

Abstract
Mechanically interlocked molecules are perhaps best known as components of molecular machines, a view further reinforced by the Nobel Prize in 2016 to Stoddart and Sauvage. Despite amazing progress since these pioneers of the field reported the first examples of molecular shuttles, genuine applications of interlocked molecular machines remain elusive, and many barriers remain to be overcome before such molecular devices make the transition from impressive prototypes on the laboratory bench to useful products. Here, we discuss simplicity as a design principle that could be applied in the development of the next generation of molecular machines with a view to moving toward real-world applications of these intriguing systems in the longer term.
Short abstract
Simplicity is discussed as a design principle that could be applied in the development of the next generation of molecular machines.
Introduction
The molecular machines employed by nature almost seem the work of science fiction, especially when represented in stylized form with the chemical detail removed.1 Using these molecular machines, living systems carry out many of the tasks essential for life including converting chemical energy from one form to another (ATP synthase),2 moving large cargoes around within cells (kinesin),3 replicating information-rich biopolymers (DNA synthase),4 carrying out complex chemical synthesis (the ribosome),5 and generating macroscopic movement (myosin).6 Typically this is achieved by coupling chemical reactions and biased Brownian motion to achieve the desired task.7
Inspired at least in part by nature’s nanotechnology, for many years, scientists have worked to develop a corresponding artificial chemical nanotechnology.8 Much of this work has focused on the use of mechanically interlocked molecules such as a rotaxanes and catenanes,9 by taking advantage of the ability of the interlocked subcomponents to undergo large amplitude relative motion. Many motivations have been proposed for this effort including using minimalist models of natural machines to aid their analysis and understanding,10 the development of an artificial molecular nanotechnology that rivals the systems found in nature in order to achieve lifelike functions (which has obvious echoes in synthetic biology) or to overcome existing chemical problems, and of course the simple aesthetic and scientific challenge of doing so—the “because it’s there” justification of mountaineers. Of these general motivations, the potential of a synthetic molecular nanotechnology to solve existing chemical challenges has become increasingly salient as the field has matured and particularly since the award of the Nobel Prize in 2016 to Feringa,11 Sauvage,12 and Stoddart,13 the latter for their work on mechanically interlocked molecular machines. Indeed, the Nobel committee highlighted that, as with computing, “miniaturization of technology can lead to a revolution”.14
Simplicity of Structure
Examination of natural protein-based molecular machines reveals extremely large molecules in which the active domain(s) often represent a relatively small fraction of the molecular mass, with much of the rest of the machine playing a structural role by protecting active sites, anchoring the complex into position, assisting in transduction of molecular motion, etc. This appears to be a luxury that nature can afford; despite the inherent cost of such large structures, as they are produced efficiently, typically using other molecular machines that have evolved for the task, the benefit of the final function renders their synthesis a sound investment.
In contrast, in the synthesis of interlocked molecular machines, molecular complexity and the consequent synthetic challenge are a real issue; many advanced interlocked molecular machines are extremely challenging to access, and thus, vanishingly small quantities of material are typically available for study. This is despite the impressive progress in methodologies for forming the mechanical bond9,15 since the first reports of synthetic rotaxanes and catenanes in the 1960s.16 In the move from prototype to genuine application, this will matter more and more as the cost/benefit relationship of a new machine will largely relate to its cost of synthesis. Furthermore, because publications in the area focus, understandably, on the properties of the final machine, synthesis is often relegated to a brief comment (and a large electronic Supporting Information) despite representing a very large proportion of the project effort.
This suggests there is a need to balance blue-skies, proof-of-principle work with subsequent optimization and simplification of structure and synthesis. The progression of Leigh’s peptide synthesizing molecular machines17 through various iterations is an excellent example of the first steps of this process. In 2013, Leigh and co-workers demonstrated that [2]rotaxane 10, in which the macrocycle bears a catalytic thiolate moiety, and the axle bears reactive phenolate esters, is able to “read” sequence information in the axle and “translate” this information into a chemical output in the form of a tripeptide with excellent sequence control (Figure 1).18 This groundbreaking result is clearly an example of proof-of-principle—the machine itself is far from the most efficient way of making a simple tripeptide! Indeed, although the synthesis of 10 was designed to be highly convergent, requiring only 10 linear steps, the yield over this shortest sequence is <1%, and the whole route requires >25 synthetic operations. Thus, the overall yield of the product peptide sequence is ∼0.1% compared with ∼50% over 4 linear steps using solution phase techniques.
Figure 1.
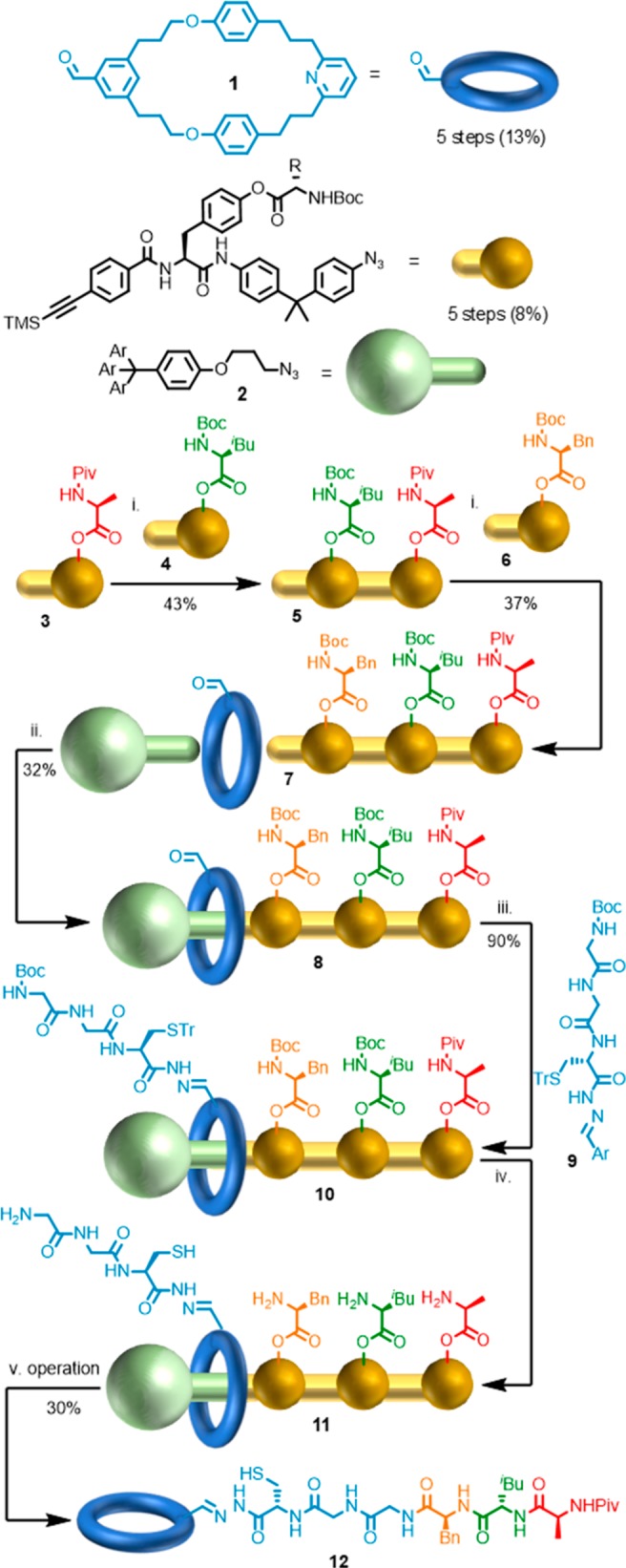
Synthesis and operation of Leigh’s first-generation peptide synthesizing molecular machine. Reagents and conditions: (i) Resin, [Cu(MeCN)4]PF6, CH2Cl2-t-BuOH 1:1, 2 days, rt. (ii) [Cu(MeCN)4]PF6, CH2Cl2-t-BuOH 2:1, rt, 4 days, 30%. (iii) PhNH2 (cat), 9, DMSO-2-(N-morpholino)ethanesulfonic acid buffer (pH 6.0) 3:1, 60 °C, 2 days, 90%. (iv) TFA, CH2Cl2. (v) iPr2NEt, TCEP-HCl, MeCN-DMF 3:1.
