the 5-HT2C target vs control.
We first prepared the apo 5-HT2C protein fused with a stabilizing partner15 without any mutation and staying in a homogeneous monomeric conformation after purification (Figure S1). The purified receptor immobilized on magnetic beads through an epitope tag was then incubated with a defined compound mixture. Ligand-bound 5-HT2C complexes were enriched by magnetic separation from the solution phase. Bound ligands were dissociated from the protein target and subjected to liquid chromatography coupled to high-resolution mass spectrometry (LC-HRMS) analysis (Figure 1E). Another purified GPCR (hydroxyl carboxylic acid receptor 2, HCA2) was immobilized and processed in the same manner to serve as a negative control (Figure S1). Quantitative comparison of the extracted ion chromatograms of individual compounds detected in the target versus the control allowed us to distinguish specific 5-HT2C ligands from nonspecific binders (Figure 1E). This method was first validated using a mixture of known 5-HT2C ligands and unrelated compounds. All agonists and antagonists with high affinity (Ki < 50 nM) were associated with the receptor in the affinity MS assay, while none of the unrelated compounds were qualified as hits (Figure S2).
Identification of New 5-HT2C Ligands from Herbal Extracts
The established affinity MS workflow was first applied to screening 5-HT2C ligands from crude extracts of AD and TR that displayed the highest potency among all natural herbs assayed. The purified receptor was incubated with either extract and underwent the same affinity MS procedure as described above. A targeted metabolomics data mining strategy previously developed by us37 was implemented to process the affinity MS screening data for individual extracts. Screening hits were selected if their mean binding indexes (BIs) were above 2.0 (P < 0.05, n = 4),44,45 and their chemical structures were assigned by matching the LC-MS features with compounds registered in a natural herb database (TCMHD).46 Unexpectedly, serotonin, the natural ligand for 5-HT receptors, and its analogue 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT) known as 5-HT2C agonist47 turned out to be top-ranking hits from screening the two extracts (Figure S3A–C, Table S1, Note S1). Furthermore, serotonin alone or serotonin combined with 5-MeO-DMT accounted for 100% of the overall 5-HT2C activity of AD and TR (Figures S3D,E and S4). As serotonin was also detected in RV and SR extracts, these four herbs were abandoned for further experimentation (Note S1).
Extract of the fifth candidate herb Stephania tetrandra (ST) with appreciable 5-HT2C agonism (EC50 = 2.13 μM) was subjected to the affinity MS screen for putative ligands (Figure 2A) which gave rise to 12 initial hits (Figure 2B, Table S2). To increase the chance of capturing ligands of low abundances in the original extract, we fractionated the extract and screened each fraction separately (Figure S5). As expected, screening three fractions altogether allowed us to interrogate a lot more constituents (1364 assigned features in total) and identify more putative ligands than screening the crude extract alone (Figure 2C). A close inspection of the assigned structures for all putative ligands from different screens revealed a cluster of aporphine alkaloids that possess a characteristic tetracyclic framework (Figure 2D, Table S3). Compounds in this alkaloid subclass have been rarely associated with 5-HT2C.48 Among the eight identified aporphine ligands, only nuciferine (compound 15856) was reported to be a 5-HT2C antagonist49 while the rest have not been associated with 5-HT2C.
Figure 2.

Identification of aporphines active at 5-HT2C from crude and fractionated extracts of Stephania tetrandra (ST). (A) Representative LC-MS chromatograms of ST crude extract, 5-HT2C target, and control. (B, C) Initial hits from screening ST crude extract (blue dots) or ST extract fractions (pink dots) by affinity MS combined with metabolomics. Aporphines are annotated with larger dots. BI, binding index. (D) Scaffold of the aporphines identified in this study (specific structures listed in Table S3). (E, F) Structural validation of 1857 and 15781 by MSMS analysis. (G) Validation of ligand binding to purified 5-HT2C by pure compounds using the affinity MS binding assay. The MS intensity of each ligand was significantly higher in the 5-HT2C target than in control (***P < 0.001, n = 4). (H, I) Competition of 5-MeO-DMT or ritanserin binding to purified 5-HT2C with increasing concentrations of each aporphine. MS intensity of 5-MeO-DMT or ritanserin bound to purified 5-HT2C was normalized to that in the absence of any aporphine. Data were obtained from two independent experiments in technical duplicate. Error bars represent SEM.
To verify their binding ability and bioactivity, we purchased pure standards for two ligands (14148 and 15856) while others are not commercially available. We then tried to isolate them directly from the extracts. Guided by the initial affinity MS screening data which pinpointed the expected retention time and accurate mass of the compounds of interest (Table S3), we were able to isolate two new putative ligands 1857 ((R)-asimilobine) and 15781 (nornuciferine) (Figure S6), and their chemical structures were confirmed by 1D and 2D NMR analysis (Note S2). Optical rotation measurement indicated that 1857 ([αD20] = −92°) is an R stereoisomer whereas 15781 ([αD] = −1.4°) is a racemic mixture. When we overlaid the MSMS spectra of the isolated compound and the putative ligand identified in the affinity MS screen, almost identical MSMS fragmentation patterns strongly corroborated the structural identity of each ligand (Figure 2E,F). We further performed CD spectroscopy analysis of four aporphine ligands to determine that 14148 is racemic, and 15856 is an R stereoisomer (Figure S7).
To confirm specific interaction of each compound with 5-HT2C, we incubated individual compounds with the purified receptor and performed the affinity MS binding assay. All four compounds showed significant binding to 5-HT2C relative to the control (Figure 2G). Furthermore, in a ligand competition experiment, all four compounds substantially reduced the binding of reference agonist and antagonist to 5-HT2C in a concentration-dependent manner (Figure 2H,I).
Discovery of a 5-HT2C Selective and Gq Biased Agonist
We subsequently assessed the four aporphines for their binding affinity to three 5-HT2 subtypes using a radiolabeled ligand displacement assay. All four ligands displayed binding affinities to the three receptors with Ki values in the medium to high nanomolar range and showed no significant preference of binding to 5-HT2C (Figure 3A–C, Table S4). Intriguingly, 1857 and 15781 possess exquisite subtype selectivity for 5-HT2C in Gq-mediated calcium signaling: both are partial agonists (1857 EC50 = 308 nM, Emax = 86.1%; 15781 EC50 = 653 nM, Emax = 65.6% relative to 5-HT) whereas none of them exhibited measurable Gq agonism for 5-HT2A or 5-HT2B (Figure 3D,E). In fact, both are weak antagonists of 5-HT2A or 5-HT2B (IC50 > 10 μM) (Figure S8A,B). In contrast to their remarkable selectivity for 5-HT2C, the approved antiobesity drug lorcaserin only displayed moderate or no selectivity for 5-HT2C over 5-HT2B or 5-HT2A (Figure 3F). The other two aporphines, 14148 and 15856, are both nonselective antagonists of Gq-mediated activity with sub-micromolar potency at three 5-HT2 subtypes (Figure S8C,D). It is worth noting that 1857 (agonist) and 14148 (antagonist) only differ by a single methyl substituent, yet they displayed opposite activity at 5-HT2C.
Figure 3.

