

Abstract

A new technology platform built on the integration of theory and experiments to enable the design of Janus colloids with precision control of surface anisotropy and amphiphilicity could lead to a disruptive transformation in the next generation of surfactants, photonic or phononic materials, and coatings. Here, we exploit molecular dynamics (MD) simulations to guide the rational design of amphiphilic polymer Janus colloids by Flash NanoPrecipitation (FNP), a method capable of the production of colloids with complex structure without the compromise of reduced scalability. Aided by in silico design, we show in experiments that amphiphilic Janus colloids can be produced using a unique blend of hydrophobic homopolymers and the addition of an amphiphilic block copolymer. The final structure of the colloids depends on the mass fraction of each homopolymer as well as the concentration and composition of the block copolymer additive. To confirm the surface activity of the colloids, we demonstrate their potential to stabilize Pickering emulsions. This hybrid approach of simulations and experiments provides a pathway to designing and manufacturing complex polymeric colloids on an industrial scale.
Short abstract
An integrated approach of molecular dynamics simulations and experiments provides a framework for the design of surface-active amphiphilic polymer colloids using a scalable fabrication technology.
Introduction
Polymeric colloids with structural complexity, including Janus colloids with two distinct surface domains, have shown promise in applications ranging from theragnostic nanomedicine to smart displays.1−5 In his 1991 Nobel lecture, Pierre-Gilles de Gennes highlighted the potential of amphiphilic “Janus grains” that have similar features to molecular surfactants.6 From this viewpoint, Janus colloids may be thought of as next generation surfactants. Homogeneous colloids may already adsorb to an oil/water interface, forming a so-called Pickering emulsion in which the colloids form a layer on the surface of the dispersed droplets.7−12 With increasing the amphiphilicity of the colloids, thereby creating a Janus structure, they more effectively reduce the interfacial tension13,14 with an adsorption energy triple that of homogeneous colloids.15,16 The large adsorption energy essentially traps amphiphilic Janus colloids at the oil/water interface. Therefore, unlike emulsions stabilized by molecular surfactants which adsorb reversibly onto a droplet surface, amphiphilic Janus colloids adsorb irreversibly at oil–water interfaces, forming a physical barrier that hinders droplet coalescence.15,17−19
Despite the manifold potential applications of amphiphilic Janus colloids, producing such surface-active colloids in sufficient quantity remains challenging, which has slowed their implementation at industrial scales.20−23 Current synthetic approaches can involve multiple complex steps, leading to low production yields and the formation of colloids with limited chemical diversity. Therefore, it is essential to develop a facile approach to produce amphiphilic Janus colloids with ease of tunability and scalability. Such an approach opens the doors to generate amphiphilic Janus colloids for Pickering emulsions and other applications at commercial scales.
Previously, we experimentally and theoretically demonstrated that the rapid mixing of a binary solution of hydrophobic homopolymers against an aqueous antisolvent stream resulted in the assembly of polymeric Janus colloids.24−26,34 Using this technique, termed Flash NanoPrecipitation (FNP), as shown schematically in Figure 1a, both colloid size and surface anisotropy were independently controlled. Unlike other precipitation-based methods, FNP induces precipitation of the polymers upon solvent exchange within a confined volume on a millisecond time scale. By controlling the polymer concentration in the feed streams and the rate of mixing, colloids with tunable sizes and with a narrow size distribution could be produced.27 Moreover, the FNP technique is capable of continuous operation—the preferred operation mode for large-scale production of polymeric colloids. In spite of the ease of operation and flexibility of the FNP technique, so far only Janus colloids comprising two dissimilar hydrophobic polymers (A and B) have been successfully produced, and the formation of amphiphilic Janus colloids via FNP remained a technical challenge.
Figure 1.

(a) Schematic illustration of the Flash NanoPrecipitation (FNP) technique for producing amphiphilic Janus colloids. (b) MD simulations and experimental design space to investigate the effects of block copolymer content and the volume fraction of the hydrophilic block on the polymer colloid structure produced via FNP.
One strategy to produce amphiphilic Janus colloids using the FNP technique, without any loss in process scalability, would be to functionalize one side of the hydrophobic Janus colloids with an amphiphilic A-b-C block copolymer (BCP). The addition of a BCP, however, introduces several parameters, which could potentially alter the assembly process of the polymers and thus influence the final structure of the precipitated colloids. Among the numerous parameters, the final morphology of the structured colloids in the presence of BCP depends strongly on the BCP concentration and volume fraction of the hydrophilic block. These factors lead to a large parameter space for producing various amphiphilic colloids using FNP and thus necessitate an extensive searching-and-screening approach that is impracticable in experiments.
To develop a feasible strategy to design amphiphilic Janus colloids, we first screened the parameter space via a series of computational studies of the FNP process. With guidance from simulations, we drastically reduced the number of experiments required to develop a formulation to produce amphiphilic Janus colloids by FNP. A design space was, therefore, developed outlining optimal ranges of BCP concentration and composition to be employed in the FNP experiments—see Figure 1b. Furthermore, the partnership between computational and experimental studies allows for a synergistic approach in understanding the mechanisms of amphiphilic Janus colloid formation as well as the molecular-scale characterization of the colloid structure. Finally, we show the potential of amphiphilic Janus colloids produced by FNP in generating Pickering emulsions.