To overcome this, and in order to allow the general principle to be studied and expanded upon, the synthesis of subsequent generations of this machine was simplified dramatically. Specifically, whereas in the first-generation device the sequence information was included in the axle by laborious iterative synthesis, third-generation device 18 uses a controlled radical polymerization process to generate functionalized axle 14 which is then joined to preformed rotaxane fragment 17 (Figure 2).19 Thus, although third-generation rotaxane 18 is synthesized in 10 linear steps, most of this effort relates to the synthesis of the macrocycle bearing the catalyst (7 steps), and the machine itself is produced in ∼10% yield with a total of 15 synthetic operations. Moreover, the operation of the machine results in the formation of an average of 6 new amide bonds, with 5% overall yield of the oligoamide product, a significant advance over first generation machine 11. The oligovaline product’s molecular weight and dispersity are determined by the functionalized polymeric axle fragment. The authors then demonstrated that the oligovaline product was a competent enantioselective catalyst, thus extending their proof-of-principle “artificial ribosome” to one capable of producing a functional product, in direct analogy with the equivalent natural molecular machine.
Figure 2.
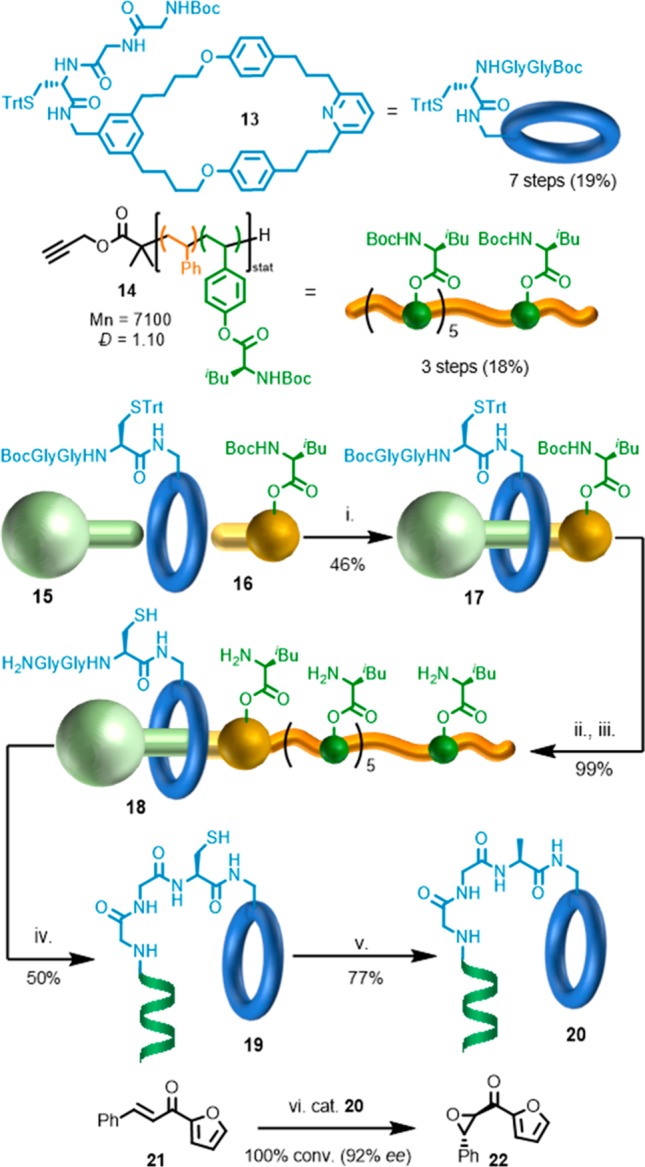
Synthesis and operation of Leigh’s third-generation peptide synthesizing molecular machine. Reagents and conditions: (i) [Cu(MeCN)4]PF6, CH2Cl2, tBuOH rt, 17 h. (ii) [Cu(MeCN)4]PF6, Tenta-Gel-TBTA CH2Cl2, tBuOH (4:1), rt, 19 h. (iii) CH2Cl2:CF3CO2H (4:1), iPr3SiH (25 equiv), rt, 2 h. (iv) NEt3 (50 equiv), PPh3 (3 equiv), DMF-d7, 65 °C, 96 h. (v) V-50, TCEP-HCl, tBuSH, NEt3, DMF, rt, 22 h. (vi) DBU, urea-H2O2, THF, rt, 18 h.
“Molecular synthesizers” 11 and 18 are extreme examples in terms of structural complexity and size, both of which are required to generate a complex outcome. The improvement in synthetic efficiency between generations is impressive and an example of the synthetic proficiency of many in the field that is not always widely recognized.
At the other end of the complexity scale are interlocked molecular shuttles. Indeed, such switches make up the vast majority of interlocked molecular machines reported to date, and a very large range of behaviors and properties have been disclosed.8 Given their relative structural simplicity compared with more advanced machines such as 11 and 18, and longer history, it is perhaps unsurprising that their synthesis is extremely well developed, particularly the mechanical bond forming step, and thus, it might be expected that simple interlocked molecular switches are closer to real-world applications.
However, even the simplest interlocked molecular shuttle runs up against a general synthetic problem; all interlocked molecular machines require at least one macrocycle. Indeed, the production of macrocycles suitable for inclusion in molecular machines is now often the most challenging aspect of their synthesis, as it was in the case of 18. Therefore, one of the key challenges when simplifying the synthesis of prototype interlocked molecular machines is simplifying the production of requisite, often functionalized macrocycles.
Perhaps the best solution to the synthetic challenge of macrocycle synthesis is to focus on examples that are essentially “free” because they are produced by natural systems. Here, the only obvious examples are the cyclodextrins (CDs) (Figure 3a), which form threaded complexes due to solvophobic effects.20 Native, unfunctionalized CDs are readily available at low cost in three sizes, and thus, if a complex molecular machine can be designed based on CDs, its synthesis is greatly simplified. However, although a great deal of elegant chemistry has been developed,21 the synthesis of functionalized CD rings remains challenging, somewhat offsetting the benefits of the availability of the native CD ring itself.
Figure 3.
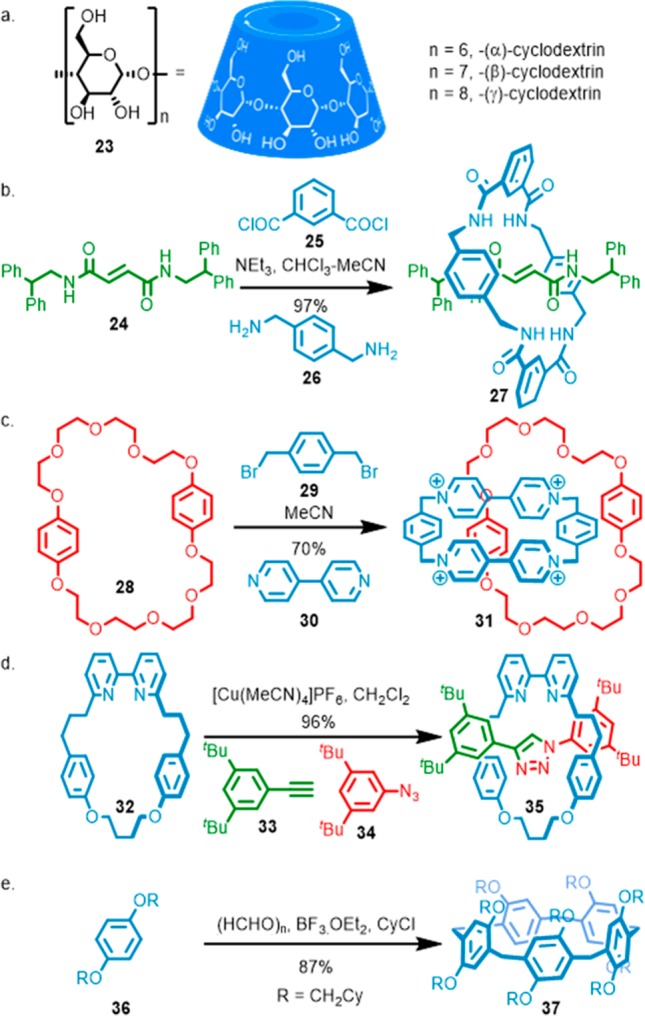
(a) Cyclodextrin family of macrocycles. (b) Leigh’s efficient synthesis of tetralactam macrocycle-based rotaxane 27. (c) Stoddart’s efficient synthesis of blue-box-based catenane 31. (d) Efficient AT-CuAAC synthesis of rotaxane 35 using Goldup’s small bipyridine macrocycle 32. (e) Ogoshi’s efficient synthesis of substituted pilar[5]arene macrocycle 37.