Pharmacological profiles of aporphine ligands. (A–C) Radiolabeled ligand binding curves for 5-HT2C, 5-HT2B, and 5-HT2A in the presence of aporphines or lorcaserin. See also Table S4 for Ki values. (D–F) Gq-mediated calcium mobilization induced by 1857, 15781, and lorcaserin. 1857 and 15781 displayed partial agonism only at 5-HT2C while lorcaserin activated three 5-HT2 receptors. (G) β-arrestin2 recruitment stimulated by 1857, lorcaserin, and ergotamine (ERG, a known β-arrestin2 biased agonist for 5-HT2C). 1857 has no measurable agonist activity following ERG treatment. (H) SFSR summary of aporphines discovered in this study. N-unsubstituted aporphine 1857 and 15781 act as selective 5-HT2C agonists. 1857 also displays Gq bias with no measurable β-arrestin activity. N-methyl substituted aporphine 14148 and 15856 act as nonselective 5-HT2 antagonists. NA, not assayed. Data represent means ± SEM of three independent experiments performed in triplicate.
For the partial agonist 1857, we then assessed its ability to recruit β-arrestin2 to 5-HT2C using a reporter gene-based Tango assay.50 Strikingly, 1857 exhibited no β-arrestin recruitment activity (Figure 3G). In fact, it acts as an antagonist of 5-HT2C related β-arrestin signaling against agonist ergotamine (Figure S8E). We also employed a bioluminescent resonance energy transfer (BRET) orthologous method that measures β-arrestin2 association with the receptor. This assay revealed that 1857 does not possess agonist activity in β-arrestin2 recruitment but acts as an antagonist for β-arrestin2 association (Figure S8F), confirming its G protein bias. In contrast, lorcaserin functioned as a full agonist at 5-HT2C in both G protein and β-arrestin pathways (Figure 3F,G).
In summary, 1857 displays a very unique pharmacological profile as a highly selective partial agonist for 5-HT2C with an exclusive bias toward Gq signaling. Moreover, our results uncovered a 5-HT2C structure–functional selectivity relationship (SFSR) for a group of aporphine alkaloids which demonstrate either subtype-selective G protein preference or nonselective antagonism, dependent on N-6 substitution (Figure 3H).
Structural Model of 5-HT2C Activation by 1857
We next predicted the binding pose of 1857 based on the solved 5-HT2C crystal structure15 (PDB ID: 6BQG) by molecular docking. Embedded deep in the orthosteric pocket of the receptor, 1857 forms a salt bridge between its protonated nitrogen and the conserved aspartate D1343.32 which is a key interaction conserved in 5-HT and other aminergic GPCRs13−15 (Figure 4A). In addition, 1857 forms extensive interactions with residues on transmembrane (TM) helices 3, 5, 6, and 7. Especially, the aporphine rings form π–π interactions with both F3276.51 and F3286.52 and hydrophobic interactions with V1353.33, A2225.46, and V3547.39 (Figure 4A). In contrast, lorcaserin, which is smaller in size than 1857, leaves more space in the pocket and makes less extensive interactions with the aforementioned residues (Figure 4B).
Figure 4.

Key interactions between 5-HT2C and 1857 specifically modulate the agonist activity. Docking poses of 1857 (A) and lorcaserin (B) in 5-HT2C. Predicted key interacting residues are in purple. (B) V3547.39 in gray does not interact with lorcaserin. Radiolabeled ligand binding curves (upper) and Gq-mediated calcium flux (lower) in cells expressing wild-type (WT) or mutant 5-HT2C in the presence of 1857 (C) or lorcaserin (D). (E) β-arrestin2 recruitment in cells expressing WT or mutant 5-HT2C in the presence of 1857 (upper) or lorcaserin (lower). Gq activity of 5-HT2C elicited by 1857 was significantly affected by mutations on five key interaction sites relative to WT, yet they hardly changed Gq activity of 5-HT2C treated by lorcaserin. See also Table S5 for IC50/EC50 values. Data represent means ± SEM of three independent experiments performed in triplicate.
Mutating F327 to L to impair the predicted π–π interaction attenuated 1857’s binding affinity and Gq-mediated agonist activity (Figure 4C), yet this mutation did not affect lorcaserin’s affinity or activity (Figure 4D). Another mutation V135L which may strengthen the hydrophobic interaction with the ligand significantly increased 1857’s affinity and promoted its Gq-mediated agonist activity, whereas slightly increased affinity and no change of Gq activity were observed on lorcaserin (Figure 4C,D). Most strikingly, V354A showed ∼300-fold increase of Gq activity (EC50 = 6.4 nM) though its binding was only enhanced by 6-fold (Figure 4C, Table S5). Mutating V354 to a bulky residue F also resulted in substantially increased Gq activity (EC50 = 54.0 nM), but affinity remained unchanged (Figure 4C, Table S5). It seems that V354 in TM7 is a critical residue specifically influencing Gq-coupled activation by 1857 but not lorcaserin (Figure 4D). Furthermore, among the mutants with increased Gq activity, 1857 only induced weak β-arrestin recruitment activity of V354A (EC50 = 2.3 μM) while the others stayed inactive in this pathway (Figure 4E). Finally, A222I substantially increased 1857’s affinity possibly due to its longer aliphatic side chain. However, this mutation almost abolished Gq activity elicited by 1857, which may be related to a locked conformation of TM5 as a result of the strengthened hydrophobic interaction (Figure 4C). All test mutants showed cell surface expression comparable to the wild-type (Figure S9). Taken together, our docking supported by mutagenesis study pinpointed key interactions specific for Gq-coupled 5-HT2C activation by 1857 and not by lorcaserin.
Agonist 1857 Suppresses Food Intake and Induces Weight Loss in Mice
Prior to evaluating 1857 in animal models, we measured its brain permeability by intravenous injection to mice (10 mg/kg; Table S6). The brain-to-plasma ratio of 1857 stayed as high as 6.4 at 4 h after dosing, indicating its excellent pharmacokinetics profile. Since 5-HT2C is a potential therapeutic target for obesity, we investigated the effects of 1857 on food intake in comparison with lorcaserin. In the first experiment, acute intraperitoneal administration of 1857 (30 mg/kg) reduced food intake from 30 min to 4 h postinjection (P < 0.01) whereas lorcaserin (10 mg/kg) suppressed food intake up to 2 h postinjection (P < 0.05) (Figure 5A,B). Thus, 1857 administered at 3 times the dose of lorcaserin produced very similar in vivo efficacy, despite that its in vitro potency for 5-HT2C activation is 2 orders of magnitude lower than that of lorcaserin (Figure 3D,F). This result could be attributed to better brain permeability and metabolic stability of 1857.
Figure 5.

In vivo antiobesity effects of 1857. Acute food intake suppression induced by lorcaserin (A) or 1857 (B). Overnight fasting mice (n = 8 each group) were treated with lorcaserin (10 mg/kg), 1857 (30 mg/kg), or vehicle 30 min before feeding. Food intake was measured at indicated time points. (C–G) 1857 inhibited food intake and showed antiobesity effects in a diet-induced obesity (DIO) mouse model. DIO mice (n = 9 each group) were treated with 1857 (30 mg/kg) or vehicle daily for 10 days. Accumulative food intake (C) and body weight change (D) were recorded during the treatment. Blood and liver were collected to measure blood glucose, serum cholesterol (TC), and triglyceride (TG) levels (E) as well as liver TC and TG levels (F). (G) The weight of white adipose tissues (WAT) was also measured, and representative tissue images are shown. Data represent means ± SEM *P < 0.05, **P < 0.01, and ***P < 0.001 (two-tailed Student’s t-test).