Results and Discussion
To design amphiphilic colloids through an approach that is both scalable and tunable, we exploited the solubility properties of homopolymer-BCP blends in the FNP technique. In both experiments and simulations, blends of two hydrophobic homopolymers and one amphiphilic BCP were incorporated into the feed stream of the FNP system, as shown in Figure 1a. The two hydrophobic homopolymers used were polystyrene (PS) and polyisoprene (PI). We used the amphiphilic BCP polystyrene-block-polyethylene glycol (PS-b-PEG). In order to efficiently survey the design space and evaluate key process parameters, we performed molecular dynamics (MD) simulations using a bead–spring polymer model in an explicit solvent27—see the methods section for more details. In particular, we systematically studied the effects of BCP content and composition on the colloid morphology. In all cases, PI formed 50% of the final colloid mass, and the other 50% of the colloid mass contained either PS or PS together with PS-b-PEG. The mass fraction of BCP relative to the total colloid mass, CBCP, was increased by replacing homopolymer PS in the system. The BCP composition, fphil, is quantified by the ratio of the volume of the PEG block (VPEG) to the total volume of a BCP chain (Vtotal):
| 1 |
Both parameters fphil and CBCP could be utilized to tune the degree of amphiphilicity of the precipitated polymer colloids, as the former controls the volume fraction of the hydrophilic block per BCP, while the latter sets the overall fraction of BCP in the system (and thus in each precipitated polymer colloid). Five BCP compositions were initially examined in our simulations, fphil = 0.09, 0.26, 0.39, 0.70, and 0.91 corresponding to 2, 6, 9, 16, and 21 hydrophilic monomers, respectively, out of 23 in the BCP. Also, five BCP contents, CBCP = 10, 20, 30, 40, and 50% were considered, while the amount of PI was kept constant at CPI = 50%, as explained earlier. We initially equilibrated the polymers in a good solvent, which was then replaced instantaneously by the antisolvent, resulting in the collapse and aggregation of the hydrophobic polymers. In all simulations, the molecular weights of the polymers were less than their entanglement Mw. Furthermore, all simulations were conducted at a temperature above the glass transition temperature of all polymers. Thus, the polymers could move freely within the polymeric colloid to optimize their arrangement. Once the systems reached equilibrium, we analyzed both the resulting morphology of the colloids as well as the spatial distribution of each of the three components in the polymeric colloidal domain.
Figure 2 shows the resulting morphology diagram from simulations, indicating that for the BCPs with the shortest hydrophilic block, fphil = 0.09, a Janus morphology was observed for the entire range of CBCP with the amphiphilic BCPs adsorbed only atop the PS domain of the Janus colloid. In this case, the influence of the hydrophilic block on the colloid morphology was negligible, and we observed the same Janus morphology as for the system of two homopolymers alone.25 For fphil = 0.26 and 0.39, similar behavior was found for CBCP ≤ 40%, until a morphology change from Janus to a transitional state occurred at CBCP = 50%. In this transitional state, colloids contained a core made of PI which was partly covered by the amphiphilic BCPs. Upon increasing fphil further to 0.70, amphiphilic Janus particles were observed up to CBCP = 20%, followed by a transition to a core–shell morphology at CBCP = 30%. In the transition state, the two homopolymers formed the core of the colloid and phase-separated into a Janus morphology, but the BCP adsorbed on the entire colloid surface regardless of the homopolymer beneath (see simulation snapshots in Figure 2). A rather different behavior was observed for the highest value of fphil = 0.91, where no core–shell morphologies developed at any value of CBCP. For this case, the behavior of the BCPs was dominated by their hydrophilic block, keeping the majority of BCPs dispersed in the solution. Only a small fraction of them adsorbed atop the colloid surface, and this fraction monotonically decreased with increasing CBCP (see Figure S2 in the Supporting Information). For all other systems in our study, all BCP chains adsorbed on the colloid surfaces.
Figure 2.

Equilibrium morphologies of simulated colloids as a function of the BCP composition, fphil, and content, CBCP. The lines in the morphology diagram are intended as guides to the eye. Representative simulation snapshots are shown at the bottom of the figure and have been rendered using Visual Molecular Dynamics v.1.9.28 PS beads are colored yellow, PI beads are colored blue, and PEG beads are colored orange. A cross-sectional view was chosen for the core–shell colloid to better visualize its morphology.