Alternatively, there are a small set of macrocycles that can be formed in situ during the mechanical bond formation from smaller, easily accessible fragments, removing the need to separately synthesize the macrocyclic component. Tetralactam macrocycles (Figure 3b)22 popularized by Leigh, and Stoddart’s “blue box” rings (Figure 3c)23 are excellent examples of these multicomponent approaches to interlocked molecule synthesis, and both have been extensively demonstrated as components of interlocked molecular machines. However, because specific noncovalent interactions are involved in kinetically favoring macrocycle formation, small structural modifications of the ring, that may be necessary to achieve a desired behavior or function in a more complex molecular device, can lead to a significant reduction in the yield of this key step.22e
Finally, one returns to the synthetic chemist’s standard solution: invest effort in optimizing the synthesis of key macrocyclic building blocks that are particularly useful.24 Simple crown ether-based macrocycles are a good example of this; metal-based templates have been employed very effectively to achieve high-yielding syntheses of some of these ubiquitous building blocks.25 However, these systems do not tolerate significant structural modification in the metal binding region as the metal templated synthesis relies on these interactions, as does the subsequent mechanical bond forming step. Similarly, cucurbituril macrocycles are extremely effective in host–guest complex formation,26 and their synthesis has now been streamlined significantly,27 although, again, despite recent advances, functionalization of these macrocycles is challenging.
The active template approach to interlocked molecules is, potentially, much more tolerant to modifications of the macrocycle structure than approaches based on thermodynamically stable complexes (Figure 3d),28 and recent efforts have extended the principle from metal-mediated reactions to metal-free organocatalytic systems.29 Bipyridine macrocycles (e.g., 32) are particularly effective in this approach, and for this reason we recently developed an optimized, flexible synthesis of these building blocks that can be readily scaled to produce gram quantities of these useful starting materials.30
Finally, perhaps the most striking example of a synthetically optimized and widely useful macrocycle is the pillararene macrocycles, introduced by Ogoshi and co-workers.31 These can be made in excellent yield from extremely simple building blocks (Figure 3e) and form threaded structures due to a combination of dipole–dipole interactions and solvophobic effects. They can also be readily functionalized for a wide range of applications and thus are now extremely widely used, particularly in the context of supramolecular polymers,32 although to date, less commonly as components of molecular machines.
In summary, although interlocked molecular machines are vastly simpler than their biological counterparts, there remains a significant need to optimize their production in the progression from prototype to application. This requires attention to all aspects of the synthesis, not just the mechanical bond forming step, and may include reverse engineering the required behavior into a minimalist structure, including replacing a bespoke, hard to access macrocycle in the prototype with a ring that is more readily available, or just “simply” optimizing the chemistry to ensure maximum yield and minimum steps: in short, the same problem that synthetic chemists working in a range of areas have been overcoming for decades.
Simplicity of Operation
The operation of many interlocked molecular switches and motors is achieved chemically by iterative addition of reagents, for instance, acid followed by base. Although this is a very effective strategy for the investigation of new systems and behaviors, it clearly presents problems in terms of waste generation over multiple cycles in the context of applications. It also requires continuous significant intervention by the operator. Thus, despite the dominance of stimuli based on the addition of reagents, photochemical and electrochemical methods seem ideal for controlling the operation of interlocked molecular machines.
The electrochemical strategy introduced by Stoddart and co-workers in their original molecular shuttles remains one of the simplest available and led to one of the first proposed applications of molecular switches to catch the imagination of the field. In 2001, Stoddart, Heath, and co-workers created tunnel junction devices in which a monolayer of molecular shuttles was trapped between the cross bars.33 Applying a write voltage switched the shuttles between the two available stations, resulting in a change in the resistance of the tunnel junction which could be read using a read voltage. In this way, Stoddart, Heath, and co-workers demonstrated the use of interlocked molecules in a “molecular memory” device with densities of up to 1011 bits/cm2.
More recently Stoddart and co-workers have demonstrated that this same simple approach can be used to control a much more complex molecular machine that they have designated a molecular pump (Figure 4).34 The operation of 38 begins by the electrochemical formation of a threaded pimer-complex between viologen macrocycle 39 and a viologen station in the “pump” region of the axle to give complex 40. This attractive interaction can then be abolished electrochemically simply by reoxidizing both macrocycle and axle to the closed-shell pyridinium forms. As this oxidation takes place, the positively charged macrocycle can either move away from the viologen binding site toward the pyridinium end of the axle, and ultimately freedom in solution, or move toward the neutral aromatic moiety. By optimizing the spacers between the viologen unit in the axle, and the pyridinium and aromatic blocking units, Stoddart and co-workers were able to kinetically bias this motion toward the neutral aromatic unit to give threaded species 41, presumably due to charge–charge repulsion between the cationic macrocycle and the cationic pyridinium moiety. Once it has gone the “wrong way”, the macrocycle becomes trapped as the dicationic viologen unit blocks its path electrostatically. The cycle completes via a slow, thermally activated slippage of the macrocycle over the neutral aromatic “speed bump” to give 42.
Figure 4.
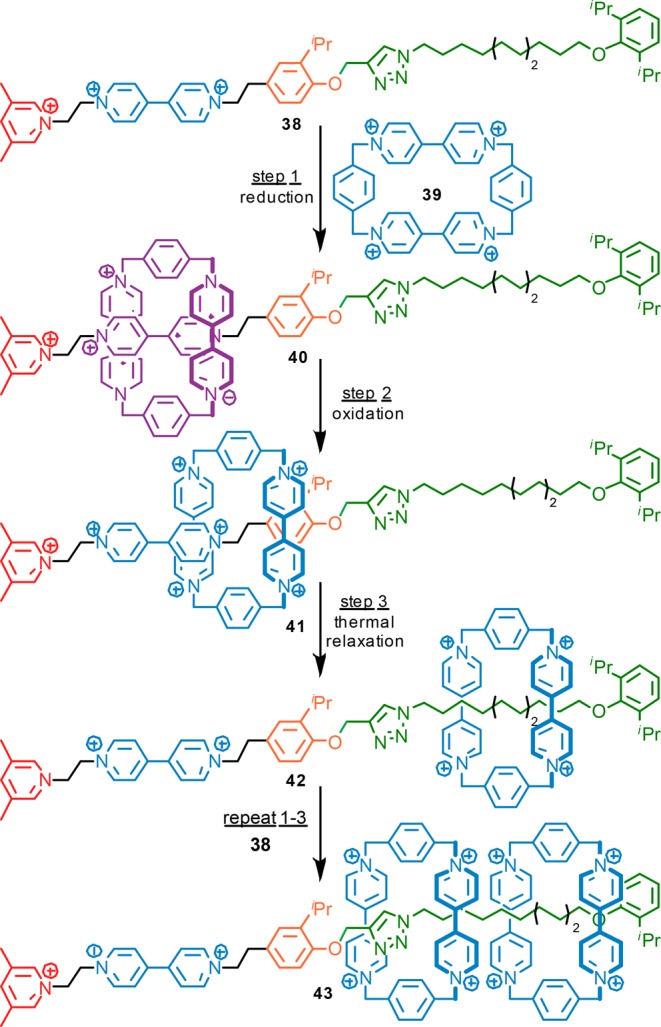
Operation of Stoddart’s molecular pump, 38.
Overall, during one complete cycle of molecular machine 38, one macrocycle is pumped from solution onto a region of the axle with which it has no significant favorable interactions, and thus, a portion of the electrochemical energy inputted is stored. Furthermore, repeating a further complete cycle allows additional rings to be pumped onto the ring collecting portion of the axle to give 43. Later versions of this machine improved the efficiency of the system and allowed the synthesis of a [5]rotaxane containing four pumped rings.35 This simple operational cycle suggests that such a machine could be used synthetically to generate unusual threaded polymers in which there is no interaction between the axle and ring, and even, as in this case, repulsive interactions between the rings themselves. Furthermore, embedding such a machine in a membrane would allow rings, and perhaps ultimately rings bearing a cargo, to be pumped against a concentration gradient using the same approach.
Light represents one of the simplest stimuli that can be applied to operate a molecular machine.36 Typically, in the context of molecular switches, this requires the application of more than one wavelength of light: one to switch in one direction, the other to reverse the effect of the first.37 Leigh and co-workers’ extensive use of fumaramide/maleamide isomerization in the development of molecular switches is an example of this approach,38 as are shuttles based on azobenzene and stilbene units.39
In some cases, as in the case of shuttles based on the isomerization of azobenzene units, spontaneous thermal reversion to the initial state can allow the switching process to reverse without the application of a second stimulus.39d Taking this concept to its logical conclusion, but using a different mechanism, Balzani, Credi, Stoddart, and co-workers demonstrated a molecular shuttle, 44, that operates continuously and autonomously under irradiation with a single wavelength of light through a photoelectron transfer process (Figure 5a).40 Upon absorption of the photon, electron transfer from the ruthenium-based stopper to the distal bipyridinium unit gives 45 in which the affinity of the macrocycle for this station is reduced, and the system relaxes toward its new equilibrium position, 46, in which the macrocycle occupies the unsubstituted bipyridinium. Spontaneous back electron transfer regenerates the original stations (47), and the macrocycle then returns to its original equilibrium position (44). Thus, over one cycle of photon absorption, electron transfer, and back electron transfer the macrocycle undergoes net displacement from one station to the other and back again.