We then examined the effect of 1857 in diet-induced obesity (DIO) mice. Mice were fed with a high-fat diet for 2 months to induce obesity before receiving a daily treatment of 1857 (30 mg/kg). Significant reduction of food intake (Figure 5C) and weight loss (Figure 5D) started to be observed in mice following treatment for 5 days and lasted until the end of this study. Consistent with weight loss, 1857 treated DIO mice had lower blood glucose (−20.9%), reduced serum total cholesterol (TC, −7.9%), and unchanged serum total triglyceride (TG) relative to vehicle (Figure 5E). The stronger lipolysis in the adipose tissues might be responsible for maintaining the serum TG level unchanged after 1857 administration, which was also observed in other antiobesity drug treatments.51 In addition, 1857 treatment decreased liver TC and TG levels (Figure 5F) as well as the total organ weight of epididymal white adipose tissue (WAT, −29.3%) and inguinal WAT (−46.4%) in DIO mice (Figure 5G). Similar effects were reported in DIO rats treated with lorcaserin.52 Therefore, our study confirmed the efficacy of 1857 in attenuating obesity which is in line with its specific activity on 5-HT2C.
Discussion
In this study, we established an affinity MS-based approach specifically for GPCR ligand discovery from natural products. To identify both agonists and antagonists for a given GPCR, the ensemble of the purified receptor used as a bait for ligand enrichment ought to comprise both active and inactive conformations. However, a large number of GPCR constructs optimized for in vitro purification and structural characterization tend to yield proteins predominantly at inactive conformational states due to the inherent flexibility and instability of functionally active states.53−57 Therefore, previous affinity MS screens of synthetic compound libraries with a purified GPCR only discovered new antagonists45,58 that readily bind to a receptor in the inactive state. To drive the heterogeneous population of 5-HT2C toward active conformations, we reversed the thermostabilizing mutation C360N7.45 originally designed for receptor crystallography,15 which increased our chances of capturing agonists. In another study of using GPCR-expressing cell membranes for the affinity MS screen, we also observed that all agonizing ligands identified with a wild-type GPCR abrogated their binding to the receptor when it incorporated multiple thermostabilizing mutations.44 Therefore, careful design of the construct to shift the receptor conformation to the active state is essential for agonist discovery using this approach.
The naturally occurring compounds 1857 ((R)-asimilobine) and 15781 (nornuciferine) discovered here possess an aporphine scaffold which represents a novel chemotype for 5-HT2C agonists. Compounds in this alkaloid subclass have been traditionally characterized as ligands for the dopamine receptor system and play potential therapeutic roles in the treatment of Parkinson’s disease and other neurological disorders.59−61 With regard to serotonin receptors, aporphines have mostly been studied as ligands for 5-HT1A and 5-HT2A receptors48 and are rarely associated with 5-HT2C. Notably, the majority of natural or synthetic aporphines exhibit antagonistic activities at serotonin receptors with no documented subtype selectivity or signaling bias.48,49,62 In contrast, 1857 is the first aporphine displaying exclusive biased G protein agonism at 5-HT2C with exquisite selectivity over 5-HT2A and 5-HT2B. The unique pharmacological profile of 1857 opens a new avenue for design of potent and functionally selective 5-HT2C ligands with great potential in the treatment of obesity, schizophrenia, and other neurological disorders.26 Moreover, 1857 could serve as a desirable probe for elucidating the structural basis of preferential G protein signaling (details in Note S3). Among the four aporphine ligands discovered in this study, 1857 and 15858 isolated from herbs are optically pure stereoisomers while the other two aporphines (15781, 14148) are racemic (Figure 3H). How the stereospecificity of different ligands affects the pharmacological properties awaits further investigation. In addition, although 1857 shows remarkable Gq-coupling selectivity at 5-HT2C, its broader selectivity among the 5-HT receptor subfamily and other aminergic GPCRs remains to be determined.
The natural product screening approach presented in this study enables rapid discovery of GPCR ligands with sophisticated pharmacological properties. Although we identified a new series of aporphine ligands for 5-HT2C, the rest of the 95 hits from screening ST extract fractions may contain more 5-HT2C modulators with novel structures and distinct bioactivities (Figure 2C, Table S2). This high-throughput and unbiased screening approach which is generalizable to other receptors would accelerate the exploration of largely untapped natural product chemical space for discovering novel and improved drug leads targeting GPCRs.
Methods
Receptor Expression and Purification
The protein expression and purification have been described previously.15,62 In brief, the 5-HT2CR-BRIL construct was subcloned into a modified pFastBac1 vector (Invitrogen). The construct was optimized with the truncation of N-terminal residues (1–39) and C-terminal residues (393–458). The residues of the third intracellular loop (IL3) from L246 to M300 were replaced by thermostabilized apocytochrome b562RIL (BRIL). No mutation sites were incorporated into this construct. The construct was then expressed in Spodoptera frugiperda (Sf9) cells with a hemagglutinin (HA) tag followed by a FLAG tag at the N terminus and a 10× His tag at the C terminus. Cells were cultured at 27 °C and harvested after 48 h postinfection. 5-HT2CR-BRIL membrane preparation and protein purification were performed with the same procedure as mentioned before.15,62 The purity and monodispersity of the 5-HT2C protein were measured by analytical size-exclusion chromatography (aSEC).
Herbal Extract Preparation
Nine herbs examined in this study are Aristolochia debilis (AD), Tetradium ruticarpum (TR), Rauvolfia verticillata (RV), Strychnos nux-vomica (SN), Stephania tetrandra (ST), Ziziphus jujuba (ZJ), Menispermum dauricum (MD), and Catharanthus roseus (CR). Each of them was first air-dried and pulverized into powder. The powder (200 g) was extracted with 500 mL of 70% ethanol by water bath ultrasonication for 30 min. The extraction was performed three times in total. Then, the organic solvent was removed by vacuum evaporation at 50 °C. The residual material was dissolved in 0.3% (v/v) hydrochloric acid and partitioned with EtOAc three times. Then, the aqueous layer was basified with 5% (v/v) ammonia to pH 9–10 and partitioned with EtOAc three times. The EtOAc phase was dried out, and the powder was stored at −80 °C. The stock solution (100 mg/mL) of each herbal crude extract was prepared by dissolving the powder with 95% DMSO and was stored at −20 °C.
Affinity MS Screening of Herbal Extracts
The method developed for GPCR ligand screening from compound libraries45 was adapted to herbal extract screening here. The purified protein 5-HT2C or HCA2 (3 μg) was immobilized on nickel agarose beads (Sigma) in the incubation buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS at 4 °C overnight. The stock of each herbal extract was diluted with the incubation buffer to a final concentration of 0.5 mg/mL. Then, the 5-HT2C beads were incubated with the diluted crude extract at 4 °C for 1 h. The supernatant was removed, and the beads were washed four times with 150 mM ammonium acetate (pH 7.5) after incubation. The compounds bound to 5-HT2C were then dissociated with 200 μL of methanol, dried out in a speed vacuum, and reconstituted in 50% methanol before LC-MS/MS analysis. The control sample was prepared with the same procedure by using HCA2 beads. All samples were prepared in four independent replicates.
Samples were analyzed on a Shimazu L30A UPLC system (Shimazu) coupled to a TripleTOF 6600 mass spectrometer (AB SCIEX) operating in the positive ion mode. Chromatographic separation was performed on a ZORBAX Eclipse Plus C18 column (3.5 μm, 2.1 × 100 mm, Agilent) at a flow rate of 300 μL/min and maintained at 40 °C with the mobile phases of water/0.1% formic acid (A) and acetonitrile/0.1% formic acid (B). The LC gradient was as follows: 0–2 min, B at 5%; 2–12 min, B at 10–30%; 12–25 min, B at 30–90%; 25–30 min, B at 90–90%, then re-equilibrate for 5 min. Full-scan mass spectra were acquired in the range 100–1500 m/z with major ESI source settings: voltage 5.0–5.5 kV; gas temperature 500 °C; curtain gas 35 psi; nebulizer gas 55 psi; and heater gas 55 psi. MSMS spectra were acquired on the top 10 precursors with collision energy set at 45 eV with a CE spread of 15 eV and other ion source conditions identical to the full scan.