On the basis of the design space explored through our MD simulations, BCPs with fphil values between 0.2 and 0.8 appear to offer the most flexibility in colloid morphology and amphiphilicity. Therefore, BCPs with fphil values of 0.27, 0.55, and 0.74 were used in our experimental studies. When blends of PS and PI together with an amphiphilic PS-b-PEG BCP with fphil = 0.74 were employed, similar to our simulation results, we observed shifts in colloid morphology as a function of BCP content—see Figure 3. With no BCP in the feed stream, Janus colloids with two hemispheres separated by a flat interface were observed. As the BCP content was increased to CBCP = 20%, the diameter of the colloids decreased; yet, the interface between the PS and PI domains remained flat. At even higher BCP content, CBCP = 40%, core–shell colloids were formed where the PI core was completely covered by the PS-b-PEG BCP. The morphologies observed experimentally are in excellent agreement with the findings of the MD simulations at the same processing conditions (see Figure 2) and indicate that the BCP was indeed adsorbing atop the PS domain.
Figure 3.

TEM images of polymer colloids prepared from mixtures with CBCP increasing from 0% to 40%, as indicated, for BCPs with fphil = 0.74. Scale bars indicate 200 nm. In the illustrations, PS is colored yellow, PI is colored blue, and PEG is colored orange.
Janus colloids prepared without BCP had a diameter of approximately 660 nm and a ζ-potential of −26 mV—see Figure 4a,b. As CBCP increased from 1% to 50%, for all BCP compositions ranging from fphil = 0.27 to 0.74, the size of the colloids decreased, and the ζ-potential of the particles became more neutral. We attribute the decrease in the size of colloids, with increasing the BCP content, to the formation of a hydrophilic corona on the colloid surface which sterically stabilized the particles against further aggregation. Similarly, the decrease in the magnitude of the colloid ζ-potential indicates that the BCP must have adsorbed atop the colloid surfaces,29 as expected from the MD simulations. Changes in ζ-potential for samples with fphil = 0.74 were more significant than those for fphil = 0.27. In the former, the isoelectric point was obtained at moderate BCP content, while in the latter, the magnitude of the ζ-potential decreased gradually to an average value of −5 mV at high BCP content. This effect might originate from the inability of the shorter PEG blocks to completely screen out the negative surface charges or from the partial PEG coverage on the colloid surface, which allows for the adsorption of negative charges on the bare PI hemisphere (see snapshots in Figure 2).
Figure 4.

(a) Average particle diameter, d, measured via dynamic light scattering (DLS) as a function of CBCP in the feed stream for three different BCP compositions, as indicated. (b) ζ-Potential of colloids shown in (a) as a function of CBCP. The magenta pentagon in both panels shows the result for purely hydrophobic Janus particles (CBCP = 0).
The location of the hydrophilic blocks in BCP on one-half of the Janus surface has been confirmed from snapshots of the MD simulations (see Figure 2). However, the amphiphilicity of the entire Janus colloid and thus its effectiveness as an emulsifier strongly depend on the density of BCP on the surface of colloids. The density of BCP, Σ, can be estimated from30
| 2 |
where Rg is the radius of gyration of the adsorbed hydrophilic block, and σ is the BCP surface density defined as σ = n/(4πRcore2). In this definition, n is the number of BCP chains adsorbed on the surface of colloids, and Rcore is the radius of the hydrophobic colloid core. The transition from a low grafting density “mushroom” state to a higher density “brush” state is defined as the point when Σ = 1.30Figure 5 shows that a brush state occurred in the simulations only for fphil = 0.70 at CBCP ≥ 30%, which coincides with the morphology transition from a Janus to a core–shell configuration; see Figure 2.
Figure 5.

BCP density, Σ, as a function of fphil for various CBCP, as indicated. The transition from the “mushroom” state to the “brush” state is indicated by the horizontal line at Σ = 1.0.
Further, we investigated the orientation of the hydrophilic blocks at the colloid surface. An analysis of the eigenvectors describing the directionality of the hydrophilic block of the BCP was performed to elucidate the conformation of the polymer with respect to the radius of the colloid core. Details of the analysis can be found in the Supporting Information, and we discuss here only the salient features. Figure 6 shows the probability distribution of the angle |cos Θ| formed between the director of the hydrophilic block, n, and its adsorption point, r, when fphil = 0.70 (a value of |cos Θ| = 1 corresponds to a stretched out brush layer, whereas a value of |cos Θ| = 0 indicates that the hydrophilic block lies flat on the colloid surface). For CBCP = 10%, we identified a maximum at |cos Θ| = 0; i.e., the hydrophilic PEG block spread onto the hydrophobic PS surface to minimize the surface tension with the surrounding liquid phase. As CBCP increased, the probability distribution became more uniform, indicating that the hydrophilic blocks started to form a more “pancake”-like layer on the PS surface due to excluded volume effects. A similar behavior was observed for BCPs with fphil = 0.39 and 0.91 (data not shown for brevity). For BCPs with fphil = 0.09, the analysis described above was not applicable since the hydrophilic BCP tail was too short.
Figure 6.

(a) Schematic describing the director of the hydrophilic block n, and its adsorption point, r. (b) Representative simulation snapshots for systems with fphil = 0.70 at CBCP = 10% and 50%. Only selected hydrophilic blocks have been rendered to improve clarity. (c) Probability density distributions, P(|cos Θ|), of the average angle between n and r in the systems where fphil = 0.70 and CBCP is varied from 10% to 50%.