Figure 5.

(a) Balzani, Credi, and Stoddart’s autonomous light-driven shuttle, 44. (b) Leigh’s photochemical information ratchet, 48. (c) Effect of an external photosensitizer on the distribution between the two compartments at photostationary state for 48.
Rotaxane 44 is a molecular switch that spontaneously switches back and forth continuously under irradiation. Leigh and co-workers demonstrated a light-driven information ratchet 48 that operates under continuous irradiation with a single wavelength of light (Figure 5b).41 Ratchet 48 operates due to the presence of two photosensitizers, one attached to the macrocycle and one exogenous sensitizer in solution. Irradiation leads to two sets of competing processes, the opening and closing of the α-methyl stilbene gate by the intramolecular sensitizer, which depends on the position of the macrocycle on the axle, and the opening and closing of the gate by the exogenous sensitizer, benzil, which does not. The interplay of these two processes leads to the macrocycle adopting a nonequilibrium distribution between the two compartments when the stilbene gate is closed (Figure 5c). When no benzil is present, irradiation leads to an equilibrium distribution between the two stations at steady state. Addition of benzil leads to a nonequilibrium distribution between (Z)-48 and (Z)-49, an effect that is enhanced in the presence of increasing equivalents of benzil.
An alternative to using simple stimuli such as electro- and photochemistry is to develop systems that can operate autonomously without external intervention; indeed, most natural molecular machines take this approach. Leigh’s synthesizing molecular machines (Figures 1 and 2) are artificial examples in that, once they are triggered, they will carry out their function without user intervention. Developing autonomous interlocked molecular motors capable of operating repetitively is challenging though, particularly those employing multiple steps that must be sequenced in time such as energy ratchet-based motors.
Recently, di Stefano and co-workers demonstrated the autonomous operation of rotaxane-based molecular switches using acidic reagents that slowly decompose to basic species, allowing pH-driven shuttles to switch through a complete cycle with a single user input.42 This concept was subsequently elaborated into an autonomous molecular motor by Leigh and co-workers by combining acid/base switched stations with acid/base labilized blocking groups, ensuring the synchronization of the key chemical steps.43 Catenane 50 (Figure 6a) is an example of an energy ratchet-based motor in which the controlled protonation of the amine to give an ammonium unit with higher affinity for the macrocycle is synchronized with removal/reintroduction of the hydrazone and disulfide gates to generate a 360° rotation.
Figure 6.

(a) Autonomous catenane motor 50 that undergoes a single revolution upon addition of trichloroacetic acid. (b) Leigh’s autonomous information ratchet catenane motor that turns continuously in the presence of Fmoc-Cl.
At high pH in the presence of NEt3, the amine station remains unprotonated, and the macrocycle preferentially occupies the triazolium station. Under these conditions the disulfide “gate” is under dynamic exchange between open and closed states through disulfide exchange. Addition of trichloroacetic acid (TCA) results in protonation of the amine station to generate a higher-affinity ammonium binding site for the macrocycle. The drop in pH shuts off the disulfide exchange reaction and initiates the dynamic exchange of the hydrazone gate. Thus, after addition of TCA the macrocycle can escape via the aldehyde branch of the large ring from the triazolium station to its new preferred ammonium binding site.
As the TCA decomposes to produce CO2 and CDCl3, the pH rises until the hydrazone exchange stops, and the disulfide exchange recommences. As the pH rises, the ammonium unit is deprotonated, and the preferred binding site is now the triazolium station. The macrocycle escapes to its new preferred equilibrium position via the thiol branch of the large macrocycle. Thus, overall, the motor undergoes a 360° rotation with each addition of the TCA “fuel”44 with CO2 and CDCl3 the only waste products, allowing the cycle to be repeated with little or no fatigue.
The motor behavior of catenane 50 is predicated on an energy ratchet mechanism that inherently relies on switching between two preferred equilibrium positions and the selective ungating/gating of the two different paths for the ring to escape to its new equilibrium position. The elegance of the operation mechanism developed is that all these steps are achieved by a simple change in pH that synchronizes both pairs of events. However, user intervention is still required at the end of each cycle, the addition of further TCA, to begin the next rotation.
Alternatively, information ratchet mechanisms can, in theory, operate autonomously and continuously if two reactions, the gating and ungating processes, can be designed to take place simultaneously, without any need to modify the affinity of the macrocycle for either compartment, by using kinetic factors to discriminate between the available pathways. Taking this approach, Leigh and co-workers designed a catenane information motor that undergoes continuous net rotation as long as a high-energy “fuel”44 remains (Figure 6b).45 The two compartments of catenane 52 contain near-identical fumaramide binding sites for the macrocycle (one station is deuterated for analytical purposes; however, the following discussion holds true even if both stations are identical). If one of the two gates is removed at random by a base-mediated elimination process, the macrocycle is free to explore both compartments and adopts an approximately 50:50 distribution between the near-isoenergetic stations. Reinstallation of the Fmoc group catalyzed by a bulky catalyst leads to kinetic discrimination in the unlinking reaction, which occurs most rapidly when the macrocycle occupies the station furthest from the alcohol functionality. In this way the small macrocycle undergoes a net half-rotation around the larger ring. Repeating this process while FmocCl remains, despite having no control over which Fmoc group is cleaved, results in continuous net rotation. Intriguingly this remains true despite the distribution of the macrocycle between the two compartments remaining at its equilibrium value of 50:50 at all times; one of the unusual features of machines like 52 is that they overcome the strictures of detailed balance, allowing continuous rotation of the motor even at steady state.
Simplicity vs Application
It should be relatively uncontroversial that for a molecule to be genuinely useful there must be a good balance of cost and benefit; to have genuine technological application, the functional value of an interlocked molecular machine must be commensurate with its operational and structural complexity, particularly when compared with other available solutions to the same problem. At the extreme, it must not be a case of using an expensive sledgehammer to crack a low-value nut. As suggested above, these problems can in part be ameliorated by the standard tools of synthetic chemistry: simplification and optimization. It is also to be expected that early prototypes will fail this cost–benefit test; all prototypes are far more expensive than the final product (witness the automotive industry). However, some awareness of the likely long-term value of a proposed application, coupled to an awareness of other, potentially simpler, approaches to achieve the same objective, might be one useful parameter in assessing the potential long-term impact of a new molecular machine behavior.
For example, although a great many molecular shuttles have been disclosed that are able to report the binding of guest analytes using an external signal (optical, electrical, etc.), and thus could be described as prototype “sensors”, to our knowledge, no examples are of genuine utility when compared with noninterlocked systems capable of achieving the same task. This is almost certainly because, even though such molecular shuttles are relatively easy to access with modern methods, they are still far more synthetically challenging than noninterlocked molecules with similar behavior; even a simple rotaxane molecular shuttle requires a macrocycle and at least two binding sites in the axle. Taking a specific example, rotaxane shuttle 56 responds to the binding of Zn2+ with an optical output (Figure 7a).46 In the presence of Zn2+ the macrocycle shuttles from the amide station to the amine station to bind the Zn2+ ion, which results in reduced photoelectron transfer (PET) from amines of the macrocycle and axle to the fluorophore and thus an optical read out. However, simple noninterlocked sensor 60 achieves the same function without requiring large amplitude molecular motion;47 binding of the metal ion into the cyclam ligand photoelectron transfer from the amine units and leads to a switch-on luminescence response. Cyclen-based sensor 60 is significantly easier to access from readily available building blocks (Figure 7b).
Figure 7.

(a) ZnII-driven shuttle 56 that reports metal binding through a switch-on fluorescence response. (b) Simple cyclam-based ZnII sensor 60 that selectively reports the presence of ZnII over other metal ions through a switch-on response.
In contrast, it is possible to design interlocked molecular shuttles that deliver functions that are hard to generate using noninterlocked systems. An elegant example is Takata and co-workers’ use of molecular switches to control polymer morphology (Figure 8a).48 Molecular shuttle 61 is composed of a polymeric axle threaded through a macrocycle that is covalently linked to one end of the chain. It has two states, one in which the macrocycle occupies a station near to its tether to the axle, and another at the end of the polymer chain. Switching between these two stations effectively switches the system between a linear polymer structure and a cyclic polymer, albeit one in which there is no covalent link between the two ends of the loop. Thus, molecule shuttle 61 presents the advantages of the relative ease of linear polymer synthesis, with the ability to generate systems that behave as if they were cyclic. Given that the synthesis of cyclic polymers remains challenging, and that their rheological properties have been the subject of some discussion for many years,49 this relatively simple system is an excellent example of balancing complexity with function.