Metabolomics Data Processing for 5-HT2C Ligand Identification
Compounds in the target and control samples were initially identified by extracting ion chromatograms (EICs) using Peakview 2.2 (AB SCIEX) based on accurate mass (<10 ppm), isotope envelop matching (<10% deviation) in accordance with the compound formula registered in TCMHD. They also need to have consistent retention times in the affinity-selected target sample and in the crude extract (<0.2 min). Binding index (BI) of each compound is defined to be the ratio of MS intensity of the compound detected in the 5-HT2C target vs control. Initial hits were selected based on a mean BI > 2 and P < 0.05 from four experimental replicates. The significant difference of each compound’s MS intensity between target and control samples was determined by a two-tailed t-test with Bonferroni correction.
Stephania tetrandra (ST) Crude Extract Fractionation
The powder of the ST crude extract (200 mg) was dissolved in methanol and then fractionated using a Sunfire C18 OBD Prep column (5 μm, 19 × 250 mm, Waters) running at a flow rate of 10 mL/min with the mobile phase of water/0.1% formic acid (A) and acetonitrile (B). The LC gradient was 0–3 min, B at 10%; 3–20 min, B at 10–90%; 20–25 min, B at 90–90%. Three fractions were collected according to the UV response and LC separation. The solvent was removed by vacuum evaporation at 50 °C, and the residue was stored at −80 °C. Each fraction was reconstituted in the same incubation buffer to the same concentration as described for the ST crude extract before the affinity MS screen.
Isolation of Two Putative Ligands from the ST Extract
Air-dried, powdered roots of Stephania tetrandra (3 kg) were extracted three times with 95% ethanol at room temperature. Then, the organic solvent was removed, and the residue underwent the same procedure as described in the natural herb extract preparation. The crude extract (100 g) was separated on a silica gel column and eluted with CHCl3–MeOH (1:0–1:1) to obtain three fractions (F1–F3). Each fraction was then analyzed with UPLC-DAD/MS to identify the putative ligands from the affinity MS screen. In Fraction F3, the detection of two peaks at m/z 268.1334 and 282.1488 which showed the same retention time as ligands 1857 and 15781 indicated the presence of the two expected aporphine alkaloids. Compounds 1857 (2.1 mg) and 15781 (10.2 mg) were then purified from Fraction F3 by Shimadzu LC-20A (Shimadzu) using Sunfire C18 column (5 μm, 19 × 250 mm, Waters) with the gradient ACN-H2O (20–50%) at a flow rate of 10 mL/min. The peaks at 8.9 and 12.1 min (for 1857 and 15781, respectively) were collected separately and dried by vacuum evaporation. The structures of the two pure compounds were elucidated with 1D (1H and 13C) and 2D (HSQC and HMBC) NMR (Avance III HD 800 MHz, Bruker) analysis, and data are shown in the Supporting Information. The configurations of the two compounds were further determined based on their optical rotation values measured with an automatic polarimeter (Autopol VI, Rudolph).
Affinity MS-Based Validation of Pure Ligand Binding to 5-HT2C
Compounds 14148 and 15856 were purchased from BioBioPha Co. Ltd. (Kunming, China) and Chengdu Herbpurify Co. Ltd. (Chengdu, China), respectively. Compounds 1857 and 15781 were in-house isolated as described above. Their structures are described in Table S3. These four aporphine alkaloids were mixed at a final concentration of 100 nM for each compound. Then, the compound mixture was incubated with purified 5-HT2C or HCA2 proteins under the same conditions of the ligand screening experiment. The receptor-associated compounds were analyzed by LC-MS/MS. In the affinity MS binding assay of this simple mixture, a short LC gradient was applied for compound separation: 0–2 min, B at 5%; 2–2.1 min, B at 5–10%; 2.1–5 min, B at 10–30%; 5–5.1 min, B at 30–90%; 5.1–7 min, B at 90%.
Specific compound peaks in target and control samples were extracted using PeakView 2.2 (AB SCIEX) based on the accurate mass measurement (<10 ppm) and RT matching with the standard (<0.2 min). BI and P values (n = 4) were determined for each compound as described in the herbal extract screening experiment to assess ligand binding specificity for 5-HT2C.
Affinity MS-Based Ligand Competition Assay for 5-HT2C
The 5-HT2C agonist 5-MeO-DMT and antagonist ritanserin were used as marker ligands in the ligand competition assay. Purified 5-HT2C protein immobilized on nickel agarose beads (Sigma) was incubated with a given marker ligand at 2 nM mixed with each test compound (aporphine ligands) at an increased concentration (0, 2, 20 μM) at 4 °C for 1 h. After incubation, the compounds were dissociated from the 5-HT2C and analyzed by LC-MS/MS using the same method in the previous pure ligand binding assay. The MS intensity of the marker ligand under different conditions was extracted from the raw data using PeakView 2.2 (AB SCIEX) based on the same criteria described in the previous pure ligand binding assay. Reduction of the marker ligand response indicated the extent of binding competition by each test compound. Two independent experiments were performed in technical duplicate under each condition.
Cloning and Mutagenesis
Mutagenesis of the 5-HT2C construct was performed according to the Q5 site-directed mutagenesis kit protocol (New England BioLabs). In brief, PCR reactions were performed using the wild-type 5-HT2C receptor cDNA (pcDNA3.1) and primers containing the mutation sites of interest to create mutant plasmids. After DpnI (New England BioLabs) digestion of the parental DNA and transformation, positive clones were selected by ampicillin resistance. DNA was prepared using the Miniprep kit (Axygen) and sequenced (Genewiz) using forward (CMV) and reverse (BGHreverse) sequence primers.
Radioligand Binding Assay
Radioligand binding assays for wild-type receptors were performed using membranes prepared from 5-HT2A/2B/2C transfected HEK293 cell lines. Radioligands used in the assays were 3H-ketansetin (PerkinElmer; specific activity = 42.5–47.3 Ci/mmol) for 5-HT2A; 3H-LSD (PerkinElmer; specific activity = 82.9–83.3 Ci/mmol) for 5-HT2B; and 3H-mesulergine (PerkinElmer; specific activity = 80.9–83.0 Ci/mmol) for 5-HT2C. The unlabeled ligands were prepared in binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) ranging from 10 μM to 30 pM. Assay plates were incubated at room temperature for 1 h, and then they were harvested using vacuum filtration onto 0.3% polyethylenimine-presoaked 96-well filter mats A (PerkinElmer) and washed three times with cold wash buffer (50 mM Tris, pH 7.4). Scintillation cocktail (Meltilex) was melted onto the dried filters, and then, the plate was read using a Wallac Trilux Microbeta counter (PerkinElmer). Data were analyzed with GraphPad Prism 7.0 (Graphpad Software Inc.) using “one site-Fit Ki” to obtain Ki. Data were normalized to the top (100%, no competitor) and bottom (0%, 10 μM clozapine for 5-HT2A, 10 μM SB206553 for 5-HT2B, 10 μM ritanserin for 5-HT2C) to represent the percent of displacement. These assays were conducted by NIMH PDSP, directed by Bryan L Roth, M.D., Ph.D., the University of North Carolina at Chapel Hill, North Carolina, and Program Officer Jamie Driscoll at NIMH, Bethesda, MD.