Amphiphilic colloids have been shown to be effective stabilizers from extensive studies of Pickering emulsions in the literature, where the most effective stabilizers were hydrophilic silica particles grafted with surfactants or BCPs.15 In this work, amphiphilic Janus colloids were prepared using FNP with a blend of 50% PI, 40% PS, and 10% amphiphilic BCP in the feed stream, as demonstrated earlier in experiments and MD simulations. To examine the ability of amphiphilic Janus colloids as emulsifiers, a vortex mixer was used to mix aqueous suspensions of the amphiphilic Janus colloids at a particle concentration of 0.05 wt % with decane at a volume ratio of 2-to-1 for aqueous-to-oil. The volume and stability of any resulting emulsion were assessed over the course of several hours. Oil/water mixtures without polymer colloids phase separated spontaneously after mixing with no emulsion phase, as shown in Figure 7a. When oil was mixed with water containing non-amphiphilic Janus colloids (i.e., bare PS/PI Janus colloids), a small emulsion phase was obtained which was stable for less than 3 h after mixing (see Figure 7b). For the same amount of time, mixtures with colloids containing amphiphilic BCPs with an fphil of 0.27, 0.55, or 0.74 exhibited a distinct emulsified phase even at low BCP content (CBCP = 10%) and low particle concentration (0.05 wt %); see Figure 7c–e. The stability of Pickering emulsions generated with amphiphilic Janus colloids is a function of the composition of the BCPs. Formulations with short hydrophilic blocks (fphil = 0.27) created a smaller emulsion phase, 38% of the total volume, whereas the colloids prepared with a more hydrophilic BCP (fphil = 0.74) resulted in a larger emulsion phase, up to 60% of the total volume; see Figure 7c–e. The higher PEG content of the more hydrophilic BCPs increased the amphiphilicity of the Janus colloid, making those colloids more effective in stabilizing Pickering emulsions than those prepared with a more hydrophobic BCP. This is direct evidence showing that the amphiphilic Janus colloids were indeed surface-active. Because amphiphilic Janus colloids adsorbed strongly to the oil/water interface, they were able to stabilize the emulsion phase better than the purely hydrophobic Janus particles.
Figure 7.

(a) Decane/water mixtures containing no colloids, (b) PS/PI Janus colloids, and (c–e) Janus colloids prepared from PS/PI/PS-b-PEG mixtures in FNP with CBCP = 10%. All images have been taken 3 h after mixing except for (a) which was right after mixing. The upper phase is decane, the lower phase is water, and the middle phase is the emulsion with the volume fraction of the emulsion indicated below the images.
Methods and Materials
Materials
Polystyrene (PS, Mn = 16.0 kg/mol, dispersity (Đ) = 1.03), polyisoprene (PI, Mn = 11.0 kg/mol, Đ = 1.06), and all polystyrene-block-polyethylene glycol (PS-b-PEG) diblock copolymers (fphil = 0.27, Mn = 5-b-2 kg/mol, Đ = 1.09; fphil = 0.54, Mn = 3.8-b-5 kg/mol, Đ = 1.06; and fphil = 0.74, Mn = 1.6-b-5 kg/mol, Đ = 1.10) were purchased from Polymer Source Inc. (Dorval, Quebec, Canada). Tetrahydrofuran (THF) was purchased from Fisher Scientific, and DI water was filtered through a 0.2 μm filter using a NANOpure Diamond filtration system. Decane was purchased from Alfa Aesar and purified over Florisil purchased from Sigma-Aldrich for 12 h.
Experimental Methods
Flash NanoPrecipitation (FNP) was performed using a confined impingement jet (CIJ) mixer with the given polymers in the desired mass ratios dissolved in THF in the feed stream at a total polymer concentration of 1 mg/mL. The feed stream was mixed with the antisolvent stream composed of DI water at a total flow rate of 2 mL/min, and the effluent was quenched in an antisolvent reservoir of DI water at a feed stream/antisolvent stream/antisolvent reservoir volume ratio of 1:1:1. The residual THF was removed from the system by rotary evaporation at a temperature of 37 °C and a pressure of 100 Torr for 30 min.
Colloids were prepared for imaging after removal of residual THF by staining with osmium tetraoxide (OsO4). A 500 μL sample of the colloidal suspension was added to 500 μL of a 0.2 wt % OsO4 aqueous solution and left for 2 h to stain at room temperature. The resulting mixture was centrifuged at 4000 rpm at 5 °C for 10 min, and a 0.8 μL drop of the supernatant was deposited on a carbon-coated copper grid (CF-200-Cu, Electron Microscopy Sciences) and air-dried for transmission electron microscopy (TEM) imaging. TEM imaging was performed on a CM200 TEM with an accelerating voltage of 200 kV.