Figure 8.

(a) Takata’s polymeric molecular switch, 61, that can be switched reversibly between linear and cyclic forms. (b) Leigh’s switchable enantioselective catalyst, 62, that can produce both hands of products in response to external stimuli.
More recently, Leigh and co-workers reported a rotaxane molecular shuttle 62 that operates as a stimuli responsive enantioselective catalyst capable of producing both hands of a product selectively (Figure 8b).50 Although the axle of rotaxane (E)-62 is chiral, the region around the catalytic pyrrolidine core is relatively symmetrical, with the first point of difference appearing at the substituents of the amidic nitrogens. Thus, the chemical space around the central catalytic pyrrolidine nitrogen is pseudosymmetrical, and unsurprisingly, the axle alone mediates the α-alkylation of aldehydes in very low ee. In rotaxane (E)-62, the macrocycle preferentially encircles the hydrazone station and effectively desymmetrizes the pyrrolidine unit, acting in effect as a large substituent of the amide carbonyl. Irradiation with light switches the hydrazone moiety from E to Z. In the preferred coconformation of rotaxane (Z)-62 the macrocycle occupies the amide station. Inspection of the structures of (E)-62 and (Z)-62 suggests that the space around the pyrrolidine unit is pseudoenantiomeric, and indeed, the two states of rotaxane 62 mediate the α-alkylation of aldehyde 63 with opposite selectivity. This general concept suggests that switchable rotaxane catalysts could be used to make either enantiomer of a given product in a stimuli responsive manner.
Rotaxanes 61 and 63 demonstrate functions that are hard to generate with noninterlocked molecules. In the case of 61 it is hard to see how the reversible isomerization from linear to cyclic polymer could be achieved without the large amplitude molecular motion provided by the interlocked system. The cost–benefit calculation for 63 is different as systems capable of controlling the stereoselectivity of catalytic reactions have been reported based on covalent hydrazone molecular switches51 and covalent molecular motors.52 However, these alternative strategies themselves require relatively complicated molecules, and this, combined with the richness of stereochemical behavior found in interlocked molecules,53 suggests that interlocked molecules have potential in this area.
However, this latter comparison raises an important point; rotaxane molecular shuttles, which are fundamentally molecular switches, must always compete with other types of switchable molecules that are, typically, structurally simpler such as azobenzenes, hydrazones, and overcrowded alkenes.54 Indeed, although rotaxane-based catalysts have been designed that switch on and off in response to external stimuli,55 similar behavior can be generated from relatively simple azobenzene and hydrazone switches,56 suggesting that, in the context of catalysis, higher value function, such as the stereocontrol demonstrated by 63, will be required to render interlocked molecules competitive. A similar comparison is worth considering with all molecular shuttle-based systems.
Finally, the reader will have noticed that we have not presented any proof-of-concept applications that take advantage of more advanced interlocked molecular machines such as motors and pumps. This is perhaps unsurprising as the synthesis and operation of these devices have only recently become more straightforward, and the structures themselves remain complex. However, it is worth highlighting that the principles embodied in such prototypes suggest long-term, high-value applications of interlocked molecule machines. Molecular pump 38, which demonstrates the ability of interlocked molecular ratchets to transport species into a region of higher chemical potential, suggests that these systems may have long-term potential in artificial active transport systems. Similarly, although a long way off, the principles embodied in synthesizing molecular machines such as 18, which at this stage inefficiently make simple peptides, could be used to address the long-standing problem of how to produce non-natural polymers with single monomer control, a particularly valuable target.57 If this promise is to be achieved, it is essential that complex molecular machines be made to operate repetitively, a key feature of their natural counterparts. If this can be achieved, then perhaps the high cost of their product can be offset by repetitively performing high-value functions.
Conclusions
The discussion above focuses, through the lens of simplicity, on the challenges and successes in the development of prototypical applications of interlocked molecular machines and their potential to progress toward being useful tools. Ultimately this can be boiled down to the relative convenience/cost of their synthesis and operation compared with other available strategies. The corollary of this is that, in addition to efforts to make their production and operation simpler, it is necessary to identify applications in which interlocked molecular machines provide a significant advantage compared with other, simpler strategies.
Given the success of nature’s molecular machines, there is a temptation to look to biology for inspiration, but there is a need to consider which of nature’s functions are of particular interest to be replicated in artificial systems and which are best addressed using other technologies. For example, nature’s use of molecular walkers to generate macroscopic motion in muscle fibers6 is impressive, but humanity has developed a large number of artificial ways to achieve macroscopic motion that were unavailable to evolving biological systems, which a biomimetic approach based on molecular walkers would have to out-compete to make a real impact. Thus, to our minds, it is worth using caution when looking to biology for inspiration, as the challenges that biology faces and has overcome by using molecular machines remain very different from those faced by humanity. In contrast, looking to biology to identify functional molecular machine mechanisms has been a very fruitful process and will surely continue.10
Finally, looking at the challenges involved in developing real-world applications of interlocked molecular machines it is tempting to become defeatist and assume that interlocked molecules are too complicated to become genuinely useful. However, that is to overlook the fact that a real-world application of molecular motion in rotaxanes has already been demonstrated: the slide-ring polymers developed by Ito and co-workers.58 These systems are not molecular machines in any sense but instead take advantage of a related structural property of the mechanical bond, the ability of the axle and macrocycle to move relative to one another in a rotaxane architecture. When these macrocycles are used as mobile cross-links between the polymer chains this mobility leads to remarkable improvements in the mechanical properties of the material and thus applications in scratch resistant paints, fatigue resistant fabric, and toughened plastics and gels. More recently, they have been investigated as binders in the anode of Li-ion batteries.59 Moreover, the macrocycles employed in Ito’s systems are readily available cyclodextrins, and the formation of the mechanical bond is achieved through simple hydrophobic threading, lowering the synthetic complexity of the system. The success of slide-ring polymers has many lessons for the development of interlocked molecular machines.
In conclusion, modern interlocked molecular machines demonstrate a range of behaviors, many of which are hard to achieve with their noninterlocked competitors. More recently, the simplification of both the synthesis and operation of devices has brought these machines closer to being useful tools. By keeping an eye on their limitations and the need to identify “killer applications”, activities that should coexist with the curiosity-driven approach that has been so successful to date, we are confident the field will continue to flourish. In his Nobel lecture, Feringa compared the development of molecular machines with the quest for powered flight at the start of the 20th century;11 although it was not obvious at the time what the long-term applications were, the development of air travel changed the world irrevocably. Given the results to date, and the importance of molecular machines in biological systems, it is reasonable to anticipate great things from future artificial molecular machines.
Acknowledgments
S.M.G. and A.W.H. thank the European Research Council (Consolidator Grant agreement 724987) and University of Southampton for funding. S.M.G. is a Royal Society Wolfson Research Fellow.
The authors declare no competing financial interest.
References
- For example, see: https://xvivo.com/blog/the-inner-life-of-the-cell-animation/ (accessed 14/10/2019).
- Junge W.; Nelson N. ATP Synthase. Annu. Rev. Biochem. 2015, 84 (1), 631–657. 10.1146/annurev-biochem-060614-034124. [DOI] [PubMed] [Google Scholar]
- Hirokawa N.; Noda Y.; Tanaka Y.; Niwa S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682. 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Maki H.; Kornberg A. The Polymerase Subunit of DNA Polymerase III of Escherichia Coli. J. Biol. Chem. 1985, 260 (24), 12987–12992. [PubMed] [Google Scholar]
- a Steitz T. A. From the Structure and Function of the Ribosome to New Antibiotics (Nobel Lecture). Angew. Chem., Int. Ed. 2010, 49 (26), 4381–4398. 10.1002/anie.201000708. [DOI] [PubMed] [Google Scholar]; b Ramakrishnan V. Unraveling the Structure of the Ribosome (Nobel Lecture). Angew. Chem., Int. Ed. 2010, 49 (26), 4355–4380. 10.1002/anie.201001436. [DOI] [PubMed] [Google Scholar]; c Yonath A. Polar Bears, Antibiotics, and the Evolving Ribosome (Nobel Lecture). Angew. Chem., Int. Ed. 2010, 49 (26), 4340–4354. 10.1002/anie.201001297. [DOI] [PubMed] [Google Scholar]
- Sweeney H. L.; Houdusse A. Structural and Functional Insights into the Myosin Motor Mechanism. Annu. Rev. Biophys. 2010, 39 (1), 539–557. 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- Astumian R. D. Trajectory and Cycle-Based Thermodynamics and Kinetics of Molecular Machines: The Importance of Microscopic Reversibility. Acc. Chem. Res. 2018, 51 (11), 2653–2661. 10.1021/acs.accounts.8b00253. [DOI] [PubMed] [Google Scholar]
- Erbas-Cakmak S.; Leigh D. A.; McTernan C. T.; Nussbaumer A. L. Artificial Molecular Machines. Chem. Rev. 2015, 115 (18), 10081–10206. 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns C. J.; Stoddart J. F.. The Nature of the Mechanical Bond: From Molecules to Machines; Wiley, 2016. [Google Scholar]
- Zhang L.; Marcos V.; Leigh D. A. Molecular Machines with Bio-Inspired Mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (38), 9397–9404. 10.1073/pnas.1712788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feringa B. L. The Art of Building Small: From Molecular Switches to Motors (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56 (37), 11060–11078. 10.1002/anie.201702979. [DOI] [PubMed] [Google Scholar]
- Sauvage J. P. From Chemical Topology to Molecular Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56 (37), 11080–11093. 10.1002/anie.201702992. [DOI] [PubMed] [Google Scholar]
- Stoddart J. F. Mechanically Interlocked Molecules (MIMs)—Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56 (37), 11094–11125. 10.1002/anie.201703216. [DOI] [PubMed] [Google Scholar]
- The Nobel Prize in Chemistry 2016. https://www.nobelprize.org/uploads/2018/06/press-36.pdf (accessed 1/1/2020).