To assay the mutants, they were first established by PCR-based site-directed mutagenesis and confirmed by DNA sequencing. The wild-type or mutant 5-HT2C was cloned into the pcDNA3.1 vector (Invitrogen) for transfection. CHO cells were washed twice 24 h after transfections and incubated with blocking buffer (F12 supplemented with 33 mM HEPES and 0.1% bovine serum albumin (BSA), pH 7.4) for 2 h at 37 °C. Subsequently, the cells were incubated in binding buffer (DMEM supplemented with 25 mM HEPES and 0.1% BSA) with a constant concentration of 3H-mesulergine (1 nM) and different concentrations of unlabeled 5-HT (1.28 nM to 100 μM), lorcaserin, and 1857 (0.64 nM to 50 μM) at room temperature for 3 h. Cells were washed three times with ice-cold PBS and lysed by 50 μL of lysis buffer (PBS supplemented with 20 mM Tris-HCl, 1% Triton X-100, pH 7.4). The plates were subsequently counted for radioactivity (counts per minute, CPM) in a scintillation counter (MicroBeta2 plate counter, PerkinElmer) using a scintillation cocktail (OptiPhase SuperMix, PerkinElmer).
Calcium Mobilization Assay
HEK293T cells stably transfected with wild-type 5-HT2A, 5-HT2B, or 5-HT2C were plated into poly(lysine) coated 384-well black clear bottom plates at a density of 15 000 cells per well with 40 μL of DMEM with 1% dialyzed FBS. For mutant activity evaluation, T-rex 293 cells (approximately 3 × 106 cells per 10 cm dish) were transfected with 5-HT2C wild-type or mutant DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocols. After 18–24 h of transfection, cells were plated at the same condition described above for stable cells. The next day, the media was decanted, and cells were incubated for 1 h at 37 °C with Fluo-4 Direct dye (Invitrogen) in FLIPR buffer (1× HBSS, 2.5 mM probenecid, and 20 mM HEPES, pH 7.4). After the dye loaded, the cells were placed in a FLIPRTETRA fluorescence imaging plate reader (Molecular Devices). Drugs were diluted at 3× final concentration in drug buffer (1× HBSS, 0.1% BSA, 20 mM HEPES, pH 7.4) and aliquoted into 384-well plates, which were then placed in the FLIPRTETRA. The fluidics module and plate reader of the FLIPRTETRA were programmed to read baseline fluorescence for 10 s (1 read/s), and then to add 10 μL of drug dilutions per well and to read for 3 min (1 read/s). Fluorescence of each well was normalized to the average of the first 10 reads. Then, the maximum-fold increase, which occurred within 60 s after drug addition, over baseline fluorescence elicited by vehicle or drug was determined. For the antagonist mode, 5-HT (3 nM) was used to activate the receptor. Data were normalized to percent 5-HT simulation, and EC50 or IC50 was analyzed in GraphPad Prism 7.0 (Graphpad) using log (agonist) vs response or log (antagonist) vs response.
Tango β-Arrestin2 Recruitment Assay
The Tango constructs of human 5-HT2C and its mutants were designed, and β-arrestin2 recruitment assay was performed as described previously.50 Briefly, HTLA cells were transfected with 5-HT2C Tango plasmid or mutants. After at least 24 h, HTLA cells were plated into poly(lysine) coated 384-well white clear bottom plates at a density of 15 000 cells per well with 40 μL of DMEM containing 1% dialyzed FBS. Six hours later, cells were simulated with 20 μL per well drug dilutions (3×) prepared in drug buffer (1× HBSS, 0.1% BSA, 20 mM HEPES, pH 7.4) and incubated at 37 °C overnight. Then, media and drug solutions were decanted and 20 μL per well of BrightGlo reagents (Promega) was added. After 20 min incubation, luminescence was read on an Envision counter (PerkinElmer). For the antagonist mode, agonist ERG EC80 (50 nM) was used to activate the receptor. Data were normalized to percent ERG simulation, and EC50 or IC50 was analyzed in GraphPad Prism 7.0 (Graphpad) using log (agonist) vs response or log (antagonist) vs response.
BRET β-Arrestin2 Recruitment Assay
We measured the effect of 1857 on the recruitment of β-arrestin2 in CHO cells stably expressing 5-HT2C-Rluc8 and β-arrestin2-Venus by bioluminescence resonance energy transfer (BRET) assay. The cells were seeded onto a 96-well plate at a density of 3 × 104 cells per well. Prior to BRET experiments, cells were rinsed twice with HBSS and then incubated with fresh HBSS for 30 min at 37 °C. After incubation with various concentrations of 1857 for 15 min, 5 μM coelenterazine-H (ThermoFisher) was added followed by 5 min incubation. Baseline BRET signals were read immediately at 470 and 535 nm for 11 cycles using an EnVision instrument (PerkinElmer). Constant concentration of the agonist lorcaserin (4 μM) was then added and detected for another 49 cycles. Data are presented as a BRET ratio, calculated as the ratio of Venus to Rluc8 signals after subtracting the lorcaserin value.
Prediction of Ligand Binding Poses by Molecular Docking
Molecular docking was performed with Scrodinger Suite 2015-4. The crystal structure of 5-HT2C with agonist ergotamine (PDB ID: 6BQG)15 was used. Processing of the protein structure was performed with the “Protein Preparation Wizard”. Converting of ligands from 2D to 3D structures was performed using “LigPrep”. Molecular docking was performed with Glide 6.9 in standard precision.
Acute Feeding Suppression
Male C57BL/6J mice were allowed to habituate to single cage housing and daily intraperitoneal injection of saline was given 1 week before starting the experiments (lights on/off 0700/1900). Food was removed at 19:00 for overnight fasting, the mice (N = 8 each group) were then treated with vehicle, lorcaserin (10 mg/kg), or 1857 (30 mg/kg) at 9:00 the next day. Food was returned 30 min thereafter. Food intake was measured at 30, 60, 90, 120, and 240 min after the presentation of food. Data represented are means ± SEM. Significant differences among groups were determined by unpaired, two-tailed Student’s t-test.
DIO Mice Feeding Suppression
Male C57BL/6J mice were fed with a high fat diet (HFD, 60% of energy from fat, Research Diets, D12492) for 8–9 weeks. The mice were housed individually, allowed ad libitum access to water, and fed with HFD. For the subchronic study (10 day treatment), DIO mice (N = 9 each group) were i.p. injected with vehicle or 1857 (30 mg/kg) daily. The body weight and food intake were monitored daily. At the end of the study, the mice were anesthetized, and blood, liver, and white adipose tissues (WAT) were collected for further analysis. Blood glucose was measured from the tail vein with a glucometer. Serum was prepared by centrifuging at 2000g for 10 min. Livers were homogenized in chloroform–methanol (2:1). The organic phase was further dried under N2 and then resolved in ethanol. Total cholesterol (TC) and total triglyceride (TG) in serum or extracted liver samples were measured with TC and TG kits (E1005-250, E1003-250, Applygen). TC and TG levels in the liver samples were normalized to the liver weight. Data represented are means ± SEM. Significant differences among groups were determined by unpaired, two-tailed Student’s t-test.
Safety Statement
No unexpected or unusually high safety hazards were encountered.