Colloid size was measured after THF removal using dynamic light scattering (DLS) on a Malvern ZetaSizer (Malvern Instruments, Malvern, UK) with a 633 nm laser and backscatter detection angle of 173°. Stability of the colloids in the presence of salt was tested using DLS by preparing aqueous KCl solutions ranging in concentration from 0.3 mM to 3 M. An 80 μL sample of the colloidal suspension to be tested was added to 160 μL of the desired salt solution in a centrifuge tube. The suspension was mixed and left to sit for at least 30 min. The sample was further diluted in DI water before measuring colloid size via DLS. ζ-Potential results were also obtained on the Malvern ZetaSizer by diluting the colloidal suspension in PBS (0.1x) to achieve sufficient conductivities for measurement in a folded capillary cell.
Emulsion testing was performed using mixtures of the aqueous colloidal suspensions after removal of the residual THF by rotary evaporation and decane that had been purified over Florisil for at least 12 h. Two milliliters of the colloidal suspension with a final colloid concentration of 0.05 wt % was added to 1 mL of decane, and the mixture was agitated on a digital vortex mixer (FisherScientific) for 1 min at 2000 rpm. After mixing, pictures were taken from the samples after 3 h sitting on the benchtop at room temperature. Image analysis was performed using ImageJ analytical software to determine the emulsion phase volume fraction. To do so, the projected side area by the emulsion was normalized by the total projected side area by all the phases (water, oil, and emulsion). There is no unexpected or unusually high safety hazards encountered with the materials or experimental procedures. Extra caution, however, must be given to handle osmium tetraoxide (OsO4) according to its safety data sheets.
Simulation Model
We performed coarse-grained molecular dynamics (MD) simulations of the FNP process using two types of hydrophobic homopolymers, polymer A (PS) and polymer B (PI), and an amphiphilic diblock copolymer A-b-C (PS-b-PEG). Our previous MD model for homopolymers in the FNP technique serves as a foundation for the current study.27 In this model, a polymer is described as a linear bead–spring chain with N beads, each with unit diameter, a, and unit mass, m. Each bead represents a Kuhn segment of the polymer chain. The PS chains employed in the experiments have a molecular weight of 16.0 kg/mol, and thus consist of N = 23 Kuhn segments given the mass of a PS Kuhn segment MK = 0.72 kg/mol.31 For simplicity, we used the same number of beads for the PI and PS-b-PEG polymers. The covalent bonds between monomers are modeled via the finitely extensible nonlinear elastic (FENE) potential.32 The standard Kremer-Grest parameters were adopted to prevent unphysical bond crossing.33 The nonbonded interactions between the beads are described via the standard Lennard-Jones (LJ) potential, ULJ:
 |
3 |
where r is the distance between the pair of particles, rcut = 3a is the cutoff radius of the potential, and εij controls the interaction strength between particles of types i and j. Three types of polymer beads were considered in the current work, and we set the intraspecies LJ interaction strength of all the types equal such that εAA = εBB = εCC = kBT ≡ ε, where kB is the Boltzmann constant and T = 1 is the temperature. The interspecies interaction strength between the two hydrophobic homopolymers, εAB = 0.9 ε, was taken from our previous simulation study of PS–PI polymer blends.25,35 We determined A–C and B–C interspecies interaction strengths, εAC and εBC, respectively, using χ-parameter estimates (i.e., χ = 0.9 and χ = 2.82 for PS-b-PEG36 and PI-b-PEG37 block copolymers, respectively) in combination with the procedure proposed by Chremos et al.38 The resulting values are εAC = 0.92 ε and εBC = 0.75 ε.
Following our previous works,25,27,35 we did not use separate particle types for the solvent and antisolvent molecules, but rather grouped those into effective LJ particles with the same size and mass as the polymer beads. In this simplified representation, solvent exchange can be mimicked by (gradually) changing the interaction between the polymer beads and all solvent particles in the system. Here, the solvent–solvent interaction strength was fixed to εSS = ε, while the interaction between solvent particles and monomers, UMS, was controlled via the dimensionless parameter, 0 ≤ λ ≤ 1:
| 4 |
where UWCA(r) is the purely repulsive Weeks–Chandler–Andersen (WCA) potential.39 In the beginning of a simulation run, the system was equilibrated at λ = 0 for all polymers, corresponding to good solvent conditions. In order to model worsening of the solvent quality during the FNP process, we instantaneously changed λ to 0.5 for the interactions between solvophobic monomers A and B and solvent particles, while keeping λ = 0 for the hydrophilic C species.27
The simulations were
performed using the HOOMD-blue simulations
package.40,41 A simulation box with an edge length of
82.7 a containing a total 373 248 particles,
consisting of solvent particles and 1024 polymers at various CBCP was adopted. For all simulations, the reduced
particle number density was fixed to ρ = 0.66 a–3, which leads to a pressure close to zero in
the pure solvent systems (P ≈ 0.02 ε/a3) and in the systems containing a polymer colloid
(P ≈ 0.07 ε/a3). A Nosé–Hoover thermostat was employed to maintain
the temperature at T = 1, and the equations of motion
were integrated using the MTTK algorithm at a time step of Δt = 0.005τ, where 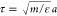 is the intrinsic MD unit of time. Each
simulation was conducted for at least 1.5 × 108 time
steps, and snapshots were saved every 30 000 time steps for
analysis. Measurements were taken only once the systems reached equilibrium
(typically after 5 × 107 time steps), which was determined
by monitoring the structure of the precipitated colloid and the potential
energy of the system over time. Figure S6 in the
Supporting Information shows the temporal evolution of the
potential energy for two selected cases. Moreover, simulation trajectories
demonstrating the dynamics of self-assembly are shown in the Supporting Information for CBCP = 10% and fphil = 0.26, and CBCP = 50% and fphil = 0.39. Unlike our previous simulations27 where an electrostatic repulsion was applied to stabilize the precipitated
colloids, such a repulsion was not used in the current study since
we are mainly interested in the equilibrium morphologies of colloids
formed in the system. In this model, the size of the final polymer
colloids is only dictated by the total number of polymers in the system,
and we found colloid diameters d ≈ 36 a in our simulations. Following the mapping established
in ref (27), this value
corresponds to a particle diameter of d ≈
54 nm, which is comparable to the average particle diameters measured
in the experiments (cf. Figure 4a).