- a Stoddart J. F. The Chemistry of the Mechanical Bond. Chem. Soc. Rev. 2009, 38 (6), 1802–1820. 10.1039/b819333a. [DOI] [PubMed] [Google Scholar]; b Beves J. E.; Blight B. A.; Campbell C. J.; Leigh D. A.; McBurney R. T. Strategies and Tactics for the Metal-Directed Synthesis of Rotaxanes, Knots, Catenanes, and Higher Order Links. Angew. Chem., Int. Ed. 2011, 50 (40), 9260–9327. 10.1002/anie.201007963. [DOI] [PubMed] [Google Scholar]; c Evans N. H.; Beer P. D. Progress in the Synthesis and Exploitation of Catenanes since the Millennium. Chem. Soc. Rev. 2014, 43 (13), 4658–4683. 10.1039/c4cs00029c. [DOI] [PubMed] [Google Scholar]; d Lewis J. E. M.; Beer P. D.; Loeb S. J.; Goldup S. M. Metal Ions in the Synthesis of Interlocked Molecules and Materials. Chem. Soc. Rev. 2017, 46 (9), 2577–2591. 10.1039/C7CS00199A. [DOI] [PubMed] [Google Scholar]; e Denis M.; Goldup S. M. The Active Template Approach to Interlocked Molecules. Nat. Rev. Chem. 2017, 1 (8), 0061. 10.1038/s41570-017-0061. [DOI] [Google Scholar]
- a Wasserman E. The Preparation of Interlocking Rings: A Catenane. J. Am. Chem. Soc. 1960, 82 (16), 4433–4434. 10.1021/ja01501a082. [DOI] [Google Scholar]; b Schill G.; Lüttringhaus A. The Preparation of Catena Compounds by Directed Synthesis. Angew. Chem., Int. Ed. Engl. 1964, 3 (8), 546–547. 10.1002/anie.196405461. [DOI] [Google Scholar]; c Harrison I. T.; Harrison S. Synthesis of a Stable Complex of a Macrocycle and a Threaded Chain. J. Am. Chem. Soc. 1967, 89 (22), 5723–5724. 10.1021/ja00998a052. [DOI] [Google Scholar]
- It could be argued that rotaxanes 11 and 18 do not conform to one of the common definitions of a molecular machine as they do not respond to external stimuli and do not consume energy to produce directed motion, what might be considered the “motor-centric” view of molecular machinery.8 However, they do conform to the iconic concept of molecular machines; they look and behave like a macroscopic machine, in this case a robotic production line. They were even highlighted in the Nobel committee’s press release as some of the most impressive examples of molecular machines produced to date. We include this point to highlight that the field remains complex, and many definitions are still evolving.
- Lewandowski B.; De Bo G.; Ward J. W.; Papmeyer M.; Kuschel S.; Aldegunde M. J.; Gramlich P. M. E.; Heckmann D.; Goldup S. M.; D’Souza D. M.; Fernandes A. E.; Leigh D. A.; D’Souza D. M.; Fernandes A. E.; Leigh D. A. Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine. Science 2013, 339 (6116), 189–193. 10.1126/science.1229753. [DOI] [PubMed] [Google Scholar]
- De Bo G.; Gall M. A. Y.; Kuschel S.; De Winter J.; Gerbaux P.; Leigh D. A. An Artificial Molecular Machine That Builds an Asymmetric Catalyst. Nat. Nanotechnol. 2018, 13 (5), 381–385. 10.1038/s41565-018-0105-3. [DOI] [PubMed] [Google Scholar]
- Harada A. Cyclodextrin-Based Molecular Mechanics. Acc. Chem. Res. 2001, 34 (6), 456–464. 10.1021/ar000174l. [DOI] [PubMed] [Google Scholar]
- a Guieu S.; Sollogoub M. Regiospecific Tandem Azide-Reduction/Deprotection to Afford Versatile Amino Alcohol-Functionalized α- and β-Cyclodextrins. Angew. Chem., Int. Ed. 2008, 47 (37), 7060–7063. 10.1002/anie.200801573. [DOI] [PubMed] [Google Scholar]; b Zaborova E.; Guitet M.; Prencipe G.; Blériot Y.; Ménand M.; Sollogoub M. An “against the Rules” Double Bank Shot with Diisobutylaluminum Hydride to Allow Triple Functionalization of α-Cyclodextrin. Angew. Chem., Int. Ed. 2013, 52 (2), 639–644. 10.1002/anie.201204974. [DOI] [PubMed] [Google Scholar]
- a Vögtle F.; Meier S.; Hoss R. One-Step Synthesis of a Fourfold Functionalized Catenane. Angew. Chem., Int. Ed. Engl. 1992, 31 (12), 1619–1622. 10.1002/anie.199216191. [DOI] [Google Scholar]; b Camara-Campos A.; Hunter C. a; Tomas S. Cooperativity in the Self-Assembly of Porphyrin Ladders. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (9), 3034–3038. 10.1073/pnas.0508071103. [DOI] [PMC free article] [PubMed] [Google Scholar]; bb Hunter C. A. Synthesis and Structure Elucidation of a New [2]-Catenane. J. Am. Chem. Soc. 1992, 114 (13), 5303–5311. 10.1021/ja00039a047. [DOI] [Google Scholar]; c Johnston A. G.; Leigh D. A.; Pritchard R. J.; Deegan M. D. Facile Synthesis and Solid-State Structure of a Benzylic Amide [2]Catenane. Angew. Chem., Int. Ed. Engl. 1995, 34 (11), 1209–1212. 10.1002/anie.199512091. [DOI] [Google Scholar]; d Vögtle F.; Dünnwald T.; Schmidt T. Catenanes and Rotaxanes of the Amide Type. Acc. Chem. Res. 1996, 29 (9), 451–460. 10.1021/ar950200t. [DOI] [Google Scholar]; e Leigh D. A.; Venturini A.; Wilson A. J.; Wong J. K. Y.; Zerbetto F. The Mechanism of Formation of Amide-Based Interlocked Compounds: Prediction of a New Rotaxane-Forming Motif. Chem. - Eur. J. 2004, 10 (20), 4960–4969. 10.1002/chem.200305662. [DOI] [PubMed] [Google Scholar]
- a Ashton P. R.; Goodnow T. T.; Kaifer A. E.; Reddington M. V.; Slawin A. M. Z.; Spencer N.; Stoddart J. F.; Vicent C.; Williams D. J. A [2] Catenane Made to Order. Angew. Chem., Int. Ed. Engl. 1989, 28 (10), 1396–1399. 10.1002/anie.198913961. [DOI] [Google Scholar]; b Trabolsi A.; Khashab N.; Fahrenbach A. C.; Friedman D. C.; Colvin M. T.; Cotí K. K.; Benítez D.; Tkatchouk E.; Olsen J.-C.; Belowich M. E.; Carmielli R.; Khatib H. A.; Goddard W. A.; Wasielewski M. R.; Stoddart J. F. Radically Enhanced Molecular Recognition. Nat. Chem. 2010, 2 (1), 42–49. 10.1038/nchem.479. [DOI] [PubMed] [Google Scholar]; c Fahrenbach A. C.; Bruns C. J.; Cao D.; Stoddart J. F. Ground-State Thermodynamics of Bistable Redox-Active Donor–Acceptor Mechanically Interlocked Molecules. Acc. Chem. Res. 2012, 45 (9), 1581–1592. 10.1021/ar3000629. [DOI] [PubMed] [Google Scholar]; d Fahrenbach A. C.; Bruns C. J.; Li H.; Trabolsi A.; Coskun A.; Stoddart J. F. Ground-State Kinetics of Bistable Redox-Active Donor-Acceptor Mechanically Interlocked Molecules. Acc. Chem. Res. 2014, 47 (2), 482–493. 10.1021/ar400161z. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Nalluri S. K. M.; Fraser Stoddart J. Surveying Macrocyclic Chemistry: From Flexible Crown Ethers to Rigid Cyclophanes. Chem. Soc. Rev. 2017, 46 (9), 2459–2478. 10.1039/C7CS00185A. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Schalley C. A. Templated versus Non-Templated Synthesis of Benzo-21-Crown-7 and the Influence of Substituents on Its Complexing Properties. Beilstein J. Org. Chem. 2010, 6, 1–8. 10.3762/bjoc.6.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lagona J.; Mukhopadhyay P.; Chakrabarti S.; Isaacs L. The Cucurbit[n]Uril Family. Angew. Chem., Int. Ed. 2005, 44 (31), 4844–4870. 10.1002/anie.200460675. [DOI] [PubMed] [Google Scholar]; b Masson E.; Ling X.; Joseph R.; Kyeremeh-Mensah L.; Lu X. Cucurbituril Chemistry: A Tale of Supramolecular Success. RSC Adv. 2012, 2 (4), 1213–1247. 10.1039/C1RA00768H. [DOI] [Google Scholar]; c Barrow S. J.; Kasera S.; Rowland M. J.; Del Barrio J.; Scherman O. A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115 (22), 12320–12406. 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Kasera S.; Rowland M. J.; Del Barrio J.; Scherman O. A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115 (22), 12320–12406. 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- a Crowley J. D.; Goldup S. M.; Lee A.-L.; Leigh D. A.; McBurney R. T. Active Metal Template Synthesis of Rotaxanes, Catenanes and Molecular Shuttles. Chem. Soc. Rev. 2009, 38 (6), 1530–1541. 10.1039/b804243h. [DOI] [PubMed] [Google Scholar]; b Denis M.; Goldup S. M. The Active Template Approach to Interlocked Molecules. Nat. Rev. Chem. 2017, 1 (8), 0061. 10.1038/s41570-017-0061. [DOI] [Google Scholar]