Acknowledgments
We thank Dr. Raymond C. Stevens for valuable advice, and Dr. Shuguang Yuan and Dr. Chu Wang (Peking University) for fruitful discussion. We also thank Xiaoyan Liu, Junlin Liu, and Fangfang Zhou from iHuman Institute, and Antao Dai and Chao Zhang from Shanghai Institute of Materia Medica for technical assistance. This work was funded by ShanghaiTech University, National Key R&D Program of China grants [2018YFA0507000 (Ming-Wei Wang), 2018YFA0507004 (Wenqing Shui), 2016YFC0905900 (Guisheng Zhong), 2017YFC1001300 (Guisheng Zhong)], National Mega R&D Program for Drug Discovery grants [2018ZX09711002-002-005 (Dehua Yang), 2018ZX09735-001 (Ming-Wei Wang)], National Natural Science Foundation of China grants [31971362 (Wenqing Shui), 81773792 (Dehua Yang), 31771130 (Guisheng Zhong)], and Novo Nordisk-CAS Research Fund grants 2017 and 2019 (Dehua Yang). We are also grateful to the staff members of animal facility at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, China, for their technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b01125.
Author Contributions
○ Bingjie Zhang and Simeng Zhao contributed equally.
Author Contributions
Bingjie Zhang performed receptor purification, affinity MS ligand screen, and binding validation experiments with the help of Haijie Cao; Bingjie Zhang and Simeng Zhao evaluated compound in vivo efficacy. Bingjie Zhang performed receptor mutagenesis and cell signaling assays, and isolated compounds with assistance from Ye Xin, Na Ye, and Yueming Xu; Dehua Yang, Xiaoqing Cai, and Wen Sun performed radiolabeled ligand binding and BRET assays. Xi-Ping Huang performed radiolabeled ligand binding assays on wild-type 5-HT2 receptors and edited the manuscript. Yiran Wu conducted molecular docking analysis supervised by Suwen Zhao. Yao Peng helped with 5-HT2C purification supervised by Zhi-Jie Liu. Guisheng Zhong and Ming-Wei Wang were involved in the overall project management and edited the manuscript. Wenqing Shui and Bingjie Zhang wrote the manuscript with edits and inputs from all authors. Wenqing Shui conceived and supervised the project.
The authors declare no competing financial interest.
Supplementary Material
References
- Meltzer H. Y.; Roth B. L. Lorcaserin and pimavanserin: emerging selectivity of serotonin receptor subtype-targeted drugs. J. Clin. Invest. 2013, 123 (12), 4986–91. 10.1172/JCI70678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Roth B. L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–42. 10.1016/j.pharmthera.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios J. M.; Pazos A.; Hoyer D. A short history of the 5-HT2C receptor: from the choroid plexus to depression, obesity and addiction treatment. Psychopharmacology (Berl) 2017, 234 (9–10), 1395–1418. 10.1007/s00213-017-4545-5. [DOI] [PubMed] [Google Scholar]
- Pogorelov V. M.; Rodriguiz R. M.; Cheng J.; Huang M.; Schmerberg C. M.; Meltzer H. Y.; Roth B. L.; Kozikowski A. P.; Wetsel W. C. 5-HT2C Agonists Modulate Schizophrenia-Like Behaviors in Mice. Neuropsychopharmacology 2017, 42 (11), 2163–2177. 10.1038/npp.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeb F. D.; Higgins G. A.; Fletcher P. J. The Serotonin 2C Receptor Agonist Lorcaserin Attenuates Intracranial Self-Stimulation and Blocks the Reward-Enhancing Effects of Nicotine. ACS Chem. Neurosci. 2015, 6 (7), 1231–40. 10.1021/acschemneuro.5b00017. [DOI] [PubMed] [Google Scholar]
- Nichols D. E.; Johnson M. W.; Nichols C. D. Psychedelics as Medicines: An Emerging New Paradigm. Clin. Pharmacol. Ther. 2017, 101 (2), 209–219. 10.1002/cpt.557. [DOI] [PubMed] [Google Scholar]
- Rothman R. B.; Baumann M. H.; Savage J. E.; Rauser L.; McBride A.; Hufeisen S. J.; Roth B. L. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 2000, 102 (23), 2836–41. 10.1161/01.CIR.102.23.2836. [DOI] [PubMed] [Google Scholar]
- Roth B. L. Drugs and valvular heart disease. N. Engl. J. Med. 2007, 356 (1), 6–9. 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- Fitzgerald L. W.; Burn T. C.; Brown B. S.; Patterson J. P.; Corjay M. H.; Valentine P. A.; Sun J. H.; Link J. R.; Abbaszade I.; Hollis J. M.; Largent B. L.; Hartig P. R.; Hollis G. F.; Meunier P. C.; Robichaud A. J.; Robertson D. W. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol. 2000, 57 (1), 75–81. [PubMed] [Google Scholar]
- Astrup A. Drug management of obesity--efficacy versus safety. N. Engl. J. Med. 2010, 363 (3), 288–90. 10.1056/NEJMe1004076. [DOI] [PubMed] [Google Scholar]
- Lorcaserin. In obesity: unacceptable risks. Prescrire Int. 2014, 23 ( (149), ), 117–20. [PubMed] [Google Scholar]
- DiNicolantonio J. J.; Chatterjee S.; O’Keefe J. H.; Meier P. Lorcaserin for the treatment of obesity? A closer look at its side effects. Open Heart 2014, 1 (1), e000173. 10.1136/openhrt-2014-000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; Shoichet B. K.; Dror R. O.; Roth B. L. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168 (3), 377–389. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Wacker D.; Wang S.; Agegnehu B.; Liu J.; Lansu K.; Tribo A. R.; Olsen R. H. J.; Che T.; Jin J.; Roth B. L. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 2018, 25 (9), 787–796. 10.1038/s41594-018-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; McCorvy J. D.; Harpsoe K.; Lansu K.; Yuan S.; Popov P.; Qu L.; Pu M.; Che T.; Nikolajsen L. F.; Huang X. P.; Wu Y.; Shen L.; Bjorn-Yoshimoto W. E.; Ding K.; Wacker D.; Han G. W.; Cheng J.; Katritch V.; Jensen A. A.; Hanson M. A.; Zhao S.; Gloriam D. E.; Roth B. L.; Stevens R. C.; Liu Z. J. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172 (4), 719–730. 10.1016/j.cell.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; McCorvy J. D.; Giguere P. M.; Zhu H.; Kenakin T.; Roth B. L.; Kozikowski A. P. Design and Discovery of Functionally Selective Serotonin 2C (5-HT2C) Receptor Agonists. J. Med. Chem. 2016, 59 (21), 9866–9880. 10.1021/acs.jmedchem.6b01194. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Giguere P. M.; Lv W.; Roth B. L.; Kozikowski A. P. Design and Synthesis of (2-(5-Chloro-2,2-dimethyl-2,3-dihydrobenzofuran-7-yl)cyclopropyl)methanamine as a Selective Serotonin 2C Agonist. Tetrahedron Lett. 2015, 56 (23), 3420–3422. 10.1016/j.tetlet.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; Giguere P. M.; Schmerberg C. M.; Pogorelov V. M.; Rodriguiz R. M.; Huang X. P.; Zhu H.; McCorvy J. D.; Wetsel W. C.; Roth B. L.; Kozikowski A. P. Further Advances in Optimizing (2-Phenylcyclopropyl)methylamines as Novel Serotonin 2C Agonists: Effects on Hyperlocomotion, Prepulse Inhibition, and Cognition Models. J. Med. Chem. 2016, 59 (2), 578–91. 