is the intrinsic MD unit of time. Each
simulation was conducted for at least 1.5 × 108 time
steps, and snapshots were saved every 30 000 time steps for
analysis. Measurements were taken only once the systems reached equilibrium
(typically after 5 × 107 time steps), which was determined
by monitoring the structure of the precipitated colloid and the potential
energy of the system over time. Figure S6 in the
Supporting Information shows the temporal evolution of the
potential energy for two selected cases. Moreover, simulation trajectories
demonstrating the dynamics of self-assembly are shown in the Supporting Information for CBCP = 10% and fphil = 0.26, and CBCP = 50% and fphil = 0.39. Unlike our previous simulations27 where an electrostatic repulsion was applied to stabilize the precipitated
colloids, such a repulsion was not used in the current study since
we are mainly interested in the equilibrium morphologies of colloids
formed in the system. In this model, the size of the final polymer
colloids is only dictated by the total number of polymers in the system,
and we found colloid diameters d ≈ 36 a in our simulations. Following the mapping established
in ref (27), this value
corresponds to a particle diameter of d ≈
54 nm, which is comparable to the average particle diameters measured
in the experiments (cf. Figure 4a).
Acknowledgments
This work was supported by the National Science Foundation (NSF) Materials Research Science and Engineering Center Program through the Princeton Center for Complex Materials (DMR-1420541) and by the German Research Foundation (DFG) under Project Number NI 1487/2-1. R.D.P. acknowledges the support of NSF PFI Grant (IIP - 1827506).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b00974.
Simulation results for the hydrophilic blocks (PEG) density and orientation on colloids surfaces. Simulation results for micelle formation. Potential energy as a function of time from selected simulations. Experimental results on the morphology of amphiphilic Janus colloids. Discussions on the stability of Pickering emulsions (PDF)
Videos of the self-assembly process for core–shell and Janus colloids (MP4-1, MP4-2)
Author Contributions
# T.I.M. and V.E.L. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Wu L. Y.; Ross B. M.; Hong S. G.; Lee L. P. Bioinspired Nanocorals with Decoupled Cellular Targeting and Sensing Functionality. Small 2010, 6, 503–507. 10.1002/smll.200901604. [DOI] [PubMed] [Google Scholar]
- Kanahara M.; Satoh H.; Higuchi T.; Takahara A.; Jinnai H.; Harano K.; Okada S.; Nakamura E.; Matsuo Y.; Yabu H. Fabrication of NIR-Excitable SERS-Active Composite Particles Composed of Densely Packed Au Nanoparticles on Polymer Microparticles. Part. Part. Syst. Charact. 2015, 32, 441–447. 10.1002/ppsc.201400191. [DOI] [Google Scholar]
- Yi Y.; Sanchez L.; Gao Y.; Yu Y. Janus Particles for Biological Imaging and Sensing. Analyst 2016, 141, 3526–3539. 10.1039/C6AN00325G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. D.; Wilson B. K.; Lim T. L.; Heinmiller A.; Prud’homme R. K. Real-Time and Multiplexed Photoacoustic Imaging of Internally Normalized Mixed-Targeted Nanoparticles. ACS Biomater. Sci. Eng. 2017, 3, 443–451. 10.1021/acsbiomaterials.6b00645. [DOI] [PubMed] [Google Scholar]
- Shahravan A.; Matsoukas T. Encapsulation and Controlled Release from Core-Shell Nanoparticles Fabricated by Plasma Polymerization. J. Nanopart. Res. 2012, 14, 668–678. 10.1007/s11051-011-0668-5. [DOI] [Google Scholar]
- de Gennes P.-G. Soft Matter (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1992, 31, 842–845. 10.1002/anie.199208421. [DOI] [Google Scholar]
- Binks B. P.; Lumsdon S. O. Pickering Emulsions Stabilized by Monodisperse Latex Particles: Effects of Particle Size. Langmuir 2001, 17, 4540–4547. 10.1021/la0103822. [DOI] [Google Scholar]