- a De Bo G.; Dolphijn G.; McTernan C. T.; Leigh D. A. [2]Rotaxane Formation by Transition State Stabilization. J. Am. Chem. Soc. 2017, 139 (25), 8455–8457. 10.1021/jacs.7b05640. [DOI] [PubMed] [Google Scholar]; b Fielden S. D. P.; Leigh D. A.; McTernan C. T.; Pérez-Saavedra B.; Vitorica-Yrezabal I. J. Spontaneous Assembly of Rotaxanes from a Primary Amine, Crown Ether and Electrophile. J. Am. Chem. Soc. 2018, 140 (19), 6049–6052. 10.1021/jacs.8b03394. [DOI] [PubMed] [Google Scholar]
- Lewis J. E. M.; Bordoli R. J.; Denis M.; Fletcher C. J.; Galli M.; Neal E. A.; Rochette E. M.; Goldup S. M. High Yielding Synthesis of 2,2′-Bipyridine Macrocycles, Versatile Intermediates in the Synthesis of Rotaxanes. Chem. Sci. 2016, 7 (5), 3154–3161. 10.1039/C6SC00011H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ogoshi T.; Kanai S.; Fujinami S.; Yamagishi T.; Nakamoto Y. Para-Bridged Symmetrical Pillar[5]Arenes: Their Lewis Acid Catalyzed Synthesis and Host-Guest Property. J. Am. Chem. Soc. 2008, 130 (15), 5022–5023. 10.1021/ja711260m. [DOI] [PubMed] [Google Scholar]; b Ogoshi T.; Yamagishi T. A.; Nakamoto Y. Pillar-Shaped Macrocyclic Hosts Pillar[n]Arenes: New Key Players for Supramolecular Chemistry. Chem. Rev. 2016, 116 (14), 7937–8002. 10.1021/acs.chemrev.5b00765. [DOI] [PubMed] [Google Scholar]
- Ogoshi T.; Yamagishi T. A.; Nakamoto Y. Pillar-Shaped Macrocyclic Hosts Pillar[n]Arenes: New Key Players for Supramolecular Chemistry. Chem. Rev. 2016, 116 (14), 7937–8002. 10.1021/acs.chemrev.5b00765. [DOI] [PubMed] [Google Scholar]
- a Green J. E.; Wook Choi J.; Boukai A.; Bunimovich Y.; Johnston-Halperin E.; Deionno E.; Luo Y.; Sheriff B. A.; Xu K.; Shik Shin Y.; Tseng H. R.; Stoddart J. F.; Heath J. R. A 160-Kilobit Molecular Electronic Memory Patterned at 1011 Bits per Square Centimetre. Nature 2007, 445 (7126), 414–417. 10.1038/nature05462. [DOI] [PubMed] [Google Scholar]; b Coskun A.; Spruell J. M.; Barin G.; Dichtel W. R.; Flood A. H.; Botros Y. Y.; Stoddart J. F. High Hopes: Can Molecular Electronics Realise Its Potential?. Chem. Soc. Rev. 2012, 41 (14), 4827–4859. 10.1039/c2cs35053j. [DOI] [PubMed] [Google Scholar]
- a Cheng C.; McGonigal P. R.; Schneebeli S. T.; Li H.; Vermeulen N. A.; Ke C.; Stoddart J. F. An Artificial Molecular Pump. Nat. Nanotechnol. 2015, 10 (6), 547–553. 10.1038/nnano.2015.96. [DOI] [PubMed] [Google Scholar]; b Goldup S. Artificial Molecular Machines: Two Steps Uphill. Nat. Nanotechnol. 2015, 10 (6), 488. 10.1038/nnano.2015.116. [DOI] [PubMed] [Google Scholar]
- a Pezzato C.; Nguyen M. T.; Cheng C.; Kim D. J.; Otley M. T.; Stoddart J. F. An Efficient Artificial Molecular Pump. Tetrahedron 2017, 73 (33), 4849–4857. 10.1016/j.tet.2017.05.087. [DOI] [Google Scholar]; b Pezzato C.; Nguyen M. T.; Kim D. J.; Anamimoghadam O.; Mosca L.; Stoddart J. F. Controlling Dual Molecular Pumps Electrochemically. Angew. Chem., Int. Ed. 2018, 57 (30), 9325–9329. 10.1002/anie.201803848. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Credi A.; Venturi M. Light Powered Molecular Machines. Chem. Soc. Rev. 2009, 38 (6), 1542–1550. 10.1039/b806328c. [DOI] [PubMed] [Google Scholar]
- Yu S.; McClenaghan N. D.; Pozzo J.-L. Photochromic Rotaxanes and Pseudorotaxanes. Photochem. Photobiol. Sci. 2019, 18 (9), 2102–2111. 10.1039/C9PP00057G. [DOI] [PubMed] [Google Scholar]
- Berná J.; Bottari G.; Leigh D. A.; Perez E. M. Amide-Based Molecular Shuttles (2001–2006). Pure Appl. Chem. 2007, 79 (1), 39–54. 10.1351/pac200779010039. [DOI] [Google Scholar]
- a Murakami H.; Kawabuchi A.; Kotoo K.; Kunitake M.; Nakashima N. A Light-Driven Molecular Shuttle Based on a Rotaxane. J. Am. Chem. Soc. 1997, 119 (32), 7605–7606. 10.1021/ja971438a. [DOI] [Google Scholar]; b Stanier C. A.; Alderman S. J.; Claridge T. D. W.; Anderson H. L. Unidirectional Photoinduced Shuttling in a Rotaxane with a Symmetric Stilbene Dumbbell. Angew. Chem., Int. Ed. 2002, 41 (10), 1769–1772. . [DOI] [PubMed] [Google Scholar]; c Qu D. H.; Wang Q. C.; Ren J.; Tian H. A Light-Driven Rotaxane Molecular Shuttle with Dual Fluorescence Addresses. Org. Lett. 2004, 6 (13), 2085–2088. 10.1021/ol049605g. [DOI] [PubMed] [Google Scholar]; d Qu D. H.; Wang Q. C.; Tian H. A Half Adder Based on a Photochemically Driven [2]Rotaxane. Angew. Chem., Int. Ed. 2005, 44 (33), 5296–5299. 10.1002/anie.200501215. [DOI] [PubMed] [Google Scholar]
- a Balzani V.; Clemente-León M.; Credi A.; Ferrer B.; Venturi M.; Flood A. H.; Stoddart J. F. Autonomous Artificial Nanomotor Powered by Sunlight. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (5), 1178–1183. 10.1073/pnas.0509011103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kay E. R.; Leigh D. A. Photochemistry: Lighting up Nanomachines. Nature 2006, 440 (7082), 286–287. 10.1038/440286b. [DOI] [PubMed] [Google Scholar]
- Serreli V.; Lee C.-F.; Kay E. R.; Leigh D. A. A Molecular Information Ratchet. Nature 2007, 445 (7127), 523–527. 10.1038/nature05452. [DOI] [PubMed] [Google Scholar]
- a Berrocal J. A.; Biagini C.; Mandolini L.; Di Stefano S. Coupling of the Decarboxylation of 2-Cyano-2-Phenylpropanoic Acid to Large-Amplitude Motions: A Convenient Fuel for an Acid-Base-Operated Molecular Switch. Angew. Chem., Int. Ed. 2016, 55 (24), 6997–7001. 10.1002/anie.201602594. [DOI] [PubMed] [Google Scholar]; b Biagini C.; Albano S.; Caruso R.; Mandolini L.; Berrocal J. A.; Di Stefano S. Variations in the Fuel Structure Control the Rate of the Back and Forth Motions of a Chemically Fuelled Molecular Switch. Chem. Sci. 2018, 9 (1), 181–188. 10.1039/C7SC04123C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbas-Cakmak S.; Fielden S. D. P.; Karaca U.; Leigh D. A.; McTernan C. T.; Tetlow D. J.; Wilson M. R. Rotary and Linear Molecular Motors Driven by Pulses of a Chemical Fuel. Science 2017, 358 (6361), 340–343. 10.1126/science.aao1377. [DOI] [PubMed] [Google Scholar]
- It is hard to reach a satisfactory definition of “fuel” in this context, particularly as no work is being done by the motors. For example, catenane 50 can be operated by sequential addition/removal of acid (e.g., TFA followed by NEt3), which would normally be described as stimuli, not fuel. Furthermore, addition of more TCA does not make the motor turn faster—only one revolution is achieved regardless of equivalents. Thus, it is not obvious that TCA should be termed a fuel, although it often is.42,43,45 In contrast, increasing the equivalents of FmocCl, in theory, causes catenane 52 to undergo more revolutions and increases the rate of the gating step. This latter behavior, at least superficially, seems more fuel-like.