10.1021/acs.jmedchem.5b01153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Cheng J.; McCorvy J. D.; Lorello P. J.; Caldarone B. J.; Roth B. L.; Kozikowski A. P. Discovery of N-Substituted (2-Phenylcyclopropyl)methylamines as Functionally Selective Serotonin 2C Receptor Agonists for Potential Use as Antipsychotic Medications. J. Med. Chem. 2017, 60 (14), 6273–6288. 10.1021/acs.jmedchem.7b00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer R. I.; Brennan P. E.; Brown A. D.; Bungay P. J.; Conlon K. M.; Corbett M. S.; DePianta R. P.; Fish P. V.; Heifetz A.; Ho D. K.; Jessiman A. S.; McMurray G.; de Oliveira C. A.; Roberts L. R.; Root J. A.; Shanmugasundaram V.; Shapiro M. J.; Skerten M.; Westbrook D.; Wheeler S.; Whitlock G. A.; Wright J. Multiparameter optimization in CNS drug discovery: design of pyrimido[4,5-d]azepines as potent 5-hydroxytryptamine 2C (5-HT(2)C) receptor agonists with exquisite functional selectivity over 5-HT(2)A and 5-HT(2)B receptors. J. Med. Chem. 2014, 57 (12), 5258–69. 10.1021/jm5003292. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery 2013, 12 (3), 205–16. 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Urban J. D.; Clarke W. P.; von Zastrow M.; Nichols D. E.; Kobilka B.; Weinstein H.; Javitch J. A.; Roth B. L.; Christopoulos A.; Sexton P. M.; Miller K. J.; Spedding M.; Mailman R. B. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320 (1), 1–13. 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Violin J. D.; Crombie A. L.; Soergel D. G.; Lark M. W. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 2014, 35 (7), 308–16. 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- McCorvy J. D.; Butler K. V.; Kelly B.; Rechsteiner K.; Karpiak J.; Betz R. M.; Kormos B. L.; Shoichet B. K.; Dror R. O.; Jin J.; Roth B. L. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 2018, 14 (2), 126–134. 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll C. C.; McKittrick B. A. Biased ligand modulation of seven transmembrane receptors (7TMRs): functional implications for drug discovery. J. Med. Chem. 2014, 57 (16), 6887–96. 10.1021/jm401677g. [DOI] [PubMed] [Google Scholar]
- Tan L.; Yan W.; McCorvy J. D.; Cheng J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61 (22), 9841–9878. 10.1021/acs.jmedchem.8b00435. [DOI] [PubMed] [Google Scholar]
- Soergel D. G.; Subach R. A.; Burnham N.; Lark M. W.; James I. E.; Sadler B. M.; Skobieranda F.; Violin J. D.; Webster L. R. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155 (9), 1829–35. 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hubner H.; Huang X. P.; Sassano M. F.; Giguere P. M.; Lober S.; Da D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537 (7619), 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (45), 18488–93. 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin J. D.; DeWire S. M.; Yamashita D.; Rominger D. H.; Nguyen L.; Schiller K.; Whalen E. J.; Gowen M.; Lark M. W. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335 (3), 572–9. 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- Urs N. M.; Bido S.; Peterson S. M.; Daigle T. L.; Bass C. E.; Gainetdinov R. R.; Bezard E.; Caron M. G. Targeting beta-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (19), E2517–26. 10.1073/pnas.1502740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charfi I.; Audet N.; Bagheri Tudashki H.; Pineyro G. Identifying ligand-specific signalling within biased responses: focus on delta opioid receptor ligands. Br. J. Pharmacol. 2015, 172 (2), 435–48. 10.1111/bph.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey A. L.; Edrada-Ebel R.; Quinn R. J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discovery 2015, 14 (2), 111–29. 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79 (3), 629–61. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Lachance H.; Wetzel S.; Kumar K.; Waldmann H. Charting, navigating, and populating natural product chemical space for drug discovery. J. Med. Chem. 2012, 55 (13), 5989–6001. 10.1021/jm300288g. [DOI] [PubMed] [Google Scholar]
- Fu X.; Wang Z.; Li L.; Dong S.; Li Z.; Jiang Z.; Wang Y.; Shui W. Novel Chemical Ligands to Ebola Virus and Marburg Virus Nucleoproteins Identified by Combining Affinity Mass Spectrometry and Metabolomics Approaches. Sci. Rep. 2016, 6, 29680. 10.1038/srep29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Liang H.; Cao H.; Zhang B.; Li J.; Wang W.; Qin S.; Wang Y.; Xuan L.; Lai L.; Shui W. Efficient ligand discovery from natural herbs by integrating virtual screening, affinity mass spectrometry and targeted metabolomics. Analyst 2019, 144 (9), 2881–2890. 10.1039/C8AN02482K. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Li X.; Chen M.; Liu F.; Han C.; Kong L.; Luo J. A strategy for screening of alpha-glucosidase inhibitors from Morus alba root bark based on the ligand fishing combined with high-performance liquid chromatography mass spectrometer and molecular docking. Talanta 2018, 180, 337–345. 10.1016/j.talanta.2017.12.065. [DOI] [PubMed] [Google Scholar]
- Song H. P.; Chen J.; Hong J. Y.; Hao H.; Qi L. W.; Lu J.; Fu Y.; Wu B.; Yang H.; Li P. A strategy for screening of high-quality enzyme inhibitors from herbal medicines based on ultrafiltration LC-MS and in silico molecular docking. Chem. Commun. (Cambridge, U. K.) 2015, 51 (8), 1494–7. 10.1039/C4CC08728C. [DOI] [PubMed] [Google Scholar]
- Song H. P.; Wu S. Q.; Qi L. W.; Long F.; Jiang L. F.; Liu K.; Zeng H.; Xu Z. M.; Li P.; Yang H. A strategy for screening active lead compounds and functional compound combinations from herbal medicines based on pharmacophore filtering and knockout/knockin chromatography. Journal of chromatography. A 2016, 1456, 176–86. 10.1016/j.chroma.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Gesmundo N. J.; Sauvagnat B.; Curran P. J.; Richards M. P.; Andrews C. L.; Dandliker P. J.; Cernak T. Nanoscale synthesis and affinity ranking. Nature 2018, 557 (7704), 228–232. 10.1038/s41586-018-0056-8. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Shipps G. W. Jr.; Cooper A.; English J. M.; Annis D. A.; Carr D.; Nan Y.; Wang T.; Zhu H. Y.; Chuang C. C.; Dayananth P.; Hruza A. W.; Xiao L.; Jin W.; Kirschmeier P.; Windsor W. T.; Samatar A. A. Discovery of novel, dual mechanism ERK inhibitors by affinity selection screening of an inactive kinase. J. Med. Chem. 2014, 57 (21), 8817–26. 10.1021/jm500847m. [DOI] [PubMed] [Google Scholar]