- Nallamilli T.; Binks B. P.; Mani E.; Basavaraj M. G. Stabilization of Pickering Emulsions with Oppositely Charged Latex Particles: Influence of Various Parameters and Particle Arrangement around Droplets. Langmuir 2015, 31, 11200–11208. 10.1021/acs.langmuir.5b02443. [DOI] [PubMed] [Google Scholar]
- Sun G.; Li Z.; Ngai T. Inversion of Particle-Stabilized Emulsions to Form High-Internal-Phase Emulsions. Angew. Chem., Int. Ed. 2010, 49, 2163–2166. 10.1002/anie.200907175. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Sun J.; Yi C.; Wei W.; Liu X. One-Step Formation of Multiple Pickering Emulsions Stabilized by Self-Assembled Poly(Dodecyl Acrylate-Co-Acrylic Acid) Nanoparticles. Soft Matter 2016, 12, 7577–7584. 10.1039/C6SM01263A. [DOI] [PubMed] [Google Scholar]
- Datta S. S.; Shum H. C.; Weitz D. A. Controlled Buckling and Crumpling of Nanoparticle-Coated Droplets. Langmuir 2010, 26, 18612–18616. 10.1021/la103874z. [DOI] [PubMed] [Google Scholar]
- Bizmark N.; Ioannidis M. A. Ethyl Cellulose Nanoparticles at the Alkane-Water Interface and the Making of Pickering Emulsions. Langmuir 2017, 33, 10568–10576. 10.1021/acs.langmuir.7b02051. [DOI] [PubMed] [Google Scholar]
- Glaser N.; Adams D. J.; Böker A.; Krausch G. Janus Particles at Liquid–liquid Interfaces. Langmuir 2006, 22, 5227–5229. 10.1021/la060693i. [DOI] [PubMed] [Google Scholar]
- Ruhland T. M.; Gröschel A. H.; Ballard N.; Skelhon T. S.; Walther A.; Müller A. H. E.; Bon S. A. F. Influence of Janus Particle Shape on Their Interfacial Behavior at Liquid–Liquid Interfaces. Langmuir 2013, 29, 1388–1394. 10.1021/la3048642. [DOI] [PubMed] [Google Scholar]
- Aveyard R.; Binks B. P.; Clint J. H. Emulsions Stabilised Solely by Colloidal Particles. Adv. Colloid Interface Sci. 2003, 100, 503–546. 10.1016/S0001-8686(02)00069-6. [DOI] [Google Scholar]
- Jiang S.; Granick S. Janus Balance of Amphiphilic Colloidal Particles. J. Chem. Phys. 2007, 127, 161102. 10.1063/1.2803420. [DOI] [PubMed] [Google Scholar]
- Schröder A.; Sprakel J.; Schroën K.; Spaen J. N.; Berton-Carabin C. C. Coalescence Stability of Pickering Emulsions Produced with Lipid Particles: A Microfluidic Study. J. Food Eng. 2018, 234, 63–72. 10.1016/j.jfoodeng.2018.04.007. [DOI] [Google Scholar]
- Tcholakova S.; Denkov N. D.; Lips A. Comparison of Solid Particles, Globular Proteins and Surfactants as Emulsifiers. Phys. Chem. Chem. Phys. 2008, 10, 1608–1627. 10.1039/b715933c. [DOI] [PubMed] [Google Scholar]
- Whitby C. P.; Wanless E. J. Controlling Pickering Emulsion Destabilisation: A Route to Fabricating New Materials by Phase Inversion. Materials 2016, 9, 626–647. 10.3390/ma9080626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S.; Granick S.. Janus Balance and Emulsions Stabilized by Janus Particles. In Janus Particle Synthesis, Self-Assembly and Applications; Jiang S., Granick S., Eds.; Royal Society of Chemistry, 2013; pp 244–256. 10.1039/9781849735100-00244. [DOI] [Google Scholar]
- Tu F.; Lee D. Shape-Changing and Amphiphilicity-Reversing Janus Particles with PH-Responsive Surfactant Properties. J. Am. Chem. Soc. 2014, 136, 9999–10006. 10.1021/ja503189r. [DOI] [PubMed] [Google Scholar]
- Razza N.; Rizza G.; Coulon P.-E.; Didier L.; Fadda G. C.; Voit B.; Synytska A.; Grützmacher H.; Sangermano M. Enabling the Synthesis of Homogeneous or Janus Hairy Nanoparticles through Surface Photoactivation. Nanoscale 2018, 10, 14492–14498. 10.1039/C8NR04239J. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Fernández D.; Liz-Marzán L. M. Metallic Janus and Patchy Particles. Part. Part. Syst. Charact. 2013, 30, 46–60. 10.1002/ppsc.201200057. [DOI] [Google Scholar]
- Grundy L. S.; Lee V. E.; Li N.; Sosa C.; Mulhearn W. D.; Liu R.; Register R. A.; Nikoubashman A.; Prud’Homme R. K.; Panagiotopoulos A. Z.; et al. Rapid Production of Internally Structured Colloids by Flash Nanoprecipitation of Block Copolymer Blends. ACS Nano 2018, 12 (5), 4660–4668. 10.1021/acsnano.8b01260. [DOI] [PubMed] [Google Scholar]