- Wilson M. R.; Solà J.; Carlone A.; Goldup S. M. S. M.; Lebrasseur N.; Leigh D. A. D. A. An Autonomous Chemically Fuelled Small-Molecule Motor. Nature 2016, 534 (7606), 235–240. 10.1038/nature18013. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Li J.; He X.; Li C.; Lv J.; Li Y.; Wang S.; Liu H.; Zhu D. A Molecular Shuttle for Driving a Multilevel Fluorescence Switch. Chem. - Eur. J. 2008, 14 (2), 754–763. 10.1002/chem.200701105. [DOI] [PubMed] [Google Scholar]
- Jobe K.; Brennan C. H.; Motevalli M.; Goldup S. M.; Watkinson M. Modular “click” Sensors for Zinc and Their Application in Vivo. Chem. Commun. 2011, 47 (21), 6036–6038. 10.1039/c1cc11213a. [DOI] [PubMed] [Google Scholar]
- Aoki D.; Aibara G.; Uchida S.; Takata T. A Rational Entry to Cyclic Polymers via Selective Cyclization by Self-Assembly and Topology Transformation of Linear Polymers. J. Am. Chem. Soc. 2017, 139 (20), 6791–6794. 10.1021/jacs.7b01151. [DOI] [PubMed] [Google Scholar]
- Bielawski C. W.; Benitez D.; Grubbs R. H. An “Endless” Route to Cyclic Polymers. Science 2002, 297 (5589), 2041–2044. 10.1126/science.1075401. [DOI] [PubMed] [Google Scholar]
- a Dommaschk M.; Echavarren J.; Leigh D. A.; Marcos V.; Singleton T. A. Dynamic Control of Chiral Space Through Local Symmetry Breaking in a Rotaxane Organocatalyst. Angew. Chem., Int. Ed. 2019, 58 (42), 14955–14958. 10.1002/anie.201908330. [DOI] [PubMed] [Google Scholar]; b Roland C. D.; Li H.; Abboud K. A.; Wagener K. B.; Veige A. S. Cyclic Polymers from Alkynes. Nat. Chem. 2016, 8 (8), 791–796. 10.1038/nchem.2516. [DOI] [PubMed] [Google Scholar]; c Roland C. D.; Li H.; Abboud K. A.; Wagener K. B.; Veige A. S. Cyclic Polymers from Alkynes. Nat. Chem. 2016, 8 (8), 791–796. 10.1038/nchem.2516. [DOI] [PubMed] [Google Scholar]
- Kassem S.; Lee A. T. L.; Leigh D. A.; Marcos V.; Palmer L. I.; Pisano S. Stereodivergent Synthesis with a Programmable Molecular Machine. Nature 2017, 549 (7672), 374–378. 10.1038/nature23677. [DOI] [PubMed] [Google Scholar]
- a Wang J.; Feringa B. L. Dynamic Control of Chiral Space in a Catalytic Asymmetric Reaction Using a Molecular Motor. Science 2011, 331 (6023), 1429–1432. 10.1126/science.1199844. [DOI] [PubMed] [Google Scholar]; b Zhao D.; Neubauer T. M.; Feringa B. L. Dynamic Control of Chirality in Phosphine Ligands for Enantioselective Catalysis. Nat. Commun. 2015, 6, 6652. 10.1038/ncomms7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson E. M. G.; Modicom F.; Goldup S. M. Chirality in Rotaxanes and Catenanes. Chem. Soc. Rev. 2018, 47 (14), 5266–5311. 10.1039/C8CS00097B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Feringa B. L., Browne W. R., Eds. Molecular Switches, 2nd ed.; WILEY-VCH Verlag & Co. KGaA: Weinheim, 2011. [Google Scholar]; b Aprahamian I. Hydrazone Switches and Things in Between. Chem. Commun. 2017, 53 (50), 6674–6684. 10.1039/C7CC02879B. [DOI] [PubMed] [Google Scholar]
- a Blanco V.; Leigh D. A.; Marcos V.; Morales-Serna J. A.; Nussbaumer A. L. A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine. J. Am. Chem. Soc. 2014, 136 (13), 4905–4908. 10.1021/ja501561c. [DOI] [PubMed] [Google Scholar]; b Blanco V.; Leigh D. A.; Marcos V.; Morales-Serna J. A.; Nussbaumer A. L. A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine. J. Am. Chem. Soc. 2014, 136 (13), 4905–4908. 10.1021/ja501561c. [DOI] [PubMed] [Google Scholar]; c Beswick J.; Blanco V.; De Bo G.; Leigh D. A.; Lewandowska U.; Lewandowski B.; Mishiro K. Selecting Reactions and Reactants Using a Switchable Rotaxane Organocatalyst with Two Different Active Sites. Chem. Sci. 2015, 6 (1), 140–143. 10.1039/C4SC03279A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco V.; Leigh D. A.; Marcos V. Artificial Switchable Catalysts. Chem. Soc. Rev. 2015, 44 (15), 5341–5370. 10.1039/C5CS00096C. [DOI] [PubMed] [Google Scholar]
- Gutekunst W. R.; Hawker C. J. A General Approach to Sequence-Controlled Polymers Using Macrocyclic Ring Opening Metathesis Polymerization. J. Am. Chem. Soc. 2015, 137, 8038. 10.1021/jacs.5b04940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ito K. Novel Cross-Linking Concept of Polymer Network: Synthesis, Structure, and Properties of Slide-Ring Gels with Freely Movable Junctions. Polym. J. 2007, 39 (6), 489–499. 10.1295/polymj.PJ2006239. [DOI] [Google Scholar]; b Mayumi K.; Ito K.; Kato K.. Polyrotaxane and Slide-Ring Materials; Monographs in Supramolecular Chemistry; Royal Society of Chemistry: Cambridge, 2015. [Google Scholar]
- Choi S.; Kwon T.-w.; Coskun A.; Choi J. W. Highly Elastic Binders Integrating Polyrotaxanes for Silicon Microparticle Anodes in Lithium Ion Batteries. Science 2017, 357 (6348), 279–283. 10.1126/science.aal4373. [DOI] [PubMed] [Google Scholar]