- Kutilek V. D.; Andrews C. L.; Richards M. P.; Xu Z.; Sun T.; Chen Y.; Hashke A.; Smotrov N.; Fernandez R.; Nickbarg E. B.; Chamberlin C.; Sauvagnat B.; Curran P. J.; Boinay R.; Saradjian P.; Allen S. J.; Byrne N.; Elsen N. L.; Ford R. E.; Hall D. L.; Kornienko M.; Rickert K. W.; Sharma S.; Shipman J. M.; Lumb K. J.; Coleman K.; Dandliker P. J.; Kariv I.; Beutel B. Integration of Affinity Selection-Mass Spectrometry and Functional Cell-Based Assays to Rapidly Triage Druggable Target Space within the NF-kappaB Pathway. J. Biomol. Screening 2016, 21 (6), 608–19. 10.1177/1087057116637353. [DOI] [PubMed] [Google Scholar]
- Qin S.; Meng M.; Yang D.; Bai W.; Lu Y.; Peng Y.; Song G.; Wu Y.; Zhou Q.; Zhao S.; Huang X.; McCorvy J. D.; Cai X.; Dai A.; Roth B. L.; Hanson M. A.; Liu Z. J.; Wang M. W.; Stevens R. C.; Shui W. High-throughput identification of G protein-coupled receptor modulators through affinity mass spectrometry screening. Chem. Sci. 2018, 9 (12), 3192–3199. 10.1039/C7SC04698G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Qin S.; Zhang B.; Dai A.; Cai X.; Ma M.; Gao Z. G.; Yang D.; Stevens R. C.; Jacobson K. A.; Wang M. W.; Shui W. Accelerating the Throughput of Affinity Mass Spectrometry-Based Ligand Screening toward a G Protein-Coupled Receptor. Anal. Chem. 2019, 91 (13), 8162–8169. 10.1021/acs.analchem.9b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.; Yan X.; Zhou J.; Xie G. Traditional Chinese medicine database and application on the Web. J. Chem. Inf. Comput. Sci. 2001, 41 (2), 273–7. 10.1021/ci0003101. [DOI] [PubMed] [Google Scholar]
- Blair J. B.; Kurrasch-Orbaugh D.; Marona-Lewicka D.; Cumbay M. G.; Watts V. J.; Barker E. L.; Nichols D. E. Effect of ring fluorination on the pharmacology of hallucinogenic tryptamines. J. Med. Chem. 2000, 43 (24), 4701–10. 10.1021/jm000339w. [DOI] [PubMed] [Google Scholar]
- Kapadia N.; Harding W. Aporphine Alkaloids as Ligands for Serotonin Receptors. Med. Chem. 2016, 6 (4), 241–249. 10.4172/2161-0444.1000353. [DOI] [Google Scholar]
- Farrell M. S.; McCorvy J. D.; Huang X. P.; Urban D. J.; White K. L.; Giguere P. M.; Doak A. K.; Bernstein A. I.; Stout K. A.; Park S. M.; Rodriguiz R. M.; Gray B. W.; Hyatt W. S.; Norwood A. P.; Webster K. A.; Gannon B. M.; Miller G. W.; Porter J. H.; Shoichet B. K.; Fantegrossi W. E.; Wetsel W. C.; Roth B. L. In Vitro and In Vivo Characterization of the Alkaloid Nuciferine. PLoS One 2016, 11 (3), e0150602. 10.1371/journal.pone.0150602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K.; Sassano M. F.; Huang X. P.; Lansu K.; McCorvy J. D.; Giguere P. M.; Sciaky N.; Roth B. L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22 (5), 362–9. 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Sun Y.; Xu D.; Yang Y.; Liu X.; Wei Y.; Chen Y.; Feng Z.; Li S.; Reyad-Ul Ferdous M.; Zhao Y.; Xu H.; Lao Y.; Ding Q. Screening of FDA-approved drugs identifies sutent as a modulator of UCP1 expression in brown adipose tissue. EBioMedicine 2018, 37, 344–355. 10.1016/j.ebiom.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen W. J.; Grottick A. J.; Menzaghi F.; Reyes-Saldana H.; Espitia S.; Yuskin D.; Whelan K.; Martin M.; Morgan M.; Chen W.; Al-Shamma H.; Smith B.; Chalmers D.; Behan D. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther. 2008, 325 (2), 577–87. 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]
- Kahsai A. W.; Wisler J. W.; Lee J.; Ahn S.; Cahill Iii T. J.; Dennison S. M.; Staus D. P.; Thomsen A. R.; Anasti K. M.; Pani B.; Wingler L. M.; Desai H.; Bompiani K. M.; Strachan R. T.; Qin X.; Alam S. M.; Sullenger B. A.; Lefkowitz R. J. Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat. Chem. Biol. 2016, 12 (9), 709–16. 10.1038/nchembio.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kim T. H.; Masureel M.; Altenbach C.; Yang Z.; Hilger D.; Lerch M. T.; Kobilka T. S.; Thian F. S.; Hubbell W. L.; Prosser R. S.; Kobilka B. K. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 2015, 161 (5), 1101–1111. 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Pu M.; Qu L.; Han G. W.; Wu Y.; Zhao S.; Shui W.; Li S.; Korde A.; Laprairie R. B.; Stahl E. L.; Ho J. H.; Zvonok N.; Zhou H.; Kufareva I.; Wu B.; Zhao Q.; Hanson M. A.; Bohn L. M.; Makriyannis A.; Stevens R. C.; Liu Z. J. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167 (3), 750–762. 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Gao Z. G.; Zhang K.; Kiselev E.; Crane S.; Wang J.; Paoletta S.; Yi C.; Ma L.; Zhang W.; Han G. W.; Liu H.; Cherezov V.; Katritch V.; Jiang H.; Stevens R. C.; Jacobson K. A.; Zhao Q.; Wu B. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 2015, 520 (7547), 317–21. 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G.; Yang D.; Wang Y.; de Graaf C.; Zhou Q.; Jiang S.; Liu K.; Cai X.; Dai A.; Lin G.; Liu D.; Wu F.; Wu Y.; Zhao S.; Ye L.; Han G. W.; Lau J.; Wu B.; Hanson M. A.; Liu Z. J.; Wang M. W.; Stevens R. C. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 2017, 546 (7657), 312–315. 10.1038/nature22378. [DOI] [PubMed] [Google Scholar]
- Whitehurst C. E.; Yao Z. P.; Murphy D.; Zhang M. X.; Taremi S.; Wojcik L.; Strizki J. M.; Bracken J. D.; Cheng C. C.; Yang X. S.; Shipps G. W.; Ziebell M.; Nickbarg E. Application of Affinity Selection-Mass Spectrometry Assays to Purification and Affinity-Based Screening of the Chemokine Receptor CXCR4. Comb. Chem. High Throughput Screening 2012, 15 (6), 473–485. 10.2174/138620712800563945. [DOI] [PubMed] [Google Scholar]
- Jenner P.; Katzenschlager R. Apomorphine - pharmacological properties and clinical trials in Parkinson’s disease. Parkinsonism Relat Disord 2016, 33 (S1), S13–S21. 10.1016/j.parkreldis.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Ye N.; Neumeyer J. L.; Baldessarini R. J.; Zhen X.; Zhang A. Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2013, 113 (5), PR123–78. 10.1021/cr300113a. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Ye N.; Zhou S.; Guo L.; Zheng L.; Liu Z.; Gao B.; Zhen X.; Zhang A. Identification of N-propylnoraporphin-11-yl 5-(1,2-dithiolan-3-yl)pentanoate as a new anti-Parkinson’s agent possessing a dopamine D2 and serotonin 5-HT1A dual-agonist profile. J. Med. Chem. 2011, 54 (13), 4324–38. 10.1021/jm200347t. [DOI] [PubMed] [Google Scholar]
- Peng Y.; Zhao S.; Wu Y.; Cao H.; Xu Y.; Liu X.; Shui W.; Cheng J.; Zhao S.; Shen L.; Ma J.; Quinn R. J.; Stevens R. C.; Zhong G.; Liu Z. J. Identification of natural products as novel ligands for the human 5-HT2C receptor. Biophys Rep 2018, 4 (1), 50–61. 10.1007/s41048-018-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.