- Li N.; Panagiotopoulos A. Z.; Nikoubashman A. Structured Nanoparticles from the Self-Assembly of Polymer Blends through Rapid Solvent Exchange. Langmuir 2017, 33, 6021–6028. 10.1021/acs.langmuir.7b00291. [DOI] [PubMed] [Google Scholar]
- Sosa C.; Liu R.; Tang C.; Qu F.; Niu S.; Bazant M. Z.; Prud’homme R. K.; Priestley R. D. Soft Multifaced and Patchy Colloids by Constrained Volume Self-Assembly. Macromolecules 2016, 49, 3580–3585. 10.1021/acs.macromol.6b00708. [DOI] [Google Scholar]
- Nikoubashman A.; Lee V. E.; Sosa C.; Prud’homme R. K.; Priestley R. D.; Panagiotopoulos A. Z. Directed Assembly of Soft Colloids through Rapid Solvent Exchange. ACS Nano 2016, 10, 1425–1433. 10.1021/acsnano.5b06890. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Pagels R. F.; Edelstein J.; Tang C.; Prud’homme R. K. Controlling and Predicting Nanoparticle Formation by Block Copolymer Directed Rapid Precipitations. Nano Lett. 2018, 18, 1139–1144. 10.1021/acs.nanolett.7b04674. [DOI] [PubMed] [Google Scholar]
- Chen W.-L.; Cordero R.; Tran H.; Ober C. K. 50th Anniversary Perspective: Polymer Brushes: Novel Surfaces for Future Materials. Macromolecules 2017, 50, 4089–4113. 10.1021/acs.macromol.7b00450. [DOI] [Google Scholar]
- Russel W. B.; Saville D. A.; Schowalter W.. Colloidal Dispersions; Cambridge University Press: Cambridge, 1989. [Google Scholar]
- Bishop M.; Kalos M. H.; Frisch H. L. Molecular Dynamics of Polymeric Systems. J. Chem. Phys. 1979, 70, 1299–1304. 10.1063/1.437567. [DOI] [Google Scholar]
- Grest G. S.; Kremer K. Molecular Dynamics Simulation for Polymers in the Presence of a Heat Bath. Phys. Rev. A: At., Mol., Opt. Phys. 1986, 33, 3628–3631. 10.1103/PhysRevA.33.3628. [DOI] [PubMed] [Google Scholar]
- Lee V. E.; Sosa C.; Liu R.; Prud’homme R. K.; Priestley R. D. Scalable Platform for Structured and Hybrid Soft Nanocolloids by Continuous Precipitation in a Confined Environment. Langmuir 2017, 33, 3444–3449. 10.1021/acs.langmuir.7b00249. [DOI] [PubMed] [Google Scholar]
- Li N.; Nikoubashman A.; Panagiotopoulos A. Z. Controlled Production of Patchy Particles from the Combined Effects of Nanoprecipitation and Vitrification. Soft Matter 2017, 13, 8433–8441. 10.1039/C7SM01896G. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Cheng S. Z. D.; Calhoun B. H.; Ge Q.; Quirk R. P.; Thomas E. L.; Hsiao B. S.; Yeh F.; Lotz B. Phase Structures and Morphologies Determined by Self-Organization, Vitrification, and Crystallization: Confined Crystallization in an Ordered Lamellar Phase of PEO-b-PS Diblock Copolymer. Polymer 2001, 42, 5829–5839. 10.1016/S0032-3861(00)00902-2. [DOI] [Google Scholar]
- Floudas G.; Ulrich R.; Wiesner U. Microphase Separation in Poly(Isoprene-b-Ethylene Oxide) Diblock Copolymer Melts. I. Phase State and Kinetics of the Order-to-Order Transition. J. Chem. Phys. 1999, 110 (1), 652–663. 10.1063/1.478122. [DOI] [Google Scholar]
- Chremos A.; Nikoubashman A.; Panagiotopoulos A. Z. Flory-Huggins Parameter χ, from Binary Mmxtures of Lennard-Jones Particles to Block Copolymer Melts. J. Chem. Phys. 2014, 140, 054909 10.1063/1.4863331. [DOI] [PubMed] [Google Scholar]
- Weeks J. D.; Chandler D.; Andersen H. C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. 10.1063/1.1674820. [DOI] [Google Scholar]
- Anderson J. A.; Lorenz C. D.; Travesset A. General Purpose Molecular Dynamics Simulations Fully Implemented on Graphics Processing Units. J. Comput. Phys. 2008, 227, 5342–5359. 10.1016/j.jcp.2008.01.047. [DOI] [Google Scholar]
- Glaser J.; Nguyen T. D.; Anderson J. A.; Lui P.; Spiga F.; Millan J. A.; Morse D. C.; Glotzer S. C. Strong Scaling of General-Purpose Molecular Dynamics Simulations on GPUs. Comput. Phys. Commun. 2015, 192, 97–107. 10.1016/j.cpc.2015.02.028. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.